
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver nanoribbons achieved by picosecond ablation using cylindrical focusing and SERS-based trace detection of TNT
Haribabu Marrapu†
a,
Ravikiran Avasarala†a,
Venugopal Rao Soma *b,
Santosh Kumar Balivadac and
Gopala Krishna Podagatlapalli*a
*b,
Santosh Kumar Balivadac and
Gopala Krishna Podagatlapalli*a
aDepartment of Electronics & Physics, GIS, GITAM deemed to be University, Visakhapatnam 530045, Andhra Pradesh, India. E-mail: gpodagat@gitam.edu
bAdvanced Centre of Research in High Energy Materials (ACRHEM), University of Hyderabad, Hyderabad 500046, Telangana, India. E-mail: soma_venu@uohyd.ac.in
cAndhra Pradesh Medtech Zone (AMTZ), Visakhapatnam 530031, Andhra Pradesh, India
First published on 12th November 2020
Abstract
We report the fabrication of silver nanoribbons by picosecond laser ablation of bulk silver (Ag) targets submerged in double distilled water (DDW) using a cylindrical focusing geometry. The laser ablation was performed by ∼2 picosecond laser pulses and the corresponding light sheet engendered by a cylindrical lens of focal length ∼4.5 cm. The input pulse energies employed at a wavelength ∼800 nm in the experiments were ∼1000 μJ, ∼1200 μJ, and ∼1400 μJ. In contrast to the case of ablation with spherical lenses, cylindrical lens ablation produced nanoparticles (NPs) and nanostructures (NSs) in 20% less time. The data obtained from the optical characterizations exemplify that localized surface plasmon resonance (LSPR) was observed at 406 nm, 408 nm, and 410 nm for the input energies of ∼1000 μJ, ∼1200 μJ, and ∼1400 μJ, respectively. Interestingly, it was observed that the ablation performed at an input energy of ∼1200 μJ demonstrated the fabrication of Ag nanoribbons rather than the formation of Ag NPs. Selected area electron diffraction (SAED) data of the nanoribbons recorded revealed their crystalline phase and linear morphology. Ag nanomaterials (NPs and ribbons) synthesized in these experiments were employed to detect the explosive molecules of 2,4,6-trinitrotoluene (TNT) at a concentration 25 nM using the technique of surface enhanced Raman scattering. The enhancement factor in the case of Ag nanoribbons (width of ∼20–30 nm, length of ∼0.6–2 μm), obtained using the cylindrical focussing geometry at input pulse energies of ∼1200 μJ, was estimated to be ∼107 for the 1362 cm−1 mode, corresponding to the symmetric NO2 stretch of TNT.
1. Introduction
Ultrafast laser ablation of metal targets submerged in liquids (ULAL) is a well-known top-down technique to produce metallic/semiconductor/ceramic nanoparticles (NPs) and nanostructures (NSs) in a single experiment dissimilar to the existing wet chemical methods.1–8 The preparation of plasmonic nanomaterials with controlled shape and size has been attractive to the scientific community due to their versatile applications in catalysis,9 optics10–13 electronics,14 biological applications,15,16 drug delivery.17,18 Unlike the traditional methods of nanomaterial synthesis, ULAL enables gram scale, green synthesis within short time scales and it does not entail the usage of surfactants to circumvent agglomeration or aggregation.19–22 ULAL is a complex laser–matter interaction which does not have a universal theory to designate the synthesis of nanomaterials. The products of the ULAL can be tweaked according to the experimental requirement in which NPs or NSs are determined by the laser parameters (wavelength, pulse energy, repetition rate, polarization etc.), liquid parameters (polarity, viscosity, refractive index) and mechanical parameters (translational stage parameters, focusing geometry etc.). The slight variation of any of the parameters stated above, by preserving the other parameters constant, results in the production of NPs/NSs with diverse physical, optical, thermodynamic, and mechanical properties.2–4 The documented conclusions in the area of ULALentitles the inherent mechanism of laser matter interaction and the scheme of the NPs or NSs generation by classifying the ablation processes arising from the irradiation of ultrafast laser pulses as spallative ablation,23–25 phase explosion ablation,26,27 spinodal decomposition,27 fragmentation and vaporization,28 plasma ablation and coulomb explosion,29 etc. In fact, the fluence of the incident laser pulse at the focus controls the process that prevail in a particular investigation. The fluence of the laser beam at the focal plane, i.e. on the metal target immersed in liquid is also determined by the sort of focusing geometry employed. Tight focusing, loose focusing or a moderate focussing of the laser pulses establish diverse ablation products. As per the existing literature, metallic/semiconductor/ceramic nanomaterials were fabricated by focusing a nanosecond/picosecond/femtosecond laser pulses with a Gaussian profile on to the metal/semiconductor/ceramic targets immersed in a liquid medium. Many researchers have utilized a direct irradiation of metal targets or 2-dimensional raster scan on the targets to be ablated those submerged in any liquid medium. However, the mentioned methodologies produce NPs and NSs in a single experiment and are time consuming.30–33 Above stated practices cannot yield regular patterns over a large area. This is due to the fluctuations in laser parameters during the very slow patterning of metal/semiconductor/ceramic targets using the focused laser beams. Cylindrical focusing geometry eradicates the complications posed by the slow patterning of the targets with laser pulses focused by the spherical lenses. Das et al.34 reported the formation low and high spatial frequency on TiO2 targets with 400 nm wavelength in cylindrical focussing geometry. Ardron et al.35 employed cylindrical lens of longer focal length to produce laser induced periodic surface structures (LIPSS) on stainless steel targets of large area with a single pass at a rate of 5 mm2 s−1. Zhang et al.36 reported the laser power density distribution at the focus of cylindrical lens was approximately four-fold larger when compared to the power density distribution by a spherical lens. They demonstrated that the uniform ablation can be expected with the cylindrical focussing geometry compared to the ablation achieved with spherical lens focussing geometry. Recently, Sindhu et al. employed cylindrical focussing geometry to produce homogeneous laser induced periodic surface structures [LIPSS] on Si using femtosecond pulses of duration ∼25 fs.37 As mentioned above, there are very few reports available on the production of NPs and NSs in very less time. The employment of a line focus, where in a Gaussian pulse is focused by a cylindrical lens, certainly result in the production of metal NPs and NSs in very short span of time. But, to the best of our knowledge, no one has so far attempted the generation NPs and NSs using laser ablation Ag target submerged in DDW with picosecond (ps) laser pulses using the cylindrical geometry. Till date, many reports have been documented in laser ablation of metal/semiconductor/ceramic nanoparticles and 99.99% of them have utilized the spherical lens focusing geometry and investigated the consequences. With cylindrical focussing geometry, there are a few works reported recently but they were performed in air ambient and mostly focused on the formation of surface/grating structures.38 The ablation of metals in liquids using cylindrical focusing lens and picosecond laser pulses has not been studied earlier. One of the main advantages of cylindrical focusing compared to spherical lens focusing is that the time required to produce an effective 1 cm × 1 cm nanostructured target. The cylindrical lens case needs <5 minutes whereas the spherical lens case requires longer time (typically >30 min). The other expected benefit is that the yield of nanoparticles/rate of generation of nanoparticles in cylindrical geometry is comparatively higher. Similarly, picosecond ablation has been proved to deliver higher yields (to the extent of few grams per hour) of nanoparticles compared to femtosecond ablation.Amongst the many applications of nanomaterials involving plasmonic metals (gold, silver, copper, platinum), surface enhanced Raman scattering [SERS] technique39–44 is a powerful tool that eliminates the poor scattering cross section [10−34 cm2 per molecule] of the spontaneous Raman process. SERS of analyte molecules adsorbed on the plasmonic metal NPs/NSs is mediated by localized surface plasmon resonances (LSPR) and propagating surface plasmon resonances (PSP) those occur in the NPs/NSs. Studies on SERS have increased exponentially during the last three decades after the establishment of various methodologies to synthesize metal/semiconductor/ceramic NPs/NSs. SERS utilizes the plasmonic nanomaterials, in which molecular probes under investigation at ultralow concentration in solution phase, adsorbed on the nanomaterials (NPs, NSs, or other shapes) and their Raman signatures can be recorded using a suitable excitation wavelength. Suitability of exciting line of Raman spectrometer plays a crucial role in the determination of the enhancement of the Raman signatures from the various vibrational modes of probe molecules adsorbed on the nanomaterials, since Raman enhancements depend on the excitation of LSPRs and propagating surface plasmons.45–47 LSPR mediated SERS has many applications which are extended in all branches of science, amongst, near-field explosives (e.g. RDX, TNT, ANTA, CL-20, FOX-7 etc.) detection is a significant one. Among the above-mentioned, well-known secondary explosive is 2,4,6-trinitrotoluene (TNT) which utilized as the probe molecule in the present study. SERS technique facilitates the detection/identification of explosive molecules which are at ultra-low concentrations.
2. Experimental details
Pure silver [Ag] targets of dimensions 1.5 × 1.5 × 0.2 cm3 [purchased from sigma Aldrich] were ultrasonicated in ethanol thrice prior to the laser ablation experiment to confiscate the surface impurities if any present on the target surface. A chirped pulse amplifier [M/s Coherent] that delivered laser pulses of Gaussian profile at a repetition rate of 1 kHz, average power 2 W (∼2 mJ per pulse), a central maximum of ∼800 nm with a pulse duration ∼2 ps was employed in the present experiment. Gaussian laser pulses with of initial beam waist (2ω0 = 10 mm) were allowed to irradiate an Ag target immersed in double distilled water (DDW) using a cylindrical lens of focal length ∼4.5 cm. Cylindrical lens results in line focus which maintains a uniform energy distribution over the focussed line. The major axis of the elliptical focus was measured to be 10 mm and minor was ∼10 μm. The Ag target was submerged in in a cylindrical vessel [Petri dish] of DDW water by ensuring the water level is ∼5 mm above the target surface. Further the Petri dish with Ag target was kept on a three-dimensional XYZ stage in which each stage is controlled by the stepper motors that provide us with a resolution of 250 nm. Placing of the Ag target immersed in DDW precisely at the focal plane of the cylindrical lens is a tedious task, since one has to ensure it from the cracking sound (resulting from surface ablation) from the Ag target when it is irradiated by a focused laser beam. If the Ag target is kept either below or above the focal plane of the cylindrical lens, we cannot accomplish an articulate ablation and, therefore, the requisite Ag NPs/NSs. Our previous reports clearly demonstrated the approaches required to keep the metal targets precisely at the focal planes of the lenses without any ambiguity.2–7,48 The XZ stage on which Ag target along with the Petri dish was mounted can be controlled by a motion controller or a computer. A Z-axis stage was utilized to ensure the position of the Ag target exactly at the focus and, X-axis translation stage was utilized to sweep the laser line focus to ablate the area of interest. XZ stages have been interfaced to ablate Ag target immersed in DDW in an area 15 × 15 mm2 in 5 minutes with the stage speed 5 mm s−1, acceleration 0.05 m s−2 along X-axis. Ablation was carried out on Ag target as a one-dimensional sweep along the X axis that covers 8 mm length. The energy per laser pulse (power) was controlled by a combination of a half wave plate (HWP) and a Brewster window [at the Ag target surface the input laser pulses had a vertical polarization (p-polarisation)]. After ablating the Ag targets, Ag colloids were collected in air-tightened glass bottles and were kept in the desiccator to avoid unwanted growth of the NPs by agglomeration and aggregation. Likewise, the Ag targets on which ablation was carried out were cleansed with double distilled water and stored in the desiccator.The synthesized Ag nano colloids were immediately characterized using UV-Visible absorption spectrometer (Jasco-V-670 spectrometer) whose recording range was in the 190–2500 nm spectral range to identify the LSPR peak position and plasmon bandwidth. Similarly, Ag suspensions were characterized by transmission electron microscope (TEM) to examine the size distribution and crystal phases of the prepared Ag colloids. Further, selected area electron diffraction (SAED) of the Ag colloids was also recorded to investigate the crystal phase of the Ag nanosuspensions. On the other hand, machined Ag targets were characterized by the field emission scanning electron microscope (FESEM) to investigate the surface morphology. Further, the Ag targets machined at 1000 μJ, 1200 μJ, and 1400 μJ were utilized to examine the SERS activity by means of the Micro Raman spectrometer (WiTec ALPHA 300 instrument) that employed 532 nm excitation. In the Raman spectrometer, a high NA objective lens (100×) focused the exciting wavelength (532 nm) on to the analyte loaded Ag nanoparticle film at a beam waist ∼650 nm. The Raman spectra were recorded for 2,4,6-trinitrotoluene (TNT) at 25 nM concentration. Prior to the Raman measurements, spectrometer was calibrated with the Silicon Raman line 520 cm−1. The Raman spectra were collected in a backscattering geometry. To achieve SERS characterization the well-known explosive molecule of 2,4,6-trinitrotoluene [C7H5N3O6] (TNT) at an ultralow concentration [25 nM] was chosen. Initially, the Ag nanoparticle films [NPF] were prepared by the Ag colloids synthesized at the energies ∼1000 μJ, ∼1200 μJ, and ∼1400 μJ in cylindrical focusing. To describe the SERS activity of the prepared nanoparticle films [NPF] they have been labelled as NPF-1000, NPF-1200, NPF-1400. Later on, the TNT analyte solution (10 μL) was drop casted on the Ag nanoparticle films [NPF] to create an analyte monolayer and allowed it to dry for some time. In the next step, the Ag NPF loaded with TNT molecules were kept under the objective lens of the micro Raman spectrometer. To compare the SERS data of TNT from these Ag NPFs, spontaneous Raman spectrum of TNT at 0.1 molar concentration was recorded from a non-plasmonic glass surface at the same conditions of the Raman experiment. In the present study, we have investigated the (a) generation of Ag NPS and NSs through laser ablation of Ag targets immersed in DDW using cylindrical focusing geometry using laser pulses of duration ∼2 ps (b) the synthesis of Ag nanoribbons at an input pulse energy 1200 μJ, (c) surface enhanced Raman studies of 2,4,6-trinitrotoluene (TNT) probe molecules at nanomolar (25 nM) concentration adsorbed Ag NPFs.
3. Results and discussion
Preceding the ablation experiments on Ag targets in DDW at input pulse energies of ∼1000 μJ, ∼1200 μJ, and ∼1400 μJ, ablation with input energies lower than 1000 μJ was attempted as well. It was observed that no significant ablation was accomplished in a short span of 5 minutes. This was evident from the coloration of the DDW in which ablation was carried out, since if at all significant ablation occurred, the released NPs in the liquid in which ablation is carried out certainly modified the color of the liquid.3.1 UV-Vis absorption characterization
The synthesized Ag colloids in DDW at low (1000 μJ), moderate (1200 μJ), and high (1400 μJ) pulse energies of the input ps laser pulses were characterized by UV-Visible spectrometer after sonicating them in hot water. Fig. 1 depicts the recorded UV-Visible absorption data and the observed localized surface plasmon resonances (LSPR) wavelengths 406 nm, 408 nm, and 410 nm for the colloids prepared at 1000 μJ, 1200 μJ, and ∼1400 μJ, respectively. Fig. 1 illustrates an increase in the absorbance as well as plasmon band width of the Ag colloids as a function of the pulse energy at which ablation was carried out. Moreover, the UV-Visible absorption spectra at each pulse energy exhibited a broad plasmon band which represents the maximum size distribution of NPs. As a function of input pulse energy, full width at half maximum [FWHM] of each spectrum was increased as shown in Fig. 1. According to the data in Fig. 1 the absorbance of the colloids increased with input pulse energy since the absorbance and the generated Ag NPs yield is proportional to the input pulse energy. Moreover, these NPs are surfactant free and do not require cleaning procedure prior to their utilization.49 Experimentally recorded FWHMs were ∼84 nm [453–369 nm], ∼118 nm [481–363 nm], and ∼146 nm [510–364 nm] for the input pulse energies ∼1000 μJ, ∼1200 μJ, and ∼1400 μJ, respectively. As Garcia et al.50 explained in effective medium theory (EMT), decrement in size of NPs leads to a red shift in the actual LSPR peak position due to combined resonances. According to the existing literature, we do expect that for one dimensional nanostructure such as nanorods two resonant peaks are observed. One peak is due to transverse surface plasmon resonance modes which do not depend on the asymmetry of the nanostructure while the other one being due to the longitudinal surface plasmon resonance modes where in the collective oscillations of conduction band electrons are along the length of the nanostructure. When the length of the nanostructure is large (e.g. nanowires or nanoribbons), surface plasmon vibrations transverse to the length of the nanowire/ribbon only sustain and the longitudinal surface plasmon peak dies off and/or is usually buried in the shoulder of the peak in the visible range. This is evident from our early reports where we observed only one SPR peak for Ag nanowires in isopropyl alcohol.51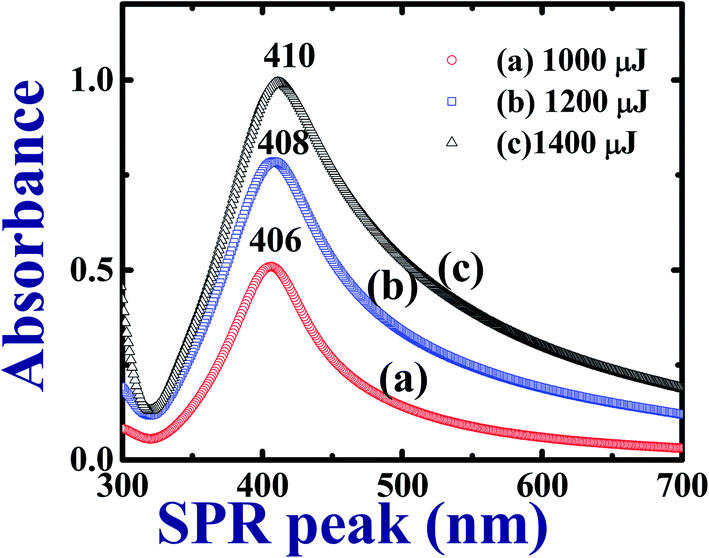 | ||
| Fig. 1 UV-Visible absorption spectra of Ag colloids in DDW prepared with cylindrical focusing using ∼2 ps laser pulses at different pulse energies. | ||
3.2 TEM characterization
TEM characterization was carried out to understand and evaluate the morphological information and the size distribution of the prepared Ag colloids. Fig. 2 illustrates the recorded TEM data of the Ag NPs prepared at 1000 μJ [Fig. 2(a)] and ∼1400 μJ [Fig. 2(b)] pulse energies. The obtained TEM data depicted monodispersed Ag NPs with an average size ∼8 nm, and ∼5 nm, respectively, as shown in the inset histograms of Fig. 2(a) and (b). To construct the histogram, roughly 500–600 nanoparticles were considered and the task was accomplished with image-J software. It was evident from the experimental data that as the input pulse energy increased the average size of the produced Ag NPs decreased which could be due to the higher rate of ablation influenced by enhanced multi-photon ionization within the metal which exerts a strong recoil pressure by the evaporation of DDW adjacent to the metallic plume. Consequently, the metallic plume is formed under the influence of line focussing. The metallic plume produced in the line focussing splashes into the particles with higher recoil pressure. Therefore, the average size of the Ag NPs fabricated at 1400 μJ were of smaller in average size compared to the Ag NPs fabricated at 1000 μJ energy. Insets in the Fig. 2(a) and (b) also depict the selected area electron diffraction (SAED) and the crystallographic phase of the Ag NPs can be drawn from the SAED patterns. SAED is a concentric ring pattern with some bright diffraction spots. SAED pattern in the insets of Fig. 2(a) and (b) demonstrate the polycrystalline nature of the Ag NPs. Concentric ring patterns in the SAED are overlapped with the dotted circles for their clear visualization. The measured diameter of the concentric ring patterns beginning from the centre are 2.25 Å, 2.1 Å, 1.6 Å corresponding to the miller indices (111), (200), (220) respectively. The measured diameter of the concentric ring pattern represents the interplanar separation of the crystallographic structure. The measured interplanar separations are well agreed with face centered cube (FCC) crystal phase of pure Ag as mentioned in PCPDF file no 01-1167.4,52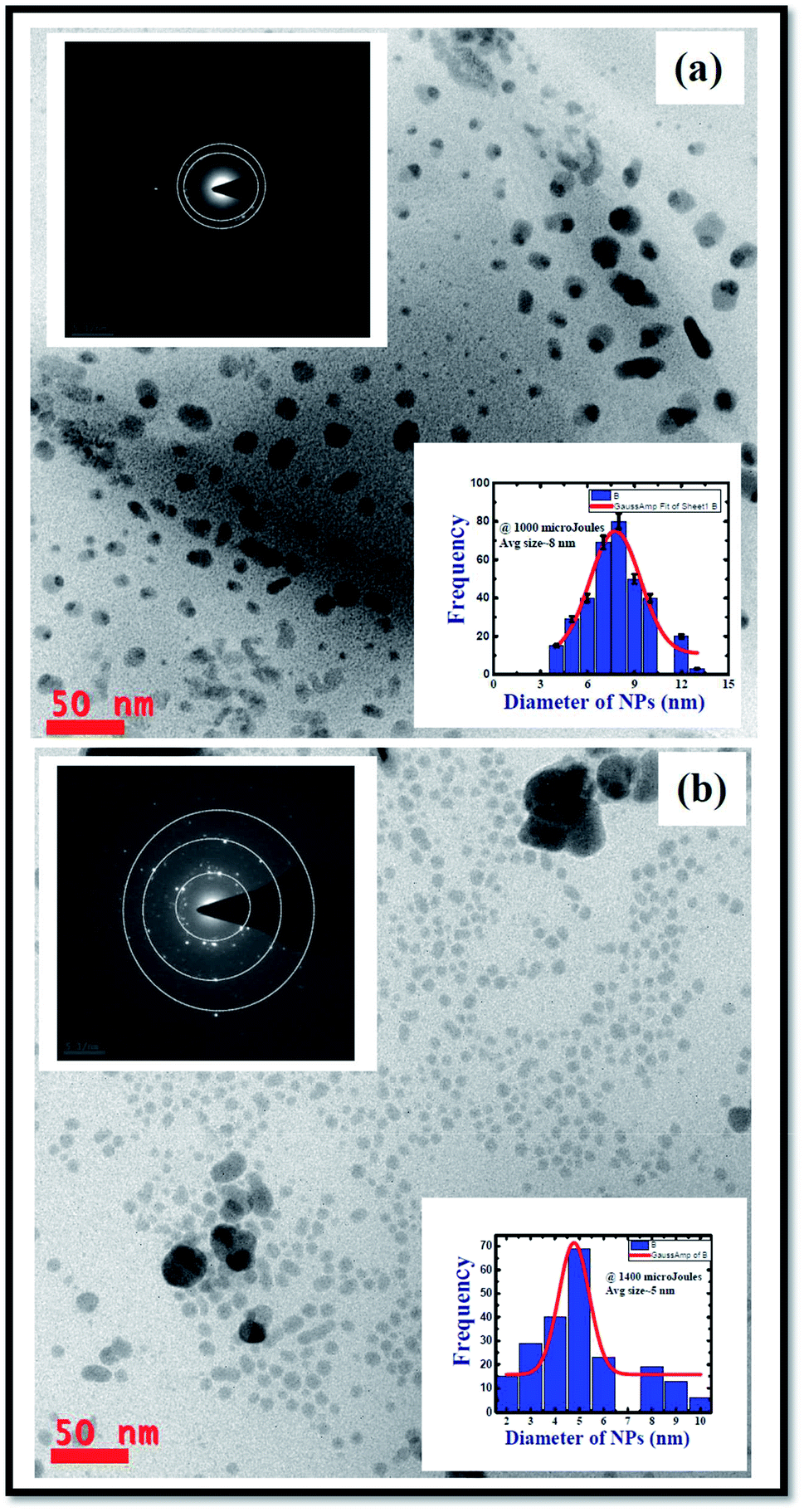 | ||
| Fig. 2 TEM images of the Ag NPs produced by cylindrical focussing geometry in laser ablation at a pulse energy of (a) 1000 μJ demonstrating the Ag nanoparticles with an average size ∼8 nm (b) 1400 μJ demonstrating the Ag nanoparticles with an average size ∼5 nm. Insets show the SAED patterns which reveal the amorphous phase of fabricated silver nanoparticles. | ||
Fig. 3 shows the TEM image of the Ag nanomaterials fabricated at ∼1200 μJ, and interestingly, we did not observe Ag nanospheres but Ag nano ribbons. In ULAL, most of the reports demonstrated the formation of nanospheres. As per the knowledge of the authors, two possibilities might cause the formation of Ag nano-ribbons. First possibility is that, the Ag nanospheres initially formed in the first nucleation process, might be under the effect of line light sheet that produced by the cylindrical focussing geometry. Consequently, light sheet might provoke the nano-welding of Ag NPs formed. Thus, the nano-welding of neighbouring Ag NPs may give rise to the chain/ribbon like structures as shown in Fig. 3. Second possibility could be attributed to the produced cavitation bubble and its oscillations in this case. An extensive theoretical as well experimental studies are essential to understand the cavitation bubble formation and the time evolution of it in the case of cylindrical focusing. In their recent work that talks about the ablation of metal targets using longer pulses (nanosecond), Wagener et al.53 revealed that the oscillations of the cavitation bubble strongly regulate the formation of the NPs and agglomerates with diverse shapes. Similarly, Lavisse et al.54 described the chain like structures in their work and they are similar to the structures that we achieved in the cylindrical focusing at a pulse energy 1200 μJ. According to their results, the shape of the produced NPs is determined by the inside pressure and temperature of the cavitation bubble. The polarity and viscosity of the liquid medium in which ablation is supposed to carry out determines the pressure inside the cavitation bubble. In due course of laser ablation, the cavitation bubble formed in the process of plasma plume expansion. Thus, formed cavitation bubble carries metallic condensates and liquid vapor inside it. The kind of interactions of the metallic condensates with the surface shock waves inside the cavitation bubble strongly influence the morphological structures of produced NPs. The production of NPs with different sizes and diverse morphologies are consequences of cavitation bubble oscillations and its collapse. The dynamics of the cavitation bubble can be explained using the famous Raleigh–Plesset (RP) equation.55 Recently DellÁglio et al.56 worked on the plasma charging effect on the NPs releasing from cavitation bubble on a silver target (immersed in liquid) ablated using nanosecond laser pulses. They demonstrated the dynamics of cavitation bubble using the technique of shadowgraphy.
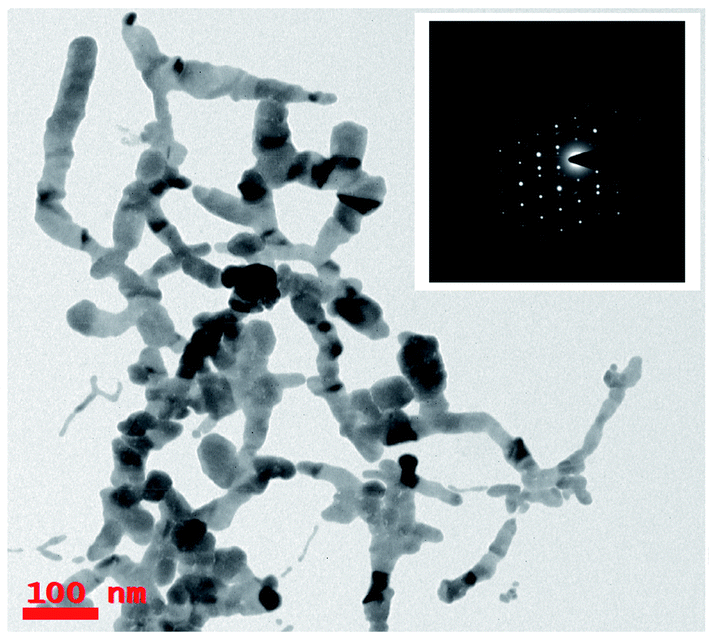 | ||
| Fig. 3 TEM images of Ag ribbons produced by cylindrical focussing geometry in laser ablation at a pulse energy ∼1200 μJ demonstrate the Ag nanowires/ribbons with average length was ∼0.5–1 μm. Inset shows the SAED patterns which reveal the crystalline phase of fabricated silver nanowires. | ||
We did not observe these kinds of structures when ablation was performed at low energy (∼1000 μJ) and high energy (∼1400 μJ) except the intermediate energy ∼1200 μJ. Furthermore, it was observed that most of the TEM images recorded for the Ag colloids prepared at ∼1200 μJ exhibited same ribbon kind of morphologies and its origin is yet to be understood. But the results obtained in the present experiment may require further theoretical studies to interpret the formation of ribbon kind of structures, particularly at this energy (1200 μJ). The measured width of the ribbons was in between 20–30 nm and with lengths varied from 0.6 μm and above. Inset of Fig. 3 depicts the SAED pattern which provided a visualization that the crystal phase of Ag nanoribbons is different when compared with the crystal phase of Ag nanospheres obtained at other two energies. Even the SAED pattern recorded for Ag nanoribbons formed in the present experiment is in close resemblance with the SAED patterns obtained for Ag nanowires that we reported earlier.51 SAED pattern of Ag nanoribbons were further analysed, as shown in Fig. 4, and those illustrated a single crystalline structure of Ag nanoribbons with an interplanar spacing ∼0.23 nm, which is in good agreement with the interplanar spacing of [111] lattice plane of Ag. In the determination of labels of each bright spot in SAED pattern, (011) was taken as the zone axis. Thus, Ag nanoribbons formed at 1200 μJ were single crystalline which is different from the poly crystalline nature of Ag nanospheres obtained at 1000 μJ, 1400 μJ.
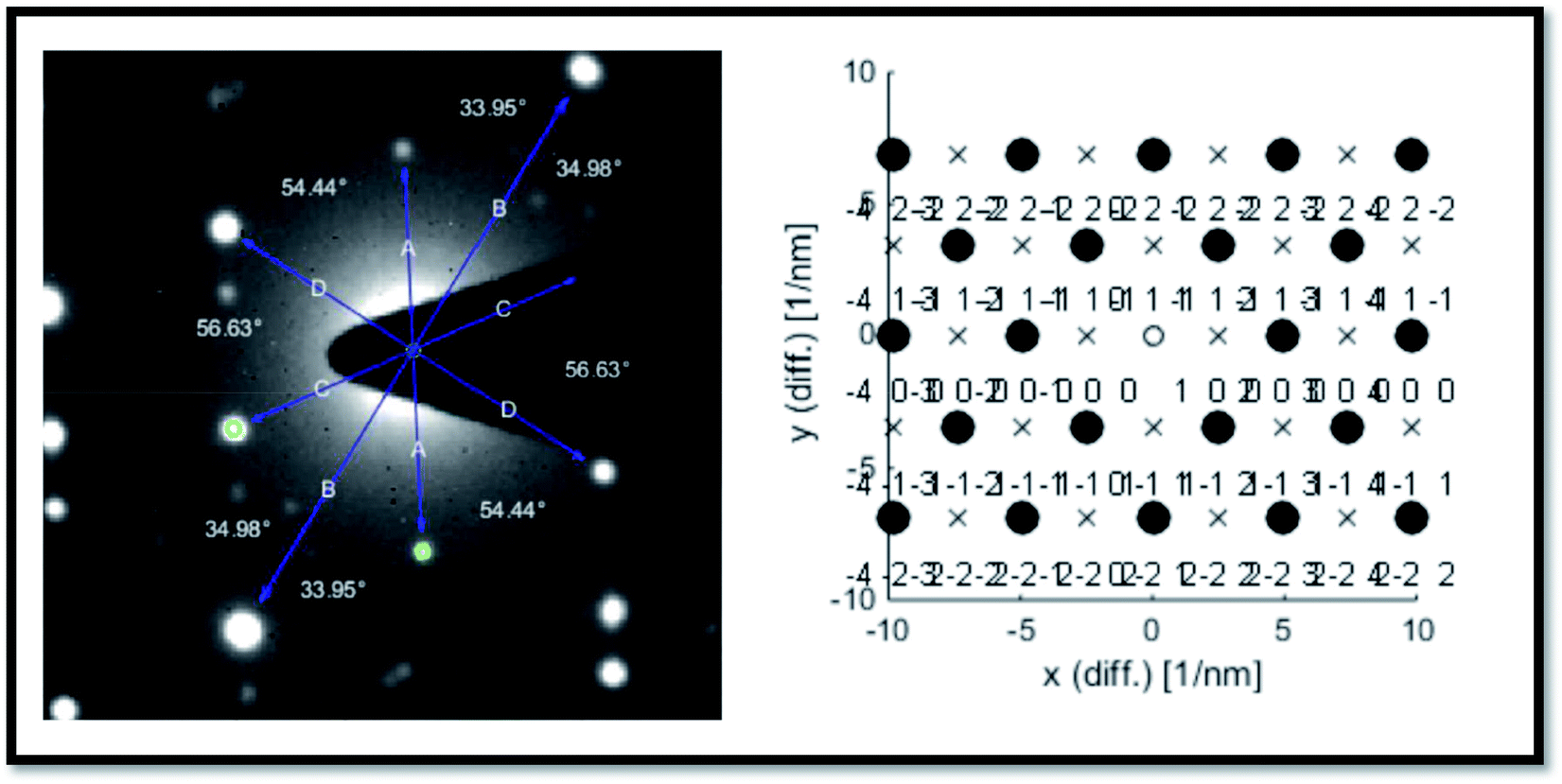 | ||
| Fig. 4 Analysis of the SAED pattern shown in the inset of Fig. 3 and the corresponding plane labels of the silver nanoribbons. | ||
We performed the silver target ablation in DDW using cylindrical focusing and after inspecting the results obtained at 1200 μJ, we initially thought it was an artifact and we repeated the experiments at this particular energy. Remarkably, we obtained a similar result i.e. ribbon structures formation rather than nanospheres. Therefore, we could believe, for the set of the parameters utilized, such as input beam diameter, polarization, liquid, height of the liquid above the target surface, focal length of the cylindrical lens, focal plane, that 1200 μJ indeed is a magic input energy though the exact reasons are yet to be ascertained. At the cylindrical focusing plane, specifically for 1200 μJ, the dynamics of the cavitation bubble might have been different compared to other two energies 1000 μJ and 1400 μJ. After achieving this result (at ∼1200 μJ) we could have tried for other input energies (e.g. 1050 μJ, 1100 μJ, 1150 μJ). We performed the ablation using an amplifier (2 ps pulse duration, 800 nm) whose repetition rate was 1 kHz. We do believe that the amplifiers do have pulse to pulse power fluctuations unlike the oscillators of higher repetition rate. In the view of this, we performed the ablation by keeping a difference of ∼200 μJ to distinguish the energies taken (1000 μJ, 1200 μJ, and 1400 μJ). Furthermore, we considered the other possibility behind the observed ribbon like structures, may be due to the aggregation of the Ag nanoparticles during the TEM grid preparation. If this was indeed the case, certainly similar structures should have been seen in the TEM grids prepared for the other two energies (1000 μJ and 1400 μJ), but we did not observe any. Consequently, we do believe these Ag ribbon structures are not formed during the TEM preparation since the water is a polar solvent which strongly opposes this kind of secondary nucleation to a certain extent due to the formation of electric double layers (EDL) on the surface of Ag spheres, which prevents the aggregation. We followed a similar procedure to prepare the TEM grids for the colloids prepared at these three energies. The results obtained for the colloids prepared at 1200 μJ were repeated and all the images those were taken for this sample exhibited same ribbon kind structures. This led us to believe that they might be formed due to the nano-welding of the Ag nanospheres under the influence of the line focusing. Though we present our experimental observations we also do believe that extensive experimental studies are required to confirm the origin of nanoribbons. Furthermore, if these Ag nanoribbons were formed while taking the TEM images due to effect of the liquid, electron diffraction patterns obtained must be same as that of the Ag nanospheres. However, we had obtained a different SAED patterns, specifically for these samples prepared at 1200 μJ, which made us trust they were indeed Ag nanoribbons. Recently, Nancy et al.57 demonstrated the properties of silver plasma formed within the liquid medium as a function input energies/fluences. They observed the aggregation of silver nanoparticles and shape variations to form chains as the fluence was increased. According to the data shown by them, Ag nano-spheres were observed at a preceding fluence where the Ag chains were observed. This shows the possibility of nano welding between the Ag nanospheres under the line focusing, to alter the shapes and forming Ag ribbon like structures. The laser ablation experiment has been carried out at 1200 μJ multiple times with the same experimental conditions and we achieved similar ribbon kind structures in the TEM analysis as shown in Fig. 5(a)–(c). Along with the ribbons, at some locations joined silver nanoparticles were also observed. After obtaining these results only we could confirm the possibility of the formation Ag ribbons at this particular energy. Fig. 2 data also confirmed that at higher energies the particle size was ∼5 nm suggesting the possibility of fragmentation. However, we still do not completely understand the origin of the formation of these Ag nanoribbons and further details experiments will be performed in future to ascertain it.
 | ||
| Fig. 5 (a)–(c). TEM images of Ag nanoribbons obtained during the ablation of Ag targets immersed in double distilled water at an input pulse energy ∼1200 μJ using 2 ps laser pulses in different experimental trials. | ||
3.3 FESEM characterization
Morphologies of laser exposed portions revealed a mere rough structure at low pulse energy and coral kind structures at a medium and at higher input pulse energies as shown in Fig. 6. It is evident from the Fig. 6(a) that the input pulse energy (∼1000 μJ) is not that enough to ablate the Ag target surface completely. Perhaps, in the line focussing, all the pulse energy is distributed along the line focussing uniformly.36 Consequently, at every point on the line focusing, energy has to be higher than the damage threshold of the Ag metal. This might not occur appropriately with the input pulse energy ∼1000 μJ, whereas at other two pulse energies this condition is fulfilled and hence a significant ablation was achieved. FESEM images demonstrated the coral structures and are increased as a function of input pulse energy. Physics behind the fabricated NPs, ribbons, and coral structures in the ablation with cylindrical beam focusing require further theoretical and experimental studies which are under progress. Further, we expected, LIPSS in the ablated portions of Ag targets using the cylindrical focussing as reported by many authors.35–37 But we did not observe any such structures at low, moderate and high energies. This may be due to the low electron-phonon coupling factor (γ) of Ag metal58,59 compared to the other plasmonic (Au, Cu), and semiconducting [Si, MOS2, etc.] determines the formation of the ripple structure. In regard of LIPSS one can express, the inherent properties of the metal targets used in ablation than the experimental conditions. Laser ablated portions of the Ag targets acts as wonderful SERS active platforms that enhance the Raman signatures of analyte molecules to a great extent. Laser ablated Ag metal targets and optical Fourier transform of them gives optical gratings with random grating period. When an analyte molecule at low concentration adsorbed on the surface and if it is irradiated by a laser light of suitable wavelength, evanescent/local fields in the proximity of analyte molecule. These machined Ag targets support the excitation of LSPs and PSPs. The magnitude of the local fields determines the enhancement of Raman signature elevations. In our earlier studies, we had extensively utilized the laser machined plasmonic metal targets as SERS active and surface enhanced fluorescence platforms (SEF).2–8,60 Though we did not pay enough attention to the left-over laser exposed portions (since our interest was with using the nano-ribbons), the recorded FESEM images (as shown in Fig. 6) demonstrate a variation in the way they got exposed. This clearly reflects in the roughness of the surfaces. Fig. 6(a) shows less roughness and 6(c) demonstrates a higher roughness, whereas Fig. 6(b) shows a moderate (intermediate) roughness. | ||
| Fig. 6 FESEM images of Ag nanostructures produced by cylindrical focussing geometry in laser ablation at a pulse energy (a) 1000 μJ (b) 1200 μJ and (c) 1400 μJ. | ||
When we started the present experiments, we tried the ablation at different energies below 1000 μJ. However, we did not achieve a considerable amount of ablation which could confirm with our naked eye too. If ablation of metal is performed in any liquid medium, nanoparticles will be released into the surrounding liquid medium. The surface plasmon effect of thus released plasmonic metal nanoparticles impart specific coloration to the liquid. Coloration is thus a good measure to describe whether the ablation was initiated or not. Depending on this observation, we could confirm the absence of ablation below 1000 μJ. This energy is too high if we has worked with a spherical lens focusing. Perhaps, the line spread of the focused light energy in cylindrical geometry required more input energy to be dumped on the target surface. As we thought 1000 μJ is the threshold energy required to achieve significant ablation, 1200 μJ is moderate and 1400 μJ is, somewhat, higher energy. We thought that if we still enhance the input pulse energy beyond 1400 μJ, it may lead to the formation of white light Continuum in the water thereby affecting the ablation mechanism. Consequently, we did not try the ablation for energies beyond 1400 μJ. In our future endeavours, definitely, we will investigate the effect of input energies beyond 1400 μJ (keeping the other parameters as they are and controlling the liquid level). Other anticipation about the ablation of Ag beyond 1400 μJ is that Ag nanospheres of average size less than ∼5 nm will be produced (since at 1400 μJ the average sizes obtained were ∼5 nm).
3.4 SERS studies of trinitrotoluene (TNT)
The SERS activity of the NPF-1000, NPF-1200, NPF-1400 was investigated by recording the Raman spectra of TNT molecules of 25 nM concentration using an exciting line 532 nm. To record the Raman spectra of TNT molecules, first master solution of 0.1 M was prepared by dissolving the ∼114 milligrams of 2,4,6-trinitrotoluene (TNT) powder was dissolved in hyper pure methanol solvent. Master solution was dissolved in 10 mL of methanol by diluting the master solution in a required proportion. To achieve this, we diluted the 0.1 M solution to prepare micromolar concentration and later on it was diluted to get 25 nM analyte solution of 2,4,6-trinitrotoluene (TNT). At first, Ag colloidal solutions fabricated at 1000 μJ, 1200 μJ, and 1400 μJ in cylindrical focusing were drop casted on three individual cover slips. The utilized quantity of Ag colloidal solution was ∼20 μJ and after drop casting, the coverslip was allowed to dry for some time. Thus, the SERS active platforms labelled NPF-1000, NPF-1200, NPF-1400 were prepared. Secondly, a monolayer of 2,4,6-trinitrotoluene (TNT) solution in a volume ∼10 μL was drop casted on the three SERS active platforms NPF-1000, NPF-1200, NPF-1400, and again allowed them to dry for some time. Unless drying up the analyte solution on SERS active platform, autofluorescence from the sample prevail the Raman signatures of the analyte molecules. While drop casting the analyte solution on the SERS active substrate, we ensured the formation of a monolayer. The usage of higher volumes of analyte, the evanescent field provided by the SERS active substrate fall down turn results in very insignificant elevation of Raman signatures of the analyte molecules. It was evident from many reports that the SERS activity of the substrate is a function of the distance between SERS active platform and the analyte molecule (typically should be few nm). When the separation between the molecule and nanoplatform is beyond the effective range of the evanescent field, Raman spectra recorded will be overlapped on the fluorescence background. 2,4,6-trinitrotoluene (TNT) sample at high concentration 0.1 M was drop casted on a non-plasmonic glass plate and the Raman spectra of the analyte solution was recorded. This Raman spectrum is considered as the reference Raman spectrum. In the next step, three SERS active platforms NPF-1000, NPF-1200, NPF-1400 loaded with analyte molecules at ultra-low concentration were kept under the WiTech Raman instrument (Alpha 300), and recoded the Raman spectra of TNT Molecules. Recorded Raman spectra of TNT at low concentration from the Ag nanomaterial films demonstrate a bit of Raman signature shifts due to the charge carrier transfer from the Ag nanoplatform and the analyte molecule. The fundamental Raman modes of TNT are tabulated (Table 1) along with the signatures obtained from the three SERS active platforms NPF-1000, NPF-1200, NPF-1400. The Raman activity of each nanoparticle film (NPF) is evaluated by estimating the analytical enhancement factor (AEF)61 as shown in eqn (1)
 | (1) |
| S. no. | Wavenumber (cm−1) | Mode assignment | |
|---|---|---|---|
| Reported | Present study | ||
| 1 | 717 | 728, 731, 746 | Asymmetric CCC in-plane ring bending deformation30 |
| 2 | 789 | — | CH out-of-plane vibrations |
| 3 | 821 | 807 | Scissoring NO2 mode |
| 920 | 914, 916, 918, 920, | — | |
| 4 | 1085 | 1020, 1070, 1076, 1086 | CH (ring) bending mode |
| 5 | 1207 | 1196, 1207, 1210, 1220 | Ring breathing |
| 6 | 1362 | 1355, 1361, 1366, 1368 | Symmetric NO2 stretching |
| 7 | 1534 | 1545, 1560 | Asymmetric NO2 stretching |
| 8 | 1617 | 1643 | Aromatic NO2 conjugation |
Fig. 7 demonstrates the Raman spectra of 2,4,6-trinitrotoluene (TNT) at a concentration of 25 nM from the Ag nanoparticle films NPF-1000, NPF-1200, and NPF-1400. It was observed that the SERS spectra of TNT molecules at nanomolar concentration recorded from the Ag nanomaterials films were different when compared with the Raman spectrum of TNT at higher concentration (0.1 M) recorded from non-plasmonic platform (Glass/Silicon surface). From the recorded SERS data of TNT, it is evident that the modes that are dormant/not elevated in the Raman spectra of TNT at 0.1 M from a non-plasmonic platform are elevated in a significant manner in the SERS spectra. The recorded Raman spectra demonstrated that a mode corresponding to 1362 cm−1 which is labelled as NO2 symmetrical stretch62 was predominantly elevated. According to the existing literature, the mode corresponding to 1362 cm−1 is very strong band but the wavenumber of the mentioned NO2 symmetrical stretch is shifted slightly to lower wavenumbers as shown in the Fig. 7. In addition to the 1362 cm−1 mode, many others such as 918 cm−1, 1020 cm−1, 1210 cm−1 modes were also observed in the SERS spectra. The reported modes of TNT molecules in the earlier reports and the modes those observed in the present study are mentioned in Table 1 along with their labels of vibration. The estimated enhancement factors (E.F) corresponding to 1362 cm−1 mode from NPF-1000, NPF-1200, and NPF-1400 were ∼3.36 × 106, ∼9.89 × 106, ∼5.0 × 106, respectively. The estimated EF for the mode corresponding to 1362 cm−1 mode from NPF-1200 was almost one order larger than the other two SERS platforms NPF-1200 which certainly confirms the different SERS activity. Consequently, NPF-1200 demonstrated a better Raman signature enhancement for NO2 symmetrical stretch mode corresponding to 1362 cm−1 mode of TNT.
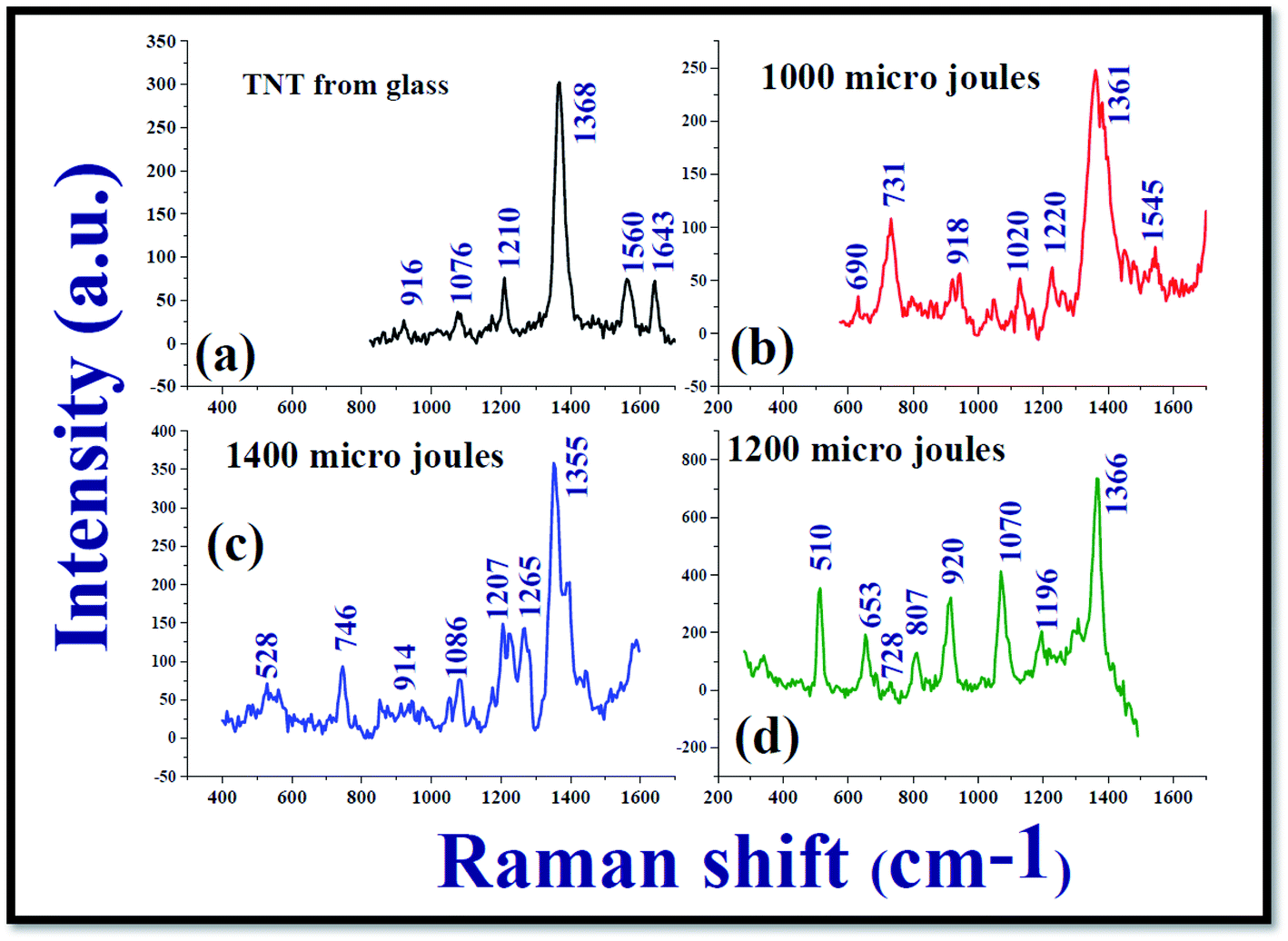 | ||
| Fig. 7 (a) Normal Raman spectrum of TNT molecules in ethanol at a concentration 0.1 M recorded from glass substrate; SERS spectrum of TNT analyte molecules at a concentration ∼25 nM recorded from (b) NPF-1000 (c) 1400 μJ and (d) 1200 μJ with an excitation wavelength of 532 nm. Time of integration for each spectrum was 5 seconds. | ||
Kneipp et al.63 attempted recording the SERS spectra of TNT molecules at a concentration 0.01–7.5 × 10−7 M in the presence of gold (Au) and silver (Ag) NPS. They achieved an enhancement factor of ∼105. Rozo et al.64,65 demonstrated high repeatability in utilizing Ag NPs as the SERS active platforms. An exclusive work on the detection of TNT molecules at ultralow concentrations using Ag and Au NPs was documented by Zapata et al.66 Chen et al.67 reported on the detection of TNT molecules both in solution and vapour phases at a concentration of 10−8 M, 10−6 M, respectively. They had taken a Silicon wafer which was decorated by Ag NPs as the SERS active platform and achieved a good limit of detection and possibility of repeatability of the detection. Ben-Jaber et al.68 exclusively reviewed the SERS studies of various active platforms to detect TNT molecules in a range of concentrations 10−5–10−9 M and could achieve enhancement factors in the range of 104–107. Most of these studies utilize the Ag/Ag NPS those prepared under standard wet chemical methods, which require vigorous cleaning procedures and the incorporation of different surfactants. In the present experiment, we achieved the same enhancement factor (107) without subjecting the SERS active platforms to chemicals and without a long-time gap, since Ag NPs (nano-ribbons too) fabricated using the cylindrical lens require very less time (typically five minutes). Further, it has been well-established that picosecond ablation provides better yield of NPs when compared to fs ablation in liquids. Additionally, we can also utilize the NSs obtained from these experiments separately for SERS measurements and also along with the NPs obtained or simple thermal evaporation of plasmonic layers, which will certainly provide additional enhancements69–71 for detection of even lower concentrations of several hazardous materials.
All the results that we presented here were confirmed to be reproducible. While working with the SERS characterization, we have prepared SERS active platforms on cover slips using the drop cast method. When we worked on the SERS, we could confirm whether the results are reproducible or not by recording the RAMAN spectra of TNT from the other cover slips in a systematic way. We could achieve a good reproducibility of the SERS measurements. We recorded the Raman spectra of TNT molecules at a concentration 25 nM from the SERS active platforms prepared by Ag nanospheres/nanoribbons prepared at 1000 μJ, 1200 μJ and 1400 μJ with an excitation wavelength of 532 nm, time of integration for each spectrum was 5 seconds. To check the uniformity of SERS activity of nanofilm fabricated by Ag nanospheres/nanoribbons, the Raman spectra of TNT were recorded from four different locations in each SERS active platform as shown in the Fig. 8 data. From the recorded Raman spectra of each SERS active platform, relative standard deviation (RSD) was measured for the prominent mode 1362 cm−1 corresponding to symmetric NO2 stretch of TNT. The estimated RSD was <3% for these three SERS active platforms prepared at 1000 μJ, 1200 μJ and 1400 μJ, confirming the reproducibility and consistency of the recorded Raman spectra of TNT. Due to experimental limitations we could not perform the Raman mapping. Our future studies will focus on the large area Raman mapping and batch-to-batch reproducibility studies for understanding the potential of these substrates for practical applications.
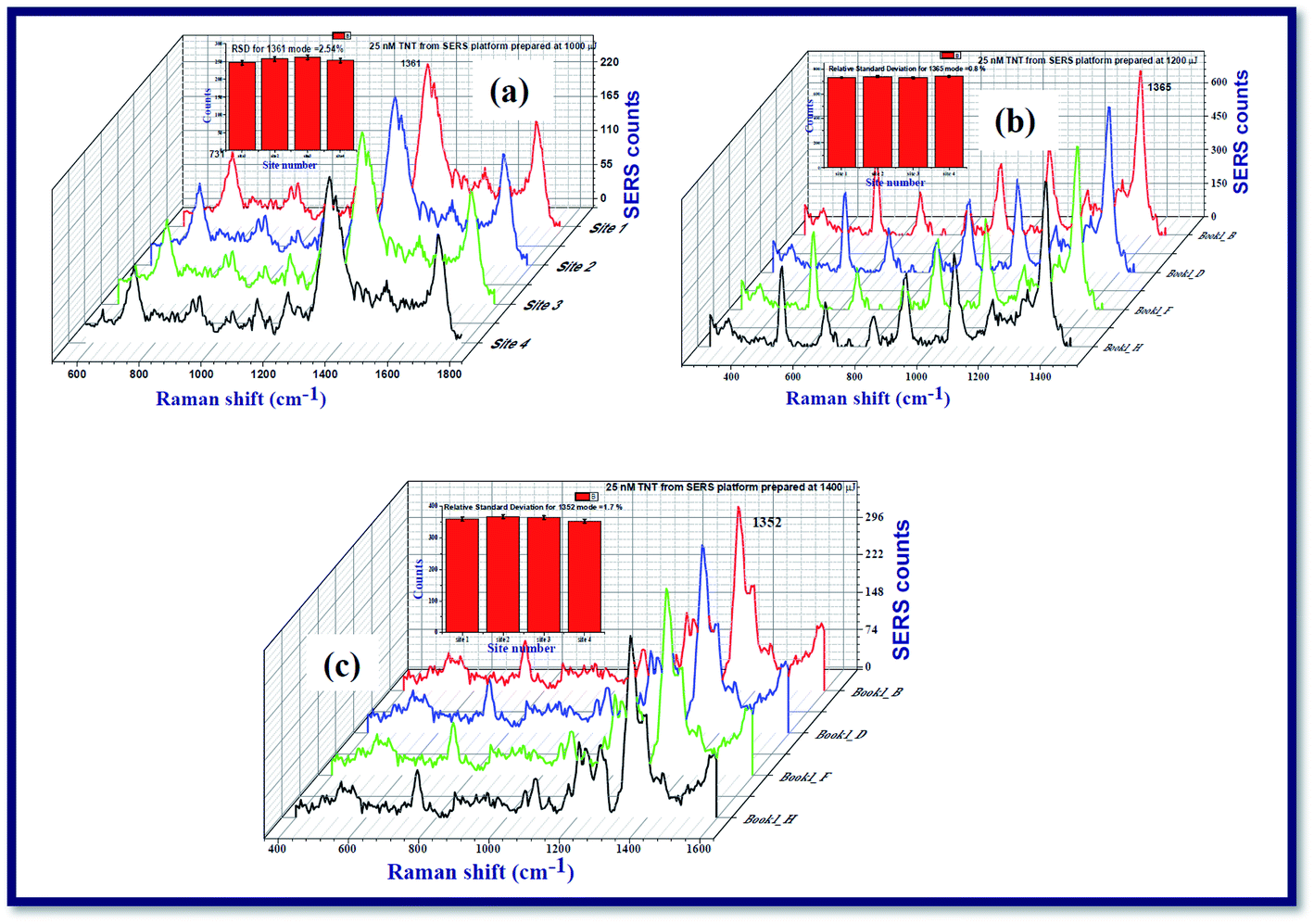 | ||
| Fig. 8 SERS spectra of TNT analyte molecules adsorbed on the films prepared using Ag nanoparticles/ribbons at four different locations of the SERS active platform. The data was recorded form the substrates fabricated at (a) 1000 μJ (b) 1200 μJ and (c) 1400 μJ which shows an RSD value of <3%. | ||
In our earlier studies, we succeeded in the detection of TNT molecules in recording the Raman spectra of micromolar concentration with good reproducibility and, hence, we tried to record/detect TNT molecules at nanomolar (25 nM) concentration with significant repeatability. For this purpose, TNT analyte solution at a concentration 25 nM in methanol was dissolved (5.67 micro grams of TNT in methanol). The Raman spectra of TNT were recorded from the SERS active platforms prepared using Ag nanoparticles/nano ribbons and drop cast method. The uniformity of the recorded data was also checked at different locations of the substrate which was found to be consistent. The utilized TNT of an amount 5.67 × 10−3 milligrams was converted into particles per billion (ppb) by utilizing the conversion unit [1 mg L−1 is ∼1 ppm (parts per million)] and obtained the detection limit of our current study was 5.67 ppb (parts per billion).
4. Conclusions
In conclusion, we investigated the fabrication of silver nanomaterials synthesized by laser ablation of Ag targets immersed in DDW with ∼2 ps pulses using cylindrical focussing geometry, at input pulse energies utilized in the experiment were ∼1000 μJ, ∼1200 μJ, and ∼1400 μJ. It was observed that the input pulse energy ∼1200 μJ demonstrated the fabrication of Ag nanoribbon (20–30 nm width, longer than 0.6 μm) like structures rather the formation of Ag NPs. Ag nanoribbons obtained at ∼1200 μJ exhibited the repeatability when the experiment was carried out for the second time. Thus, the fabricated Ag nanomaterials were utilized to detect the 2,4,6-trinitrotoluene (TNT) explosive molecules at ∼25 nM concentration via surface enhanced Raman scattering studies. The estimated enhancement factor demonstrated that the films prepared by the Ag nanomaterials obtained at energy ∼1200 μJ was ∼107 for 1362 cm−1 mode, corresponding to symmetric NO2 stretch of TNT molecules. The estimated enhancement factor is one order higher for the SERS active platforms prepared with Ag nano-ribbons than the Ag nanospheres. The estimated limit of detection in the present case was ∼6 ppb.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the Advanced Centre of Research in High Energy Materials (ACRHEM), Hyderabad, India funded by DRDO, India, for providing laboratory facilities and assistance. We thank the CNF, University of Hyderabad for assistance in acquiring the TEM images. Venugopal Rao Soma thanks DRDO for financial support through project # ERIP/ ER/ 1501138/ M/01/ 319/ D(R&D) dated February 27, 2017.References
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC.
- G. K. Podagatlapalli, S. Hamad, S. Sreedhar, S. P. Tewari and S. V. Rao, Chem. Phys. Lett., 2012, 530, 93–97 CrossRef CAS.
- G. K. Podagatlapalli, S. Hamad, S. P. Tewari, S. Sreedhar, M. D. Prasad and S. V. Rao, J. Appl. Phys., 2013, 113, 073106 CrossRef.
- G. K. Podagatlapalli, S. Hamad, M. A. Mohiddon and S. V. Rao, Appl. Surf. Sci., 2014, 303, 217–232 CrossRef.
- S. Hamad, G. K. Podagatlapalli, V. S. Vendamani, S. V. S. Nageswara Rao, A. P. Pathak and S. V. Rao, J. Phys. Chem. C, 2014, 118, 7139–7151 CrossRef CAS.
- S. Hamad, G. K. Podagatlapalli, M. A. Mohiddon and V. R. Soma, Appl. Phys. Lett., 2014, 104, 263104 CrossRef.
- S. V. Rao, G. K. Podagatlapalli and S. Hamad, J. Nanosci. Nanotechnol., 2014, 14, 1364–1388 CrossRef CAS.
- S. Hamad, S. S. B. Moram, B. Yendeti, G. Krishna Podagatlapalli, S. V. S. Nageswara Rao, A. P. Pathak, M. A. Mohiddon and V. R. Soma, ACS Omega, 2018, 3, 18420–18432 CrossRef CAS.
- A. Wagner and W. Pruss, J. Phys. Chem. Ref. Data, 2002, 31, 387–535 CrossRef.
- G. Mie, Ann. Phys., 1908, 4, 1–69 Search PubMed.
- V. Kotaidis, C. Dahmen, G. von Plessen, F. Springer and A. Plech, J. Chem. Phys., 2006, 124, 184702 CrossRef CAS.
- A. Plech, P. Leiderer and J. Boneberg, Laser Photonics Rev., 2009, 3, 435–451 CrossRef CAS.
- M. I. Stockman, Opt. Express, 2011, 19, 22029–22106 CrossRef.
- D. Tan, S. Zhou, J. Qiu and N. Khusro, J. Photochem. Photobiol., C, 2013, 17, 50–68 CrossRef CAS.
- V. Talanquer and D. W. Oxtoby, J. Chem. Phys., 1994, 100, 5190–5200 CrossRef CAS.
- V. E. Gruzdev, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 205106 CrossRef.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS.
- K. Park, S. Lee, E. Kang, K. Kim, K. Choi and I. C. Kwon, Adv. Funct. Mater., 2009, 19, 1553–1566 CrossRef CAS.
- W. J. Galush, S. A. Shelby, M. J. Mulvihill, A. Tao, P. Yang and J. T. Groves, Nano Lett., 2009, 9, 2077–2082 CrossRef CAS.
- R. W. Schoenlein, W. Z. Lin, J. G. Fujimoto and G. L. Eesley, Phys. Rev. Lett., 1987, 58, 1680–1683 CrossRef CAS.
- R. R. Letfullin, T. F. George, G. C. Duree and B. M. Bollinger, Adv. Opt. Technol., 2008, 2008, 1–8 CrossRef.
- R. H. M. Groeneveld, R. Sprik and A. Lagendijk, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 5079 CrossRef.
- D. Perez and L. J. Lewis, Phys. Rev. B, 2003, 67, 184102 CrossRef.
- M. Gill-Comeau and L. J. Lewis, Phys. Rev. B, 2011, 84, 224110 CrossRef.
- M. E. Povarnitsyn, T. E. Itina, P. R. Levashov and K. V. Khishchenko, Phys. Chem. Chem. Phys., 2013, 15, 3108 RSC.
- K. Sokolowski-Tinten, J. Bialkowski, A. Cavalleri, D. von der Linde, A. Oparin, J. Meyer-ter-Vehn and S. I. Anisimov, Phys. Rev. Lett., 1998, 81, 224 CrossRef CAS.
- M. E. Povarnitsyn, T. E. Itina, P. R. Levashov and K. V. Khishchenko, Phys. Chem. Chem. Phys., 2013, 15, 3108 RSC.
- D. Perez and L. J. Lewis, Phys. Rev. Lett., 2002, 89, 255504 CrossRef.
- X. Z. Zeng, X. L. Mao, R. Greif and R. E. Russo, Proc. SPIE, 2004, 5448, 1150 CrossRef CAS.
- A. M. Kietzig, S. Hatzikiriakos and P. Englezos, Patterned, Langmuir, 2009, 25, 4821–4827 CrossRef CAS.
- M. Gedvilas, J. Miksys and G. Raciukaitis, RSC Adv., 2015, 5, 75075–75080 RSC.
- A. Borowiec and H. K. Haugen, Appl. Phys. Lett., 2003, 82, 4462 CrossRef CAS.
- J. Bonse and J. Kruger, Femtosecond laser-induced periodic surface structures, J. Laser Appl., 2012, 24, 042006 CrossRef.
- S. K. Das, K. Dasari, A. Rosenfeld and R. Grunwald, Nanotechnology, 2010, 21(15), 155302 CrossRef.
- M. Ardron, N. Weston and D. Hand, Appl. Surf. Sci., 2014, 313, 123–131 CrossRef CAS.
- Y. Zhang, G. Zou, L. Liu, Y. Zhao, Q. Liang, A. Wu and Y. N. Zhou, Appl. Surf. Sci., 2016, 389, 554–559 CrossRef CAS.
- M. S. Sindu, P. Munjal and K. P. Singh, Appl. Phys. A, 2018, 124, 46 Search PubMed.
- A. H. Qiao, J. Yang, J. Li, Q. Liu, J. Liu and C. Guo, Materials, 2018, 11, 2380 CrossRef.
- D. L. Jeanmaire and R. P. V. Duyne, Annu. Rev. Phys. Chem., 1977, 84, 1–20 Search PubMed; M. Fleischman, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- A. Crookell, M. Fleischmann, M. Hanniet and P. J. Hendra, Chem. Phys. Lett., 1988, 149, 123–127 CrossRef CAS.
- D. B. Chase and B. A. Parkinson, Appl. Spectrosc., 1988, 42, 1186–1187 CrossRef CAS.
- S. M. Angel, L. F. Katz, D. D. Archibald and D. E. Hongis, Appl. Spectrosc., 1989, 43, 367–372 CrossRef CAS.
- K. Kniepp, Exp. Tech. Phys., 1990, 38, 3–28 Search PubMed.
- P. Mulvaney, Surface Plasmon Spectroscopy of Nanosized Metal Particles, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS.
- Y. Yokata, K. Ueno and H. Misawa, Chem. Commun., 2011, 47, 3505–3507 RSC.
- G. Krishna Podagatlapalli, S. Hamad and S. V. Rao, Laser Phys. Lett., 2015, 12, 036003 CrossRef.
- R. M. Tilaki, A. Iraji Zad and S. M. Mahdavi, Appl. Phys. A, 2006, 84, 215–219 CrossRef CAS.
- M. A. Garcia, J. Phys. D: Appl. Phys., 2011, 44, 283001 CrossRef.
- A. Rambabu, B. Dipanjan, A. Ravi Kiran, S. Hamad, S. V. Rao and G. Krishna Podagatlapalli, Opt. Mater., 2019, 96, 109305 CrossRef.
- E. R. Jette and F. Foote, J. Chem. Phys., 1935, 3, 605–616 CrossRef CAS.
- P. Wagener, S. Ibrahimkutty, A. Menzel, A. Plech and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3068–3074 RSC.
- L. Lavisse, J. L. Le Garrec, L. Hallo, J. M. Jouvard, S. Carles, J. Perez, J. B. A. Mitchell, J. Decloux, M. Girault, V. Potin, H. Andrzejewski, M. C. Marco de Lucas and S. Bourgeois, Appl. Phys. Lett., 2012, 100, 164103 CrossRef.
- A. De Bonis, M. Sansone, L. D'Alessio, A. Galasso, A. Santagata and R. Teghil, J. Phys. D: Appl. Phys., 2013, 46(44), 445301 CrossRef.
- M. DellÁglio and A. D. Giocomo, Appl. Surf. Sci., 2020, 515, 146031 CrossRef.
- P. Nancy, J. James, S. Valluvadasan, R. A. V. Kumar and N. Kalarikkal, Nano-Struct. Nano-Objects, 2018, 16, 337–346 CrossRef CAS.
- Z. Lin and L. V. Zhigilei, Phys. Rev. B, 2008, 77, 075133 CrossRef.
- W. Li, Phys. Rev. B, 2015, 92, 075405 CrossRef.
- S. Hamad, G. Krishna Podagatlapalli, Md A. Mohiddon and S. V. Rao, Chem. Phys. Lett., 2015, 621, 171–176 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, J. Chem. Phys., 2009, 130, 181101–181104 CrossRef CAS.
- P. M. Fierro-Mercado and S. P. Hernandez-Rivera, Int. J. Mass Spectrom., 2012, 2012, 716527 Search PubMed.
- K. Kneipp, Y. Wang, R. R. Dasari, M. S. Feld, B. D. Gilbert, J. Janni and J. I. Steinfeld, Spectrochim. Acta, Part A, 1995, 51, 2171–2175 CrossRef.
- J. I. Jerez-Rozo, A. M. Chamoun, S. L. Pena and S. P. Hernandez-Rivera, Proc. SPIE, 2007, 6538, 653824 CrossRef.
- J. I. Jerez-Rozo, O. M. Primera-Pedrozo, M. A. Barreto-Caban and S. P. Hernandez-Rivera, IEEE Sensors, 2008, 8, 974–982 CAS.
- F. Zapata, M. López-López and C. García-Ruiz, Appl. Spectrosc. Rev., 2016, 51(3), 227–262 CrossRef CAS.
- N. Chen, P. Ding, Y. Shi, T. Jin, Y. Su, H. Wang and Y. He, Anal. Chem., 2017, 89(9), 5072–5078 CrossRef CAS.
- S. Ben-Jaber and I. P. Parkin, Sci. Rev. Chem. Commun., 2018, 8(2), 1–10 Search PubMed.
- C. Byram, S. S. B. Moram and V. R. Soma, J. Raman Spectrosc., 2019, 50, 1103–1113 CrossRef CAS.
- B. Chandu, M. S. S. Bharati and S. V. Rao, Analyst, 2019, 144, 2327–2336 RSC.
- C. Byram and V. R. Soma, Nano-Struct. Nano-Objects, 2017, 12, 121–129 CrossRef CAS.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |