
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A biradical oxo-molybdenum complex containing semiquinone and o-aminophenol benzoxazole-based ligands†
Mina Nasibipoura,
Elham Safaei *a,
Andrzej Wojtczakb,
Zvonko Jagličićc,
Agustín Galindod and
Marzieh Sadat Masoumpoure
*a,
Andrzej Wojtczakb,
Zvonko Jagličićc,
Agustín Galindod and
Marzieh Sadat Masoumpoure
aDepartment of Chemistry, College of Sciences, Shiraz University, 71454, Shiraz, Iran. E-mail: e.safaei@shirazu.ac.ir
bNicolaus Copernicus University, Faculty of Chemistry, 87-100 Torun, Poland
cInstitute of Mathematics, Physics and Mechanics & Faculty of Civil and Geodetic Engineering, University of Ljubljana, Jadranska 19, Ljubljana, Slovenia
dDepartamento de Química Inorgánica, Facultad de Química, Universidad de Sevilla, Aptdo. 1203, 41071 Sevilla, Spain
eDepartment of Chemistry, Estahban Higher Education Center, Estahban 74519-44655, Iran
First published on 9th November 2020
Abstract
We report a new mononuclear molybdenum(IV) complex, MoOLBISLSQ, in which LSQ (2,4-di-tert-butyl o-semibenzoquinone ligand) has been prepared from the reaction of the o-iminosemibenzoquinone form of a tridentate non-innocent benzoxazole ligand, LBIS, and MoO2(acac)2. The complex was characterized by X-ray crystallography, elemental analysis, IR and UV-vis spectroscopy and magnetic susceptibility measurements. The crystal structure of MoOLBISLSQ revealed a distorted octahedral geometry around the metal centre, surrounded by one O and two N atoms of LBIS and two O atoms of LSQ. The effective magnetic moment (μeff) of MoOLBISLSQ decreased from 2.36 to 0.2 μB in the temperature range of 290 to 2 K, indicating a singlet ground state caused by antiferromagnetic coupling between the metal and ligand centred unpaired electrons. Also, the latter led to the EPR silence of the complex. Cyclic voltammetry (CV) studies indicate both ligand and metal-centered redox processes. MoOLBISLSQ was applied as a catalyst for the oxidative cleavage of cyclohexene to adipic acid and selective oxidation of sulfides to sulfones with aqueous hydrogen peroxide.
Introduction
Scientists have discovered in recent decades that some enzymes with special ligands are able to perform redox processes at both their metal centers and their coordinated ligands. The variation of oxidation states of such ligands is a special and important feature that causes their bound metals to change their oxidation states. These are known as non-innocent ligands. The term (redox) “non-innocent ligand” is used in the scientific literature to reflect the unclear oxidation state of a ligand in a metal complex. This characteristic leads to obscurity in the exact defined oxidation state of the bound metal center. Hence complexes containing non-innocent ligands are known as redox active metal complexes. This feature of the ligands is important for the catalytic activity of the broadly investigated complexes in which redox reactions could be ligand or metal localized. In this way, metal complexes of non-innocent ligands are now undergoing a renaissance in synthetic chemistry because they are good structural and functional models for the mentioned enzymes.1Molybdenum is used in some groups of oxotransferases or hydrolases enzymes, in which, they catalyze important and vital reactions like oxygen atom transfer reactions (OAT) and water transfer reactions to substrates, respectively. These enzymes, based on their oxidized active center structure are divided into three different categories referred to sulfite and xanthine oxidase and DMSO reductase. All these enzymes contain a mononuclear molybdenum-oxo center.2
Due to the importance of these reactions the active sites of oxotransferases have been structurally mimicked by a large number of molybdenum complexes.3 They catalyze a variety of reactions, especially oxidation of several organic substrates with various oxidants, such as tert-butyl hydroperoxide (TBHP) and H2O2.4 It is worth mentioning that during the last decades, scientists paid attention to H2O2 as a clean oxidant and appropriate alternative to the environmentally hazardous ones for metal-catalyzed oxidation of the organic substrate.5 However, use these complexes is sometimes affected by problems like a small rate of active oxygen preparation and easy decomposition. Therefore, scientists have focused on effective catalysts that can activate hydrogen peroxide without any decomposition. Complexes with V, VI, and VII group transition metals, especially molybdenum, are one of the best candidates to prevail in these limitations.
Adipic acid (AA) or 1,6-hexanedioic acid, is an essential raw compound in chemical industries, such as nylon 6 production. Oxidation of KA (ketone–alcohol) oil (cyclohexanone–cyclohexanol) mixture by nitric acid is the current industrial way to produce adipic acid.6 Generating of a large amount of nitrous oxide (N2O), i.e. environmentally harmful gas, is the major defect of this process.7 One-step production of adipic acid via oxidative cleavage of cyclohexene using H2O2 has been presented as a green alternative method (Scheme 1).8
 | ||
| Scheme 1 Proposed routes for the synthesis of adipic acid (AA).9 | ||
It is worth saying that in recent years a rapid AA production via high temperatures and pressure,10 via membrane reactors,11 and microwave settings12 has been described.
On the other hand, there is air pollution and acid rain because of the sulfur pollutants in fuels as an anthropogenic source of atmospheric SOx. Therefore, to decrease the sulfur oxide emissions, the hydrodesulfurization of petroleum on molybdenum-containing catalysts in high temperature and high H2 pressure conditions has long been one of the major catalytic methods used in the industry.13
Only a few sulfur compounds can be unchanged in this process and have slow desulfurization with excessive conditions what makes the process expensive. Especially, some fused-ring thiophenes, e.g. dibenzothiophene (DBT) are challenging compounds.
Following the idea that the redox transformations could be modulated by the o-aminophenol as an “electron reservoir” ligand,1c,d our present research is committed to the development of the Mo complex containing redox-active ligand.
In the paper, we describe the preparation and characterization of oxo molybdenum complex coordinated by the redox-active o-amino phenol ligand providing N,O additional donors capable of two and tridentate coordination and apply this complex for AA production under mild and safe conditions (low temperature, eluding the H2O2 decomposition, one-step reaction, cheap catalyst, and atmospheric pressure). Furthermore, this paper presents the oxidation of some sulfides including DBT under mild conditions.
The ligand HLBAP, its one electron oxidized radical form of LBIS and its semiquinone hydrolyzed form LSQ, 3,5-di-tert-butyl benzosemiquinone are given in Fig. 1.
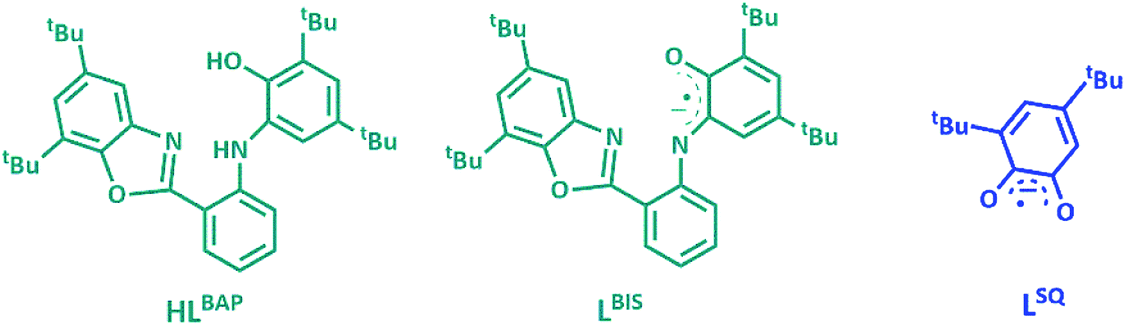 | ||
| Fig. 1 The ligand HLBAP, its one electron oxidized radical forms of LBIS and its semiquinone hydrolyzed form LSQ. | ||
Results and discussion
Synthesis of MoOLBISLSQ complex
The redox-active aminophenolate ligand H2LBAP was synthesized from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of 2-aminobenzyl amine and 3,5-DTBQ.14 The synthesis of MoOLBISLSQ complex was performed in 40% yield by refluxing CH2Cl2 solution of a ligand H2LBAP and Et3N with MoO2(acac)2 (Scheme 2). The complex was purified by crystallization from CH3OH/CH2Cl2 1:1 mixture and single crystals suitable for X-ray analysis were obtained after several recrystallizations.
:2 molar ratio of 2-aminobenzyl amine and 3,5-DTBQ.14 The synthesis of MoOLBISLSQ complex was performed in 40% yield by refluxing CH2Cl2 solution of a ligand H2LBAP and Et3N with MoO2(acac)2 (Scheme 2). The complex was purified by crystallization from CH3OH/CH2Cl2 1:1 mixture and single crystals suitable for X-ray analysis were obtained after several recrystallizations.
 | ||
| Scheme 2 Synthesis MoOLBISLSQ complex from HLBAP, Mo(O)2(acac)2 and Et3N, (the LSQ ligand in the resulted complex result from the hydrolysis of HLBAP). | ||
Elemental analysis data of the complex are consistent with theoretical ones and confirm the 1:1:1:1 molar ratio of Mo:O:LBIS:LSQ. The free ligand H2LBAP shows characteristic IR bands at 3415 (νO–H), 3258 (νN–H), 1047 (C–N stretching), 1595 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching), and 1542 cm−1 (CN stretching), while tert-butyl group bands appear at 2962 cm−1 (Fig. S1†). In the IR spectrum of the complex, a sharp band is observed at 1026 cm−1, which is characteristic of the νMoO stretch,15 and the sharp and strong νO–H and νN–H absorptions of the ligand disappear, which confirms their coordination to the Mo(IV) center. All the vibrations of the ligand observed in the IR spectrum of the complex confirm the presence of the ligand in the structure (Fig. S2†).
C stretching), and 1542 cm−1 (CN stretching), while tert-butyl group bands appear at 2962 cm−1 (Fig. S1†). In the IR spectrum of the complex, a sharp band is observed at 1026 cm−1, which is characteristic of the νMoO stretch,15 and the sharp and strong νO–H and νN–H absorptions of the ligand disappear, which confirms their coordination to the Mo(IV) center. All the vibrations of the ligand observed in the IR spectrum of the complex confirm the presence of the ligand in the structure (Fig. S2†).
X-ray analysis
Crystallographic data of the complex are shown in Table 1. The Selected bond distances and angles are given in Table 2 and complete bond distances and angles and torsion angles [°] are presented in ESI Tables S1 and S2,† respectively.| Empirical formula | C49H64MoN2O5 |
|---|---|
| Formula weight | 856.96 |
| Crystal system | Triclinic |
| Space group | P−1 |
| Unit cell dimensions | a = 10.6437(4), b = 14.3165(10), c = 16.2402(11) |
| α = 96.269(6), β = 104.683(5), γ = 96.637(5) | |
| Volume | 2353.0(3) Å3 |
| Z | 2 |
| Temperature | 293(2) K |
| Density (calculated) | 1.210 Mg m−3 |
| Crystal size | 0.531 × 0.385 × 0.167 mm3 |
| Absorption coefficient | 0.322 mm−1 |
| Reflections collected | 17020 |
| Independent reflections | 10489 [R(int) = 0.1080] |
| Goodness-of-fit on F2 | 1.039 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0887, wR2 = 0.2033 |
| R indices (all data) | R1 = 0.1396, wR2 = 0.2544 |
| Mo1–O4 | 1.993(4) | O5–Mo1–O3 | 159.75(15) |
| Mo1–O5 | 1.686(4) | N1–Mo1–N2 | 83.35(16) |
| Mo1–O1 | 1.955(4) | O3–Mo1–N2 | 79.43(16) |
| Mo1–N1 | 2.037(4) | O5–Mo1–O4 | 92.12(18) |
| Mo1–O3 | 2.091(4) | O4–Mo1–O1 | 86.52(15) |
| Mo1–N2 | 2.148(4) | O4–Mo1–N2 | 106.36(19) |
| C2–C3 | 1.383(9) | O1–Mo1–N2 | 158.35(16) |
| C4–C5 | 1.385(9) | N1–Mo1–O3 | 86.68(17) |
| O1–C1 | 1.340(7) | O1–Mo1–N1 | 78.86(16) |
| C6–N1 | 1.399(7) | O4–Mo1–O3 | 75.54(15) |
The asymmetric unit of the structure contains the MoOLBISLSQ molecule (Fig. 2). The Mo(IV) oxidation form is assigned based on the presence of O2− oxo, LBIS 1− and LSQ 1− (Fig. 1). The central Mo(IV) has a distorted octahedral coordination MoN2O4 sphere formed by tri-dentate LBIS ligand (the oxidized form of LAP), the bi-dentate LSQ and the oxo ligand. Similar coordination environment was also observed in complex VOLBIS(SQ).16
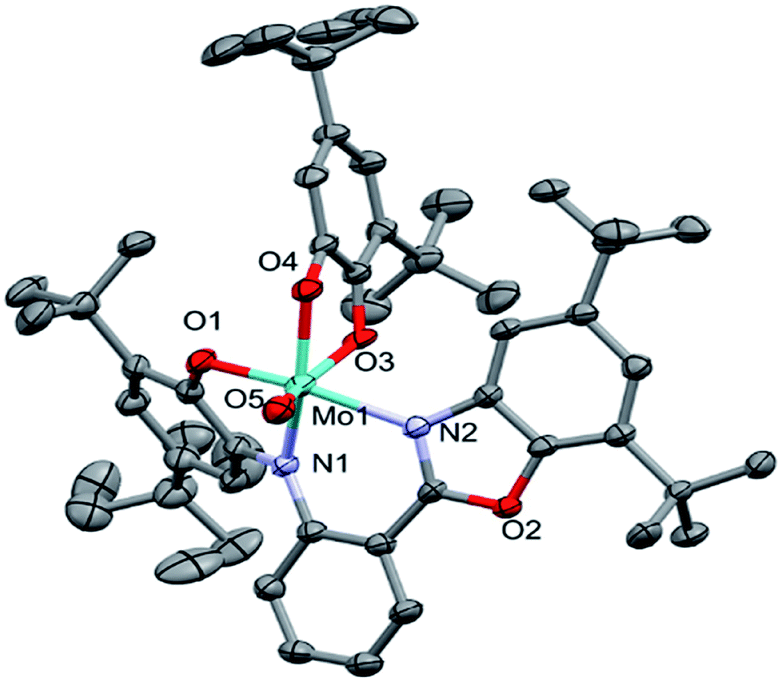 | ||
| Fig. 2 Molecular structure of MoOLBISLSQ, hydrogen atoms have been omitted for clarity. The ADPs are plotted at 30% probability level. | ||
In the Mo coordination sphere, the shortest bond is the Mo1–O5 distance of 1.686(4) Å for the oxo ligand, while those of Mo1–O1, Mo1–O4 and Mo1–O3 are significantly longer, with the respective distances of 1.955(4), 1.993(4) and 2.091(4) Å. The Mo–N bonds formed by bridging N1 and the benzoxazole N2 are 2.037(4) and 2.148(4) Å, respectively. The LSQ is coordinated to Mo in the hydrophobic cleft defined by di-tBu-phenolic and di-tBu-benzoxazole moieties of LBIS. Such a complex architecture significantly affects the geometry of the Mo coordination sphere, resulting in a distorted octahedral geometry.
In the LBIS ligand, the C2–C3 and C4–C5 bond lengths are close to 1.38 Å, and are shorter than other endocyclic C–C bonds in the phenolic ring, which range from 1.40 to 1.42 Å (Table 2), suggesting at least partially localized double bonds. Both bonds involving imine N1 are similar in length and differ by approximately 1σ. Also, the O1–C1 and C6–N1 bonds are significantly shorter than single bonds.
Concerning the LSQ ligand, both O3–C36 and O4–C41 are double bonds with the respective distances of 1.320(6) and 1.326(7) Å, while the C37–C38 and C39–C40 bonds are slightly shorter than the other endocyclic C–C bonds of the ligand (Table 2). All that suggests that LBIS ligand is found in the semi-quinone form with some coupling between the phenolic and imine moieties.
Magnetic susceptibility measurements
Variable-temperature magnetic susceptibility measurement for the crystalline samples of MoOLBISLSQ was performed with an applied magnetic field of 10000 Oe in the temperature range 2–290 K (Fig. 3). The effective magnetic moment (μeff) for MoOLBISLSQ at 290 K is 2.36 μB, which is slightly lower than the spin-only value expected for two S = 1/2 radical spins. This value drops to almost zero (0.2 μB) along with the decreasing temperature to 2 K indicating the ground singlet spin state (Stotal = 0) and a dominant antiferromagnetic coupling in LBIS–Mo(IV)–LSQ where spin alignment seems to be [(↓)–(↓↑)–(↑)] (Scheme 3). However, the temperature dependence of μeff is rather linear. Magnetic analysis by applying the spin system of two S = 1/2 was unsuccessful. These data are consistent with the X-ray structural data described above. Similar behavior was observed for the metal complex with a non-innocent ligand.16 Such behavior may be responsible for an unclear oxidation state of the Mo ion with redox-active ligands.
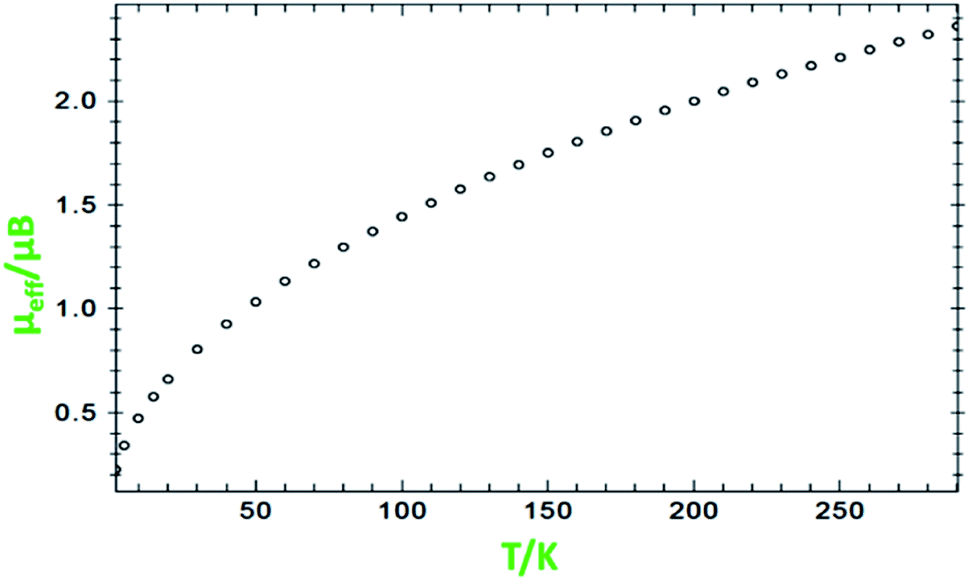 | ||
| Fig. 3 Variation of effective magnetic moment (μeff) with variation in temperature for MoOLBISLSQ. | ||
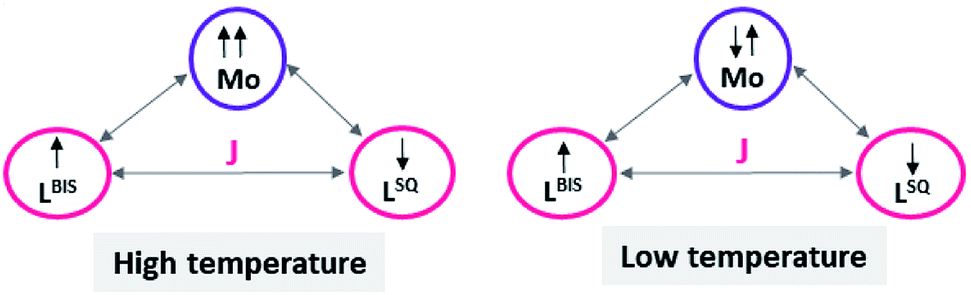 | ||
| Scheme 3 The schematic representation of spin distribution in MoOLBISLSQ at high and low temperature. | ||
Electrochemistry
The electrochemical properties of the MoOLBISLSQ complex were recorded in CH2Cl2 by cyclic voltammetry (CV) at a low temperature (233 K). Before starting the measurement, GC electrodes were polished with 0.1 mm alumina powder and washed with distilled water. CV reveals that there are three quasi-reversible redox peaks for the complex (Fig. 4). The complex underwent two quasi-reversible ligand centered one-electron redox processes, iminosemibenzoquinone LBIS/iminobenzoquinone LBIQ and o-semibenzoquinone LSQ/o-benzoquinone LQ redox couples at positive potentials, and MoIV/MoIII at negative potential as seen in eqn (1)–(3) (Scheme 4).17| MoIVLBISLSQ ↔ MoIVLBIQLSQ | (1) |
| MoIVLBIQLSQ ↔ MoIVLBIQLQ | (2) |
| MoIVLBIQLQ ↔ MoIIILBIQLQ | (3) |
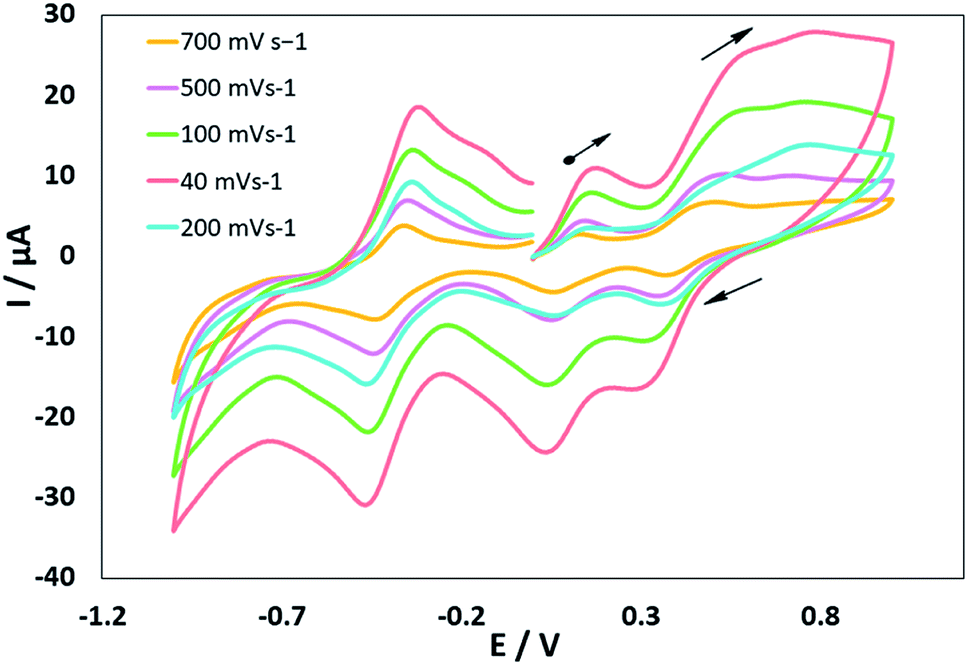 | ||
| Fig. 4 Cyclic voltammograms of MoOLBISLSQ. Conds: 1 mM complex, 0.1 M NBu4ClO4, CH2Cl2, 298 K. | ||
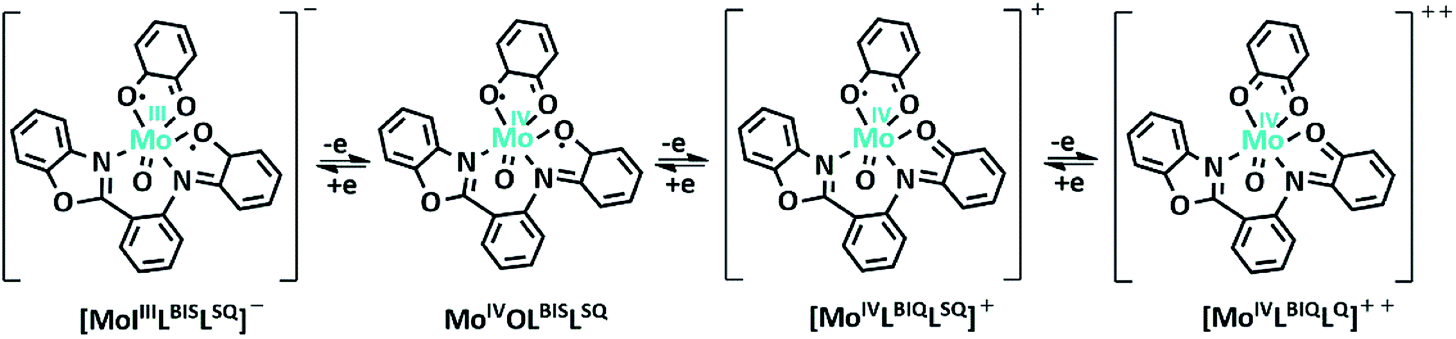 | ||
| Scheme 4 Schematic representation of MoOLBISLSQ complex oxidation state variation (tBu groups are omitted for clearly). | ||
Electronic spectroscopy
The electronic absorption (UV-vis/NIR) spectrum of the MoOLBISLSQ complex in dichloromethane is shown in Fig. 5. The spectra were recorded at room temperature (25 °C) in the range of 230–850 nm. The intense absorption bands in 246 and 296 nm in the higher energy and near-UV regions (below 320 nm) are resulted by π → π* transitions that involve iminosemiquinone, LBIS, and LSQ, units. The broad electronic absorption band in the region around 344–380 and 483–542 nm are consistent with iminosemiquinone ligands (LBIS and LSQ)-to-Mo, (π)-to-Mo-(dπ*), the ligand to metal charge-transfer (LMCT). The absorption band that appeared at lower energy (788 nm) is due to d–d charge transfer.17a The investigation of the theoretical analysis of the spectrum has been given below.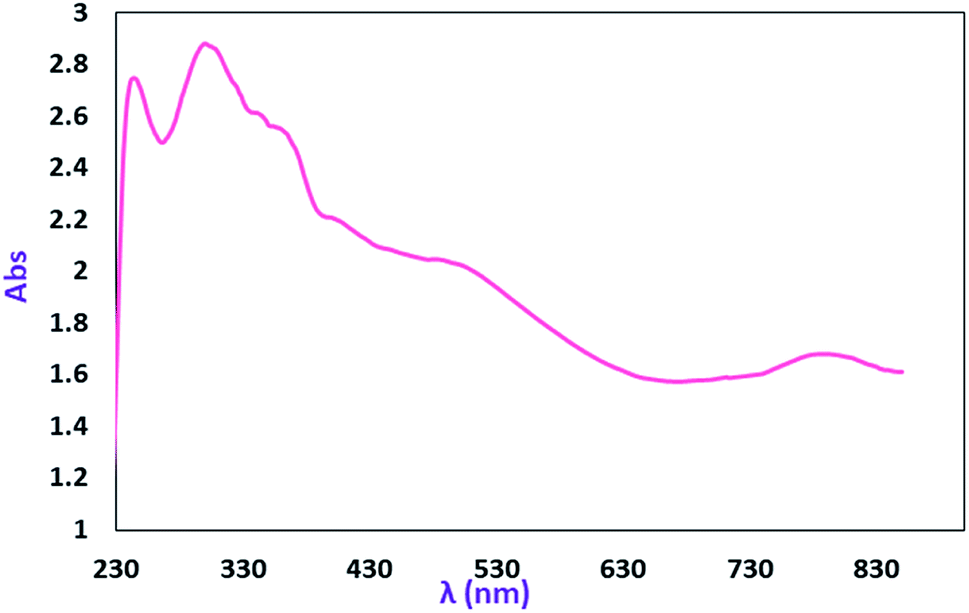 | ||
| Fig. 5 Electronic spectra of 2 mM CH2Cl2 solutions of MoOLBISLSQ. | ||
DFT studies
To gain deeper understanding of complex [MoOLBISLSQ], this species, the H2LBAP compound and (LBIS)n− and (LSQ)n− ligands (n = 2, 1, 0), were theoretically analyzed by using the Density Functional Theory (DFT) approach at the B3LYP-LANL2DZ/6-311++G** level. The resulting optimized structure and HOMO of the precursor ligand H2LBAP are shown in Fig. S3.† The theoretical description fits well with the X-ray experimental data previously reported for this compound.14 The HOMO of this species is centered on the π phenyl system and the distinctive π3* character is observed in the o-aminophenol moiety. This MO is responsible for the non-innocence behavior when it acts as ligand due to the π-donation from this orbital to the metal. Usually, this specific part is typically found as the main contribution to the SOMO of o-aminophenolate (−1) and benzosemiquinone (−1) type ligands. In fact, the characteristic antibonding combinations of C–N and C–O bonds and the C–C bonding combination were clearly observed in the HOMO of optimized (LBIS)2− and (LSQ)2− ligands or SOMO of (LBIS)− and (LSQ)− ligands (Fig. S4 and S5,† respectively). For a six-coordinated oxo-molybdenum(IV) complex, the LUMO description would be an empty dxy orbital. Thus, the π-donation to this orbital from the (LBIS)1− and (LSQ)1− ligands is foreseeable as it is schematically shown in Fig. 6. This fits well with the observed bending of the metallacycle Mo–O1–C1–C6–N1 along the O1–N1 vector in the X-ray structure. This takes place for maximizing the overlap between the dxy and π3* orbitals (white lobes in Fig. 6, center), as it has been previously noted in related ligands.18 Coordinates of the optimized compounds are listed in Table S3.† | ||
| Fig. 6 π-Donation to dxy Mo orbital from the π3* orbital of (LBIS)1− and (LSQ)1− ligands. | ||
Prior to the DFT study of MoOLBISLSQ, free ligands (LBIS)n− and (LSQ)n− (n = 2, 1, 0) were analyzed. HOMO, SOMO and LUMO of these ligands are similar and their respective MO energies decreases from the dianionic to neutral species. Consequently, the dianionic ligand is the most suited for the π-donation to molybdenum dxy orbital. Concerning the structural parameters, as expected, an enlargement of CO and CN– bond distances was observed going from neutral to dianionic ligand with a concomitant modification of phenyl C–C bonds (Fig. S4 and S5†). Based on these data and that of X-ray within the LBIS and LSQ ligands, the radical-anion form of these ligands was considered.19 Therefore, geometry optimization has been carried out assuming the presence of two unpaired electrons (S = 1) on ligands. The optimized MoOLBISLSQ complex describes the experimental structural parameters reasonably well (Fig. 7 and Table S4†). Fig. 8 displays the SOMO and SOMO−1 of this complex, in which SOMO−1 is mainly composed of LBIS and LSQ ligands (46 and 52%, respectively). SOMO involves the LBIS ligand (55%) with 28% of LSQ− and a small contribution of molybdenum (16%). Accordingly, both SOMOs are mostly centered on the (LBIS)− and (LSQ)− ligands confirming the proposed formulation. Additionally, the molecule was also optimized for S = 0 state, corresponding with the antiferromagnetic coupling observed at low temperature, and no significant structural changes were observed (see ESI†).
 | ||
| Fig. 7 Optimized structure of complex MoOLBISLSQ (hydrogen atoms omitted for clarity). | ||
 | ||
| Fig. 8 SOMO and SOMO−1 of complex MoOLBISLSQ (hydrogen atoms omitted for clarity). | ||
TD-DFT calculation
To gain detailed insight into the absorption spectra and charge transitions, TD-DFT calculations were performed at the B3LYP level. The calculated excitation wavelength, oscillator strengths, and their assignment are given in Table S5.† The energies and compositions of molecular orbitals with a major contribution to charge transitions, are listed in Table S6.† Contour plots of these orbitals are shown in Table S7.† The six highest occupied molecular orbitals (HOMO, HOMO−2, HOMO−3, HOMO−4, HOMO−6, HOMO−8) of the Mo complex have large contributions (>90%) from LBIS and LSQ ligand π-bonding orbitals, while HOMO−1 consist of 25% Mo d-orbitals. As shown in Table S6,† the contribution of LBIS and LSQ ligands to the four lowest unoccupied orbitals (LUMO, LUMO+1, LUMO+2, LUMO+3) decrease and show great mixing of oxygen non-bonding, LBIS and LSQ ligands π* and Mo d-orbitals, while LUMO+1 mainly consist of LBIS π* orbitals. The complex shows sharp bands at 246 and 296 nm in dichloromethane. These are mainly LBIS ligand π-bonding to Mo d-orbitals/oxygen non-bonding orbitals charge transfer (ligand to metal or ligand to ligand (LLCT)). A shoulder appearing in the region around 344–380 nm can be assigned to the LBIS to Mo/oxygen charge-transfer transition, while the low energy broad band located at 450–542 nm can be assigned predominately to the LBIS ligand π-bonding orbitals to metal transition (Tables S5 and S6 of ESI†).Oxidation of cyclohexene to adipic acid. The catalytic behaviour of the MoOLBISLSQ complex in the oxidative cleavage of cyclohexene to adipic acid was investigated. A simple method was developed using 25% H2O2 as a green oxidant. After some screening experiments (Table 3), the highest yield of adipic acid was achieved after 10 h at 75 °C in the presence of MoOLBISLSQ (5 mol%) and H2O2 (5 eq.) (Table 3, entry 10). Under these conditions, the resulting white crystalline solid, was isolated by filtration and isolated, was identified as pure adipic acid (IR and mp at 151–152 °C). High selectivity was obtained according to the 1H NMR spectrum of the crude product which did not show any detectable by-products.
|
||||||
|---|---|---|---|---|---|---|
| Catalyst (mol%) | Solvent | H2O2 (eq.) | Time (h) | Temp. (°C) | Yieldb (%) | |
| a Reaction condition: cyclohexene (1 mmol), H2O2 (25%), catalyst (MoOLBISLSQ), solvent (2 mL).b Isolated yield. | ||||||
| 1 | 3 | Free | 3 | 6 | 60 | — |
| 2 | 3 | H2O | 3 | 5 | 60 | — |
| 3 | 3 | H2O/EtOH | 3 | 6 | 60 | 10 |
| 4 | 3 | EtOH | 4 | 8 | 60 | 25 |
| 5 | 3 | CH2Cl2 | 4 | 8 | 60 | 35 |
| 6 | 3 | CH3CN | 4 | 8 | 60 | 44 |
| 7 | 4 | CH3CN | 5 | 8 | 60 | 56 |
| 8 | 4 | CH3CN | 5 | 8 | 70 | 60 |
| 9 | 5 | CH3CN | 5 | 8 | 70 | 70 |
| 10 | 5 | CH3CN | 5 | 10 | 75 | 84 |

A suggested mechanism for the oxidation of cyclohexene to adipic acid is presented in Scheme 5.20,21 This mechanism consists of: (a) transformation of cyclohexene to 1,2-epoxycyclohexane, (b) 1,2-cyclohexanediol formation, (c) oxidation of the resulted diol to 2-hydroxycyclohexanone and then 7-hydroxyoxepan-2-one, and (d) adipic anhydride formation is the final intermediate which can simply go through the ring-opening in the acidic environment to produce adipic acid.
 | ||
| Scheme 5 Proposed reaction pathway for cleavage oxidation of cyclohexene to adipic acid. | ||
The proposed mechanism of MoOLBISLSQ for the oxidation of cyclohexene to adipic acid is shown in Scheme 6. The reaction can be catalyzed by in situ development of peroxomolybdenum species from MoOLBISLSQ in acidic media. This active peroxo complex could undergo oxygen transfer to the substrate and produce epoxycyclohexane that in turn transforms to adipic acid via the steps which are shown in Scheme 5. After these steps, the recovered catalyst was subjected for another catalytic cycle.20c
 | ||
| Scheme 6 Proposed pathway for the formation of adipic acid. | ||
Table 4 represents the comparison between the current work and some other previously reported literature procedures for the oxidative cleavage of cyclohexene to adipic acid by using different catalysts and reaction conditions.20b,22–34 Comparison shows that in other systems the reaction was performed using more expensive catalysts, longer time or stronger oxidant.
| Catalyst | Oxidant | Time (h) | Temp. (°C) | AA (%) | Ref. |
|---|---|---|---|---|---|
| MoOLBISLSQ | H2O2 | 10 | 60 | 84 | This work |
| Ag2WO4–IL 1 | H2O2 | 18 | 75 | 85 | 22 |
| [C16H33N(CH3)3]2W2O3(O2)4 | H2O2 | 20 | 90 | 78 | 23 |
| SSA@[BMIm]WO42− | H2O2 | 18 | 75 | 87 | 24 |
| SBA@Ti–Al | t-BuOOH | 48 | 80 | 80 | 25 |
| MIL-101 | H2O2 | 8 | 70 | 90 | 26 |
| InCl3 | t-BuOOH | 9 | 90 | 92 | 27 |
| RuCl3 | NaIO4 | 0.5 | RT | 90 | 28 |
| [LSO3H]WO42− | H2O2 | 12 | 87 | 85 | 20b |
| Na2WO4·2H2O [CH3(n-C8H17)3N]HSO4 | H2O2 | 8 | 75–90 | 90 | 29 |
| Na2WO4 + ILs | H2O2 | 10 | Reflux | 100 | 30 |
| H2WO4 + IL | H2O2 | 12 | 73–87 | 85–96 | 31 |
| H4SiW12O40 | H2O2 | US (25 kHz) | 4 | 92 | 32 |
| Ti-MMM-2Ce-SBA-15 | H2O2 | 72 | 82 | 10–30 | 33 |
| Ti-AlSBA-15 | H2O2 | 24 | 70 | 80 | 34 |
Sulfide oxidation. The catalytic activity of the complex MoOLBISLSQ in the oxidation of some sulfides at room temperature under the reaction conditions stated in the Experimental section was also studied (Scheme 7).
 | ||
| Scheme 7 Selective oxidation of sulfides to sulfoxides or sulfones catalyzed by MoOLBISLSQ. | ||
First, we optimized conditions (catalyst amount, oxidant, and solvent) for the transformation of sulfides to sulfoxide and sulfone, using the oxidation of methyl phenyl sulfide as a model reaction (Table 5).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | H2O2 (eq.) | Time (h) | Conversiona (%) | TONb |
| a Sulfide to sulfone conversions were determined by using GC.b TON = (substrate/catalyst) × conversion. | ||||||
| 1 | 1 | EtOH | 2 | 5 | 50 | 50 |
| 2 | 1 | EtOH | 3 | 5 | 70 | 70 |
| 3 | 2 | EtOH | 3 | 4 | 94 | 47 |
| 4 | 2 | EtOH | 4 | 4 | 100 | 50 |
| 5 | 2 | H2O | 3 | 6 | 15 | 75 |
| 6 | 2 | EtOH/H2O | 3 | 6 | 30 | 15 |
| 7 | 2 | Toluene | 3 | 4 | 45 | 22.5 |
| 8 | 2 | Free | 3 | 4 | 50 | 25 |
| 9 | 2 | Free | 4 | 4 | 56 | 28 |
| 10 | 3 | Free | 4 | 5 | 76 | 25.3 |
| 11 | 2 | Acetone | 3 | 4 | 100 | 50 |

The effect of several solvents, such as EtOH, H2O, etc., was studied revealing that acetone and ethanol (Table 5, entry 4, and 11) appear to be the most favorable solvents. The free solvent condition was then investigated and a good result was also achieved (Table 5, entry 10).
We also checked the amount of H2O2 for the oxidation of methyl phenyl sulfide at room temperature and realize that the best catalytic performance was achieved by using 3 and 4 eq. of H2O2 in the case of acetone and ethanol respectively (Table 5, entries 4 and 11). Then we studied the effect of the amount of catalyst between 1 mol% and 3 mol% and the best results were reached when the amount of the catalyst was 2 mol%. As it is obvious in Table 5, the catalytic activity of the MoOLBISLSQ depends on the amount of catalyst and oxidant considerably.
After determining under optimized reaction conditions for sulfoxidation of methyl phenyl sulfide (substrate: 1 mmol, catalyst: 2 mol%, acetone 2 mL, H2O2: 3 eq., time: 4 h and room temperature), the catalytic activity of the MoOLBISLSQ was studied in the oxidation of different sulfides under these optimized parameters (Table 6).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time (h) | Conversionb (%) | Selec. to sulfonec (%) | TONd |
| a Reaction conditions: catalyst:sulfide:H2O2 = 0.02:1:3 in 2.5 mL acetone.b Sulfide to sulfone conversions were determined by using GC.c Selectivity to sulfone = [A%/(A + B%)] × 100.d TON = (moles of substrate/moles of catalyst) × conversion.e Isolated yield. |
|||||
| 1 |  |
4 | 100 | >99 | 50 |
| 2 |  |
4 | 100 | >99 | 50 |
| 3 |  |
4 | 100 | >99 | 50 |
| 4 |  |
5 | 66 | >99 | 33 |
| 5 |  |
4 | 100 | >99 | 50 |
| 6 |  |
4 | >99 | >99 | 49 |
| 7 |  |
5 | 84 | >99 | 42 |
| 8 |  |
4 | >99 | >99 | 49.5 |
| 9 |  |
5 | 88 | >99 | 44 |
| 10 |  |
5 | >99 | >99 | 49.5 |
| 11 |  |
5 | 91 | >99 | 45.5 |
| 12 |  |
6 | 50e | >99 | 25 |

The catalyst performance is good for various sulfides. It has to be stated that the oxidation of DBT (dibenzothiophene) did not happen in any measurable amount in the optimized conditions. New reactions for DBT with a stronger oxidant and TBHP (tert-butyl hydroperoxide or t-BuOOH) were studied. These results are summarized in Table 7. We can see the oxidation of DBT using MoOLBISLSQ with the efficient conversion of 90% and selectivity of >99 and TON of 30 (Table 7, entry 10).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Oxidant (eq.) | Time (h) | Conversiona (%) | Selec. to sulfoneb (%) | TONc |
| a The conversions were determined by GC.b Selectivity to sulfone = [A%/(A + B%)] × 100.c TON = (substrate/catalyst) × conversion. | |||||||
| 1 | 2 | Acetone | H2O2 (3) | 5 | 10 | >99 | 5 |
| 2 | 2 | Acetone | H2O2 (4) | 5 | 10 | >99 | 5 |
| 3 | 3 | Acetone | H2O2 (4) | 5 | 15 | >99 | 5 |
| 4 | 3 | EtOH | H2O2 (4) | 4 | 15 | >99 | 5 |
| 5 | 2 | Acetone | H2O2 (excess) | 4 | 15 | >99 | 7.5 |
| 6 | 2 | Acetone | TBHP (2) | 4 | 75 | >99 | 37.5 |
| 7 | 2 | Free | TBHP (2) | 4 | 50 | >99 | 25 |
| 8 | 2 | EtOH | TBHP (2) | 4 | 60 | >99 | 30 |
| 9 | 2 | Toluene | TBHP (2) | 5 | 80 | >99 | 40 |
| 10 | 3 | Toluene | TBHP (2) | 5 | 90 | >99 | 30 |
| 8 | 3 | Toluene | TBHP (3) | 5 | 90 | >99 | 30 |

Finally, the role of the catalyst and also the effect of metal–ligand synergistic effects proved by some blank tests with H2O2, MoO2(acac)2/H2O2, and H2LBIS/H2O2 catalytic systems and the results are collected in Table S8.†
To improve the understanding of the reaction mechanism, sulfide was added to the stirring solution of MoOLBISLSQ. No obvious and noticeable change was observed in the reaction mixture even after 2 h of stirring (Table S8,† entry 5). This result proved the essential presence of H2O2 to activate the complex.
We proposed a plausible mechanism for this oxidation reaction which is presented in Scheme 8. In the first step, the peroxo complex of the catalyst MoOLBISLSQ was achieved in the presence of H2O2, and these active spices are involved in the sulfoxidation reaction. The oxotransfer from the peroxo complex could be achieved in two ways. One is by direct oxygen transfer and the other is the coordination of the sulfide to the molybdenum. We proposed that due to the absence of free coordination position in the complex, the second idea seems to be more plausible. Thus, the oxygen transfer took place and the former complex recovered. Finally, sulfone could be obtained by another oxygen transfer to the resulted sulphoxide.35
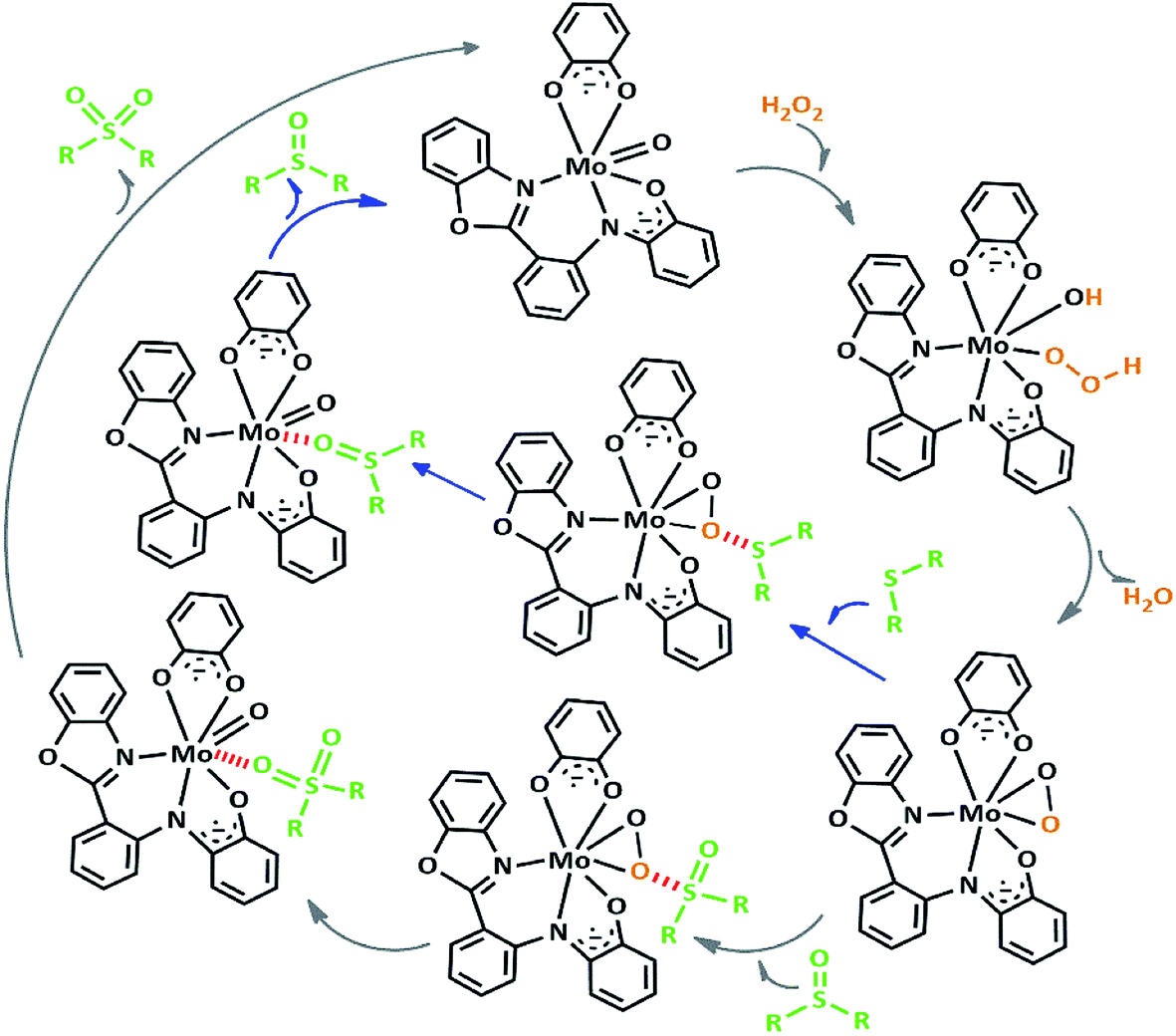 | ||
| Scheme 8 Proposed pathway for the formation of sulfoxide and sulfone. | ||
Some literature data for the oxidation of sulfides with homogeneous and heterogeneous Mo-based catalysts are listed in Table 8. Comparison proves that the developed catalyst shows a good affinity for sulfide oxidation at mild condition comparing other reports.35–47
| Catalyst (mol%) | Sulfide | Reaction condition: oxidant (mmol)/T (h)/temp. (°C) | AA (%) | Ref. | |
|---|---|---|---|---|---|
| 1 | [MoO2(CH3OH)H2O(CH3OH)] (5) | Thioanisole | H2O2 (1)/13 h/0 | 83 | 2011 (ref. 35) |
| 2 | [MoO(O2)2(H2O)n]/HLiPr/[PPh4]Br (2.5) | Thioanisole | H2O2 (1)/1 h/0/reactor | 95 | 2018 (ref. 36) |
| 3 | Mo(O2)L (5) L = 4,6-O-ethylidene-N-(2-hydroxybenzylidene)-β-D-glucopyranosylamine | Thioanisole | UHP (1)/15 h/RT | 86 | 2016 (ref. 37) |
| 4 | MoO2(acac)2 | Thioanisole | TBHP/—/50–70 | 98 | 1966 (ref. 38) |
| 5 | MoO2Cl2 (15) | Aryl sulfides | H2O2 (4)/8 h/RT | 93 | 2006 (ref. 39) |
| 6 | MoO2Cl2 (4,4′-di-tert-butyl-2,2′-bipyridine) | S in diesel | H2O2/3 h/50 | 76 | 2016 (ref. 40) |
| 7 | [MoO2(L) (CH3OH)] (5) | Thioanisole | UHP/0.5 h/RT | 92 | 2009 (ref. 41) |
| 8 | (NH4)6Mo7O24·4H2O (10) | Aliphatic and aryl sulfides | H2O2 (4)/45 min/RT | 95 | 2009 (ref. 42) |
| 9 | [(n-C4H9)4N]4(α-Mo8O26) | Thioanisole | H2O2 (1)/10 min/RT | 99 | 2009 (ref. 43) |
| 10 | Na3[CrMo6O24H6]·8H2O (2) | Thioanisole | H2O2 (2)/10 h/60 | 94 | 2010 (ref. 44) |
| 11 | (PyH)(H3PMo11VO40) (1) | Thioanisole | H2O2 (20)/2.5 h/40 | 95 | 2011 (ref. 45) |
| 12 | (CTA)2[MoO(O2)2(C2O4]·H2O (2.5) | Thioanisole | H2O2 (1)/30 min/95 | 95 | 2012 (ref. 46) |
| 13 | Mo@imine-Z | Diphenyl sulfide | H2O2 (2.5)/30 h/RT | 95 | 2018 (ref. 47) |
| 14 | MoOLBISLSQ (2) | Thioanisole | H2O2 (4)/4 h/RT | 100 | This work |
Experimental
Materials
All the chemical compounds and solvents were purchased from commercial companies and used as received, except those for electrochemical measurements. 3,5-Di-tert-butylcyclohexa-3,5-diene-1,2-dione (3,5-DTBQ) was synthesized according to the literature procedure.48Synthesis of H2LBAP
Ligand (H2LBAP), 2,4-di-tert-butyl-6-(2(5,7-di-tert-butylbenzo[d]oxazol-2-yl) phenylamino) phenol, was synthesized with the procedure reported in the literature.14 Yield: 0.418 g (79%). Anal. calcd (found) for C35H46N2O2: C 79.71 (79.56), H 8.74 (8.51), N 5.30 (5.36). νmax (KBr)/cm−1: 3415 (O–H), 3258 (N–H), 2959 (C–H), 1592 (CC), 1542 (CN), 1262 (C–O), 1047 (C–N) (Fig. S1†).
Synthesis of MoOLBISLSQ
To a solution of H2LBAP (0.262 g; 0.5 mmol) in CH2Cl2 (5 mL), [Mo(O)2(acac)2] (0.164 g; 0.5 mmol) was added under an argon atmosphere and refluxed for 3 hours. Then the solution was stirred in air at room temperature for 1 hour. The resulting dark violet solution was then mixed with CH3OH and filtered. Dark violet micro crystals obtained after two days and single crystals suitable for X-ray analysis were obtained after several recrystallizations from CH3OH/CH2Cl2 1:1 mixture. Yield: 0.350 g (40%). Anal. calcd (found) for C49H64MoN2O5: C 68.71 (67.91), H 7.47 (7.71), N 3.26 (3.46). νmax (KBr)/cm−1: 2955 (C–H), 1650 (CC), 1519 (CN), 1365 (C–O), 1164 (C–N) (Fig. S2†).
General procedure for investigating the catalytic activity of MoOLBISLSQ complex in the cyclohexene oxidation
In a typical procedure: MoOLBISLSQ (4 mol%, 0.034 g), cyclohexene (1 mmol) and 4 eq. of 25% H2O2 were mixed in a 25 mL round bottomed flask. Then the mixture was vigorously stirred at room temperature for 10 minutes and stirred at 70 °C for required time. The white precipitate was separated via filtration and then washed with a little volume of cold water to give adipic acid which was identified by 1H NMR (400 MHz, DMSO-d6): 1.50 (t, 4H), 2.25 (t, 4H), 12.10 (s, 2H) (Fig. S6†).20General procedure for determination the catalytic activity of MoOLBISLSQ complex in the sulfide oxidation
In a typical experimental procedure, a mixture of sulfide (1 mmol), H2O2 (3 eq., 25%), MoOLBISLSQ (2 mol%, 0.017 g) were firstly added to a 25 mL round bottom flask and stirring was continued for the required time (Table 3) at room temperature. TLC monitored the progress of the reaction (n-hexane:ethyl acetate, 5:1). After completion of the reaction, the conversion of sulfide and selectivity were determined by GC.
In the case of dibenzothiophene sulfone isolated yield has been reported. 1H NMR (400 MHz, chloroform-d) δ 7.90–7.79 (m, 4H), 7.67 (td, J = 7.60, 1.19 Hz, 2H), 7.56 (td, J = 7.58, 1.07 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 121.60, 122.20, 130.40, 131.62, 133.91, 137.71 (Fig. S7 and S8†).16
The conversion of the reaction was then calculated from the sulfide, sulfoxide and sulfone area signals by following formula:
% conversion = 100 × ([product])/([substrate] + [product]).
Measurements
Dark violet crystals of MoOLBISLSQ were grown from the CH3OH:CH2Cl2 1:1 solution. Diffraction data for this complex were collected with the Oxford Sapphire CCD diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) at 292(2) K, by the ω–2θ method. The structure was solved using the Patterson method and refined by the full-matrix least-squares method on F2 with the SHELX2017 program package.49 Analytical absorption correction was applied by the RED171 package of programs50 Rigaku OD, 2015, and the minimum and maximum transmission of 0.957 and 0.905. Hydrogen atoms were positioned with electron density maps and constrained in the refinement.
Elemental analyses (C, H, and N) were done by Elementar Vario EL III. Fourier transform infrared spectroscopy on KBr pellets was performed with FT IR Bruker Vector 22 instrument. 1H NMR spectra were performed on a Bruker DRX instrument in CDCl3 at 400 MHz. The chemical shifts were referred to as TMS by residual signals from the solvent. Cyclic voltammetry (CV) was acquired on a PAR-263A potentiometer analyzer equipped with 0.1 M NBu4ClO4 solutions in CH2Cl2 as supporting electrolyte and with an Ag wire as the reference electrode, a glassy carbon as the working electrode, a Pt counter electrode, and also ferrocene internal standard. UV-vis absorbance spectra were recorded on a CARY 100 Bio spectrophotometer. The magnetic measurements were performed using a Quantum Design SQUID magnetometer MPMS-XL between 1.8 and 300 K. Measurements were done on a polycrystalline sample of 35 mg for MoOLBISLSQ.
Computational details
The electronic structure and geometry optimized of MoOLBISLSQ complex, H2LBAP compound, and (LBIS)n− and SQn− ligands (n = 2, 1, 0) were computed using density functional theory at the B3LYP level.51 The Mo atom was described with the LANL2DZ basis set52 while the 6-31G** basis set was initially used for C, O, N and H atoms. Optimized geometries of all the compounds were characterized as energy minima by the nonexistence of imaginary frequencies (NImag = 0) in the diagonalization of the analytically computed Hessian (vibrational frequencies calculations). The resulting geometries were further optimized at the LANL2DZ/6-311++G** level of theory for obtaining a better MO description. DFT calculations were achieved using the Gaussian 09 suite of programs.53 Coordinates of the optimized compounds are given in Table S3 (ESI†).The solvent was included in the geometry optimization and TD-DFT computations utilizing the Polarizable Continuum Model (PCM)54 using dichloromethane (CH2Cl2) as a solvent with dielectric constant ε = 8.94.
Conclusion
In this work, complex MoOLBISLSQ was synthesized and characterized. This complex is a distorted octahedral mononuclear molybdenum(IV) complex with the MoN2O4 sphere and with the coordinating ligands LBIS and LSQ present in the one-electron oxidized o-iminobenzosemiquinone [(ISQ)1−] forms. The complex acts as an efficient and environmentally friendly catalyst under mild conditions for oxidative cleavage of cyclohexene using H2O2 as an oxidant. One of the interesting features of this work lies with high selectivity toward adipic acid. A comparison of the adipic acid production with the catalyst reported here and other complexes shows the good activity of this complex. The catalyst was also efficient and selective for the oxidation of sulfides, especially DBT. The surrounding non-innocent ligands have a strong effect on the total electronics and catalytic reactivity of the complex.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to Shiraz, Nicolaus Copernicus, Ljubljana and Sevilla Universities for support. E. Safaei greatly appreciated Dr Eckhardt Bill for his valuable help. Special thanks to Dr Atsushi Okazawa and Dr Saeed Zakavi for nice discussions and comments. A. Wojtczak acknowledges the partial support of the BRAIN Center of the Excellence in Research at the N. Copernicus University.References
- (a) W. Kaim, Inorg. Chem., 2011, 50, 9752 CrossRef CAS; (b) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS; (c) B. de Bruin, P. Gualco and N. D. Paul, Ligand Design in Metal Chemistry: Reactivity and Catalysis, John Wiley & Sons, 2016, pp. 176–204 Search PubMed; (d) D. L. Broere, R. Plessius and J. L. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886 RSC; (e) P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS; (f) B. De Bruin, D. G. Hetterscheid, A. J. Koekkoek and H. Gruetzmacher, Prog. Inorg. Chem., 2007, 55, 247 CAS; (g) P. J. Chirik, Inorg. Chem., 2011, 50, 9737 CrossRef CAS; (h) J. W. Whittaker, Chem. Rev., 2003, 103, 2347 CrossRef CAS; (i) J. K. Klein and L. Que Jr, Encyclopedia of Inorg. Bioinorg, Chem., 2011, pp. 1–22 Search PubMed; (j) M. Costas, K. Chen and L. Que Jr, Coord. Chem. Rev., 2000, 200, 517–544 CrossRef; (k) J. Bautz, P. Comba, C. Lopez de Laorden, M. Menzel and G. Rajaraman, Angew. Chem., Int. Ed., 2007, 46, 8067 CrossRef CAS; (l) S. P. de Visser, M. G. Quesne, B. Martin, P. Comba and U. Ryde, Chem. Commun., 2014, 50, 262 RSC; (m) R. Van Eldik and J. Reedijk, Advances in Inorganic Chemistry: Homogeneous Biomimetic Oxidation Catalysis, Elsevier, 2006 Search PubMed; (n) S. Fukuzumi, Y. M. Lee and W. Nam, J Porphyrins Phthalocyanines, 2020, 24, 21 CrossRef CAS; (o) S. P. de Visser, J. S. Valentine and W. Nam, Angew. Chem., Int. Ed., 2010, 49, 2099 CrossRef CAS; (p) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS; (q) X. Shan and L. Que Jr, J. Inorg. Biochem., 2006, 100, 421–433 CrossRef CAS; (r) D. H. Jo and L. Que Jr, Angew. Chem., 2000, 112, 4454 CrossRef; (s) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939 CrossRef CAS; (t) L. R. Widger, Y. Jiang, M. A. Siegler, D. Kumar, R. Latifi, S. P. de Visser, G. N. Jameson and D. P. Goldberg, Inorg. Chem., 2013, 52, 10467 CrossRef CAS; (u) S. P. de Visser, S. Shaik, P. K. Sharma, D. Kumar and W. Thiel, J. Am. Chem. Soc., 2003, 125, 15779 CrossRef CAS; (v) S. Y. T. Dakhili, S. A. Caslin, A. S. Faponle, P. Quayle, S. P. de Visser and L. S. Wong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5285 CrossRef.
- (a) R. Hille, Chem. Rev., 1996, 96, 2757 CrossRef CAS; (b) G. Yin, Coord. Chem. Rev., 2010, 254, 1826 CrossRef CAS; (c) R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963 CrossRef CAS.
- (a) J. H. Enemark, J. J. A. Cooney, J. J. Wang and R. H. Holm, Chem. Rev., 2004, 104, 1175 CrossRef CAS; (b) G. Lyashenko, G. Saischek, M. E. Judmaier, M. Volpe, J. Baumgartner, F. Belaj, V. Jancik, R. Herbst-Irmer and N. C. Mösch-Zanetti, Dalton Trans., 2009, 29, 5655 RSC.
- (a) J. Haber, in Studies Inorganic Chemistry, Elsevier, 1994, vol. 19 Search PubMed; (b) A. Szymańska, W. Nitek, M. Oszajca, W. Łasocha, K. Pamin and J. Połtowicz, Catal. Lett., 2016, 146, 998 CrossRef; (c) C. Djordjevic, B. C. Puryear, N. Vuletic, C. J. Abelt and S. J. Sheffield, Inorg. Chem., 1988, 27, 2926 CrossRef CAS; (d) M. H. Dickman and M. T Pope, Chem. Rev., 1994, 94, 569 CrossRef CAS; (e) W. R. Thiel, J. Mol. Catal., 1997, 117, 449 CrossRef CAS; (f) M. Jia and W. R. Thiel, Chem. Commun., 2002, 2392 RSC; (g) C. C. Pereira, S. S. Balula, F. A. Almeida Paz, A. A. Valente, M. Pillinger, J. Klinowski and I. S. Gonçalves, Inorg. Chem., 2007, 46, 8508 CrossRef CAS; (h) J. A. Brito, H. Teruel, G. Muller, S. Massou and M. Gómez, Inorg. Chim. Acta, 2008, 361, 2740 CrossRef CAS; (i) M. Abrantes, R. R. Amarante, M. M. Antunes, S. Gago, F. A. Almeida Paz, I. Margiolaki, A. E. Rodrigues, M. Pillinger, A. A. Valente and I. S. Gonçalves, Inorg. Chem., 2010, 49, 6865 CrossRef CAS; (j) J. Pisk, D. Agustin, V. Vrdoljak and R. Poli, Adv. Synth. Catal., 2011, 353, 2910 CrossRef CAS; (k) J. Pisk, B. Prugovecki, D. Matkovic-Calogovic, R. Poli, D. Agustin and V. Vrdoljak, Polyhedron, 2012, 33, 441 CrossRef CAS; (l) T. R. Amarante, P. Neves, F. A. A. Paz, M. Pillinger, A. A. Valente and I. S. Gonçalves, Inorg. Chem. Commun., 2012, 20, 147 CrossRef CAS; (m) S. Rayati, N. Rafiee and A. Wojtczak, Inorg. Chim. Acta, 2012, 386, 27 CrossRef CAS; (n) M. Masteri-Farahani and Z. Kashef, J. Magn. Magn. Mater., 2012, 324, 1431 CrossRef CAS; (o) G. Grivani and A. Akherati, Inorg. Chem. Commun., 2013, 28, 90 CrossRef CAS; (p) C. Djordjevic, N. Vuletic, B. A. Jacobs, M. Lee-Renslo and E. Sinn, Inorg. Chem., 1997, 36, 1798 CrossRef CAS; (q) N. Gharah, S. Chakraborty, A. K. Mukherjee and R. Bhattacharyya, Chem. Commun., 2004, 2630 RSC; (r) M. M. W. Plass, Inorg. Chem. Commun., 2007, 10, 677 CrossRef; (s) F. Madeira, S. Barroso, S. Namorado, P. M. Reis and B. Royo, Inorg. Chim. Acta, 2012, 83, 152 CrossRef; (t) C. J. Carrasco, F. Montilla, E. Álvarez, M. Herbert and A. Galindo, Polyhedron, 2013, 54, 123 CrossRef CAS.
- (a) C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, R. Soc. Chem., Cambridge, 1999, vol. 5, p. 65 Search PubMed; (b) G. Strukul, Catalytic Oxidations with Hydrogen Peroxide as Oxidant, Kluwer Academic, Dordrecht, The Netherlands, 1992 CrossRef; (c) R. A. Sheldon and J. K. Kochi, Metal Catalyzed Oxidations of Organic Compounds, Academic Press, New York, NY, USA, 1981 Search PubMed; (d) A. Goti and F. Cardona, in Green chemical reactions, Springer, Dordrecht, 2008, pp. 191–212 Search PubMed; (e) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC; (f) R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374 CrossRef CAS; (g) M. Rasouli, M. A. Zolfigol, M. H. Moslemin and G. Chehardoli, Green Chem. Lett. Rev., 2017, 10, 117 CrossRef CAS.
- (a) S. Van de Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465 RSC; (b) R. A. Mayer, The 100 Most Important Chemical Compounds, 1st edn, Greenwood Press, London, 2007 Search PubMed; (c) M. T. Musser, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed; (d) C. Aellig, C. Girard and I. Hermans, Angew. Chem., Int. Ed., 2011, 50, 12355 CrossRef CAS.
- R. E. Dickinson and R. J. Cicerone, Nature, 1986, 319, 109 CrossRef CAS.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646 CrossRef CAS.
- J. Pisk, D. Agustin and R. Poli, Molecules, 2019, 24, 783 CrossRef.
- (a) M. Damm, B. Gutmann and C. O. Kappe, ChemSusChem, 2013, 6, 978 CrossRef CAS; (b) M. Shang, T. Noel, Y. Su and V. Hessel, Ind. Eng. Chem. Res., 2016, 55, 2669 CrossRef CAS; (c) I. Vural-Gursel, Q. Wang, T. Noel, V. Hessel and J. T. Tinge, Ind. Eng. Chem. Res., 2013, 52, 7827 CrossRef; (d) M. Shang, T. Noël, Q. Wang, Y. Su, K. Miyabayashi, V. Hessel and S. Hasebe, Chem. Eng. J., 2015, 260, 454 CrossRef CAS; (e) Y. Wen, X. Wang, H. Wei, B. Li, P. Jin and L. Li, Green Chem., 2012, 14, 2868 RSC; (f) U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. Da Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1 CrossRef CAS.
- M. G. Buonomenna, G. Golemme, M. P. De Santo and E. Drioli, Org. Process Res. Dev., 2010, 14, 252 CrossRef CAS.
- J. Freitag, M. Nüchter and B. Ondruschka, Green Chem., 2003, 5, 291 RSC.
- (a) P. Grange, Catal. Rev.: Sci. Eng., 1980, 21, 135 CrossRef CAS; (b) H. Tops and B. S. Clausen, Catal. Rev.: Sci. Eng., 1984, 26, 395 CrossRef; (c) R. R. Chianelli, Catal. Rev.: Sci. Eng., 1984, 26, 361 CrossRef CAS; (d) R. J. Angelici, Polyhedron, 1997, 16, 3073 CrossRef CAS; (e) I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607 CrossRef CAS; (f) A. Rothlisberger and R. Prins, J. Catal., 2005, 235, 229 CrossRef; (g) E. R. Skov and D. C. England, Hydrocarbon Eng., 2007, 12, 33 CAS.
- S. E. Balaghi, E. Safaei, L. Chiang, E. W. Wong, D. Savard, R. M. Clarke and T. Storr, Dalton Trans., 2013, 42, 6829 RSC.
- C. W. Kee, J. Chem., 2015, 2015, 439270 Search PubMed.
- R. Saeedi, E. Safaei, Y. I. Lee and J. Lužnik, Appl. Organomet. Chem., 2019, 33, 4781 CrossRef.
- (a) P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213 CrossRef CAS; (b) J. I. van der Vlugt, Chem.–Eur. J., 2019, 25, 2651 CrossRef CAS; (c) J. Jacquet, K. Cheaib, Y. Ren, H. Vezin, M. Orio, S. Blanchard, L. Fensterbank and M. Desage El Murr, Chem.–Eur. J., 2017, 23, 15030 CrossRef CAS; (d) A. Gushchin, Y. A. Laricheva, P. A. Abramov, A. V. Virovets, C. Vicent, M. N. Sokolov and R. Llusar, Eur. J. Inorg. Chem., 2014, 2014, 4093 CrossRef CAS; (e) K. G. Matz, R. P. Mtei, R. Rothstein, M. L. Kirk and S. J. N. Burgmayer, Inorg. Chem., 2011, 50, 9804 CrossRef CAS.
- (a) A. Galindo, A. Ienco and C. Mealli, New J. Chem., 2000, 24, 73 RSC; (b) A. Galindo, A. Ienco and C. Mealli, Comments Inorg. Chem., 2002, 23, 401 CrossRef CAS; (c) C. Mealli, A. Ienco, A. D. Phillips and A. Galindo, Eur. J. Inorg. Chem., 2007, 2556 CrossRef CAS.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251 CrossRef CAS.
- (a) A. Lesbani, F. Fitriliana and R. Mohadi, Indones. J. Chem., 2015, 15, 64 CrossRef CAS; (b) M. Vafaeezadeh and M. M. Hashemi, Chem. Eng., 2013, 221, 254 CrossRef CAS; (c) M. Vafaeezadeh and M. M. Hashemi, Catal. Commun., 2014, 43, 169 CrossRef CAS.
- S. Van de Vyver and Y. Roman-Leshkov, Catal. Sci. Technol., 2013, 3, 1465 RSC.
- M. Vafaeezadeh and M. M. Hashemi, RSC Adv., 2015, 5, 31298 RSC.
- W. Zhu, H. Li, X. He, Q. Zhang, H. Shu and Y. Yan, Catal. Commun., 2008, 9, 551 CrossRef CAS.
- M. Vafaeezadeh, M. Mahmoodi Hashemi and M. Shakourian-Fard, Catal. Commun., 2012, 26, 54 CrossRef CAS.
- G. Lapisardi, F. Chiker, F. Launay, J. P. Nogier and J. L. Bonardet, Catal. Commun., 2004, 5, 277 CrossRef CAS.
- Z. Saedi, S. Tangestaninejad, M. Moghadam, V. Mirkhani and I. Mohammadpoor-Baltork, Catal. Commun., 2012, 17, 18 CrossRef CAS.
- B. C. Ranu, S. Bhadra and L. Adak, Tetrahedron Lett., 2008, 49, 2588 CrossRef CAS.
- F. Zimmermann, E. Meux, J. L. Mieloszynski, J. M. Lecuire and N. Oget, Tetrahedron Lett., 2005, 46, 3201 CrossRef CAS.
- K. Sato, M. Aoki and R. A. Noyori, Science, 1998, 281, 1646 CrossRef CAS.
- J. Gui, D. Liu, X. Cong, X. Zhang, H. Jiang, Z. Hu and Z. Sun, J. Chem. Res., 2005, 8, 520 CrossRef.
- B. Wang, Z. Zhang, X. Zhang, S. Sun, L. Wu and R. Xing, Chem. Pap., 2018, 72, 643 CrossRef CAS.
- Z. P. Lin and L. Wan, Asian J. Chem., 2013, 25, 6008 CrossRef CAS.
- M. N. Timofeeva, O. A. Kholdeeva, S. H. Jhung and J. S. Chang, Appl. Catal., A, 2008, 345, 195 CrossRef CAS.
- F. Chiker, F. Launay, J. P. Nogier and J. L. Bonardet, Chem. Lett., 2003, 1, 117 CrossRef CAS.
- R. D. Chakravarthy, K. Suresh, V. Ramkumar and D. K. Chand, Inorg. Chim. Acta, 2011, 376, 57 CrossRef CAS.
- C. J. Carrasco, F. Montilla and A. Galindo, Molecules, 2018, 23, 1595 CrossRef.
- N. Baig, V. K. Madduluri and A. K. Sah, RSC Adv., 2016, 6, 28015 RSC.
- L. Kuhnen, Angew. Chem., Int. Ed., 1996, 5, 893 CrossRef.
- K. Jeyakumar and D. K. Chand, J. Chem. Sci., 2009, 121, 111 CrossRef CAS.
- D. Julião, A. C. Gomes, M. Pillinger, R. Valença, J. C. Ribeiro, I. S. Gonçalves and S. S. Balula, Dalton Trans., 2016, 45, 15242 RSC.
- I. Sheikhshoaie, A. Rezaeifard, N. Monadi and S. Kaafi, Polyhedron, 2009, 28, 733 CrossRef CAS.
- K. Jeyakumar, R. D. Chakravarthy and D. K. Chand, Catal. Commun., 2009, 10, 1948 CrossRef CAS.
- C. Yang, Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. Deng, Green Chem., 2009, 11, 1401 RSC.
- A. R. Supale and G. S. Gokavi, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 725 CrossRef CAS.
- G. P. Romanelli, P. I. Villabrille, C. V. Cáceres, P. G. Vázquez and P. Tundo, Catal. Commun., 2011, 12, 726 CrossRef CAS.
- R. D. Chakravarthy, V. Ramkumar and D. K. Chand, Green Chem., 2014, 16, 2190 RSC.
- A. Dadashi Hadigavabar, K. Tabatabaeian, M. A. Zanjanchi and M. Mamaghani, J. Chil. Chem. Soc., 2018, 63, 3829 CrossRef.
- T. Khomenko, O. Salomatina, S. Kurbakova, I. Il’ina, K. Volcho, N. Komarova, D. Korchagina, N. Salakhutdinov and A. Tolstikov, Russ. J. Org. Chem., 2006, 42, 1653 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr. Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3 Search PubMed.
- CrysAlisPro 1.171.38.43 software, Rigaku OD, 2015 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- (a) T. H. Dunning Jr and P. J. Hay, Modern Theoretical Chemistry, Plenum, New York, 1976, p. 1 Search PubMed; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision B.01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2013761. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06351g |
| This journal is © The Royal Society of Chemistry 2020 |