
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Bromoanthraquinone as a highly efficient photocatalyst for the oxidation of sec-aromatic alcohols: experimental and DFT study†
Shengfu Liao ab,
Jianguo Liua,
Long Yana,
Qiying Liuab,
Guanghui Chenc and
Longlong Ma*ab
ab,
Jianguo Liua,
Long Yana,
Qiying Liuab,
Guanghui Chenc and
Longlong Ma*ab
aBiomass Catalytic Conversion Laboratory, Guangzhou Institute of Energy, Chinese Academy of Sciences, Guangzhou, Guangdong 510640, China. E-mail: mall@ms.giec.ac.cn; Fax: +86-20-87057673; Tel: +86-20-87057673
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemistry, Shantou University, Shantou 515063, Guangdong, PR China
First published on 7th October 2020
Abstract
Anthraquinones are recognized as high efficiency photocatalysts which can perform various redox reactions in aqueous or organic phases. We have experimentally proven that 2-BrAQ can undergo hydrogen transfer with an alpha-aromatic alcohol under light conditions, thereby efficiently oxidizing the aromatic alcohol to the corresponding product. The yield of 1-phenethanol to acetophenone can reach more than 96%. In subsequent catalyst screening experiments, it was found that the electronegativity of the substituent at the 2 position of the anthraquinone ring and the acidity of the solvent affect the photocatalytic activity of anthraquinones. After using various aromatic alcohol substrates, 2-BrAQ showed good conversion and selectivity for most aromatic alcohols, but showed C–C bond cleavage and low selectivity with non-α-position aromatic alcohols. In order to explore the mechanism of the redox reaction of 2-BrAQ in acetonitrile solution, the corresponding free radical reaction pathway was proposed and verified by density functional theory (DFT). Focusing on calculations for 2-BrAQ during the reaction and the first-step hydrogen transfer reaction between the 2-BrAQ triplet molecule and the 1-phenylethanol molecule, we recognized the changes that occurred during the reaction and thus have a deeper understanding of the redox reaction of anthraquinone compounds in organic systems.
1 Introduction
Aromatic alcohol oxidation has been one of the most widely used reactions in organic chemistry.1–3 1-Phenylethanol has the typical monomer structure of a secondary aromatic alcohol in the lignin structure and its efficient oxidation to acetophenone is the research goal of many scholars.4,5 By oxidizing the secondary alcohol hydroxyl group in the lignin β-O-4 linkage to the corresponding ketocarbonyl group, the bond energy of the lignin β-O-4 bond can be effectively reduced and the depolymerization of lignin can be accelerated.4 Therefore, study of the oxidation of secondary aromatic alcohols is very useful for development in the chemical industry.Photocatalytic oxidation is a mild and green oxidation method compared to traditional thermal catalysis and has been popular in the oxidation of aromatic compounds since it was discovered by Masamichi Fujihira.6 Many photocatalysis systems have been formed for alcohol oxidation, but homogeneous catalysis is recognized for its higher activity and dispersion. The most used catalysts in homogeneous photocatalysis include ruthenium tri(bipyridine),7 iridium tris(phenylpyridine), eosin, and rhodamine-6G. However, the expense of noble metals, undesired overoxidation, use of co-catalysts (such as TEMPO) and cumbersome syntheses remain large obstacles for the practical application of these approaches in a cost-effective fashion. Consequently, choosing a highly selective and non-precious photocatalyst is a general trend for catalysis applications.
Anthraquinone (AQ) and its derivatives are well-recognized for their exceptional redox abilities in the chemical industry.8,9 Anthraquinone is widely used in the pharmaceutical and chemical industries, in peroxide production, and in anti-cancer, anti-inflammatory, antinociceptive activities, among others.10–13 In addition, AQs are the most abundant group of dyes apart from azo dyes. As one of the cheapest organic dyes, AQs have been developed very quickly during the past decades14 for their wide range of colors. A typical example of their application in redox reactions is 2-ethylanthraquinone (EAQ) catalyzing hydrogen peroxide (H2O2) generation from hydrogen (H2) and oxygen (O2) on an industrial scale.15 In this process, the EAQ was first reduced to 2-ethylanthrahydroquinone (EAQH2) by H2. EAQH2 possesses moderate reduction potentials16,17 and is reduced to EAQ by O2 in the next step. At the same time, H2O2 was produced.18,19 However, in many photocatalytic oxidation studies, anthraquinone, as an organic dye, is often degraded.20,21 In recent years, with the application of anthraquinone in photocatalytic bromination, iodination, and hydrogen peroxide production,22–24 the excellent photocatalytic performance of anthraquinone has begun to receive close attention.
In this work, we used 2-bromoanthraquinone as a photocatalyst to promote the transfer of protons from alcohols to the hydrogen acceptor (nitrobenzene). As is well known, oxygen (O2) is the most used oxidant due to its strong oxidizing properties and easy availability. However, due to its free radical reaction characteristics, the oxidation process is uncontrollable and over-oxidation occurs.25,26 Therefore, we chose nitrobenzene as a hydrogen acceptor. This process is divided into three steps: first, anthraquinone molecules were excited by light to generate excited singlet states. The singlet molecules were unstable and rapidly formed triplet molecules through intersystem crossing (ISC) in a solvent. Second, the triplet anthraquinone molecules were reduced by alcohol to generate hydroanthraquinone molecules and the hydroxyl group was simultaneously converted to the corresponding aldehyde or ketone group. Thirdly, hydroanthraquinone molecules were oxidized by nitrobenzene, generating anthraquinone molecules and aniline.
In recent years, in order to explore the photoredox mechanism of anthraquinone catalysts, many theoretical and experimental studies have been published. Density functional theory is one of the most popular theoretical research methods and is favored by many scholars. The B3LYP hybrid functional research method has been the most widely used in quantum chemistry since it was proposed in 1994, due to its universal applicability and high cost performance. In 2012, David Lee Phillips and colleagues discussed the redox reaction of 2-hydroxyethylanthraquinone in aqueous solutions using the B3LYP method.27 They also used time-resolved spectroscopy and density functional theory to unravel the photodeprotection mechanism of anthraquinon-2-ylmethoxycarbonyl-caged alcohols.28 In both studies, anthraquinone molecules formed a hydroanthraquinone structure, proving that this is a key intermediate in the anthraquinone molecule's redox reaction. Therefore, this study will also explore the key intermediates of anthraquinone molecules in organic solvents to reveal the anthraquinone redox cycle under free radical reaction conditions.
2 Experimental
2.1 Materials
30 W purple LED lights (wavelength range around 400 nm) were purchased from Shenzhen Leixi Lighting Co. Ltd. 2-Bromoanthraquinone, 2-chloroanthraquinone, 2-methylanthraquinone, anthraquinone, and anthraquinone-2-carboxylic acid were obtained from Sarn Chemical Technology Co., Ltd. 1-Phenylethanol was purchased from Adamas Reagent Co., Ltd. Acetonitrile was purchased from Shanghai Macklin Biochemical Technology Co., Ltd. Nitrobenzene, benzyl alcohol and other alcohols were obtained from Shanghai Aladdin Bio-Chem Technology Co., Ltd. All chemicals were used without further purification.2.2 Photocatalytic oxidation of alcohols
2.3 Density functional theory (DFT) calculation
Density function theory (DFT) calculations were performed in the Gaussian 09W D01 program package. In all calculations, acetonitrile was used as the default solvent model. In the geometric optimization calculations, Becke's three-parameter hybrid method with Lee–Yang–Parr correlation function approximation with D3 dispersion correction (B3LYP-D3BJ) with a 6-311G* basis set was employed. Calculation of the excited state of 2-BrAQ was carried out under the time-dependent (TD) UB3LYP-D3BJ and 6-311+G* basis set. TD-DFT methodology was also used to compute the low-lying excited states of transient species of interest. The relative energy in the 2-BrAQ photoredox cycle was calculated using the M06-2X density functional with the def2-TZVP basis set. The calculation results were viewed with GaussView 5.0 software.3 Result and discussion
3.1 Molar extinction coefficient of anthraquinones
The molar extinction coefficient of anthraquinones was measured using a UV-visible spectrophotometer. It can be seen from Fig. 1 that the absorption spectrum of anthraquinone compounds is concentrated in the wavelength band before 420 nm. This shows that, in order to achieve good photocatalytic effect, the light source must also have a band before 420 nm. Therefore, we chose a 30 W purple LED lamp with a wavelength of about 400 nm.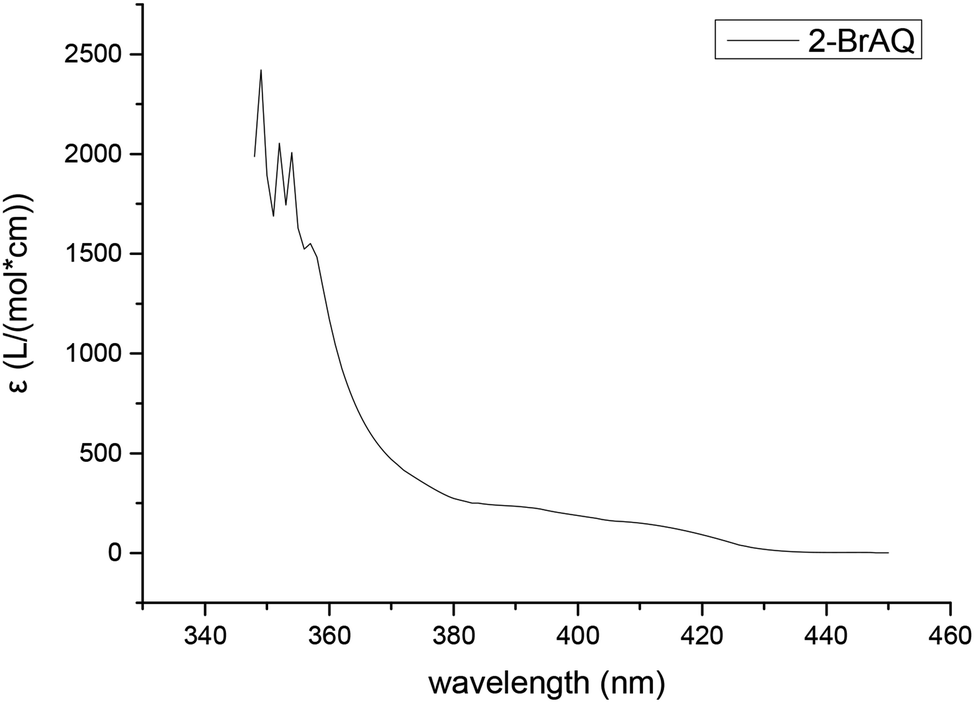 | ||
| Fig. 1 Molar extinction coefficient of 2-BrAQ photocatalyst. Conditions: 0.5 g L−1 2-bromoanthraquinone in acetonitrile solution, optical path length 1 cm. | ||
3.2 Photocatalytic oxidation of aromatic alcohols
In order to test the reactivity of different anthraquinone catalysts, we selected several anthraquinones with different substituents. It can be seen from Table 1 that when the hydrogen atom at position 2 in anthraquinone is replaced by a halogen atom, the reactivity is increased and that the bromine substitution is stronger than the chlorine substitution. This can be explained by the anthraquinone photocatalytic reaction pathway. In the photophysical process of anthraquinone molecules, an excited anthraquinone molecule excited by light of a specific wavelength undergoes an intersystem crossing process and becomes a triplet molecule. The triplet anthraquinone molecule is considered to have a strong intramolecular electron transfer ability with electron enrichment on the carbonyl oxygen atom and is an active molecule in the redox reaction.29,30 When a hydrogen atom in the molecule is substituted by a halogen atom, the quantum-yield ratio of phosphorescence to fluorescence increases.31,32 This indicates an increase of the intersystem crossing coefficient, leading to a relatively large number of triplet molecules and resulting in enhanced photocatalytic reactivity. The larger the molecular weight of the substituted halogen atom, the stronger this effect. It may explain why 2-bromoanthraquinone is the most reactive photocatalyst among these catalysts. When anthraquinone has amino substitution, there is no reactivity. This is because the excited 2-aminoanthraquinone molecule has a large deactivation constant in acetonitrile,30 leading to the decrease in the reactivity of the molecules. In theory, one molecule of nitrobenzene completely reduced to aniline can accept six hydrogen atoms and one molecule of 1-phenylethanol converted to acetophenone can provide two hydrogen atoms. Therefore, the conversion ratio of 1-phenylethanol to nitrobenzene should be 3/1. However, according to entry 2 of Table 1, the conversion ratio of 1-phenylethanol to nitrobenzene in the experiment was 2.6/1, which indicates that nitrobenzene was consumed in excess in this process and not completely reduced to aniline. This is why the yield of aniline is lower than the conversion rate of nitrobenzene. In fact, there are many paths in the reduction process of nitrobenzene in which some incomplete reduction products33 (nitrosobenzene, N-phenylhydroxylamine) or polymerization products34 will be produced.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Reaction time (h) | Conversion (%) | Yieldb (%) | ||
| 1 | 2 | 3 | 4 | |||
| a General conditions: 0.1 mmol 1-phenylethanol, 0.1 mmol nitrobenzene, 0.01 mmol catalyst, 3 mL acetonitrile, 30 W LED illuminated for 5 h, 30 °C water bath.b Yield was determined by gas chromatography. | ||||||
| 1 | 2-Cl-AQ | 5 | 70 | 31 | 70 | 20 |
| 2 | 2-Br-AQ | 5 | 76 | 29 | 76 | 20 |
| 3 | 2-Me-AQ | 5 | 49 | 25 | 48 | 9 |
| 4 | 2-NH2-AQ | 5 | 5 | 0 | 5 | 0 |
| 5 | 2-COOH-AQ | 5 | 57 | 14 | 56 | 9 |
| 6 | AQ | 5 | 64 | 30 | 62 | 9 |
| 7 | 1,4-2NH2-AQ | 5 | 0 | 0 | 0 | 0 |
| 8 | Eosin Y | 5 | 10 | 0 | 10 | 0 |
| 9 | Rhodamine 6G | 5 | 0 | 0 | 0 | 0 |
Next, we investigated the effects of solvents on the oxidation of 1-phenylethanol. As shown in Table 2, acetonitrile is considered the best solvent for this reaction. According to entries 1, 2, 9 and 10, there is no large difference between acetone and acetonitrile. We chose acetonitrile as the standard solvent for subsequent experiments. The conversions of 1-phenylethanol using other solvents such as EtOH, toluene, THF and DMF (entries 5–8) are very low, but the corresponding conversion of nitrobenzene was very high. This is because these solvents could reduce nitrobenzene, thereby preventing the oxidation of 1-phenylethanol. The GC spectra after the reaction are shown in ESI Fig. S9–S12.† When 1-phenylethanol is oxidized to acetophenone, nitrobenzene is also partially reduced to aniline. This is probably because the protons on the alcohol are transferred to the nitrobenzene, accompanied by the reduction of nitrobenzene.
| Entry | Solvent | Reaction time (h) | Conversion (%) | Yieldb (%) | ||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| a General conditions: 0.1 mmol 1-phenylethanol, 0.1 mmol nitrobenzene, 0.01 mmol 2-Br-AQ, 3 mL solvent, 30 W LED illuminated for specified time, 30 °C water bath.b Yield was determined by gas chromatography. | ||||||
| 1 | Acetonitrile | 5 | 76 | 29 | 76 | 20 |
| 2 | Acetone | 5 | 75 | 37 | 75 | 14 |
| 3 | Ethyl acetate | 5 | 63 | 37 | 63 | 15 |
| 4 | DCM | 5 | 64 | 28 | 61 | 7 |
| 5 | Ethanol | 5 | 8 | 100 | 8 | 0 |
| 6 | Toluene | 5 | 0 | 87 | 0 | 27 |
| 7 | THF | 5 | 4 | 100 | 4 | 12 |
| 8 | DMF | 5 | 1 | 91 | 1 | 26 |
| 9 | Acetonitrile | 20 | 90 | 38 | 90 | 15 |
| 10 | Acetone | 20 | 88 | 45 | 88 | 11 |
Then, we explored the effect of reaction time on the reaction. It can be seen from Fig. 2 that when the reaction proceeds to 20 h, the acetophenone yield reaches more than 90%. The reaction proceeded very rapidly in the first 8 hours, but after 8 hours, became slow and the various reactants and products tended to plateau.
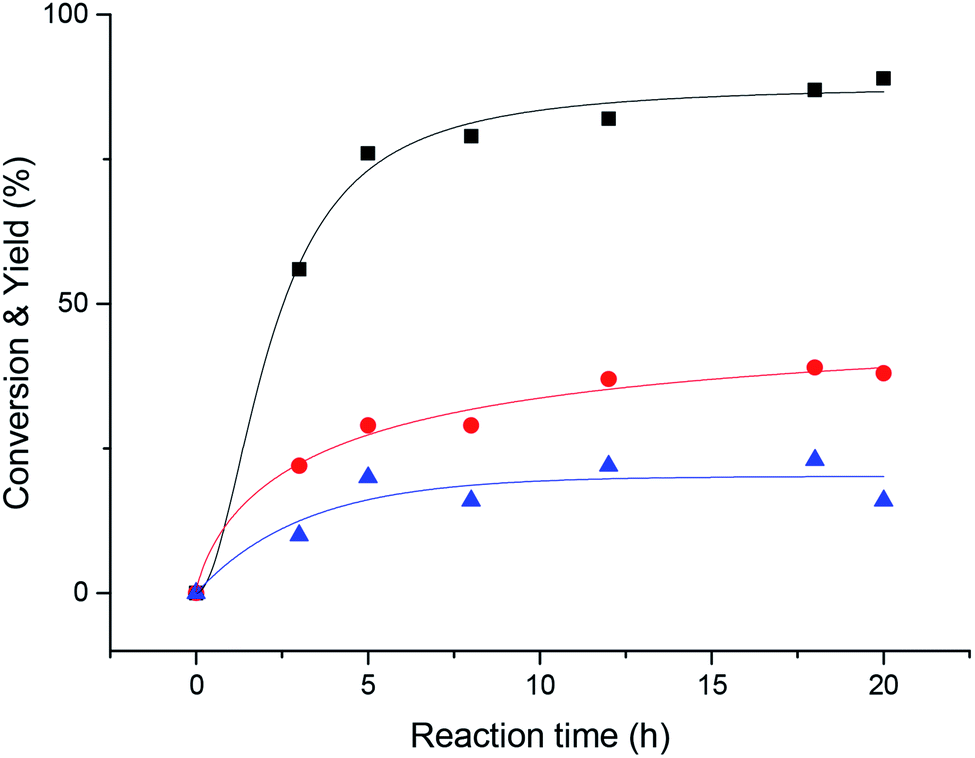 | ||
| Fig. 2 Time course plots of each main reactant and product. Black, 1-phenylethanol and acetophenone; red, nitrobenzene; blue, aniline. General conditions: 0.1 mmol 1-phenylethanol, 0.1 mmol nitrobenzene, 0.01 mmol 2-Br-AQ, 3 mL acetonitrile, 30 W LED illuminated for specified time, 30 °C water bath. | ||
To prove that this reaction is light-driven, we performed a series of control experiments. From entries 1 and 2 in Table 3, we can see that this reaction gave 76% conversion using light while there is no reactivity when it is carried out without light. When there is no catalyst, the reaction rarely gave conversion. When nitrobenzene was missing, the reaction conversion and selectivity were reduced. When the amount of catalyst or nitrobenzene was reduced, the reaction conversion rate decreased about 10%. When the molar ratio of nitrobenzene to 1-phenylethanol was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the 1-phenylethanol conversion and acetophenone yield only increased by 3%. Therefore, using a catalyst equivalent of 0.1 and a nitrobenzene equivalent of 1 achieves a reasonable yield.
:1, the 1-phenylethanol conversion and acetophenone yield only increased by 3%. Therefore, using a catalyst equivalent of 0.1 and a nitrobenzene equivalent of 1 achieves a reasonable yield.
| Entry | Catalyst amount (equiv.) | Nitrobenzene amount (equiv.) | Conversion of alcohol (%) | Yield of ketone (%) |
|---|---|---|---|---|
| a General conditions: 0.1 mmol 1-phenylethanol, nitrobenzene, and 2-Br-AQ, 3 mL of solvent, 30 W LED illuminated for 5 h, 30 °C water bath.b Reaction proceeded in the dark. | ||||
| 1 | 0.1 | 1 | 76 | 76 |
| 2b | 0.1 | 1 | 0 | 0 |
| 3 | 0 | 1 | 5 | 5 |
| 4 | 0.1 | 0 | 41 | 28 |
| 5 | 0.05 | 1 | 65 | 63 |
| 6 | 0.1 | 0.5 | 66 | 65 |
| 7 | 0.1 | 2 | 79 | 79 |
Under standard reaction conditions, we tested the conversion and selectivity of various alcohol substrates. For pyridine aromatic alcohols, the oxidation results are not satisfactory, which may be caused by the electron-deficiency of the pyridine ring. For entries 4, 8, 10, 11, 15, 22, 23 and 24 in Table 4, the reactions all showed rather high conversion and selectivity, which may be due to the relatively stable ketone structure formed. For entries 5, 7, 9, 13, 16, 17, 19 and 20 in Table 4, the reaction showed rather high conversion but low selectivity. This may be due to the unstable products produced or unnecessary side reactions in the system. It can be seen from Table S1 and Fig. S1† that when a product containing an aldehyde group is formed, dehydration condensation occurs with the generated aniline, which has been confirmed by Itoh et al.35 If the alcoholic hydroxyl group and benzene ring in the substrate are not on the same carbon atom, such as in entries 18 and 25 in Table 4, C–C bond cleavage occurs after oxidation and the selectivity is low. This indicates that 2-bromoanthraquinone has a strong regioselectivity for the oxidation of α-position aromatic alcohols.
| Entry | Substrate | Conversion (%) | Yieldb (%) |
|---|---|---|---|
| a General conditions: 0.1 mmol substrate, 0.1 mmol nitrobenzene, 0.01 mmol 2-Br-AQ, 3 mL solvent, 30 W LED illuminated for 20 h, 30 °C water bath.b Yield was determined by 1H NMR using 1,4-dimethoxybenzene as internal standard.c Yield was determined by gas chromatography. | |||
| 1 | 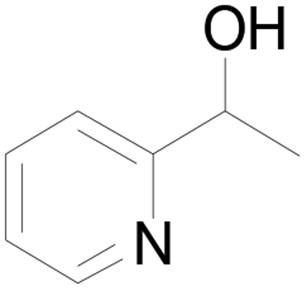 |
71 | 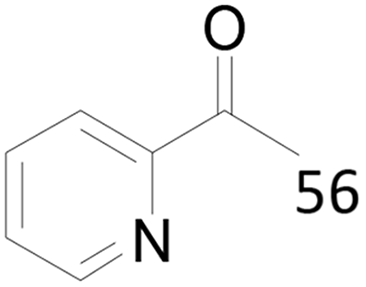 |
| 2 | 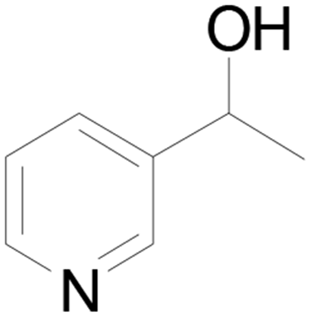 |
0 | 0 |
| 3 | 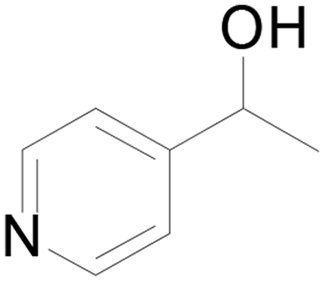 |
0 | 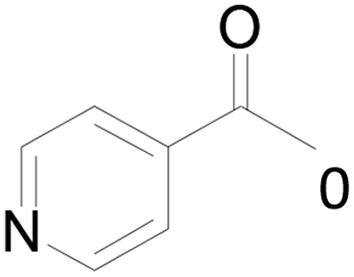 |
| 4 | 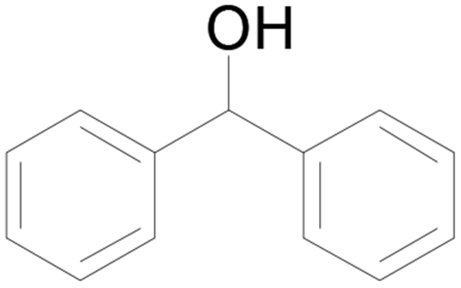 |
100 | 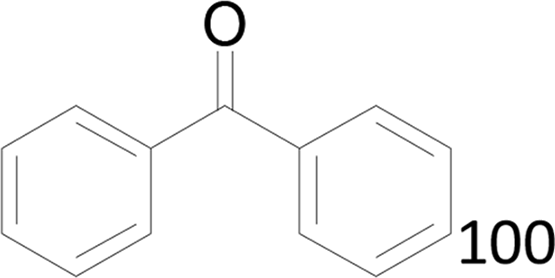 |
| 5 | 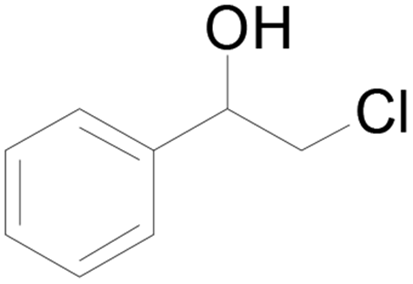 |
44 | 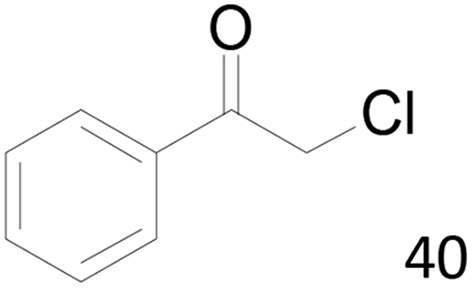 |
| 6 | 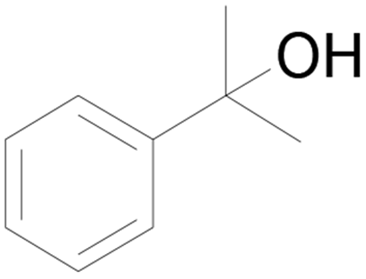 |
6 | 0 |
| 7 | 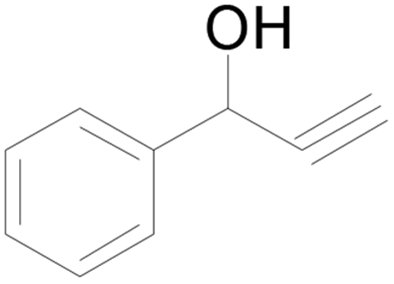 |
48 | 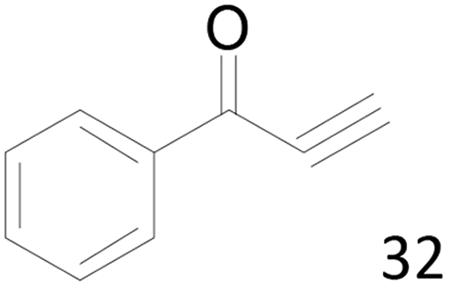 |
| 8 | 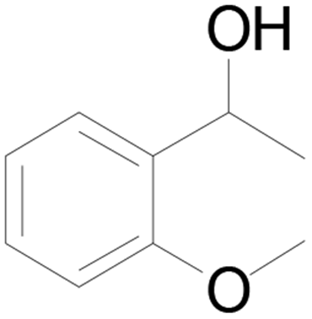 |
86 | 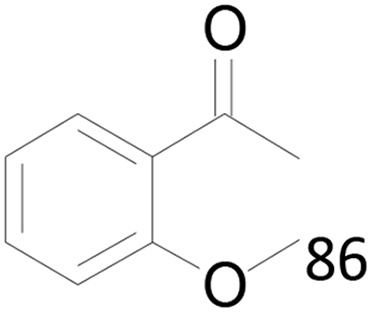 |
| 9 | 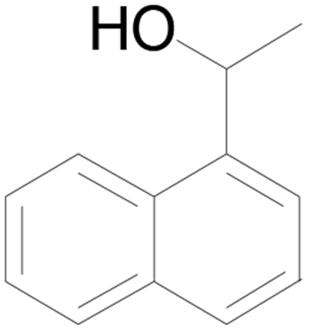 |
65 | 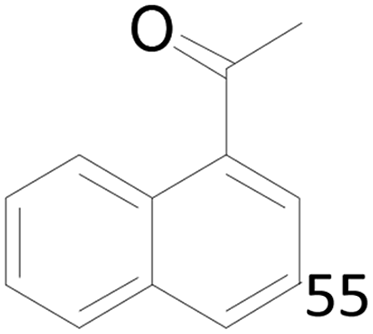 |
| 10 | 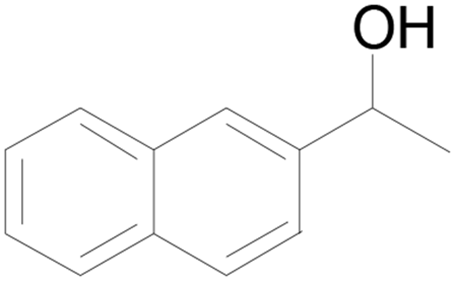 |
92 | 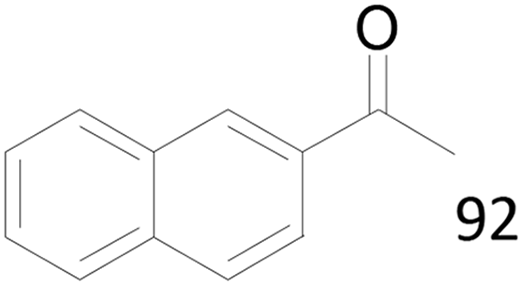 |
| 11 | 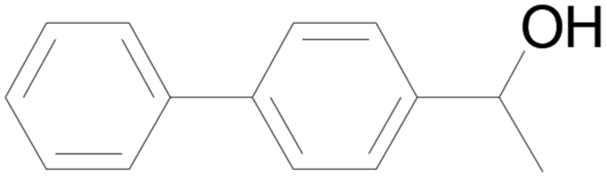 |
100 | 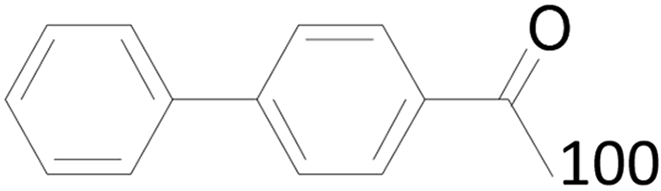 |
| 12 | 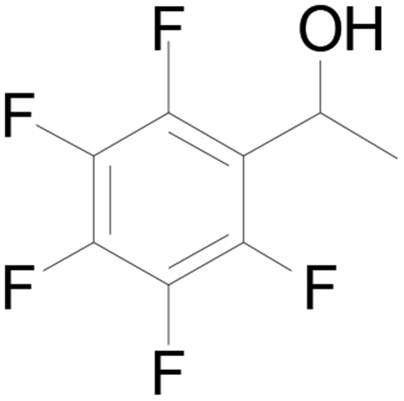 |
12 | 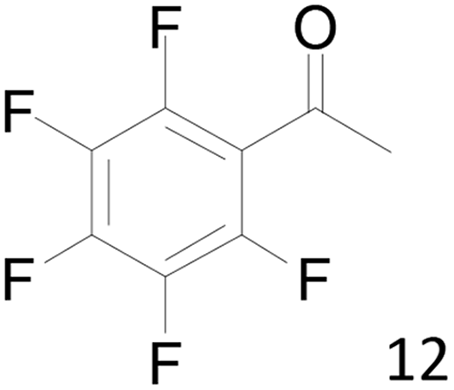 |
| 13 | 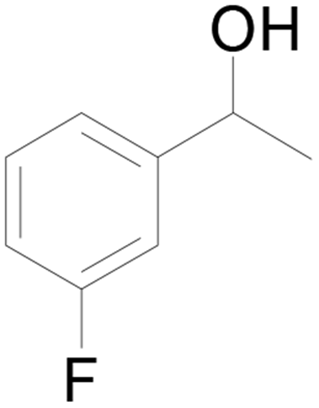 |
71 | 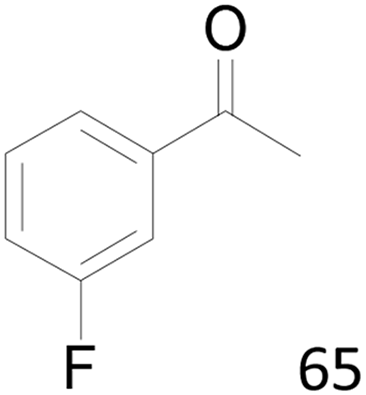 |
| 14 | 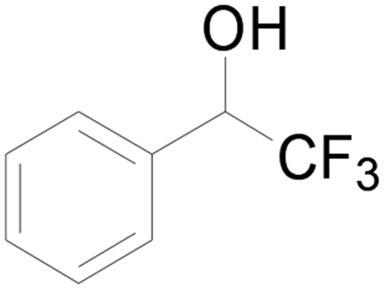 |
0 | 0 |
| 15 | 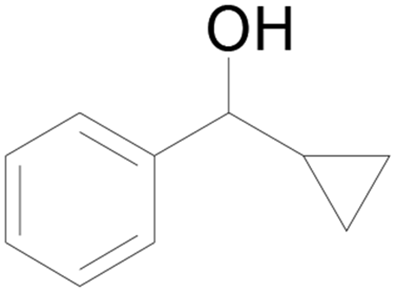 |
96 | 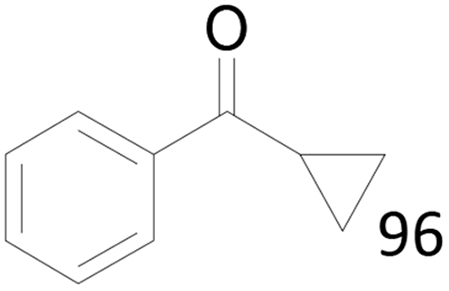 |
| 16 | 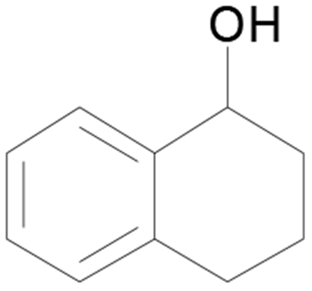 |
100 | 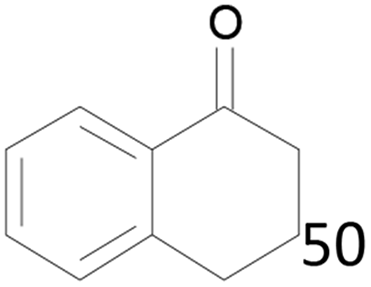 |
| 17c | 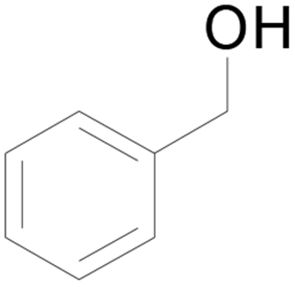 |
99 | 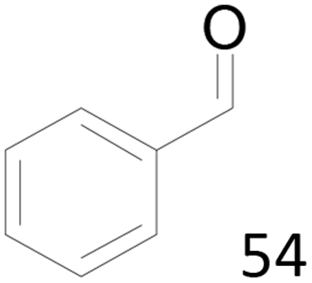 |
| 18c | 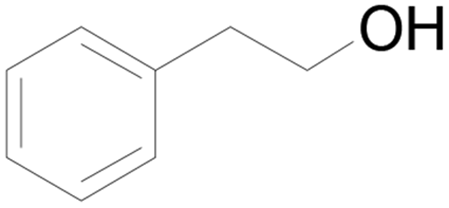 |
66 | 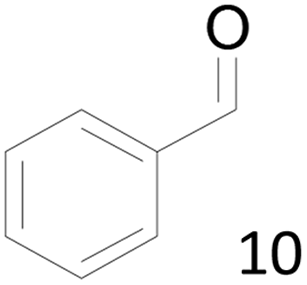 |
| 19c | 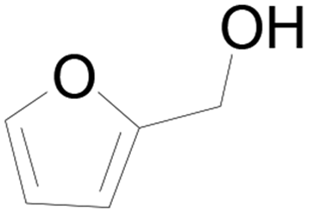 |
96 | 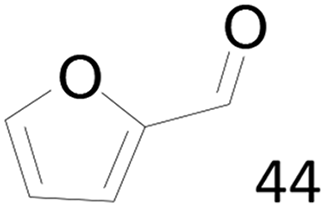 |
| 20c | 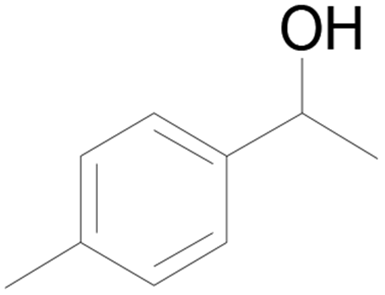 |
100 | 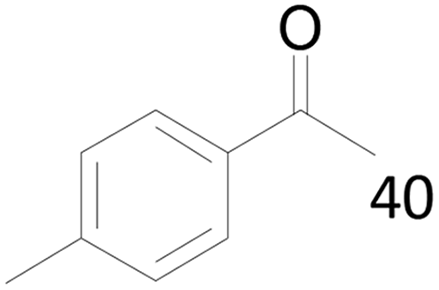 |
| 21c | 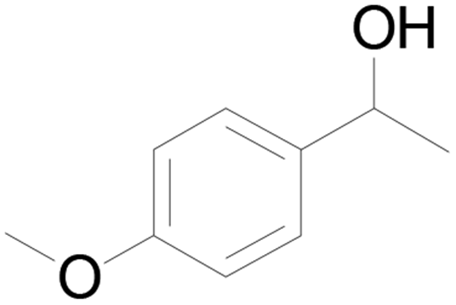 |
100 | 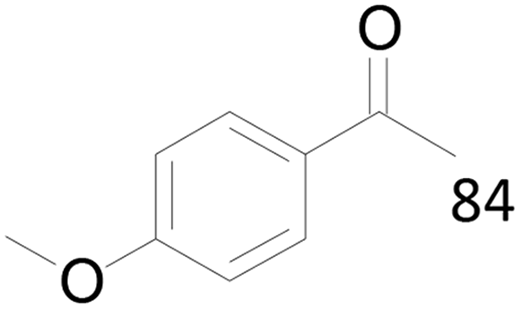 |
| 22c | 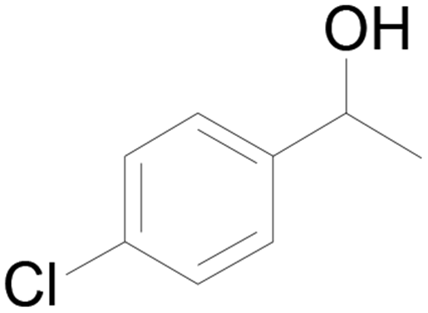 |
87 | 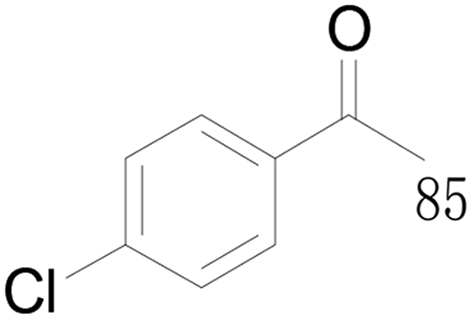 |
| 23c | 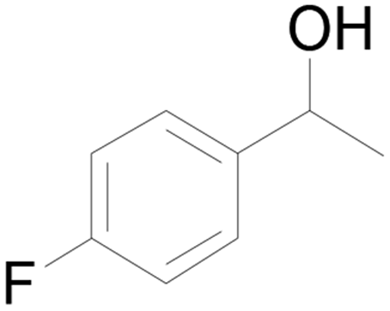 |
90 | 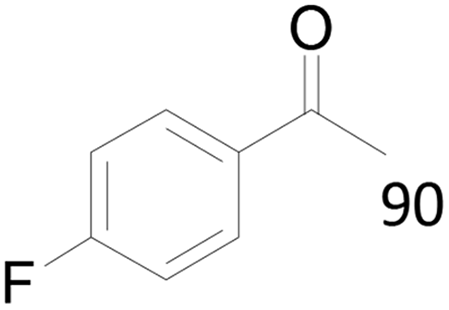 |
| 24c | 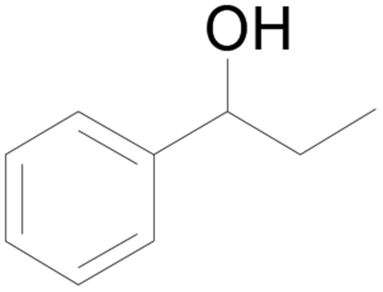 |
100 | 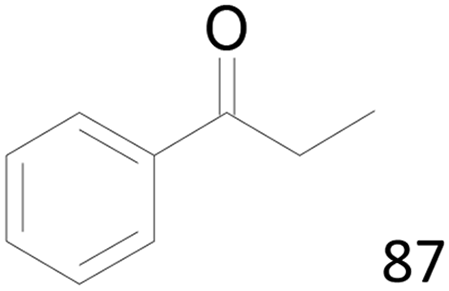 |
| 25c |  |
67 |  |
 |
|||
3.3 Possible reaction mechanism of 1-phenylethanol photocatalytic oxidation on 2-BrAQ
In order to understand the reaction mechanism in depth, we first calculated the surface charges of the catalyst and the 1-phenylethanol molecule using Gaussian 09W. The results are shown in Fig. 3. As can be seen from Fig. 3, the negative charge of the anthraquinone molecule is concentrated at two oxygen atoms and the negative charge of the upper oxygen atom is slightly higher than the other oxygen atom. The electron density of the oxygen atom on the excited 2-BrAQ molecule is slightly lower than that on the ground state molecule. This may be due to the relative dispersion of the charge caused by the electron transition from the HOMO energy level to the LUMO energy level. The positive charge on the 1-phenylethanol molecule is concentrated on the hydrogen atom on the hydroxyl group. This shows that the first step of the reaction is likely to be the transfer of a hydrogen atom from the 1-phenylethanol hydroxyl group to an oxygen atom from 2-BrAQ. As shown in Scheme 1, first, the 2-BrAQ molecule is excited by light to form an excited singlet state. The excited singlet state is unstable and has a low lifetime. It quickly becomes ground or triplet molecules through internal conversion, fluorescence emission, and intersystem crossing. The triplet molecule is then excited to collide with the 1-phenylethanol molecule to generate a 2-BrAQH radical and a 1-phenylethanol radical. 1-Phenylethanol radicals continue to react with the 2-BrAQ molecule, generating 2-BrAQH molecules and acetophenone. Finally, the 2-BrAQH molecule is oxidized by the nitrobenzene to 2-BrAQ and the nitrobenzene is simultaneously reduced to aniline.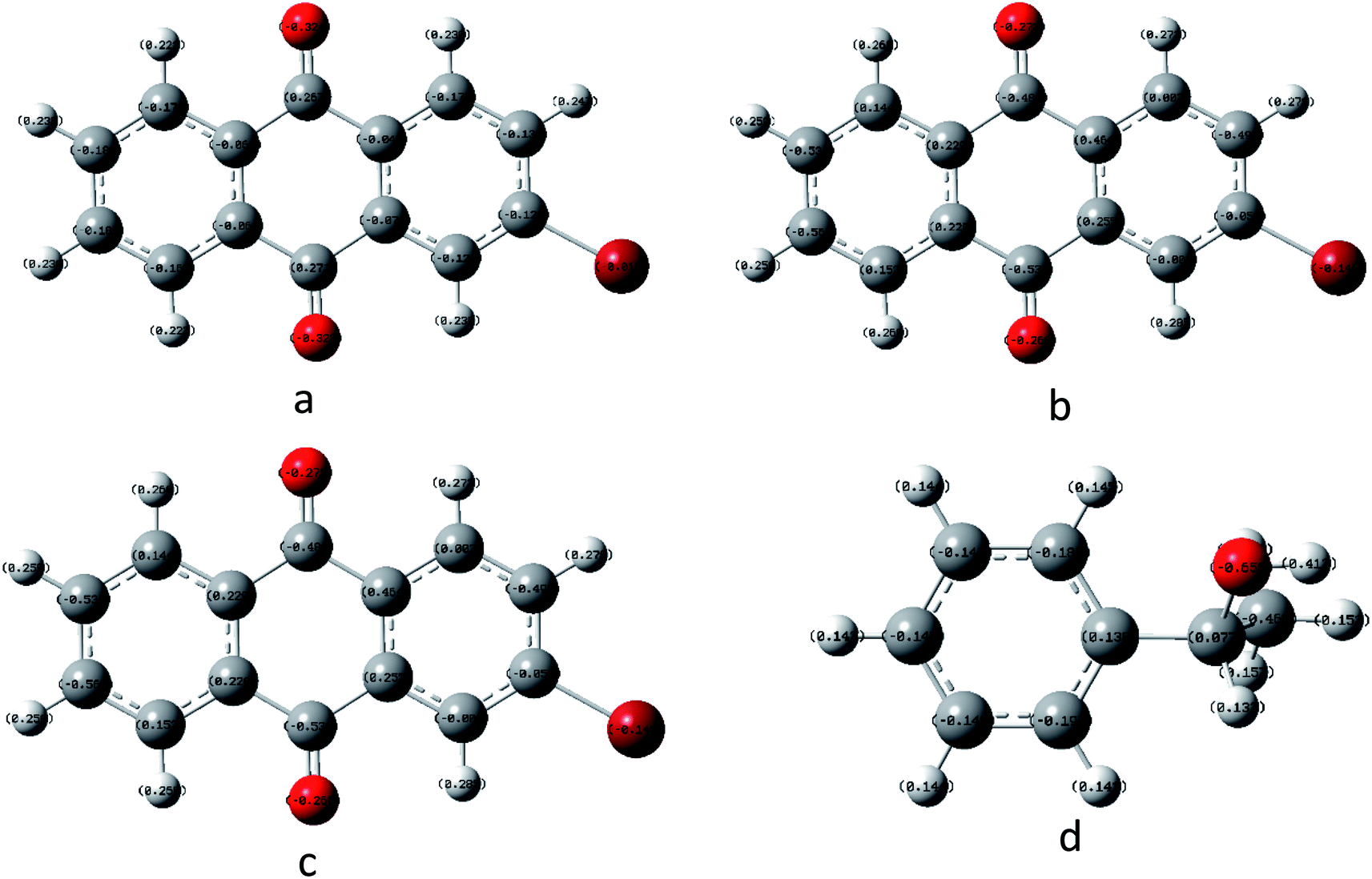 | ||
| Fig. 3 Catalyst and reactant molecular charge distributions using B3LYP/6-311+G*. (a) 2-BrAQ ground state, (b) 2-BrAQ excited singlet state, (c) 2-BrAQ excited triplet state and (d) 1-phenylethanol. | ||
 | ||
| Scheme 1 Possible reaction mechanism of 1-phenylethanol photocatalytic oxidation. | ||
In order to experimentally verify the radical process of the reaction, a series of radical capture experiments was carried out. We used butylated hydroxytoluene (BHT) as a free radical scavenger to scavenge free radicals generated during the reaction. The results are shown in Fig. 4. When 1 equivalent of BHT was used, the reaction conversion and yield were reduced to 12%. This indicates that the use of BHT inhibits the progress of the reaction by scavenging free radicals in the reaction. When the amount of BHT was increased to 5 equivalents, the 1-phenylethanol in this system did not undergo any oxidation. In addition, experiments using N-tert-butyl-α-phenylnitrone (PBN) as a free radical scavenger (Fig. S1†) show that when free radicals in the reaction are trapped, 1-phenylethanol oxidation proceeds with difficulty. Through these experimental results, the free radical path of the reaction was verified.
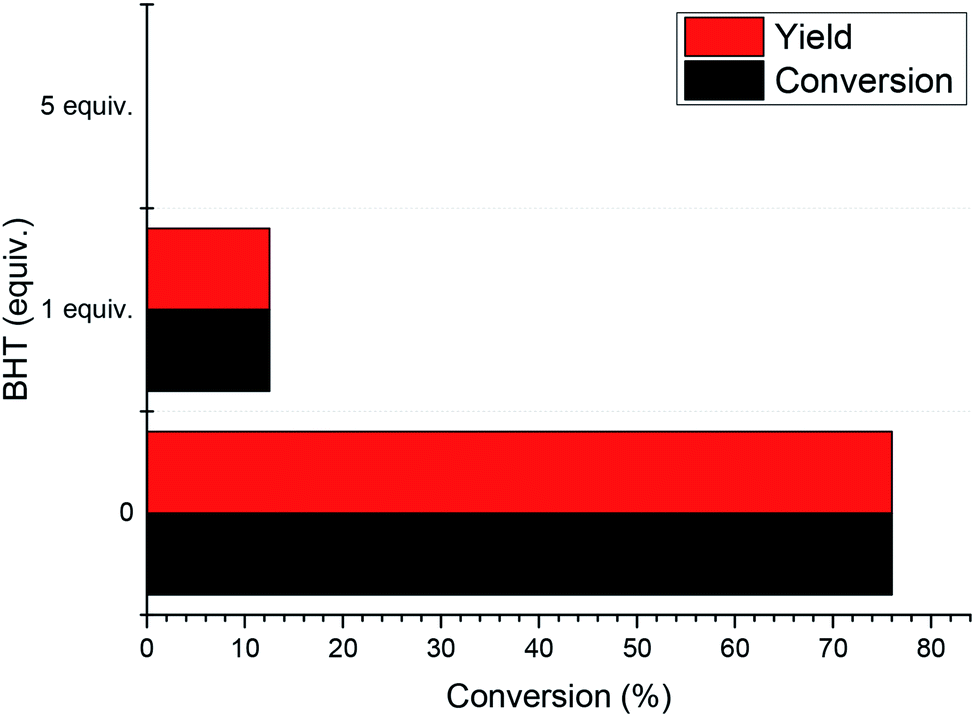 | ||
| Fig. 4 Radical capture experiments of 2-BrAQ photocatalytic oxidation. General conditions: 0.1 mmol 1-phenylethanol, 0.1 mmol nitrobenzene, 0.01 mmol 2-Br-AQ, butylated hydroxytoluene (BHT), 3 mL acetonitrile, 30 W LED illuminated for specified time, 30 °C water bath. | ||
4 DFT and TD-DFT calculation
4.1 Calculation of 2-BrAQ excited state
In order to explore the spectral characteristics and energy data of 2-BrAQ molecules from a theoretical perspective, this study uses TD-DFT calculations to calculate the excited state of 2-BrAQ molecules. Based on the calculation results, we have drawn a graph of the energy of 2-BrAQ in the reaction; see Scheme 2 for details. The 2-BrAQ molecule needs 3.026 eV to transition from the ground state (S0) to the first excited singlet state (S1), corresponding to an excitation wavelength of 410 nm. This transition has a 96.3% chance of the electron moving from the electron-occupied orbit 70 to LUMO orbit 72. The corresponding electron transition energies of S2, S3, S4 and S5 are 3.309, 3.318, 3.672 and 3.750 eV, respectively. The energy corresponding to the T1 state is 2.620 eV. More detailed calculation data are listed in the ESI (Table S4†).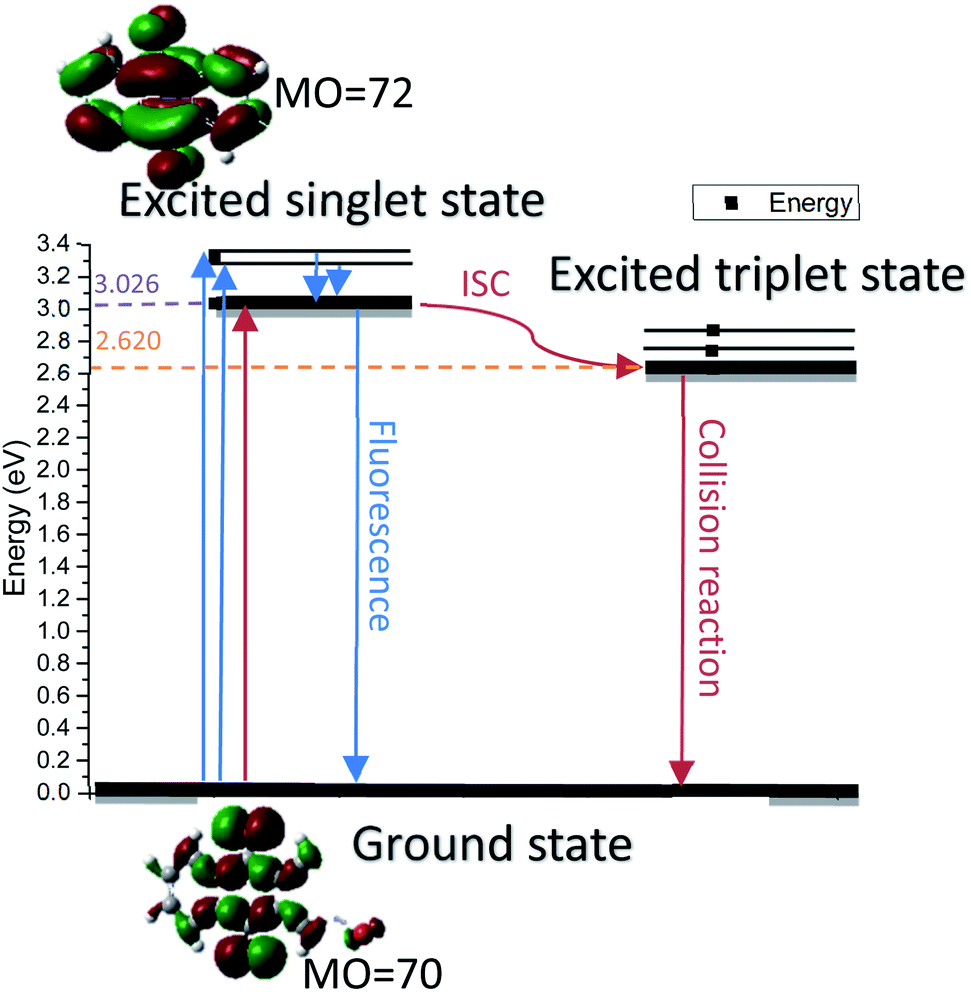 | ||
| Scheme 2 Diagram of 2-BrAQ orbital transition. Calculated by TD-B3LYP/6-311G*-D3(BJ). | ||
4.2 DFT calculation for reaction process analysis
In order to better understand the photo-redox reaction of 2-BrAQ in acetonitrile solution, DFT calculations were used to check the intermediates and activation energy that the reaction may produce. The reaction history of anthraquinone photocatalyst in acid aqueous solution is well understood.27 As can be seen in Scheme 3, first, 2-HEAQ molecules become excited triplet molecules through photoexcitation and intersystem migration. Then, the triplet 2-HEAQ molecules undergo protonation and isomerization in an acidic aqueous solution and become hydrogenation intermediates. Finally, the hydrogenation intermediate is oxidized to form the original 2-HEAQ molecule, completing a redox cycle. The anthraquinone molecules follow the ionic reaction mechanism in acidic aqueous solution. The high hydrogen ion concentration in acidic aqueous solution has a quenching effect on the triplet state.36 However, in neutral acetonitrile, the levels of various ions are very low, so anthraquinone molecules should follow the mechanism of the free radical reaction.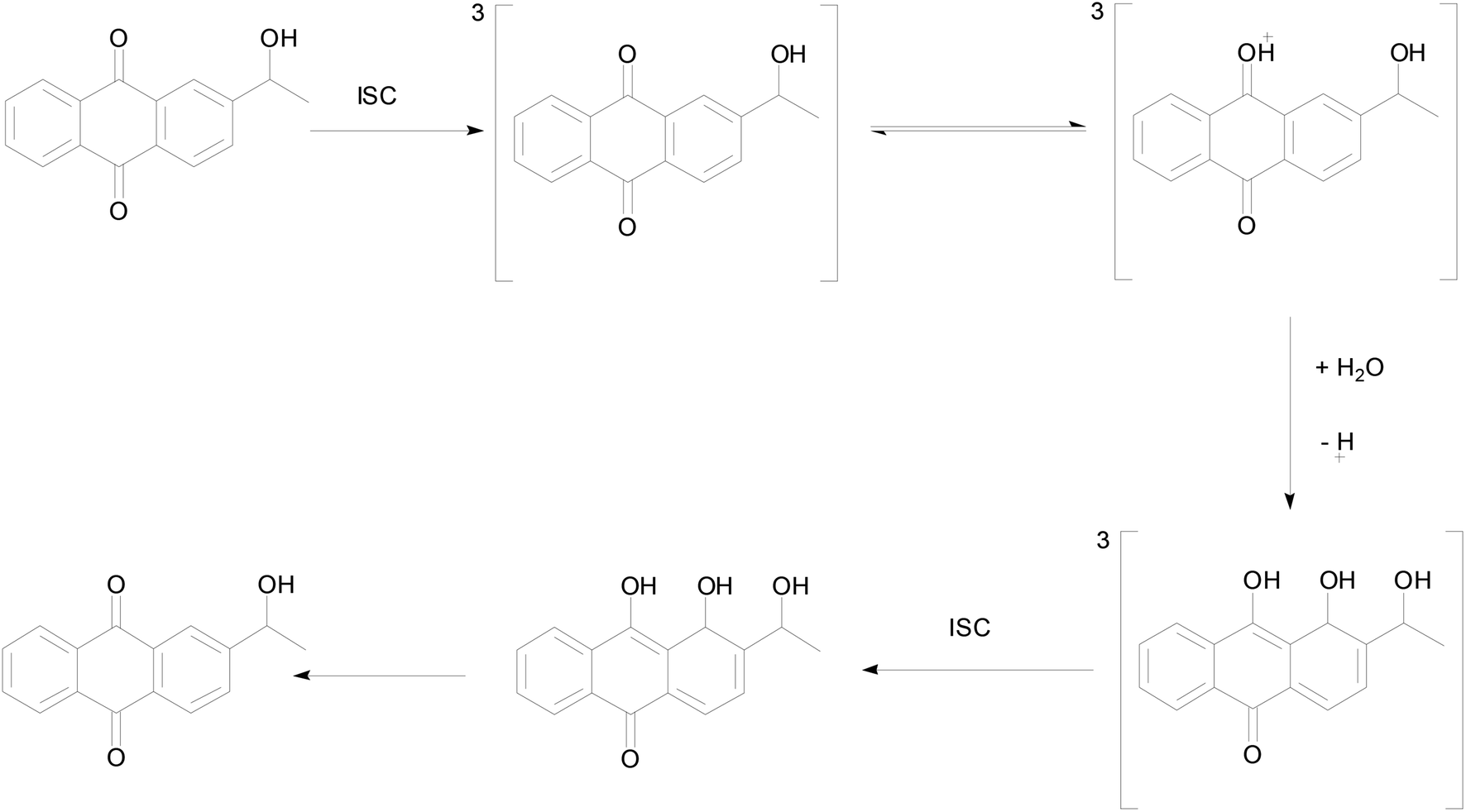 | ||
| Scheme 3 Proposed photohydration reaction mechanism for 2-HEAQ in acidic aqueous solution, from David Lee Phillips and co-workers.27 | ||
After undergoing processes such as photoexcitation and intersystem crossing, the triplet 2-BrAQ molecule is a good hydrogen atom acceptor. Note that photohydration takes place at both the ortho and meta positions.27 According to Gaussian 09W structure optimization and single-point energy calculations, the ortho-hydrogenated 2-BrAQ molecule (generate HT1) showed an energy about 1.4 kcal mol−1 lower than that of meta-hydrogenated 2-BrAQ molecule (generate HT2). This would yield a transient with a longer lifetime.36,37 Therefore, ortho-hydrogenated 2-BrAQ (HT1) was the most likely hydrogenation intermediate in this reaction. Based on this, we optimized the most likely structure of 2-BrAQ in the reaction. After the 2-BrAQ molecule was excited by light, as shown in Scheme 4, the molecular unsaturation and structure were changed due to the electrons in the occupied molecular orbital transitioning to the LUMO orbital. During this process, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length and molecular dipole moment were increased. In addition, the triplet T1 molecule has a relatively long life, which allows it to easily undergo a hydrogen transfer reaction with 1-phenylethanol molecules to generate hydro anthraquinone transition state (HT) molecules. The electrons in the HT molecules were rearranged, resulting in lower energy hydro anthraquinone ground state (HS) molecules. Finally, the HS molecule was oxidized by the oxidant, lost the hydrogen atom and became the ground state (S0) molecule, ending the redox cycle.
O bond length and molecular dipole moment were increased. In addition, the triplet T1 molecule has a relatively long life, which allows it to easily undergo a hydrogen transfer reaction with 1-phenylethanol molecules to generate hydro anthraquinone transition state (HT) molecules. The electrons in the HT molecules were rearranged, resulting in lower energy hydro anthraquinone ground state (HS) molecules. Finally, the HS molecule was oxidized by the oxidant, lost the hydrogen atom and became the ground state (S0) molecule, ending the redox cycle.
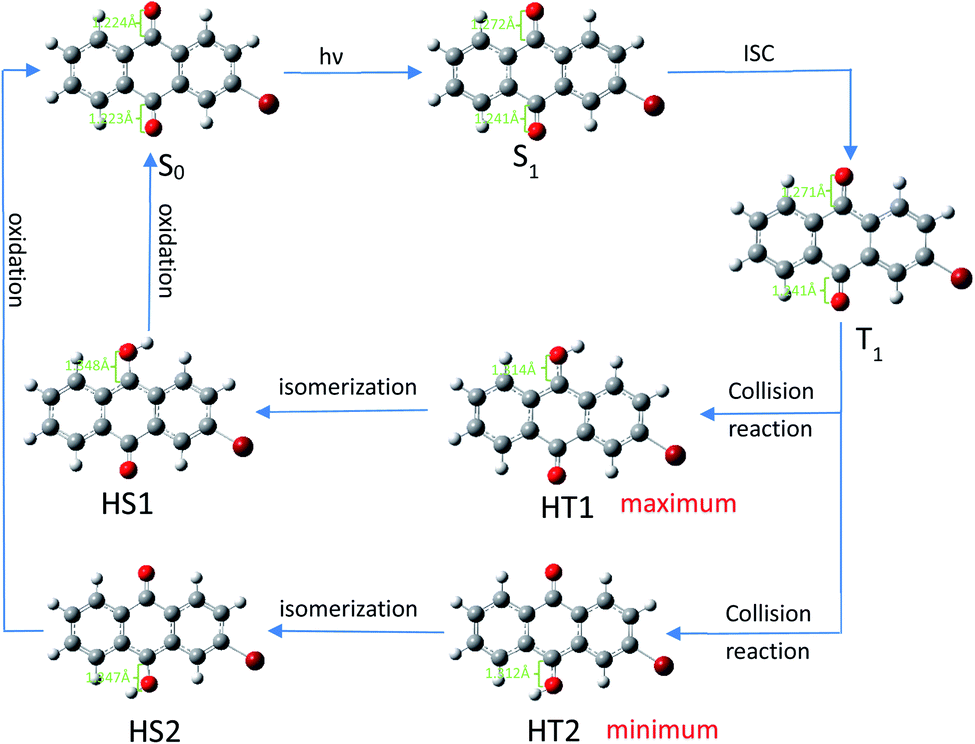 | ||
| Scheme 4 Redox cycle of 2-BrAQ molecule in acetonitrile solution. Optimization calculated by (TD)B3LYP/6-311G*-D3(BJ). | ||
Fig. 5 shows the relative energy curve of the 2-bromoanthraquinone redox cycle. Inspection of Fig. 5 indicates that the initial excitation of 2-bromoanthraquinone requires 63.6 kcal mol−1 of energy. When the triplet state T1 is reached, the relative energy is reduced by about 9 kcal mol−1. When the ortho-hydrogenated 2-BrAQ molecule transition state (HT1) is formed through the hydrogen transfer process, the relative energy is reduced to −317.3 kcal mol−1, which indicates that 2-bromoanthraquinone transfers up to 371.6 kcal mol−1 of energy through the collision reaction with 1-phenylethanol to activate 1-phenylethanol. About 48.7 kcal mol−1 of energy is dissipated in the isomerization of hydro anthraquinone molecules.
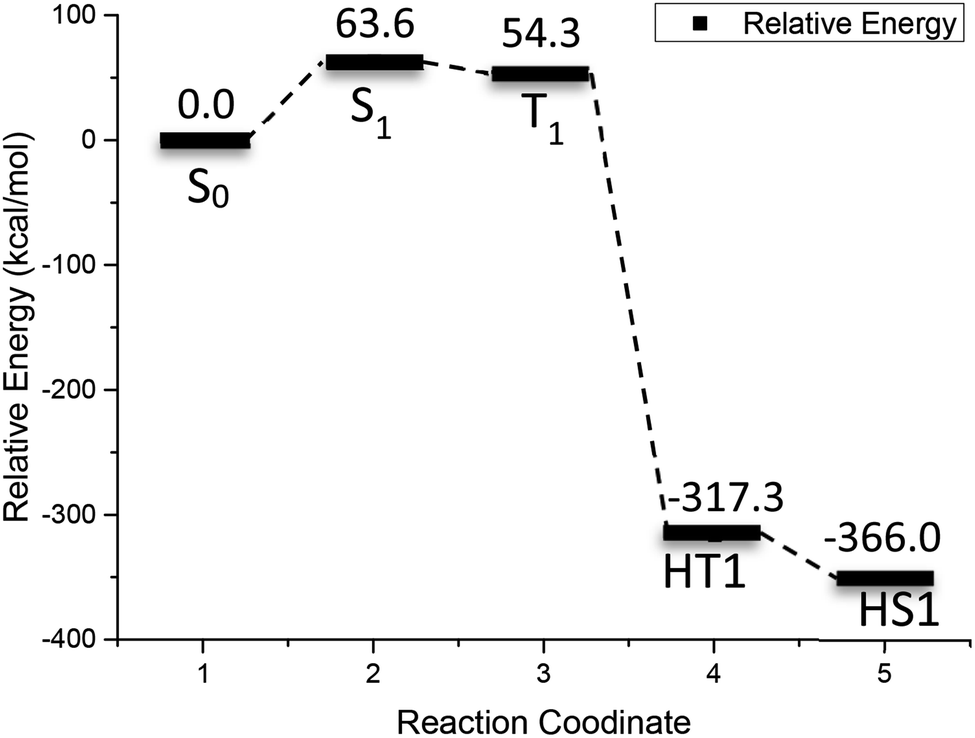 | ||
| Fig. 5 Reactive energy profile obtained from (U)M06-2X/def2-TZVP calculations after optimization for the photo redox reaction of 2-BrAQ. | ||
5 Conclusion
In summary, we have found a new strategy to efficiently oxidize α-position aromatic alcohols. In the presence of nitrobenzene, under specific light conditions, the use of hydrogen transfer between 2-bromoanthraquinone and an α-position aromatic alcohol can oxidize the α-position aromatic alcohol to the corresponding ketone with high selectivity. By applying the organic small molecule dye anthraquinone to the photocatalytic oxidation of secondary aromatic alcohols, we have discovered a new green catalytic method that makes selective alcohol oxidation conditions less demanding. In addition, it may be possible to apply this selective oxidation to lignin pretreatment and depolymerization due to it being rich in Cα–OH.In order to fill the theoretical photo redox gap of anthraquinone molecules in organic solvents, we used density functional theory to describe the anthraquinone molecules in this reaction and proposed the free radical theory of anthraquinone photocatalysis. This brings us to a further understanding of the catalytic mechanism of organic photocatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Key R&D Program of China (2018YFB1501402), the Natural Science Foundation of Guangdong Province (2017A030308010), the National Natural Science Foundation of China (51576199, 51976225 and 51536009), the DNL Cooperation Fund, CAS (DNL180302 and DNL201916), the “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA21060102), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01N092). In addition, special thanks to Department of Chemistry of Shantou University for providing calculation support.References
- W. Zhang, M. Liu, H. Wu, J. Ding and J. Cheng, Tetrahedron Lett., 2008, 49, 5336–5338 CrossRef CAS.
- A. Sedrpoushan, M. Heidari and O. Akhavan, Chin. J. Catal., 2017, 38, 745–757 CrossRef CAS.
- L. Wang, X. Zhang, L. Yang, C. Wang and H. Wang, Catal. Sci. Technol., 2015, 5, 4800–4805 RSC.
- J. Luo and J. Zhang, J. Org. Chem., 2016, 81, 9131–9137 CrossRef CAS.
- D. O. Mountfort, D. White and R. A. Asher, Appl. Environ. Microbiol., 1990, 56, 245–249 CrossRef CAS.
- M. Fujihira, Y. Satoh and T. Osa, Nature, 1981, 293, 206–208 CrossRef CAS.
- F. Nastasi, A. Santoro, S. Serroni, S. Campagna, N. Kaveevivitchai and R. P. Thummel, Photochem. Photobiol. Sci., 2019, 18, 2164–2173 RSC.
- H. Bai, X. Fang, C. Peng, Q. Liu, W. Xie, L. Jia and Z. Song, J. Mol. Liq., 2019, 289, 111122 CrossRef CAS.
- F. Gao, X. Li, Y. Zhang, C. Huang and W. Zhang, ACS Omega, 2019, 4, 13721–13732 CrossRef CAS.
- Y. Li and J.-G. Jiang, Food Funct., 2018, 9, 6063–6080 RSC.
- K. Panthong, S. Hongthong, C. Kuhakarn, P. Piyachaturawat, K. Suksen, A. Panthong, N. Chiranthanut, P. Kongsaeree, S. Prabpai, N. Nuntasaen and V. Reutrakul, Phytochemistry, 2020, 169, 112182 CrossRef CAS.
- J. G. Park, S. C. Kim, Y. H. Kim, W. S. Yang, Y. Kim, S. Hong, K. H. Kim, B. C. Yoo, S. H. Kim, J. H. Kim and J. Y. Cho, Mediators Inflammation, 2016, 2016, 1903849 Search PubMed.
- J. Garcia-Serna, T. Moreno, P. Biasi, M. J. Cocero, J. P. Mikkola and T. O. Salmi, Green Chem., 2014, 16, 2320–2343 RSC.
- A. U. Chaudhari, D. Paul, D. Dhotre and K. M. Kodam, Water Res., 2017, 122, 603–613 CrossRef CAS.
- I. V. Kolesnichenko, P. R. Escamilla, J. A. Michael, V. M. Lynch, D. A. Vanden Bout and E. V. Anslyn, Chem. Commun., 2018, 54, 11204–11207 RSC.
- R. Wightman, J. Cockrell, R. W. Murray, J. Burnett and S. B. Jones, J. Am. Chem. Soc., 1976, 98, 2562–2570 CrossRef CAS.
- X. Lu, M. Zhou, Y. Li, P. Su, J. Cai and Y. Pan, Electrochim. Acta, 2019, 320, 134552 CrossRef CAS.
- Y. Cheng, L. Wang, S. Lü, Y. Wang and Z. Mi, Ind. Eng. Chem. Res., 2008, 47, 7414–7418 CrossRef CAS.
- E. Santacesaria, M. Di Serio, A. Russo, U. Leone and R. Velotti, Chem. Eng. Sci., 1999, 54, 2799–2806 CrossRef CAS.
- J. Theurich, D. W. Bahnemann, R. Vogel, F. E. Ehamed, G. Alhakimi and I. Rajab, Res. Chem. Intermed., 1997, 23, 247–274 CrossRef CAS.
- S. U. Ying-ying, Y. U. Yan-qing, Y. Pei-shan, W. Xin-ting and Z. H. U. Xiao-bin, China Environ. Sci., 2009, 29, 1171–1176 Search PubMed.
- D. Petzold and B. Konig, Adv. Synth. Catal., 2018, 360, 626–630 CrossRef CAS.
- H.-i. Kim, Y. Choi, S. Hu, W. Choi and J.-H. Kim, Appl. Catal., B, 2018, 229, 121–129 CrossRef CAS.
- R. Narobe, S. J. S. Düsel, J. Iskra and B. König, Adv. Synth. Catal., 2019, 361, 3998–4004 CrossRef CAS.
- I. M. Denekamp, M. Antens, T. K. Slot and G. Rothenberg, ChemCatChem, 2018, 10, 1035–1041 CrossRef CAS.
- J. Zhao, D. Wu, W. Y. Hernández, W.-J. Zhou, M. Capron and V. V. Ordomsky, Appl. Catal., A, 2020, 590, 117277 CrossRef.
- J. Ma, T. Su, M.-D. Li, W. Du, J. Huang, X. Guan and D. L. Phillips, J. Am. Chem. Soc., 2012, 134, 14858–14868 CrossRef CAS.
- Y. Guo, Q. Song, J. Wang, J. Ma, X. Zhang and D. L. Phillips, J. Org. Chem., 2018, 83, 13454–13462 CrossRef CAS.
- K. Tickle and F. Wilkinson, Trans. Faraday Soc., 1965, 61, 1981–1990 RSC.
- H. Inoue, M. Hida, N. Nakashima and K. Yoshihara, J. Phys. Chem., 1982, 86, 3184–3188 CrossRef CAS.
- M. Gronkiewicz, B. Kozankiewicz and J. Prochorow, Chem. Phys. Lett., 1976, 38, 325–328 CrossRef CAS.
- B. Valeur, in Molecular Fluorescence: Principles and Applications, John Wiley & Sons, Inc., 2001, ch. 3, pp. 34–71 Search PubMed.
- Y. F. Ji, T. Fan and Y. Luo, Phys. Chem. Chem. Phys., 2020, 22, 1187–1193 RSC.
- Y. Q. Huang and J. Lessard, Electroanalysis, 2016, 28, 2716–2727 CrossRef CAS.
- H. Inagawa, S. Uchida, E. Yamaguchi and A. Itoh, Asian J. Org. Chem., 2019, 8, 1411–1414 CrossRef CAS.
- M. Ramseier, P. Senn and J. Wirz, J. Phys. Chem. A, 2003, 107, 3305–3315 CrossRef CAS.
- Y. Du, J. Xue, M. Li and D. L. Phillips, J. Phys. Chem. A, 2009, 113, 3344–3352 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06414a |
| This journal is © The Royal Society of Chemistry 2020 |