
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic magnetic core–shell nanoparticles for DNA extraction†
Tammar Hussein Ali ab,
Amar Mousa Mandalc,
Thorsten Heidelberg*a,
Rusnah Syahila Duali Hussena and
Ean Wai Goha
ab,
Amar Mousa Mandalc,
Thorsten Heidelberg*a,
Rusnah Syahila Duali Hussena and
Ean Wai Goha
aChemistry Department, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: heidelberg@um.edu.my
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Al-Muthanna University, 66001 Samawah, Al Muthanna, Iraq. E-mail: tammar86@gmail.com; tammar@mu.edu.iq
cCollege of Basic Education, Science Department, Al-Muthanna University, 66001 Samawah, Al Muthanna, Iraq
First published on 23rd October 2020
Abstract
Magnetic nanoparticles with specific surface features are interesting materials for biomedical applications. The combination of molecular interactions on small particles with macroscopic cohesion forces offers unique opportunities. This work reports the synthesis of magnetic core–shell nanoparticles with alkylimidazolium coated surface for effective DNA extraction. A magnetic Fe2O3 core was coated with a silica shell and functionalized with an organic halide. This enabled a surface coating with organic cations to mediate effective molecular interactions with polyanionic DNA. The large surface area of the ∼20 nm small particles with a magnetization of 25 emu g−1 enabled high DNA particle loading of 1/30 m% with easy isolation based on an external magnetic field. Moreover, the coating of the particles stabilized DNA against ultrasound initiated fragmentation.
Introduction
Magnetic nanoparticles (MNPs) have huge application potential in various fields, covering industrial use as well as medical diagnostics1 and even therapy.2 The simple and reversible switch between a submicron-scaled dispersion, with high surface area and almost unlimited accessibility, and pseudo-bulk material, with easy and selective collection opportunity, provides almost revolutionary benefits for separations, provided that the particle surface can be designed for selective chemical transformations.3 Particularly interesting chemical applications involve the selective isolation of specific compounds, e.g. from renewable resources,4 as well as organic synthesis, especially in the design of chemical libraries for drug development.5Conjugation of MNPs with biological molecules, in particular nucleic acids, allows designing various nanobiohybrid systems that possess unique magnetic properties and biological selectivity to improve the efficiency of diagnosis and therapy of diseases.4 Various approaches for the conjugation of nucleic acids with MNPs have been proposed:6 A nucleic acid molecule can either directly bind to MNPs, or the formation of chemical bonds requires preliminary modification of the surface of MNPs and/or biomolecules.7 Currently nonspecific interactions between nucleic acid molecules and nanoparticles are typically avoided in nanobiofabrication.8,9
There have been a number of recent studies demonstrating the potential of magnetic nanoparticale as a mean of extraction or delivery of bio-molecules.10–19 Lee et al. systematically investigated an ultrasound enhanced siRNA delivery using a nanocomposite comprising of a deoxycholic acid–chitosane coated perfluoroakane droplets with attached magnetic nanoparticles. The exposure to ultrasound did not affect the siRNA-nanocomposite complex, which was found to be stable at 37 °C for up to 4 h in the serum. Moreover, they confirmed the potential for magnetic localization of the complexes using an externally magnetic field.10 In another paper Xiong et al. used magnetic core–silica shell nanoparticles with large radial mesopores for siRNA delivery. The functionalized nanoparticles combined a high siRNA loading capacity of 2 wt% with strong response to external magnetic fields. The protection of siRNA increased upon coating the core–shell nanoparticles with tannic acid. A corresponding siRNA-loaded dispersion could be applied in a pH-responsive releasing switch.20 In 2020, core–shell magnetic molecularly imprinted polymers were applied for the extraction of patulin from juice samples. The magnetic dispersion of the solid-phase provided a good selectivity for the adsorption.21 In same year two different studies proposed magnetite-based core–shell nanoparticles for solid-phase extraction of antibiotics and heavy metals due to high sensitivity, low-cost and simple operation.22,23 These results demonstrate the promising potential of magnetic core–silica shell nanoparticles with an organic coating for extraction and delivery applications. This prompted us to develop a simple adsorbent for low cost solid-phase extraction of DNA based on functionalization of magnetic core–shell nanoparticles with alkyl imidazoles.
Materials and characterization
Chemicals were obtained from various commercial sources and used without purification. Purification applied column chromatography on silica gel 35–60 mesh using the flash technique. TLC was performed on pre-coated plates of silica gel 60 (GF254). Visualization of compounds applied treatment with KMnO4 and subsequent heating.IR spectra were recorded on a PerkinElmer Frontier ATR FT-IR spectrometer. Dried nanoparticles were subjected to thermal gravimetric and differential thermal analysis (TG-DTA) with 20 °C min−1 ramping. Transmission Electron Microscopy (TEM) was performed with a FEI Tecnai G2-F20 operating at 200 kV with 0.2 nm resolution. Samples were prepared by dispersing the nanoparticles in ethanol and sonicate for 5 min. Prior to deposition on an amorphous carbon-coated 300 mesh copper grid before allowing the solvent to evaporate. The surface morphology of the samples was analyzed by a field emission scanning electron microscopy (SEM) on a Hitachi SU8200 equipped with an energy dispersive X-ray spectrometer EDX (QUANTAX FlatQUAD, Bruker AXS) operated at an accelerating voltage of 20 kV and a working distance of 13 mm. A few milligrams of dried sample were added into a FESEM cell and subsequently scanned by the electronic microscope. The XRD pattern of the particles was recorded on a Bruker D8 Advance X-ray diffraction AXS using Cu Kα radiation (d = 1.54 Å) at a voltage of 40 kV and current of 40 mA at ambient temperature. The magnetic properties were investigated by using a Lakeshore 7400 series, 7407 model with 7 inch electromagnet vibrating sample magnetometer (VSM). The analysis was conducted at room temperature in the field of ±10 kOe. Structural identities are based on NMR spectra (1H and 13C, recorded on a Bruker AVN-400 MHz spectrometer).
Methods
Synthesis of core–shell magnetic nanoparticles (NpFeSi)
Magnetic nanoparticles (MNPs) were prepared by chemical co-precipitation of Fe3+ and Fe2+ ions at a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 according to modified reported method:24,25 Anhydrous FeCl3 (6.0 g, 37 mmol) was added in small portions to a solution of FeCl2·4H2O (4.9 g, 24 mmol) in aqueous ammonia (30%, 100 mL). The reaction mixture with pH ∼ 10 was stirred at 600 rpm for 1 h at room temperature followed by the addition of 15 wt% TEOS (1.6 g, 7.8 mmol). Vigorous stirring was continued for 24 h to generate a black colored precipitate. The resulting silica coated nanoparticles were separated by centrifugation and washed repeatedly with deionized water and HCl (1 N). Washing with acetone was followed by drying at 60 °C overnight to provide NpFeSi (7.0 g).
:1 according to modified reported method:24,25 Anhydrous FeCl3 (6.0 g, 37 mmol) was added in small portions to a solution of FeCl2·4H2O (4.9 g, 24 mmol) in aqueous ammonia (30%, 100 mL). The reaction mixture with pH ∼ 10 was stirred at 600 rpm for 1 h at room temperature followed by the addition of 15 wt% TEOS (1.6 g, 7.8 mmol). Vigorous stirring was continued for 24 h to generate a black colored precipitate. The resulting silica coated nanoparticles were separated by centrifugation and washed repeatedly with deionized water and HCl (1 N). Washing with acetone was followed by drying at 60 °C overnight to provide NpFeSi (7.0 g).
Synthesis of chloride-functionalized magnetic nanoparticles (NpFeSiCl)
Functionalized magnetic nanoparticles were prepared by treating a dispersion of the silica coated nanoparticles NpFeSi (3.0 g) in toluene (20 mL) with (3-chloropropyl)triethoxysilane (CPTES) (3.0 g, 12.5 mmol). The reaction mixture was stirred at 90 °C for 24 h, before the product was isolated by centrifugation. It was washed with toluene and acetone and finally dried at 40 °C for 6 h providing dark brown colored chloride-functionalized magnetic nanoparticles (NpFeSiCl, 4.0 g).General procedure for the tosylation of Guerbet alcohols26
A solution of the Guerbet alcohol, i.e. 2-butyloctanol (2.0 g, 10.7 mmol) or 2-hexyldecanol (2.0 g, 8.2 mmol), in CH2Cl2 (40 mL) was treated with pTsCl (3.2 g, 17 mmol) at 5 °C. NEt3 (1.7 g, 17 mmol) was added dropwise to the reaction mixture and stirring was continued at 5 °C for 1 h, before the reaction was allowed to warm to room temperature overnight. The reaction mixture was poured into NaHCO3 aq (10%) and extracted three times with CH2Cl2. The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated to furnish 2-butyl-octyl tosylate (3.4 g, 93%) and 2-hexyl-decanyl tosylate (3.1 g, 95%), respectively, as yellow oil.General procedure for the synthesis of Guerbet imidazoles27–29
KOH (2.0 g, 36 mmol) was added to a solution of imidazole (2.0 g, 29 mmol, 2 eq.) in DMSO (30 mL). The mixture was stirred at room temperature for 30 min before the Guerbet tosylate (5.0 g C12/5.8 g C16, 15 mmol) was added dropwise. Stirring was continued overnight. The reaction mixture was extracted with Et2O (3× 25 mL) and the combined organic phase was washed with water and dried over anhydrous MgSO4. Evaporation of the solvent at reduced pressure provided the Guerbet imidazole (3.3 g, 95% C12/4.3 g, 95% C16) as yellowish oil.Synthesis of magnetic nanoparticle supported alkyl imidazolium (NpFeSiImR)
Silyl chloride-functionalized magnetic nanoparticles (2.0 g) were dispersed in MeCN (50 mL) and ultrasonicated for 20 min. Alkyl imidazole (2.0 g) [13-14] was added and the reaction mixture was stirrer at 55 °C for 2 days. After cooling to room temperature the product was isolated by magnetic decantation. It was washed with acetone (3× 20 mL), and dried for 6 h at 45 °C to provide NpFeSiImR (∼2.8–3.2 g) as dark brown solid.DNA attachment
Genomic DNA was direct extraction and characterization from human whole blood. DNA–Np complexes were prepared with different weight ratios of magnetic Np to DNA range from 10, 20, 30, 40 to 50. DNA (30 μg mL−1) dissolved in nuclease-free water was mixed with magnetic Np. After brief vortexing, the mixture was incubated for 30 min at 4 °C to allow the formation of carrier/DNA complex. To confirm the binding of DNA to the nanoparticles, electrophoresis (1% agarose gel) was carried out at 110 V for 15 min in TBE buffer. The band was stained with ethidium bromide (EthBr) included in the agarose gel. The resulting migration patterns were visualized under UV irradiation (G-BOX, SYNGENE). NpFeSiImR (C6:10) was used to determine the optimum ratio of Np/DNA, which was found to be 30 Np/DNA. The latter has been applied to five different alkyl chains attached to magnetic Np.DNA release
DNA was purified by the addition of 50 μL of elution buffer AW1, which contains a high concentration of chaotropic salt, and incubated at 70 °C for 10 minutes to dissociate the bonded DNA from the magnetic nanoparticles. Subsequently the mixture was vortexed until the pellet was (visually) completely re-suspended. After centrifugation for 30 seconds at approximately 14000 rpm, the supernatant was transferred to the spin column (containing silica membrane). DNA was washed twice with buffer (containing absolute ethanol) then centrifuge for 1 min at 14000 rpm. After the centrifugation the DNA filament attached to the silica membrane and the nanoparticles was removed from the silica to the collection tube. The spin column was subsequently re-centrifuged for 3 min at 14000 rpm to dry the silica membrane containing the DNA from ethanol. After that 60 μL of elution buffer was added to the center of the spin column tube and it was centrifuged for 1 min at 14000 rpm. With this approach the DNA could be isolated and purified from nanoparticles and stored at 4 °C until use. The DNA concentration was verified by agarose gel electrophoresis.
DNA stability
DNA was suspended in buffer and exposed to ultrasound. The sample was aligned at the focus of a 60 kHz single-element ultrasound transducer (model WiseClean) in a tank containing deionized water at 30 °C for periods ranging from 0 to 40 s in 10 s increments. Electrophoresis (1% agarose gel) were then carried out at 110 V for 15 min in TBE buffer to determine whether the DNA had been structurally damaged or not. The band was stained with EthBr included in the agarose gel. Subsequently DNA–NpFeSiImR-complexes (based on Guerbet C16) were exposed to ultrasound using the same conditions and subjected to gel electrophoresis to assess the release of DNA.Extraction of DNA from blood
Transfer 300 μL of whole blood to 1.5 mL tube containing 900 μL RBC Lysis Solution. Mix toughly by vortexing and subsequently incubate for 5 minutes at room temperature. Centrifuge at 13000 rpm for one minute. Remove supernatant except the white cell pellet are remain, 20 μL of proteinase K and 300 μL of BB3 (Cell Lysis Solution) were mixed inside a micro-centrifuge tube by vortexing for 15 seconds and subsequently incubated at 56 °C for 15 minutes. Add 1.5 μL RNase a solution to the cell lysate and incubate at 37 °C for 15–30 minutes, chill mixture to room temperature, then add 100 μL PPT (protein precipitation) buffer to the cell lysate and vortex vigorously at high speed for 20 seconds. Add 3 mg (300 μL) of nano-material with 300 μL of binding buffer to the cell lysate and incubate the reaction for 15 minutes at room temperature with occasional gentle mixing. The nano-material with DAN attached was collected by magnetic separation and discard the rest of lysate content. Add 1 mL of 70% ethanol to the DNA–nanoparticle and do pipetting up and down several times to wash the DNA–nanoparticle. The nano-material with DNA attached was collected after washing by applying an external magnetic field and discard the rest of the solution. Repeat this step one more time to insure that the DNA molecules washed perfectly. The hot eluted (65 °C) was added to the DNA–nanoparticle mixture and incubate for one min at room temperature, which was disassemble the DNA–nanoparticle connected. The nanoparticle molecules magnetically separate from the DNA and the elution buffer with DNA transfer to the new sterile micro centrifuge tube. For long-term storage, store the purified DNA at −20 °C.
Results and discussion
Particle synthesis
Ionic magnetic nanoparticles were synthesized using a multistep strategy, as shown in Fig. 1. The magnetic nanoparticle core (Np) was prepared by a chemical co-precipitation of ferrous and ferric ions in aqueous ammonia (pH 9) at a stoichiometric ratio of 3:2, leading to the formation of Fe2O3 hematite nanoparticles.4,7,30 A layer of SiO2 was coated on the nanoparticles using a modified Stober method. The purpose for this coating was to avoid possible oxidation or aggregation of the magnetic nanoparticles and to provide silanol groups on the particle surface for further functionalization.7,8,30 Treatment of the latter with CPTES grafted an organic halide on the nanoparticle surface for the final coupling with a series of different alkyl imidazoles, which were prepared following a reported approach.28 The immobilization of the imidazoles on NpFeSiCl provided positive charges on the nanoparticle surface, which were balanced by electrostatically bonded chloride anions. The result was magnetic core–shell nanoparticles with a cationic alkyl-imidazolium-derived surface, suitable for the interaction with anionic biomolecules, like DNA.
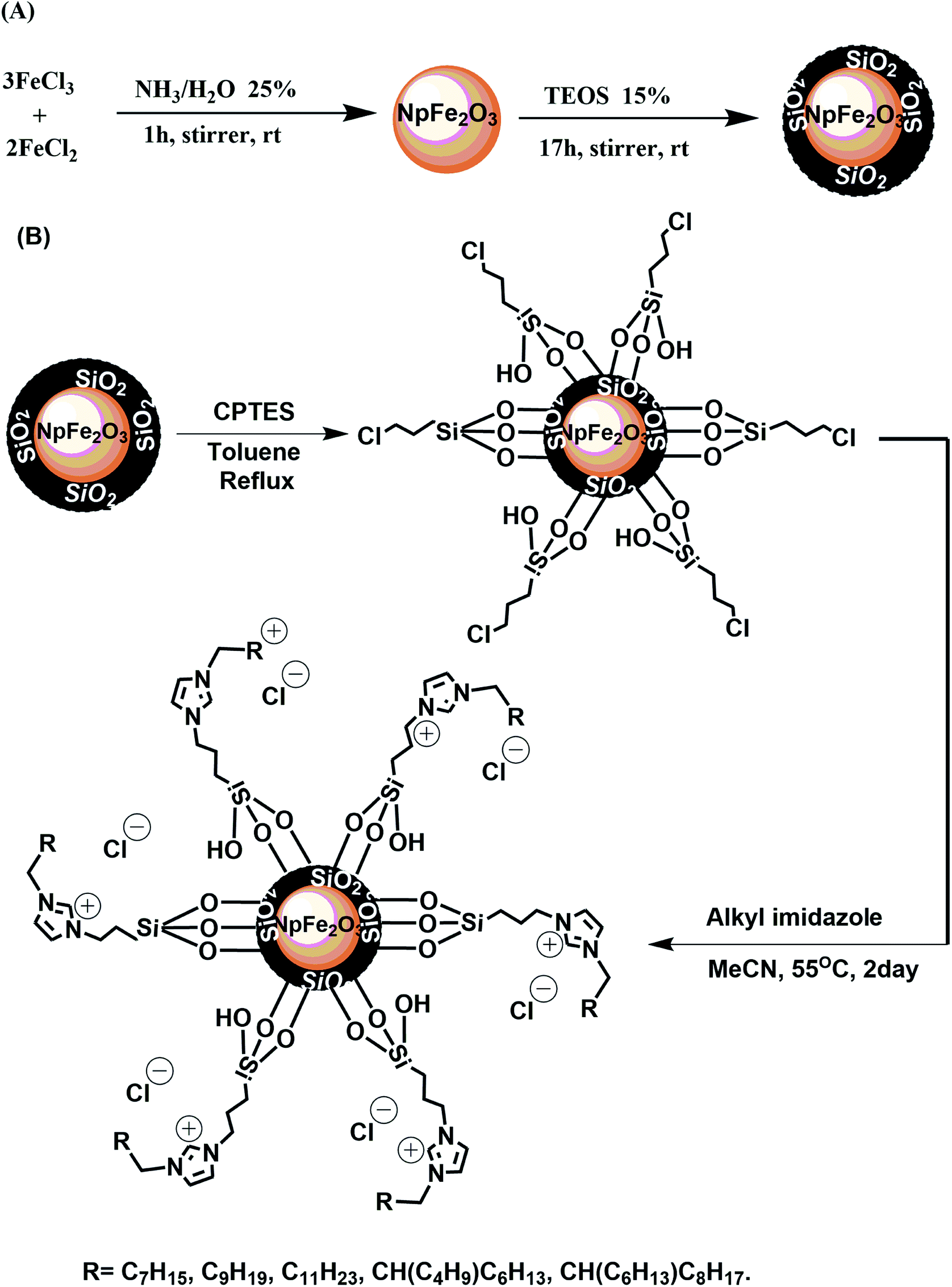 | ||
| Fig. 1 Synthetic pathways for the synthesis of ionic magnetic nanoparticles: (A) synthesis of core–shell magnetic nanoparticles, (B) grafting and immobilization of magnetic nanoparticles. | ||
Nanoparticle characterization
The phase of the particles were determined by powder X-ray diffraction. Fig. 2 shows the XRD patterns for different stages of the nanoparticles. The initial core–shell particle NpFeSi was identified as a combination of hematite (Fe2O3) and quartz (SiO2) based on the diffraction peaks at 25.3, 35.4, 38.7, 46.3, 56.1, 62.6, and 63.2°. These correspond to 30%, 70%, 30% and 30% of Fe2O3 on the (012), (110), (214) and (300) crystallographic planes, respectively (PDF Ref Cod: 033-0664, match 60%), while the SiO2 diffractions reflect the (101), (102), (201) and (103) crystallographic planes with 100%, 8%, 4% and 2%, respectively (PDF Ref Cod: 033-1161, match 37%). The sharp diffraction peaks indicate high crystallinity and particle sizes in the upper nano-range.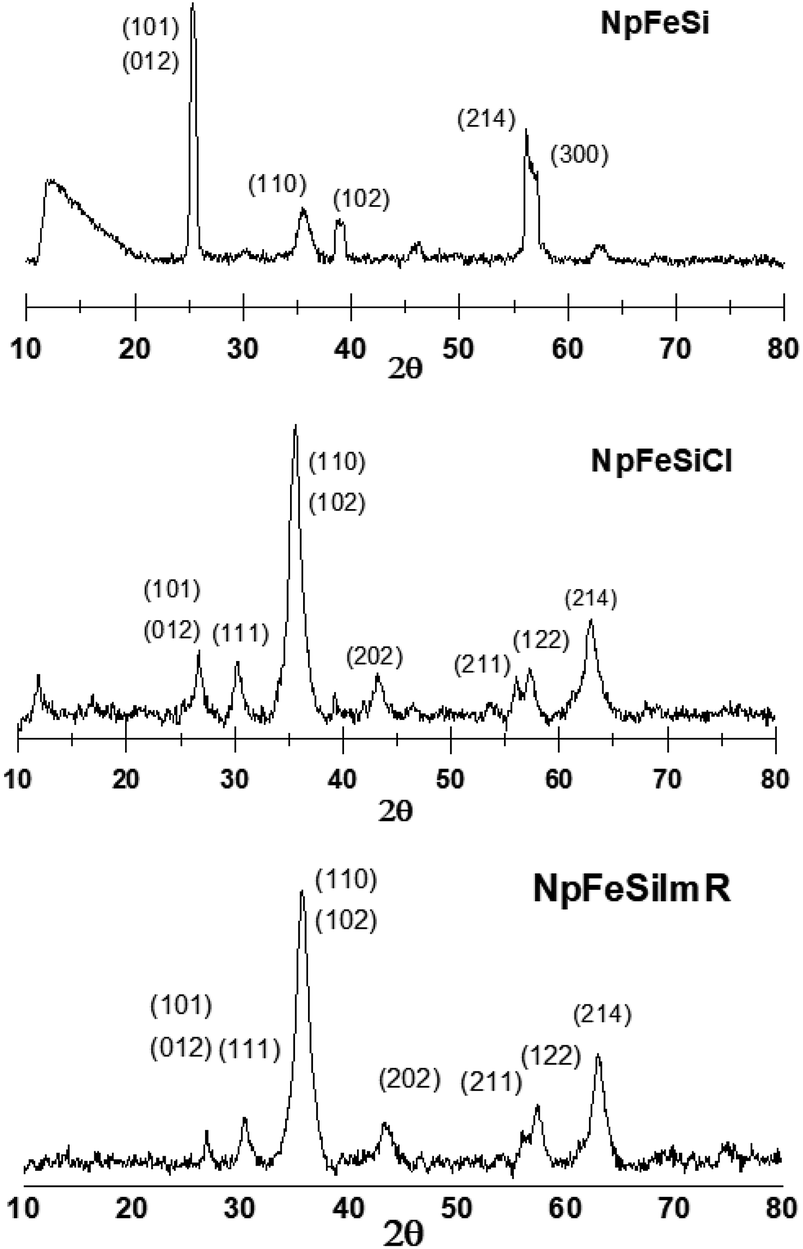 | ||
| Fig. 2 XRD patterns of magnetic nanoparticles. | ||
The surface modification by CPTES alters the diffraction pattern of NpFeSiCl, while the grafting of the imidazole on the nanoparticle surface has practically no impact on the crystal structure, as reflected in an unchanged diffraction pattern for NpFeSiImR. The XRDs of NpFeSiCl and NpFeSiImR are dominated by SiO2, reflecting an increase of the shell due to the treatment with CPTES. The broader peaks compared to NpFeSi reflect a less regular structure of the silica-shell due to the partial alkylation of the silicon. Besides, partial agglomeration owing to cross linking of particles during the reaction could also account for crystal inhomogeneity. The new diffraction peaks are in line with reference patterns for SiO2 (PDF Ref Cod: 033-0664, match 69% and PDF Ref Cod: 033-1161, match 72%).
Fig. 3 shows FESEM images of the final magnetic nanoparticles NpFeSiImR. The particles form large agglomerates with a regular pattern. The image suggests a rather homogenic size of particles. However, owing to the agglomeration, this impression cannot be confirmed. In view of the disperibility of the particles, the agglomerates are expected to reflect mostly assemblies of isolated particles, although partial fusion of particles during the surface functionalization is expected as well.
 | ||
| Fig. 3 FESEM images of NpFeSiImR. | ||
Images from a high resolution transmission electron microscopic (HRTEM) investigation of NpFeSiImR are shown in Fig. 4. They provide information about the atomic structure and morphology of the nanoparticles. The lattice fringes of the magnetic core can be clearly observed. The estimated lattice d-spacing were found to be ∼0.49, 0.27, and 0.25 nm, which correspond to (100), (111) and (110), respectively. Other atomic planes, particularly reflecting the silica shell, are not visible in the image. This probably reflects the lower contrast of the lighter metal atoms. The particle core appeared as an ellipsoid with an estimated diameter ratio of about 3:2. They appear to differ only moderately in size with a diameter of about up to 16 nm along the longer axis. Larger sizes, ∼23 nm, were attributed to agglomeration (Fig. 4C). The reason behind the latter could drying of the specimen on the grid for the TEM observations. The relative contribution of the magnetostatic interaction appeared to increase, leading to ordering and self-assembly.31
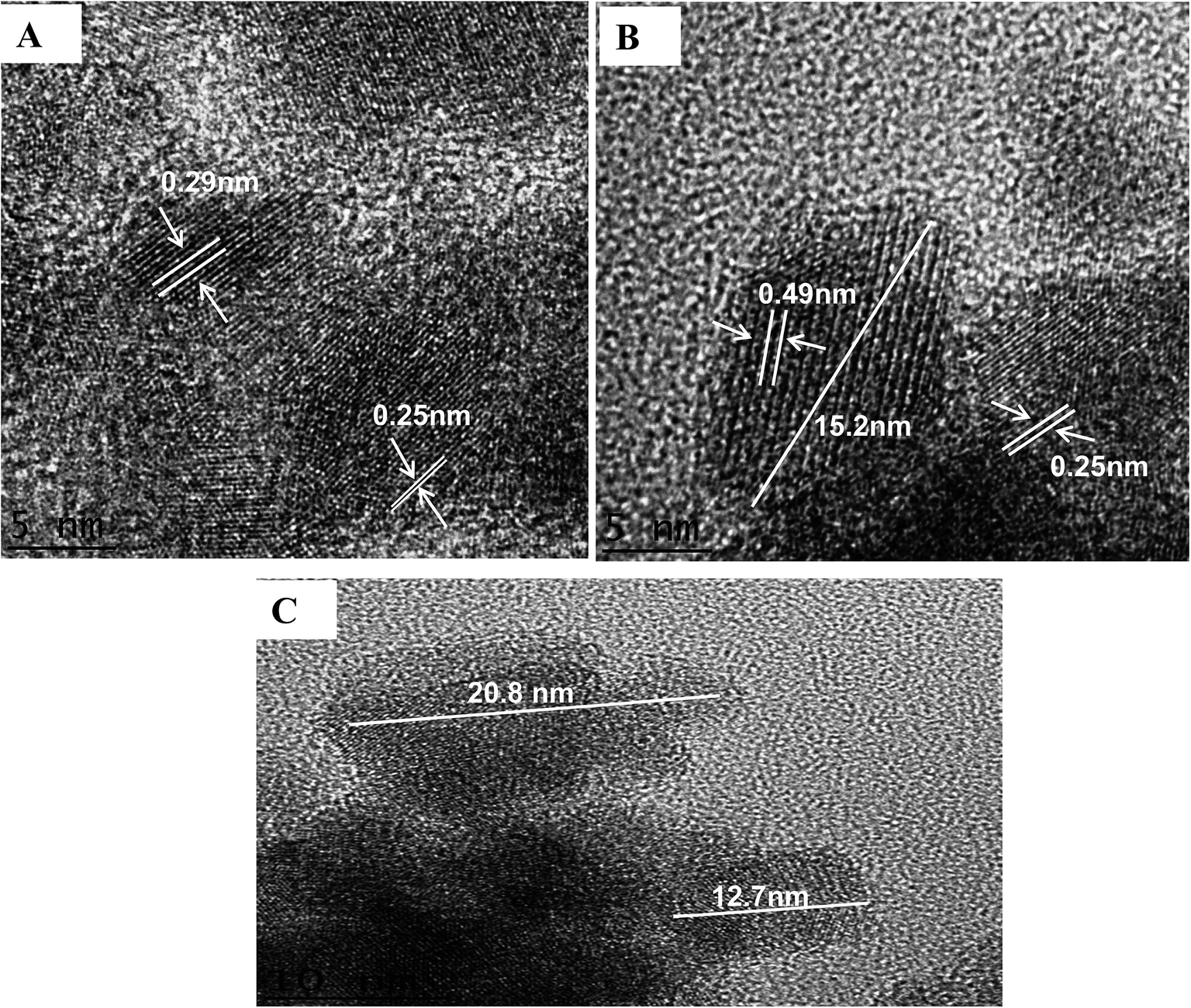 | ||
| Fig. 4 (A) and (B) HRTEM image of NpFeSiImR nanoparticles with lattice fringes display. (C) The partical size of NpFeSiImR nanoparticles. | ||
The elemental composition of the nanoparticles was studied by energy dispersive X-ray microscopy (EDX). The analysis for the particles NpFeSi, displayed in Fig. 5, indicates an average molar ratio of the hematite core and the silica shell of 3.6:1. This corresponds to a mass content of 86% for the magnetic core. The slightly mismatching light intensities for the respective atomic radiation reflects minor differences in the particle composition, due to inhomogen growth of the silica shell on the hematite particle cores.
 | ||
| Fig. 5 EDX analysis of core–shell nanoparticles NpFeSi. | ||
The presence of chloride as a new element in the NpFeSiCl sample, as shown in Fig. 6a, confirmed the successful surface anchoring of CPTES. This is also reflected in the increased ratio of Si and Fe (1:6.0 in NpFeSiCl compared to 1:7.2 in NpFeSi). The comparison of the elemental radiation maps suggested a homogeneous activation of the particle surface.
 | ||
| Fig. 6 EDX analysis of surface functionalized nanoparticles NpFeSiCl and NpFeSiImR. | ||
The ratio of Si and Fe remained unchanged upon anchoring of the imidazole on the particle surface, indicating no changes in the hard core–shell structure. The reduced chlorine content is associated with the presence of nitrogen as a new element. The content of the latter, however, exceeds the surface functionalization content significantly, as can be seen by comparing the molar ratio of Si and N. It is assumed that some of the imidazole attaches physically to the particle surface, thereby causing the unexpected high N-content, which does not reflect the expected ratio of 2:1 for N and Cl either. The comparison of the radiation maps for different elements in Fig. 6b confirmed a homogeneous surface modification.
The grafting of organic reagent on the nanoparticle surface was also monitored by IR-spectroscopy. The spectra in Fig. 7 document organic reagents in both NpFeSiCl and NpFeSiImR. While the intense absorption above 3200 cm−1 reflects remaining hydroxyl groups on the particle surface, the stretching vibrations slightly below 3000 cm−1 represent hydrocarbon chains on the surface. The increase of these absorption upon introduction of the imidazole reflects the alkylation of the latter. Corresponding bending vibrations were observed at 1439 cm−1. The band at 1623 cm−1 could be assigned to the bending vibration of trapped water molecules within the silica matrix.32 The Si–C stretching vibration was observed at 1271 cm−1, while the bands at 1114 and 1025 cm−1 were attributed to Si–O–Si stretching vibrations. These values confirm the formation of amorphous silica matrix.33 The C–Cl stretching of the capping reagent (CPTES) was found at 796 cm−1. The intensity decreased after surface grafting of the alkyl imidazole. However, the IR suggests an incomplete conversion of the chloride surface reagent in the final ionic magnetic nanoparticles. All NpFeSiImR with different straight and branch alkyl chains showed very similar IR spectra.
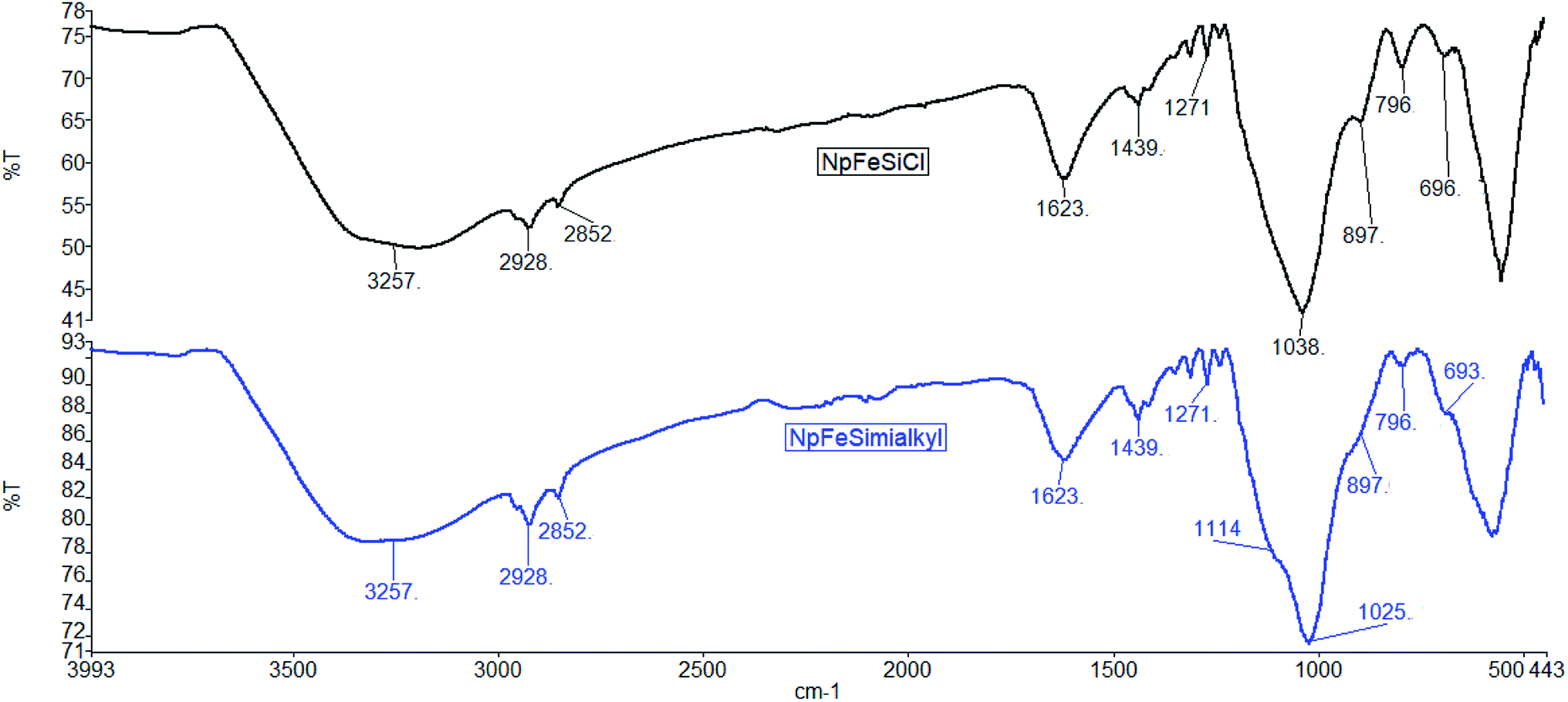 | ||
| Fig. 7 ATR FT-IR spectra of NpFeSiPrCl and NpFeSiImR. | ||
A quantitative analysis of the anchoring of organic contents on the particle surface was performed by thermogravimetric analysis (TGA). Mathematical data-processing provided with differential thermogravimetry (DTG) spectra that enables an easier analysis. The TGA-DTG curves of NpFeSi showed one major characteristic decomposition stage at 259 °C with mass loss of 5.7%, which can be assigned to physisorbed water inside the nanopartical matrix and further condensation of the hydroxyl group of the silica shell Q3 (Si(OSi)3(OH)) to result Q4 (Si(OSi)4). The peak disappeared upon functionalization of the magnetic nanoparticles with the chloropropyl group owing to the reflux condition, which allowed full access to the hydroxyl groups on the shell. A new region of mass loss between 400–422 °C (3.9%) was attributed to the decomposition of the chloropropyl group attached to the silica in NpFeSiCl. The imidazolium functionalized nanoparticles with single alkyl chains NpFeSiImR, with R = C8, C10 and C12, exhibited a peak at around 450 °C, probably reflecting the degradation of the imidazolium. The mass loss was higher for the longer chain length (18% for C12, 15% for C10), which is in line with a different mass contribution to the overall particle. For the C8 compound an additional peak was found slightly above 300 °C, which could reflect associated N-octyl-imidazole, which is not covalently bonded. The nanoparticles incorporating Guerbet alkyl chains exhibited showed a considerable mass loss at significantly lower temperature, i.e. around 300 °C, reflecting a reduced thermal stability of the imidazolium salt near the chain branching.
Magnetic properties of NpFeSiCl and NpFeSiImR nanoparticles were investigated at room temperature as a function of magnetic field (H), as these properties reflect a key feature of the material, differentiating them from conventional nanoparticles. The analysis of magnetic property for the precursors NpFe and NpFeSi was neglected, because they have already been investigated in previous studies20,33 The saturation magnetization (Ms) of the nanoparticles droped from 29 emu g−1 for NpFeSiCl to 25 emu g−1 for NpFeSiImR, as shown in Fig. 8. This drop could reflect the weight fraction of the alkyl imidazole in the composite particles and the coating of the surface by long alkyl chains. The decrease in Ms is an additional indicator for the successful incorporation of the alkyl imidazole into the magnetic nanoparticles. The magnetization relates to a coercivity of 15.5 G coercivity for NpFeSiImR, which is significantly lower than 27.2 G for NpFeSiCl. Very small hysteresis loops were observed for both NpFeSiCl and NpFeSiImR nanoparticles at low magnetic field with weak remnant magnetization (Mr) of 1.47 and 0.62 emu g−1, respectively. These values are conspicuously characteristic for super-paramagnetic behavior of the nanoparticles.
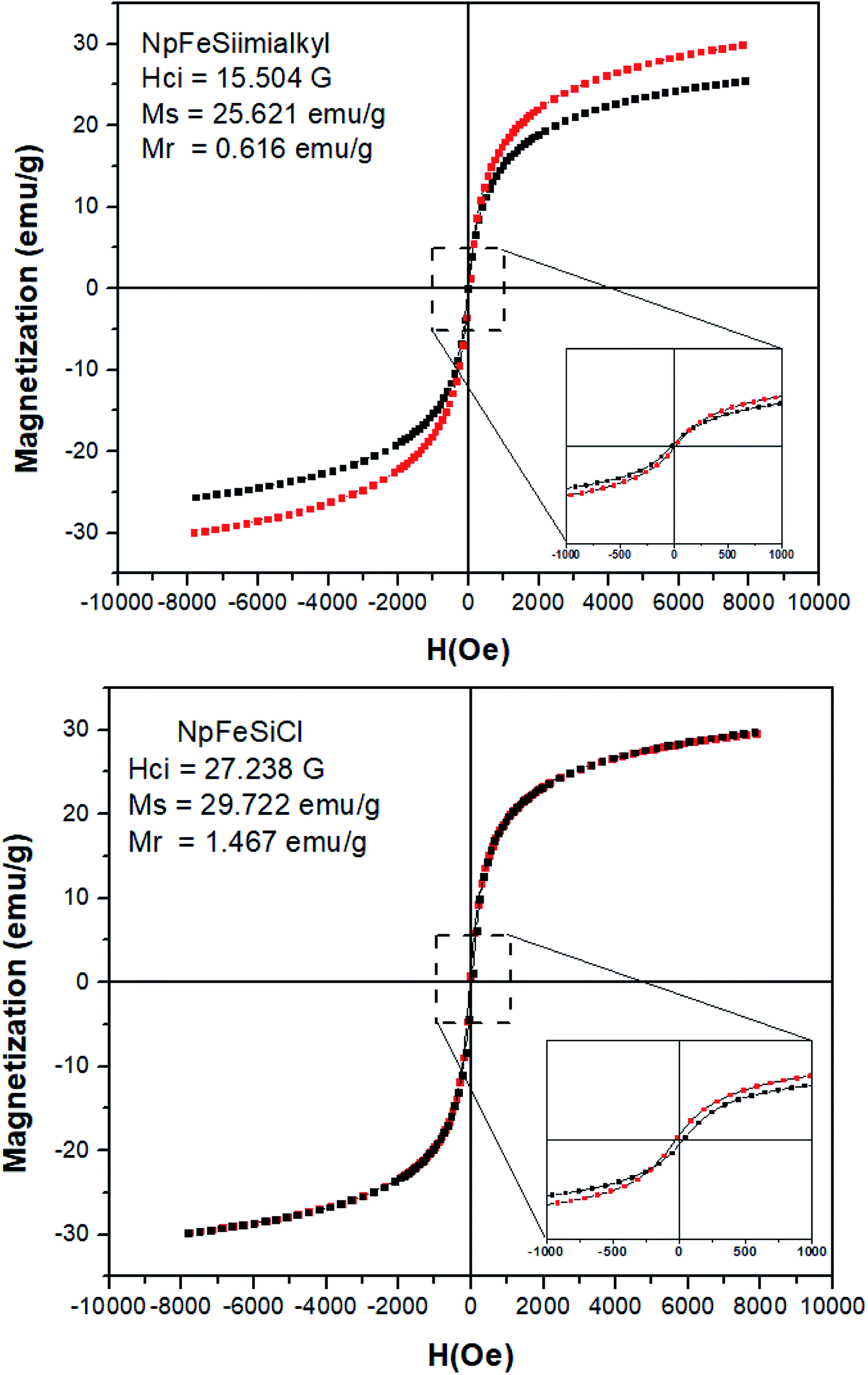 | ||
| Fig. 8 Super-paramagnetic hysteresis loops for NpFeSiCl and NpFeSiImR nanoparticles. | ||
Nanoparticle interaction with DNA
Different strategies have been developed for DNA extraction methods in order to produce a high recovery of DNA and removal of impurities and inhibitors. The most common DNA extraction strategies can be grouped into three methods, i.e. organic extraction, solid-phase extraction and ionic chelating resins.34,35 These methods consistently yield isolated DNA, but they differ in both, quality and quantity of the obtained DNA. All along this article, basic principles and specific procedures for ionic magnetic core–shell as a solid-phase extraction system are described. In view of recent studies on an eutectic composite based on chitosan, Fe3O4 and PEG coated multi-walled carbon nanotubes enabling the isolation of 79% of DNA without conformational damage13 the performance expectations on new DNA extraction methods are high. However, the method suffers from the drawbacks of lower desorption rate. Our magnetic nanoparticle approach, however, ensures a high desorption rate (10 min.) with more than 86% of unchanged DNA loading.The main cohesion forces between the magnetic nanoparticles and DNA are believed to be ionic interactions of the imidazolium cation on the nanoparticle surface and the phosphorester anion on DNA. Practically, DNA replaces some of the halide counter ions in NpFeSiImR. The largest negative charge is localized on the phosphate groups present on the surface of the DNA molecule and in the minor groove, although other sites have been reported to exhibit stronger molecular interactions based on hydrogen bonding.36,37 However, in view of the absence of both acidic hydrogen and electron-donor capacity of the imidazolium ion, the interactions with the nanoparticles are probably solely of electrostatic nature. Additional hydrophobic interactions, mediated by organic residues can strengthen the conjugation, particularly for nanoparticles with longer and possibly branched alkyl chains.
Despite the strong interaction of DNA with the magnetic nanoparticles, the DNA–nanoparticle complex can be separated to recover the DNA. Fig. 9 shows electrophoretic DNA profiles for eluates from different stages of the chromatographic DNA recovering process, indicating the DNA recovery in a selective fraction.
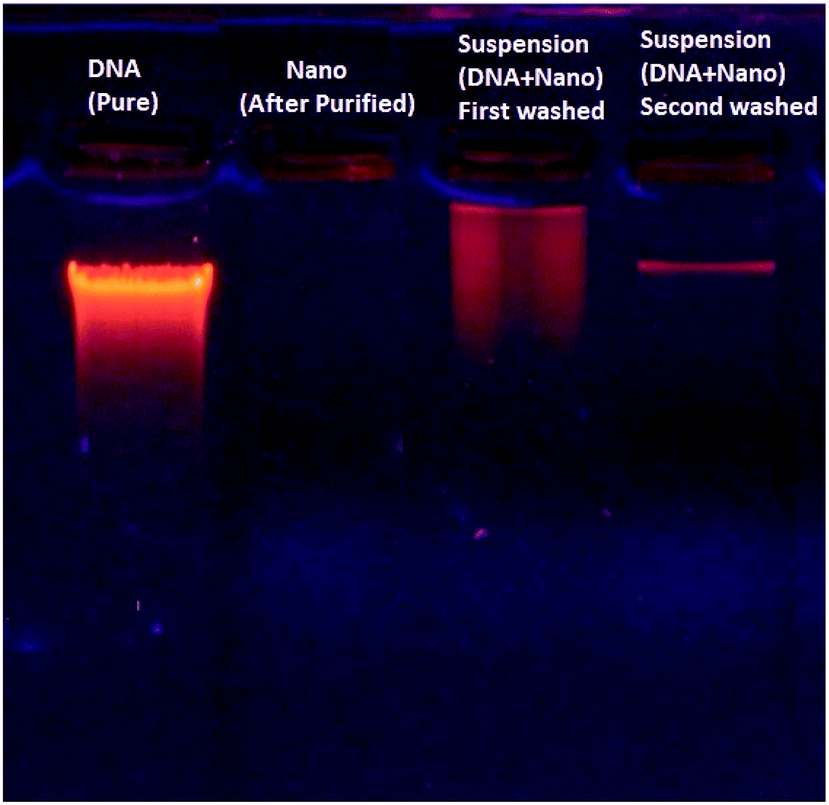 | ||
| Fig. 9 Shows electrophoretic DNA profiles for recovering process. | ||
One concern associated with ultrasound mediated delivery is the potential for damaging therapeutic molecules. The impact of ultrasound on naked DNA was therefore investigated at different ultrasound exposure times ranging from 0 to 40 s. Following the exposure, an agarose gel retardation assay was used to detect DNA degradation or structural changes. Fig. 10A indicated gradual degradation of DNA, reflected in a changed electrophoretic mobility on agarose gel, thereby indicating damage of the macromolecule upon ultrasound exposure. DNA complexes with the magnetic nanoparticles, on the other hand, remained majorly intact over an exposure of almost 30 s, as shown in Fig. 10B. This indicates that the magnetic nanoparticle protects the DNA against ultrasonic damage, although not for unlimited exposure time.
 | ||
| Fig. 10 Gel retardation assay of ultrasound-exposed DNA after increasing exposure times [s]. (A) Scrambled DNA; (B) DNA–MNPs complexes. | ||
To determine the most effective alkyl chain for the imidazolium on the magnetic nanoparticles NpFeSiImR, gel electrophoresis were conducted with DNA–Nps complexes containing different alkyl chains at the optimum weight ratio of 10 Np/DNA. The results are shown in Fig. 11. Lowest migrations were observed for the most hydrophobic compounds NpFeSiImC12 and NpFeSiIm C10:C6, reflecting highest DNA binding efficiency. Therefore, these nanoparticles were chosen as most promising candidates. No DNA interaction and loading were observed for NpFeSi and NpFeSiCl (Fig. 12). Therefore the presence of the ionic charge plays an important role for the strength of the interaction of the complex.
 | ||
| Fig. 11 Gel retardation assay of DNA–NPs complex with different alkyl chain. | ||
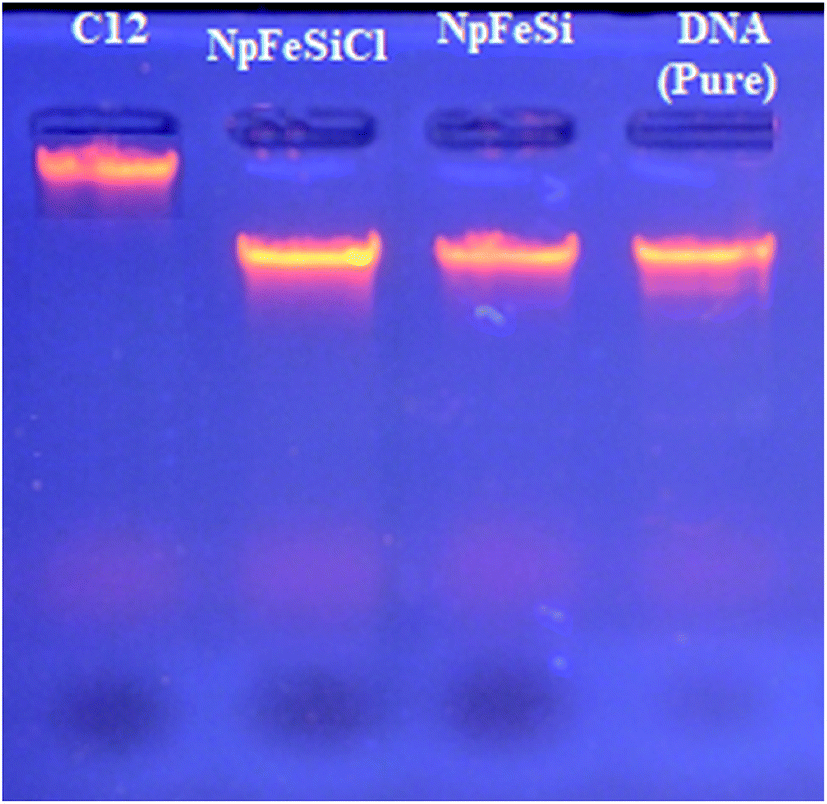 | ||
| Fig. 12 Gel retardation assay of DNA–NPs complex with NpFeSiImC12, NpFeSiCl, and NpFeSi. | ||
The DNA loading capacity was evaluated by agarose gel electrophoresis. The investigation was applied on only two different magnetic Nps: On of them involved a straight C12 chain, while the other contained a C16-Guerbet residue, reflecting a branched C10:C6 hydrocarbon domain. The weight ratio of nanoparticles and DNA was varied from 10:1 to 50:1. Fig. 13 indicates almost complete retard migration for all ratios. However, minor amounts of non-complexed DNA are still recognizable for ratios up to 20:1, whereas no peaks can be found for a nanoparticle DNA ratio of 30:1 or above. For optimum efficiency a ratio of 30:1 is, hence, recommended. The complex found to be stable enough for more than three months without dissociation from the particles when stored at 4 °C.
 | ||
| Fig. 13 Complexation test of magnetic Nps and DNA in water. Each DNA–Nps complex was prepared at a weight ratio (10, 20, 30, 40 and 50) of MNPs to DNA. | ||
The NpFeSiImalkyl composites have been applied for the extraction of DNA from blood to evaluate their practical applicability. The recovered DNA was examined by agarose gel electrophoresis and the results are promising as shown in Fig. 14.
 | ||
| Fig. 14 Gel retardation assay of extraction of DNA from blood sample (DNA–NPs complex with NpFeSiImR.). | ||
Conclusion
In summary, alkyl imidazole based magnetic core–shell nanoparticle composites were synthesized and employed for the magnetic solid-phase extraction of DNA for the first time. The synthesized particles were successfully conjugated to control DNA and the DNA conjugates were found to be stable at room temperature. The functionalized nanoparticles exhibit a particle size ∼20 nm and magnetization of 25 emu g−1 with 15.5 G coercivity. In additional, a very small hysteresis loop for NpFeSiImR nanoparticles at low magnetic field with weak remnant magnetization (Mr) of 0.62 emu g−1 was observed. These values are conspicuously characteristic of super-paramagnetic behavior of the nanoparticles. As a result, these nanoparticles exhibit both high loading capacity of DNA: particle ratio 1:30 wt% and significantly enhanced the extraction of DNA samples with the aid of an external magnetic field. The approach can be considered as a simple, rapid, effective and environmentally friendly separation procedure. NpFeSiImC12 and NpFeSiIm C10:C6, reflected the highest DNA binding efficiency. The results suggest the above ionic magnetic nanoparticles with alkyl imidazole coating as a promising platform for future therapeutic delivery applications. Future work can be anticipated by exploring potential multifunctionality of these nanoparticles, such as magnetic extraction of several basic pharmaceutical compounds and magnetic resonance imaging.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors of this paper gratefully acknowledge financial support from the University of Malaya under Research Grants GPF-061-2018 and RG383-17AFR, as well as from the Al Muthanna University.References
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CAS.
- J. Dobson, S. C. McBain and H. H. P. Yiu, Int. J. Nanomed., 2008, 3, 169–180 CrossRef.
- S. C. Halim and W. J. Stark, Chimia, 2008, 62, 13–17 CrossRef CAS.
- Hybrid Nanocomposites for Nanotechnology, ed. L. Merhari, Springer US, New York, 2009 Search PubMed.
- M. A. Bodaghifard, M. Hamidinasab and N. Ahadi, Curr. Org. Chem., 2018, 22, 234–267 CrossRef CAS.
- S. B. Sant, Mater. Manuf. Processes, 2012, 27, 1462–1463 CrossRef CAS.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS.
- N. Geerts and E. Eiser, Soft Matter, 2010, 6, 4647–4660 RSC.
- R. M. Patil, P. B. Shete, S. M. Patil, S. P. Govindwar and S. H. Pawar, RSC Adv., 2015, 5, 88375–88381 RSC.
- J. Y. Lee, C. Crake, B. Teo, D. Carugo, M. de Saint Victor, A. Seth and E. Stride, Adv. Healthcare Mater., 2017, 6, 1–9 Search PubMed.
- S. Liu, S. Li, W. Yang, F. Gu, H. Xu, T. Wang, D. Sun and X. Hou, Talanta, 2019, 194, 514–521 CAS.
- S. Xu, J. Li and L. Chen, J. Mater. Chem., 2011, 21, 4346–4351 CAS.
- K. Xu, Y. Wang, H. Zhang, Q. Yang, X. Wei, P. Xu and Y. Zhou, Microchim. Acta, 2017, 184, 4133–4140 CrossRef CAS.
- J. Li, S. Wang, L. Chen, W. Lu, A. Wu, J. Choo and L. Chen, Sens. Actuators, B, 2014, 193, 857–863 CrossRef.
- X. Fu, D. Zhu, L. Huang, X. Yan, S. Liu and C. Wang, Microchem. J., 2019, 150, 104169 CrossRef CAS.
- D. Li, T. Li, L. Wang and S. Ji, J. Chromatogr. A, 2018, 1581–1582, 1–7 CrossRef CAS.
- S. Jamshidi, M. K. Rofouei and G. Thorsen, J. Sep. Sci., 2019, 42, 698–705 CrossRef CAS.
- D. D. Zhou, H. Zhang, Q. Zhang, Z. M. Qian, W. J. Li, C. H. Li, F. Q. Yang and H. Chen, J. Chromatogr. A, 2019, 1591, 24–32 CrossRef CAS.
- Z. H. Deng, X. Wang, X. L. Wang, C. L. Gao, L. Dong, M. L. Wang and R. S. Zhao, Microchim. Acta, 2019, 186, 0–8 CAS.
- L. Xiong, J. Bi, Y. Tang and S. Z. Qiao, Small, 2016, 12, 4735–4742 CrossRef CAS.
- M. Zhao, H. Shao, J. Ma, H. Li, Y. He, M. Wang, F. Jin, J. Wang, A. M. Abd El-Aty, A. Hacımüftüoğlu, F. Yan, Y. Wang and Y. She, J. Chromatogr. A, 2020, 1615, 460751 CrossRef CAS.
- Y. Zhao, R. Wu, H. Yu, J. Li, L. Liu, S. Wang, X. Chen and T.-W. D. Chan, J. Chromatogr. A, 2020, 1610, 460543 CrossRef CAS.
- N. Kobylinska, L. Kostenko, S. Khainakov and S. Garcia-Granda, Microchim. Acta., 2020, 187, 289 CrossRef CAS.
- A. Halilu, T. H. Ali, A. Y. Atta, P. Sudarsanam, S. K. Bhargava and S. B. Abd Hamid, Energy Fuels, 2016, 30, 2216–2226 CAS.
- A. Halilu, T. H. Ali, P. Sudarsanam and S. K. Bhargava, Symmetry, 2019, 11, 524 CrossRef CAS.
- A. Ossola, E. Macerata, E. Mossini, M. Giola, M. C. Gullo, A. Arduini, A. Casnati and M. Mariani, J. Radioanal. Nucl. Chem., 2018, 318, 2013–2022 CrossRef CAS.
- N. N. Al-Mohammed, R. S. Duali Hussen, T. H. Ali, Y. Alias and Z. Abdullah, RSC Adv., 2015, 5, 21865–21876 RSC.
- N. N. Al-Mohammed, R. S. D. Hussen, Y. Alias and Z. Abdullah, RSC Adv., 2015, 5, 21869–21881 Search PubMed.
- N. N. Al-Mohammed, Y. Alias and Z. Abdullah, RSC Adv., 2015, 5, 92602–92617 RSC.
- S. B. Primrose; and R. M. Twyman, Principles of Gene Manipulation and Genomics, Blackwell Publishing, Malden, 7th edn, 2006 Search PubMed.
- Y. Takeno, Y. Murakami, T. Sato, T. Tanigaki, H. S. Park, D. Shindo, R. M. Ferguson and K. M. Krishnan, Appl. Phys. Lett., 2014, 105, 0–5 CrossRef CAS.
- K. Mohammed, F. Adam and T. Hussein, J. Taiwan Inst. Chem. Eng., 2014, 45, 134–142 CrossRef.
- F. Adam, K. M. Hello and T. H. Ali, Appl. Catal., A, 2011, 399, 42–49 CrossRef CAS.
- K. M. Elkins, in Forensic DNA Biology, Academic Press, San Diego, 2013, pp. 39–52 Search PubMed.
- J. M. Butler, Forensic DNA typing: biology, technology, and genetics of STR markers, Elsevier Academic Press, Amsterdam; Boston, 2nd edn, 2005 Search PubMed.
- B. Schneider, K. Patel and H. M. Berman, Biophys. J., 1998, 75, 2422–2434 CrossRef CAS.
- R. Lavery and B. Pullman, Nucleic Acids Res., 1981, 9, 4677–4688 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05933a |
| This journal is © The Royal Society of Chemistry 2020 |