
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and photophysical studies of an indigo derivative: N-octyl-7,7′-diazaindigo†
Gustavo de Miguel *a,
Andrés Garzón-Ruizb,
Amparo Navarroc and
Eva M. García-Frutos*d
*a,
Andrés Garzón-Ruizb,
Amparo Navarroc and
Eva M. García-Frutos*d
aInstitute of Fine Chemistry and Nanochemistry, Department of Physical Chemistry and Applied Thermodynamics, University of Cordoba, Campus Universitario de Rabanales, Edificio Marie Curie, Córdoba, E-14014, Spain. E-mail: q62mirog@uco.es
bDepartment of Physical Chemistry, Faculty of Pharmacy, Universidad de Castilla-La Mancha, Cronista Francisco Ballesteros Gómez, 1, E02071 Albacete, Spain
cDepartment of Physical and Analytical Chemistry, Faculty of Experimental Sciences, Universidad de Jaén, Campus Las Lagunillas, E23071 Jaén, Spain
dInstituto de Ciencia de Materiales de Madrid (ICMM), CSIC, Cantoblanco, Madrid, E-28049, Spain. E-mail: emgfrutos@icmm.csic.es; Tel: +34 91 334 9038
First published on 18th November 2020
Abstract
In this paper, we explore the synthesis, characterization, and photophysical properties of a novel indigo derivative, N-octyl-7,7′-diazaindigo, being the first time that diazaindigos have been studied as photophysically-active chemical entities. Reduction of the neutral “keto-form” to the so-called “leuco-form” changes the global spectroscopic and photophysical behaviors. Both species have been investigated by different photophysical studies, such as analysis of absorption and emission spectra, fluorescence quantum yields (ΦF) and lifetimes. Finally, to appraise in depth the deactivation of the excited state of the keto form, femtosecond transient absorption (TA) experiments and Density Functional Theory (DFT) and Time Dependent (TD)-DFT calculations were performed. In an organic aprotic solvent (N,N-dimethylformamide), TA experiments showed a fast deactivation channel (τ1 = 2.9 ps), which was ascribed to solvent reorganization, and a longer decay component (τ2 = 86 ps) associated with an internal conversion (IC) process to the ground-state, in opposition to the excited state proton transfer (ESPT) mechanism that takes place in the indigo molecules but in protic solvents. A comparative study was also carried out on the parent molecule, 7,7′-diazaindigo, corroborating the previous conclusions obtained for the alkyl derivative. In agreement with experimental observations, DFT and TD-DFT calculations revealed that the deactivation of the S1 state of the keto form takes place through an internal conversion process.
Introduction
Indigo1,2 is one of the oldest and most famous natural dyes, isolated from certain plant leaves and used along all of human history.3–5 It was already known in the ancient Egyptian civilization and its use has continued throughout the centuries.6 It has been mainly used for inks and paints, from war skin paintings in the Roman Empire Period, to the oil paintings by some of the great masters of 17th and 18th centuries. Indigo dye is an organic compound with an intense blue colour, which presents good stability and longevity as a colorant. Its interest has reached the 21st century, as it is not only used as a blue colorant, for instance to give colour for the production of denim cloth for the worldwide famous blue jeans,6,7 but also as centre of new innovations related with green chemistry, where researchers study the production of indigo by microorganisms.8,9Beside indigo, other well-known chemical derivatives, characterized by substitutions at the indigo benzene rings, have been used, such as the Tyrian purple (6,6′-dibromoindigo),1,10 secreted by a common Mediterranean snail. This analogues appears in some ancient Phoenician texts dated by the 14th century B.C., which explain an important purple dye industry from shellfishes.11 The importance of indigo and its derivatives during all of these centuries has given rise to the study of different aspects of their chemistry, photochemistry, photophysics, as well as dye stability. For instance, it has been studied how the incorporation of indigo to form organic–inorganic hybrid using clay palygorskite (Maya Blue)12 or zeolites13 affords stability.
The proof of its potential along human history is that a huge amount of new structures based on indigo platform, called indigoids, have been described in the last decades. These indigoids contain very low toxicity and good chemical stability. For this reason, they have also been widely employed as textile dyes, food colorants, and pigments for contact lenses. In addition, indigo and some indigoids dyes have been described as good active materials for battery applications14,15 and organics electronics, due to their good stability and electrical properties. Indigo, isoindigo and their derivatives have demonstrated a great success as an electron-accepting scaffold in the development of new materials for various organic electronic applications, in particular focusing on organic field effect transistors (OFETs) and organic photovoltaics (OPVs).16–22
Other highlighted indigo derivatives have been widely described in this area, such as thioindigo,23 (where the two NH groups are substituted by S atoms), or isoindigo18,24 (linked by methylene bridges at the 3-position), including thienoisoindigo,25 fluorinated isoindigo26 and diazaisoindigo (with two nitrogen atoms at different positions).27–30 In this last derivative, the diazaisoindigo, the replacement of two carbons in the benzene rings by two nitrogen atoms in positions 7 and 7′ clearly imposes a higher planarity compared to the isoindigo counterpart.27 The incorporation of these nitrogen atoms to the platform modifies the photophysical properties of the final dye.
In the last years, the photophysical studies of indigo and related molecules have been studied, because the presence of organic functional groups, such as –NH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, favours numerous inter- and intramolecular hydrogen bonds. These groups generate in indigo and its derivatives two types of species, the neutral form known as keto species, and the dianionic leuco form, which is obtained when these dyes are treated with a reducing agent (Fig. 1).
O, favours numerous inter- and intramolecular hydrogen bonds. These groups generate in indigo and its derivatives two types of species, the neutral form known as keto species, and the dianionic leuco form, which is obtained when these dyes are treated with a reducing agent (Fig. 1).
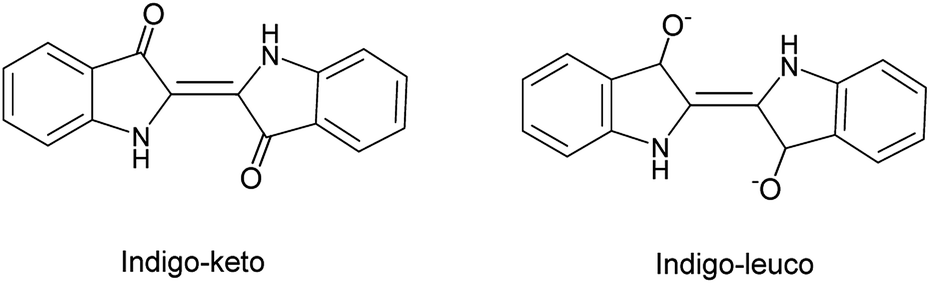 | ||
| Fig. 1 Structure of the keto and leuco forms of indigo. | ||
In this manuscript, we develop the synthesis of a novel indigo derivative, the N-octyl-7,7′-diazaindigo, which presents two nitrogens in the positions 7 and 7′ of indigo. Absorption and emission spectra, fluorescence quantum yield (ΦF), and lifetimes of its keto and leuco forms have been studied and compared to its parent compound 7,7′-diazaindigo. Additionally, femtosecond transient absorption (TA) studies and Density Functional Theory (DFT) calculations were carried out to investigate in depth the deactivation of the excited state of the keto form.
Experimental section
Materials and instrumentation
All reagents for the synthesis of products 1 and 2 were purchased from Merck/Sigma-Aldrich. Solvents were purchased from Scharlab. All reactions were monitored by silica gel thin layer chromatography using a UV light (254 nm). Column chromatography was carried out on silica gel (200–300 mesh). 1H and 13C NMR spectra were obtained on a Bruker AVANCE 300 spectrometer, 300 MHz (1H) or at 75.4 MHz (13C) at 25 °C, in 5 mm tubes at room temperature with CDCl3 as deuterated solvent. Chemical shifts (δ) are quoted in parts per million (ppm), referenced to the residual trace of non-deuterated solvent. Mass spectra were confirmed using Matrix Assisted Laser Desorption (MALDI) mass spectrometer in positive ion reflector mode.UV-visible absorption spectra were measured on a Cary 100 Bio UV-visible spectrophotometer. Steady-state and time-resolved fluorescence measurements were performed on FLS920 Fluorimeter (Edinburgh Instrument Ltd, Livingston, UK).
Femtosecond transient absorption measurements were performed using a broadband pump–probe transient absorption spectrometer (HELIOS, Ultrafast Systems) which is described in detail elsewhere.31 The pump light was generated by using a regenerative Ti:sapphire amplifier (Spitfire Ace, Spectra Physics, 1 kHz) that provides ultra-short pulses of high power at 800 nm. This system requires a seed laser to provide the input pulses and a pump laser to energize the amplifier. The regenerative amplifier is seeded using a Ti:sapphire femtosecond laser (MaiTai SP, Spectra Physics, 80 MHz) and pumped by a diode-pumped Q-switched laser of Nd:YLF (Empower 45, Spectra Physics, 1 kHz). The amplified radiation (800 nm, 100 fs, 5 W) is divided into two beams. The first one is used to pump a tunable optical parametric amplifier (TOPAS prime, Spectra Physics) in the range 290–1600 nm that generates the excitation of the sample and the second one is employed to generate white light for the absorption of the sample. In the Helios spectrometer, the UV-vis white light continuum generated by focusing the fundamental light from the amplifier to a thin CaF2 crystal after the controlled optical delay (3.3 ns), was used as a probe beam, directed to the sample and detected with a CMOS sensor. The pump pulse was chopped by a mechanical chopper synchronized to one-half of the laser repetition rate, resulting in a pair of the spectra with and without the pump, from which the absorption change induced by the pump pulse was estimated.
Computational details
Density Functional Theory (DFT) calculations were performed on the Gaussian 09 package (version D.01)32 at the PBE0 level of theory33,34 along with the 6-31+G* basis set. The molecular geometry of the ground (Sn) and first excited (S1) states were fully optimized. The long alkyl chains –C8H17 were substituted by –C2H5 to reduce the computational cost. The polarizable continuum model (PCM) was used to include the effect of the solvent (N,N-dimethylformamide, DMF) as implemented in the Gaussian package.35,36 Time-dependent DFT calculations (TD-PBE0/6-31+G*) were performed to compute vertical electronic transitions in DMF solution. The fluorescence emission energy from the first excited state (S1) was calculated from the equation| ΔE(S1 → S0) = E(S1)G(S1) − E(S0)G(S1) | (1) |
| ΔE(S1 → Sn) = E(Sn)G(S1) − E(S1)G(S1). | (2) |
Results and discussion
Synthesis and characterization
The synthetic routes for the preparation of the different compounds are illustrated in Scheme 1.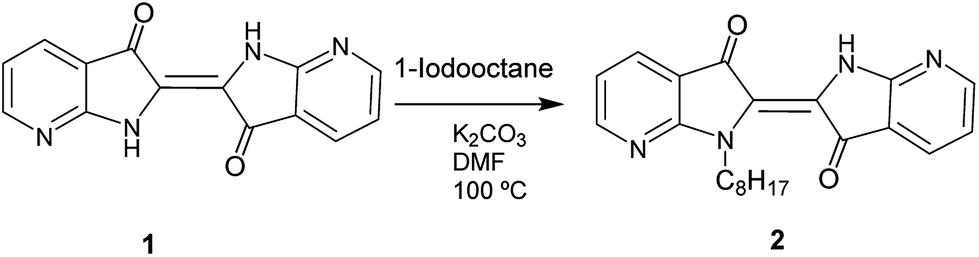 | ||
| Scheme 1 Synthesis of diazaindigo derivative 2 from 1. | ||
Compound 7-aza-3-indoxyl acetate and 1 were synthesized according to the reported literature (Scheme S1†).39 The 7-aza-3-indoxyl acetate was prepared from commercially available 7-azaindole by oxidation with thallium(III) acetate in acetic acid. The synthesis of 7,7′-diazaindigo 1 was carried out by homocoupling of 7-aza-3-indoxyl acetate in presence of 25% aq. ammonium hydroxide (Scheme S1†). Finally, the synthesis of the N-octyl-7,7′-diazaindigo molecule (2, Scheme 1) was achieved by the alkylation of 1 with 1-iodooctane in presence of K2CO3 or KOH as base in dimethylformamide (DMF), at 110 °C for 2 h. Then, the compound was purified by column chromatography using hexane:acetone mixtures as eluent, affording 2 in good yield. The compound 1 has poor solubility in common organic solvents, probably due to intermolecular hydrogen bonding. However, the incorporation of the octyl alkyl chain on one of the nitrogens in 2 contributed to enhance the solubility in such common solvents such as chloroform, dicloromethane or tetrahydrofuran. Moreover, the N-substitution of 1 derivative is an important key parameter to help the processability and the study of this new material.
A deep blue high-quality single crystal of 2 is analyzed by X-ray diffraction (see crystallographic data in Tables S1, S2 and Fig. S1†). The crystals of compound 2 are very fine blue-coloured needles and they are obtained from diethyl ether/diclorometane slow evaporation. The analysis of monocrystalline structure of 2 revealed the non-planarity of the nucleus and a significant intermolecular interaction between adjacent platforms. The non-planarity is observed with an angle of about 10° of torsion in the molecule. Compound 2 crystallizes in the monoclinic space group P1211. The coupling of the molecules in the crystal is achieved by means of π–π interactions that give rise to columns of stairs, parallel in one direction, where the adjacent molecules are at a distance of approximately 3.67 Å with respect to each other. Alkyl chains at the nitrogens are located in the same direction, favouring the interaction between them by van der Waals forces.
Photophysical studies
Solutions of the 7,7′-diazaindigo (1) and N-octyl-7,7′-diazaindigo (2) in dimethylformamide (DMF) afforded the keto forms of both compounds, while the addition of some drops of an aqueous solution of sodium dithionate/sodium hydroxide to the previous DMF solution produced a reduction reaction to generate the leuco forms. Fig. 2A and S2A† show the normalized absorption and emission spectra (λexc = 520 and 535 nm) of the keto forms or 1 and 2 in DMF, respectively. The UV-vis absorption spectrum of compound 2 displays a very narrow peak centred at 315 nm, together with a broad and asymmetric absorption band at 600 nm, with a shoulder in the high-energy region (550 nm). The spectrum of compound 1 is similar to that of 2 but with a blue shift of the low energy peak (λabs = 565 nm). The molar absorption coefficients were also calculated for the visible absorption peaks (see Table 1), resulting values of 2400 M−1 cm−1 (565 nm) and 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 580 M−1 cm−1 (600 nm) for 1 and 2 which are comparable to those measured in the indigo molecule.40 The visible absorption signal in 1 is shifted to the blue compared with indigo (λabs = 619 nm), which indicates the effect of the diaza substitution in the energy levels of the molecule.41 Moreover, the N-alkyl substitution in the indigo molecule typically produces a red shift of the main absorption band, which is also observed in the azaindigo derivative. The emission spectra of the keto forms of 1 and 2 (Fig. 2A and S2A†) display an asymmetric peak centred at 610 nm and 640 nm, respectively, which are mirror images of the visible absorption signals. The position of the emission peaks are blue-shifted with respect to those in indigo (λem = 653 nm), similarly to the absorption peaks. Absorption and emission measurements at different concentrations have been also done showing no changes in the shape of spectra, which rules out the formation of aggregates.
580 M−1 cm−1 (600 nm) for 1 and 2 which are comparable to those measured in the indigo molecule.40 The visible absorption signal in 1 is shifted to the blue compared with indigo (λabs = 619 nm), which indicates the effect of the diaza substitution in the energy levels of the molecule.41 Moreover, the N-alkyl substitution in the indigo molecule typically produces a red shift of the main absorption band, which is also observed in the azaindigo derivative. The emission spectra of the keto forms of 1 and 2 (Fig. 2A and S2A†) display an asymmetric peak centred at 610 nm and 640 nm, respectively, which are mirror images of the visible absorption signals. The position of the emission peaks are blue-shifted with respect to those in indigo (λem = 653 nm), similarly to the absorption peaks. Absorption and emission measurements at different concentrations have been also done showing no changes in the shape of spectra, which rules out the formation of aggregates.
 | ||
| Fig. 2 Normalized absorption and emission spectra of the keto (A) and leuco (B) forms of 2 in DMF. | ||
| Compound | λabs/nm | λem/nm | εmax/M−1 cm−1 | ΦF | τF/ns | krad/ns−1 | knrad/ns−1 | |
|---|---|---|---|---|---|---|---|---|
| Keto form | 1 | 565 | 610 | 2400 | 2.7 × 10−3 | 0.140 | 1.88 × 10−2 | 6.93 |
| 2 | 600 | 640 | 22580 |
1.5 × 10−3 | 0.078 | 1.92 × 10−2 | 12.8 | |
| Leuco form | 1 | 445 | 505 | 1500 | 0.03 | 6.3 | 4.76 × 10−3 | 0.15 |
| 2 | 470 | 585 | 10800 |
0.02 | 0.73 | 2.74 × 10−2 | 1.34 | |
Fig. 2B and S2B† exhibit the normalized absorption and emission spectra (λexc = 410 and 475 nm) of the leuco forms of 1 and 2 compounds. The visible absorption signals of the leuco forms are blue shifted with respect to the keto form, appearing at λabs = 445 nm (1) and 500 nm (2), which becomes an evidence of the formation of the reduced form as it occurs in other indigo-based dyes, like the tyrian purple, indigocarmine or indirubin.40 The absorption coefficients result in a value of 1500 and 10800 M−1 cm−1 for compounds 1 and 2, respectively. The emission spectra of the leuco forms exhibits a broad peak centred at λem = 505 nm (1) and 585 nm (2) with a shoulder in the long wavelength region.
The fluorescence quantum yields (ΦF) of both keto and leuco forms in DMF for both compounds were also measured and are shown in Table 1. The low ΦF values determined for the keto form of 1 and 2 agree well with that reported for the indigo molecule (ΦF = 2.3 × 10−3) or with that from the N-hexylindigo in DMSO (ΦF = 0.0008).41 In case of the leuco forms, the ΦF values determined for 1 and 2 are more than 10-fold lower than that of indigo (ΦF = 0.348), which would indicate that a fast non-radiative recombination pathway is active. A similar low ΦF value has also been reported for other indigo derivatives that might be ascribed to the formation of a triplet state through an intersystem crossing process.42
The fluorescence decays were also recorded for both keto and leuco forms of 1 and 2 compounds (see Table 1). The lifetimes of the keto forms are comparable to that of indigo (0.135 ns) and N-hexylindigo in DMSO (0.064 ns). Thus, the calculated values for the radiative (krad) and non-radiative (knrad) rate constants are in the same order of magnitude to those found for the indigo molecule, indicating that the presence of the two diaza groups does not modify the deactivation scheme respect to the indigo molecule. The fluorescence lifetime of compound 1 in the leuco form is comparable to that of indigo (τF = 3.15 ns).40 In line with the reduced quantum yield of the leuco form of 1, the radiative rate constant, krad, is ∼23-fold smaller than that of indigo (krad = 0.111 ns−1) while similar values for the non-radiative rate constant, knrad, are found for both compounds.40 In case of the leuco form of 2 compound, both krad and knrad are higher, in consonance with the shorter fluorescence lifetime determined for this species.
Flash photolysis measurements of the leuco forms were also performed in DMF to investigate the formation of long-lived transient species, such as the triplet excited state. However, no transient signals have been detected in opposition to the results previously obtained for several indigo derivatives.40 Therefore, we attribute the absence of triplet transient absorption to a lower intersystem crossing triplet yield, as a consequence of a more efficient non-radiative deactivation pathway.
Femtosecond transient absorption (TA) experiments
To in-depth investigate the excited state deactivation of the keto form, femtosecond transient absorption (TA) experiments were performed in DMF. Fig. 3A and S3A† display the transient absorption spectra at different time delays after excitation at λexc = 540 and 625 nm for compounds 1 and 2, respectively. Spectra exhibit positive TA bands in the 400–525 nm and 650–750 nm ranges, similar to those reported for the indigo and its monohexyl derivative. These signals have previously been assigned to the photoinduced absorption (PIA) of the singlet excited state. Moreover, the negative signal in the 525–600 nm range is attributed to the ground state bleaching (GSB) and, unlike the case of indigo, no stimulated emission (SE) is observed in compounds 1 or 2. A global fit analysis of the transient kinetics has been performed to evaluate the photophysical deactivation scheme. Fig. 3B and S3B† show the TA decays at 580–590 and 690–700 nm together with the best fit results. The experimental decays are well fit with the sum of two exponential functions with decay times of τ1 = 1.3 ps and τ2 = 79 ps for compound 1 and τ1 = 2.9 ps and τ2 = 86 ps for compound 2. The longer decay component is in good agreement with the fluorescence decay time obtained in the time-correlated single photon counting experiments, τfl = 114 and 78 ps for 1 and 2, respectively.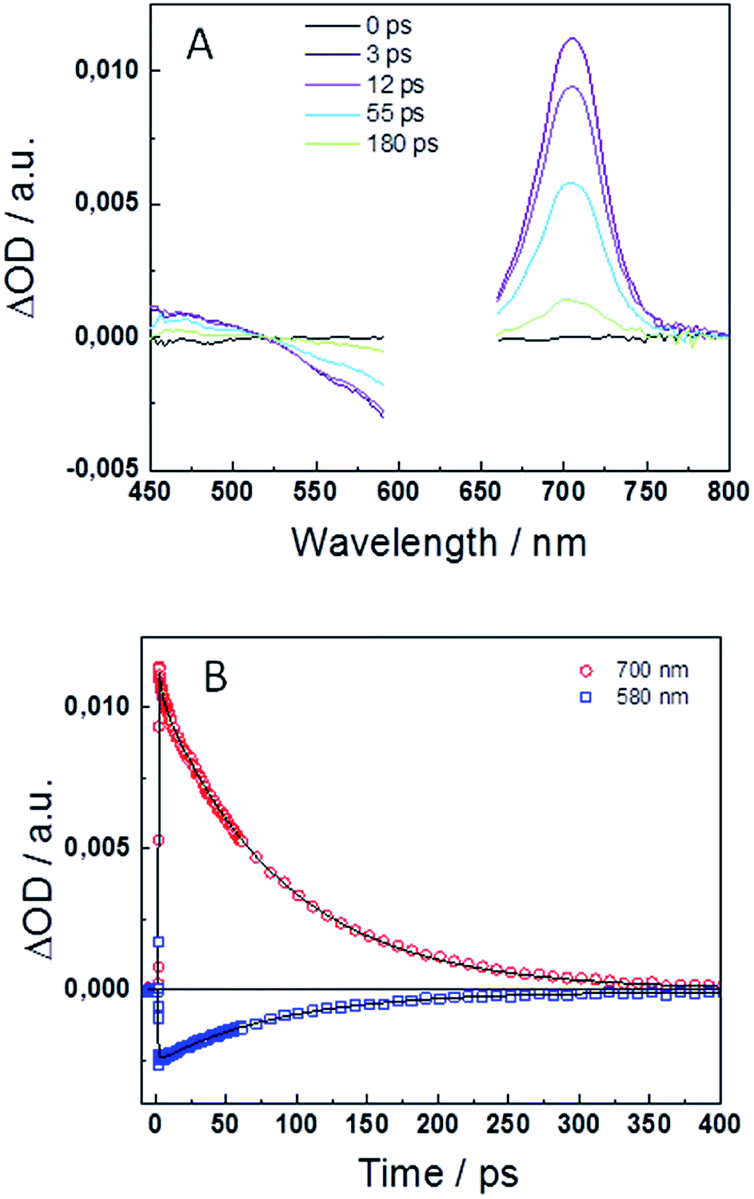 | ||
| Fig. 3 Transient absorption spectra at different time delays (A) and time decays at λprobe = 700 nm and 580 nm (B) for the keto form of 2 in DMF with λexc = 625 nm. The solid lines in (B) are the best fits of the experimental data obtained from the global analysis. | ||
The fast deactivation of the excited state of the highly soluble indigo carmine derivative has been studied in detail in previous articles.43,44 The deactivation dynamics is strongly dependent on the aprotic/protic nature of the solvent with an excited state proton transfer (ESPT) as the main deactivation mechanism mediated by the formation of intermolecular H-bonds in protic solvents as H2O. In case of organic aprotic solvents, such as DMSO or DMF, the intramolecular H-bonds retain the molecular planarity, preventing the ESPT process.44 Considering the previous discussion, the deactivation of the keto form of compounds 1 and 2 in DMF is expected to occur through an internal conversion (IC) process to the ground state, without passing by the enol form. DFT calculations were carried out to corroborate this assumption (vide infra). The insolubility of the keto form of the studied compounds in H2O prevented us from performing the femtosecond transient absorption experiments in this protic media. In line with this deactivation mechanism, the fast decay times (τ1 = 1.3–2.9 ps) measured in the femtosecond transient absorption measurements can be attributed to solvent reorientation.43,45 The longer decay component (τ2 = 79–86 ps) is associated to the non-radiative IC process to the ground-state.
Theoretical insights on the excited state deactivation mechanism
DFT calculations were carried out to shed light on the excited state deactivation of compounds 1 and 2. We were particularly interested in determining if ESPT mechanism occurs in an organic aprotic solvent as DMF. ESPT mechanism would lead to an interconversion from the form keto tautomer to enol form in the excited state. PBE0/6-31+G* calculations showed that the keto form of molecule 2 is slightly twisted with an interring dihedral angle (τ) of 8.2° while 1 is totally planar (see Fig. S4†). The NH⋯OC distances (≤2.30 Å) are smaller than the sum of the van der Waals radii of the atoms, suggesting the existence of intramolecular hydrogen bonds (see Fig. S4†).46 In the ground state, we assume that the keto form is predominant in solution because the enol form is higher in energy than the former, i.e. ΔEen–kt(S0) is 1.79 eV for 1 and 0.51 eV for 2 (ΔEen–kt is the difference of the computed energies for the enol and keto form). In addition, the energy calculated for the vertical transition S0 → S1 of both molecules in keto form (at the TD-PBE0/6-31+G* level of theory) matches better with the experimental UV-vis absorption data than the corresponding calculations in enol form (see Tables 2, S3 and Fig. S5† for details). According to the oscillator strength values, the next likely electronic transition involves the S6 excited state in the keto form (and S5 state in enol form). For both the keto and enol forms of 2, the vertical energies calculated from S1 to the upper excited states are close between them (ΔE = 0.04 eV) and in good accordance with the transient absorption spectrum. A larger energy difference was obtained between the keto and enol form (ΔE = 0.09 eV) in the calculation of the corresponding vertical transitions of molecule 1 (see Table 2). The transient absorption data obtained for molecule 1 matches better with the upward vertical transition calculated for the keto form (ΔEe–c = 0.21 eV) than for the enol form (ΔEe–c = 0.30 eV) (ΔEe–c is the difference in energy between experimental and calculated vertical energies) (see Fig. 4). This result, as well as the higher thermodynamic stability of the keto forms in the S1 state (ΔEen–kt(S1) is 0.92 eV for 1 and 0.24 eV for 2), suggests that the excited state deactivation of the studied 7,7′-diazaindigo derivatives occurs from the keto form in organic aprotic solvents.| Compound | Form | ΔE(S0 → S1)/nm [eV] | ΔE(S1 → S0)/nm [eV] | ΔE(S1 → S6/5)a/nm [eV] | |||
|---|---|---|---|---|---|---|---|
| Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | ||
| a Energy calculated for the S1 → S6 transition of the keto form and S1 → S5 transition of the enol form. | |||||||
| 1 | Keto | 534.4 [2.32] | 563.6 [2.20] | 576.7 [2.15] | 607.8 [2.04] | 784.7 [1.58] | 692.6 [1.79] |
| Enol | 873.1 [1.42] | — | — | — | 832.1 [1.49] | — | |
| 2 | Keto | 566.1 [2.19] | 599.0 [2.07] | 639.1 [1.94] | 639.1 [1.94] | 712.6 [1.74] | 700.5 [1.77] |
| Enol | 619.9 [2.00] | — | — | — | 696.5 [1.78] | — | |
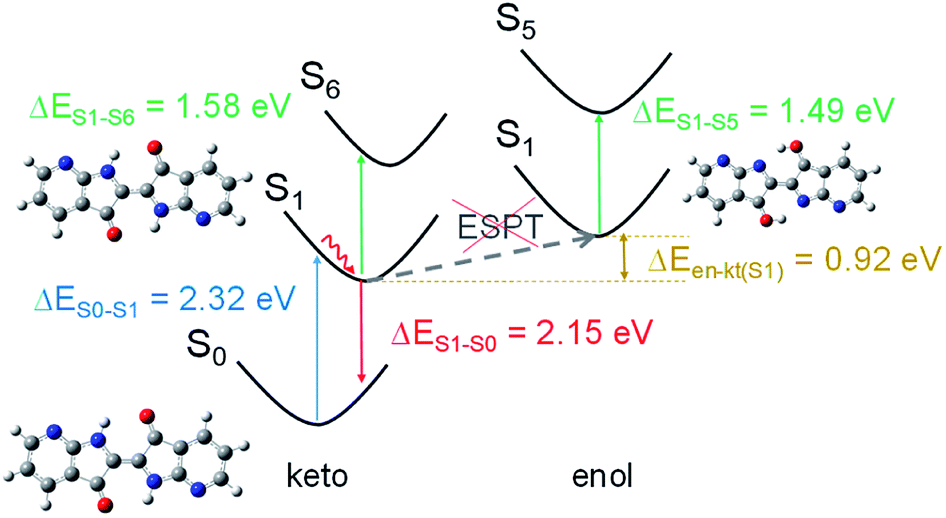 | ||
| Fig. 4 Scheme of different vertical transition calculated for compound 1 in DMF solution. | ||
Conclusions
The replacement of the C atoms in the 7 and 7′positions of the indigo ring by N atoms does not modify largely the photophysical scheme and kinetics of the excited state. In the keto form, the absorption and emission peaks are slightly shifted to the blue respect to indigo, but the fluorescence quantum yield and lifetimes are comparable to those observed in the reference molecule. Femtosecond transient absorption experiments and DFT calculations suggest that the deactivation of the excited state in organic aprotic solvent occurs through an internal conversion process, in opposition to the ESPT mechanism that takes place in protic solvents like H2O. In the leuco form, the absorption and emission maxima are blue shifted with respect to those determined for the keto form. The fluorescence quantum yield also increases about 10-times with respect to the keto form associated to a slower non-radiative rate constant.Conflicts of interest
Authors declare that there are no conflicts of interest.Acknowledgements
This work was financially supported by the Ministry of Economy and Competitiveness of Spain through projects (CTQ2017-84221-R and CTQ2017-84561-P). G. M. thanks to the Ministry of Economy and Competitiveness for a “Ramón y Cajal” contract (RYC-2013-12772).Notes and references
- R. J. H. Clark, C. J. Cooksey, M. A. M. Daniels and R. Withnall, Endeavour, 1993, 17, 191–199 CrossRef CAS.
- E. S. B. Ferreira, A. N. Hulme, H. McNab and A. Quye, Chem. Soc. Rev., 2004, 33, 329–336 RSC.
- D. Cardon, Le monde des teintures naturelles, Berlin, 2003 Search PubMed.
- H. Schweppe, Handbuch der Naturfarbstoffe, Nikol, Hamburg, 1993 Search PubMed.
- R. Prasad, Indian J. Hist. Sci., 2018, 296–301 Search PubMed.
- J. Balfour-Paul, Indigo, British Museum Press, London, 1998 Search PubMed.
- J. W. Davis and L. Strauss, USPTO, 139121, 1873.
- A. N. Fabara and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2020, 104, 925–933 CrossRef CAS.
- A. Berry, T. C. Dodge, M. Pepsin and W. Weyler, J. Ind. Microbiol. Biotechnol., 2002, 28, 127–133 CrossRef CAS.
- C. Cooksey, Molecules, 2001, 6, 736–769 CrossRef CAS.
- I. Karapanagiotis, Sustainability, 2019, 11, 3595 CrossRef CAS.
- C. Ouellet-Plamondon, P. Aranda, A. Favier, G. Habert, H. van Damme and E. Ruiz-Hitzky, RSC Adv., 2015, 5, 98834–98841 RSC.
- P. Woodtli, S. Giger, P. Müller, L. Sägesser, N. Zucchetto, M. J. Reber, A. Ecker and D. Brühwiler, Dyes Pigm., 2018, 149, 456–461 CrossRef CAS.
- M. Yao, K. Kuratani, T. Kojima, N. Takeichi, H. Senoh and T. Kiyobayashi, Sci. Rep., 2015, 4, 3650 CrossRef.
- M. Yao, M. Araki, H. Senoh, S. Yamazaki, T. Sakai and K. Yasuda, Chem. Lett., 2010, 39, 950–952 CrossRef CAS.
- I. V. Klimovich, L. I. Leshanskaya, S. I. Troyanov, D. V. Anokhin, D. V. Novikov, A. A. Piryazev, D. A. Ivanov, N. N. Dremova and P. A. Troshin, J. Mater. Chem. C, 2014, 2, 7621–7631 RSC.
- N. M. Randell and T. L. Kelly, Chem. Rec., 2019, 19, 973–988 CrossRef CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef.
- E. D. Głowacki, D. H. Apaydin, Z. Bozkurt, U. Monkowius, K. Demirak, E. Tordin, M. Himmelsbach, C. Schwarzinger, M. Burian and R. T. Lechner, et al., J. Mater. Chem. C, 2014, 2, 8089–8097 RSC.
- A. K. Rajan and L. Cindrella, Opt. Mater., 2019, 88, 39–47 CrossRef CAS.
- D. Franchi, M. Calamante, C. Coppola, A. Mordini, G. Reginato, A. Sinicropi and L. Zani, Molecules, 2020, 25(15), 3377 CrossRef CAS.
- M. Hosseinnezhad, S. Moradian and K. Gharanjig, Dyes Pigm., 2015, 123, 147–153 CrossRef CAS.
- J.-L. Li, J.-J. Cao, L.-L. Duan and H.-L. Zhang, Asian J. Org. Chem., 2018, 7, 2147–2160 CrossRef CAS.
- P. Josse, S. Dabos-Seignon, S. M. McAfee, G. C. Welch, P. Blanchard and C. Cabanetos, Dyes Pigm., 2017, 145, 7–11 CrossRef CAS.
- J. Miao, H. Xu, B. Meng, J. Liu and L. Wang, Chin. J. Chem., 2018, 36, 411–416 CrossRef CAS.
- G. de Miguel, L. Camacho and E. M. García-Frutos, J. Mater. Chem. C, 2016, 4, 1208–1214 RSC.
- M. Moral, A. Navarro, A. Garzón-Ruiz and E. M. García-Frutos, J. Phys. Chem. C, 2019, 123, 153–164 CrossRef CAS.
- A. Garzón, A. Navarro, D. López, J. Perles and E. M. García-Frutos, J. Phys. Chem. C, 2017, 121, 27071–27081 CrossRef.
- Y. Lu, Y. Liu, Y.-Z. Dai, C.-Y. Yang, H.-I. Un, S.-W. Liu, K. Shi, J.-Y. Wang and J. Pei, Chem.–Asian J., 2017, 12, 302–307 CrossRef CAS.
- S.-Y. Kim, Y.-J. Cho, A.-R. Lee, H.-J. Son, W.-S. Han, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 426–435 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 09 (version D.01), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- R. Improta, J. Chem. Phys., 2006, 125, 054103 CrossRef.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef.
- X. Cheng, K.-H. Merz, S. Vatter, J. Christ, S. Wölfl and G. Eisenbrand, Bioorg. Med. Chem., 2014, 22, 247–255 CrossRef CAS.
- J. Seixas de Melo, A. P. Moura and M. J. Melo, J. Phys. Chem. A, 2004, 108, 6975–6981 CrossRef CAS.
- J. Pina, D. Sarmento, M. Accoto, P. L. Gentili, L. Vaccaro, A. Galvão and J. S. Seixas de Melo, J. Phys. Chem. B, 2017, 121, 2308–2318 CrossRef CAS.
- R. Rondão, J. S. Seixas de Melo and G. Voss, ChemPhysChem, 2010, 11, 1903–1908 Search PubMed; J. S. S. de Melo, R. Rondão, H. D. Burrows, M. J. Melo, S. Navaratnam, R. Edge and G. Voss, ChemPhysChem, 2006, 7, 2303–2311 CrossRef.
- Y. Nagasawa, R. Taguri, H. Matsuda, M. Murakami, M. Ohama, T. Okadaa and H. Miyasaka, Phys. Chem. Chem. Phys., 2004, 6, 5370–5378 RSC.
- P. P. Roy, J. Shee, E. A. Arsenault, Y. Yoneda, K. Feuling, M. Head-Gordon and G. R. Fleming, J. Phys. Chem. Lett., 2020, 11, 4156–4162 CrossRef CAS.
- M. L. Homg, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef.
- S. S. Batsanov, Van der Waals Radii of Elements, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, single crystal X-ray diffraction data for 2, 1H NMR and 13C NMR spectra data. CCDC 2013556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06451c |
| This journal is © The Royal Society of Chemistry 2020 |