
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew perspective of a nano-metal preparation pathway based on the hexahydro-closo-hexaborate anion
Jun Liua,
Xue Zhao*ab and
Haibo Zhang *b
*b
aCollege of Chemistry and Materials Engineering, Hunan University of Arts and Science, Changde 415000, P. R. China. E-mail: xuezhao0208@gmail.com
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China. E-mail: haibozhang1980@gmail.com
First published on 10th September 2020
Abstract
Today, metal-based nanomaterials play an increasingly important role in the energy, environment, medical and health fields. In order to meet the needs of various fields, it is necessary to continuously develop advanced technologies for preparing metal-based materials. Inspired by previous research, the results of a proof-of-concept experiment show that the hexahydro-closo-hexaborate anion (closo-[B6H7]−) in the borane cluster family has properties similar to NaBH4. Closo-[B6H7]− can not only convert common precious metal ions such as Au3+, Pd2+, Pt4+ and Ag+ to the corresponding zero-valence state, but also convert some non-precious metals such as Cu2+ and Ni2+ to the zero-valent or oxidation state. Closo-[B6H7]− moderate reduction to cause rapid aggregation of metal-based materials is not easy compared with NaBH4. Compared with closo-[B12H12]2−, closo-[B6H7]− achieves the conversion of Pt4+ to Pt0 under ambient conditions, and its reduction performance extends to non-precious metals. The excellent stability and easy modification characteristics determine the universality of the closo-[B6H7]− reduction strategy for metal ions.
Introduction
Bulk metals severely limit their surface exposure due to their low specific surface area, and the development of metal nanoparticles has promoted rapid changes in the chemical world. Today, the excellent performance of nanometals is driving rapid progress in different fields such as molecular catalysis, biomedicine, energy storage, optics, and magnetism, and researchers from various regions are constantly exploring the multifunctional applications of diverse nanometals.1–4 Traditional methods for preparing nanometals usually include thermal decomposition of metal ions, chemical reduction, electrodeposition, and mechanical grinding. The thermal decomposition of metal ions is usually done in hydrothermal (or solvothermal) kettles and tube furnaces (or muffle furnaces), where the principle is that the chemical reducing agent (such as ethylene glycol or H2) converts the metal from the ionic state to the zero-valent state or oxide.5–7 The implementation of this method requires the support of special equipment, and is often accompanied by potential safety hazards and large energy consumption. Electrodeposition is a new way of preparing zero-valent metals, which converts metal ions to a zero-valent state by applying a certain potential (depending on the standard electrode potential of the metal ions).8,9 On the one hand, the electrodeposition method needs to strictly control the reduction potential, and the solution must not contain other substances that are easily converted. On the other hand, because the reaction occurs on the electrode, this limits the possibility of large-scale preparation of zero-valent nanometals, and the nanometals obtained by electrodeposition can only be supported on specific electrode carriers. The mechanical grinding method is the most traditional way to crush a bulk metal body from top to bottom to reach a specified size, and its realization is usually carried out in a ball mill or mortar.10 The mechanical grinding method can increase the specific surface area of the metal to a certain extent, but it is usually accompanied by uneven particle size of the metal particles and air oxidation pollution caused by long-term grinding. The chemical reduction method is the most widely used method for preparing the corresponding zero-valent nanometals from metal ions, and is characterized by being fast, simple and practical. In general, sodium borohydride (NaBH4) is the most widely used chemical reducing agent, and can reduce most metal ions due to its strong reducing ability.11 However, the strong reducing power of NaBH4 results in the reaction being completed instantaneously. The result is severe aggregation or large particle size distribution of the nanometals. Based on this, it is of practical significance to continue to develop new borohydrides with appropriate reducing power as chemical reducing agents for the preparation of nanometals.Recently, our team has proved that closo-[B12H12]2− can reduce Au3+, Pd2+, Pt4+ and Ag+ noble metal ions to the zero valence state, and except for Pt4+, which needs to be reduced at 80 °C, the other reduction reactions can be successfully achieved at room temperature.12–20 The development of this technology has opened up a brand-new route for the preparation of precious metal nanoparticles (including supported nanometals), which benefits from the excellent modification properties of the new closo-[B12H12]2− reducing agent. For example, constructing a boron organic polymer or metal boron organic polymer frame material with closo-[B12H12]2− can realize the pre-fixation of the reducing agent in the frame, and precious metal ions can be converted into the corresponding zero-valent metals in situ (by replacing the closo-[B12H12]2− position). This is an innovative strategy for preparing metal-based catalysts. However, the properties of other closed borohydrides of the closo-[BnHn]2− family have not been developed and reported in this regard.
Here, we report a closed borohydride reductant with a reduction capacity between NaBH4 and closo-[B12H12]2−, closo-[B6H6]2− (except in strong alkali environments, where it exists in the form of closo-[B6H7]−), which can not only reduce noble metal ions to the corresponding zero-valent metals, but also reduce some non-noble metal ions. On the one hand, closo-[B6H6]2− has a moderate reduction capacity compared to NaBH4, which can avoid excessive violent reaction. On the other hand, closo-[B6H6]2− has the same modifiable characteristics as closo-[B12H12]2−, so it can be used in various fields, which will add a wide range of uses and lay a solid foundation for this new boron polyhedron-stabilized metal nanocatalyst.
Experimental
Chemicals and materials
Cs[B6H7] was synthesized according to the literature method,21 and other chemical reagents were purchased from Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). Among them, NaBH4, HAuCl4, Na2PdCl4, H2PtCl6, AgNO3, Ni(NO3)2·6H2O and Cu(NO3)2·3H2O are all analytical grade. Except for HAuCl4, Na2PdCl4 and H2PtCl6, which were formulated into a solution of specified concentration, the other chemical reagents were used directly as they are. Milli-Q ultrapure water (18 MΩ cm−1) was used to prepare all solutions.Characterization method
In this study, X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi, USA) and powder X-ray diffraction (PXRD, Rigaku Miniflex600, Japan) were mainly used to identify the valence state and crystal phase composition of the metal after the reaction. Field emission scanning electron microscopy (FE-SEM, Zeiss SIGMA, UK) and transmission electron microscopy (TEM, HITACHI H-7000FA, Japan) were used to observe the micro morphology of Au, Pd, Pt and Ag driven by closo-[B6H7]−. Among them, SEM adopted the InLens mode and the acceleration voltage was 5 kV, and the voltage and current of TEM were 75 kV and 4 A, respectively.Experimental procedures
Results and discussion
The scope of application of nanomaterials is constantly expanding, and their preparation strategies are also being constantly enriched. Previous studies have shown that closo-[B12H12]2− can transform Au3+, Pd2+, Pt4+ and Ag+ noble metal ions into the corresponding zero valence states in a mild manner, where only Pt4+ to Pt0 requires an additional condition of 80 °C. This indicates that the closed borane anion cluster has properties similar to NaBH4. However, apart from the reports of our team, few other studies have been reported. In order to explore the potential of closo-[B6H7]− in the preparation of metal-based materials, we carried out a detailed exploration with NaBH4 as a reference.Fig. 1 shows the PXRD of metal-based materials prepared with closo-[B6H7]− and NaBH4 as reducing agents, respectively, which can show the crystalline phase composition in the material. Among them, Fig. 1a–f are the results of a reducing agent acting on Au3+, Pd2+, Pt4+, Ag+, Ni2+ and Cu2+; closo-[B6H7]− and NaBH4 can reduce them to the zero valence state, and they present a face-centered cubic structure. In the case of closo-[B6H7]− and NaBH4 as reducing agents, the PXRD results of the metal-based materials obtained are almost consistent, indicating that the mechanism of action of closo-[B6H7]− is consistent with that of NaBH4. Although PXRD can judge the composition state of the material to a certain extent from the crystal phase composition, some compositions exist in a disordered state. In order to further understand the evolution of Au3+, Pd2+, Pt4+, Ag+, Ni2+ and Cu2+ under the action of closo-[B6H7]−, the valence state of the composition in the material was analyzed by XPS (Fig. 2). In the XPS after Au3+ evolution, only two separate signal peaks appear, among which the two peaks near the bond energy of 84.0 eV and 87.7 eV can be attributed to Au4f7/2 and Au4f5/2. In the evolution of Pd2+, there are signal peaks that can be split further. The two narrower and prominent signal peaks around 335.0 eV and 340.3 eV were attributed to the contribution of zero-valent Pd (Pd3d5/2 and Pd3d3/2), while the two weaker wide peaks around 337.0 eV and 342.0 eV were attributed to PdO3d5/2 and PdO3d3/2, respectively. In the XPS after Pt4+ evolution, only two independent signal peaks appear at 71.6 eV and 74.95 eV, corresponding to the binding energy of Pt4f7/2 and Pt4f5/2. Similarly, in the XPS after Ag+ evolution, only two independent signal peaks appear at 368.3 eV and 374.3 eV, corresponding to the contribution of Ag3d5/2 and Ag3d3/2. XPS shows that Ni2+ evolved into two forms of Ni and NiO. The signal peak of 852.5 eV is the characteristic binding energy of Ni2p3/2, and the signal peak of the corresponding salt appears at 861.3 eV. The XPS signal peak of NiO appears near 855.5 eV, and the signal peak of its corresponding salt appears near 880.0 eV. It is difficult to analyze the evolution of Cu2+ in XPS, because the binding energies of Cu, Cu2O and CuO are similar (all around 932.5 eV), but combined with the PXRD results, it can be well judged that Cu2+ has evolved into zero-valent Cu and CuO. In summary, the analysis of the XPS results is: (1) Au3+, Pt4+ and Ag+ are almost completely converted to the zero-valent state (Au0, Pt0, Ag0); (2) the vast majority of Pd2+ is converted to Pd0, and a small part is in the state of PdO, some of which may come from the inevitable oxidation of the surface; (3) most of the Ni2+ is converted into the form of NiO, and a small part is in the zero valent state; (4) Cu2+ is converted into two forms of Cu and CuO.
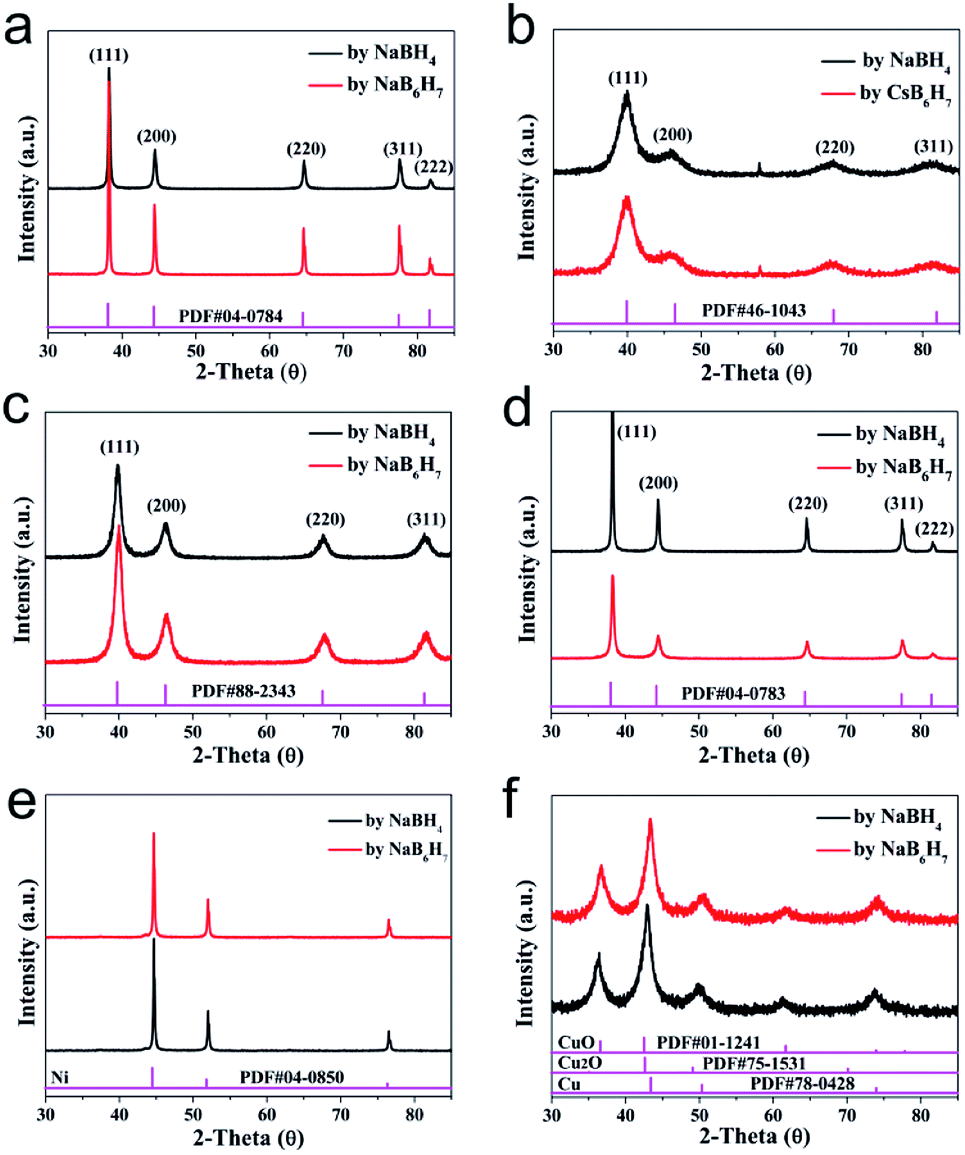 | ||
| Fig. 1 PXRD results of metal materials prepared with Cs[B6H7] and NaBH4 as reducing agents. (a) Converted Au3+; (b) converted Pd2+; (c) converted Pt4+; (d) converted Ag+; (e) converted Ni2+; and (f) converted Cu2+. | ||
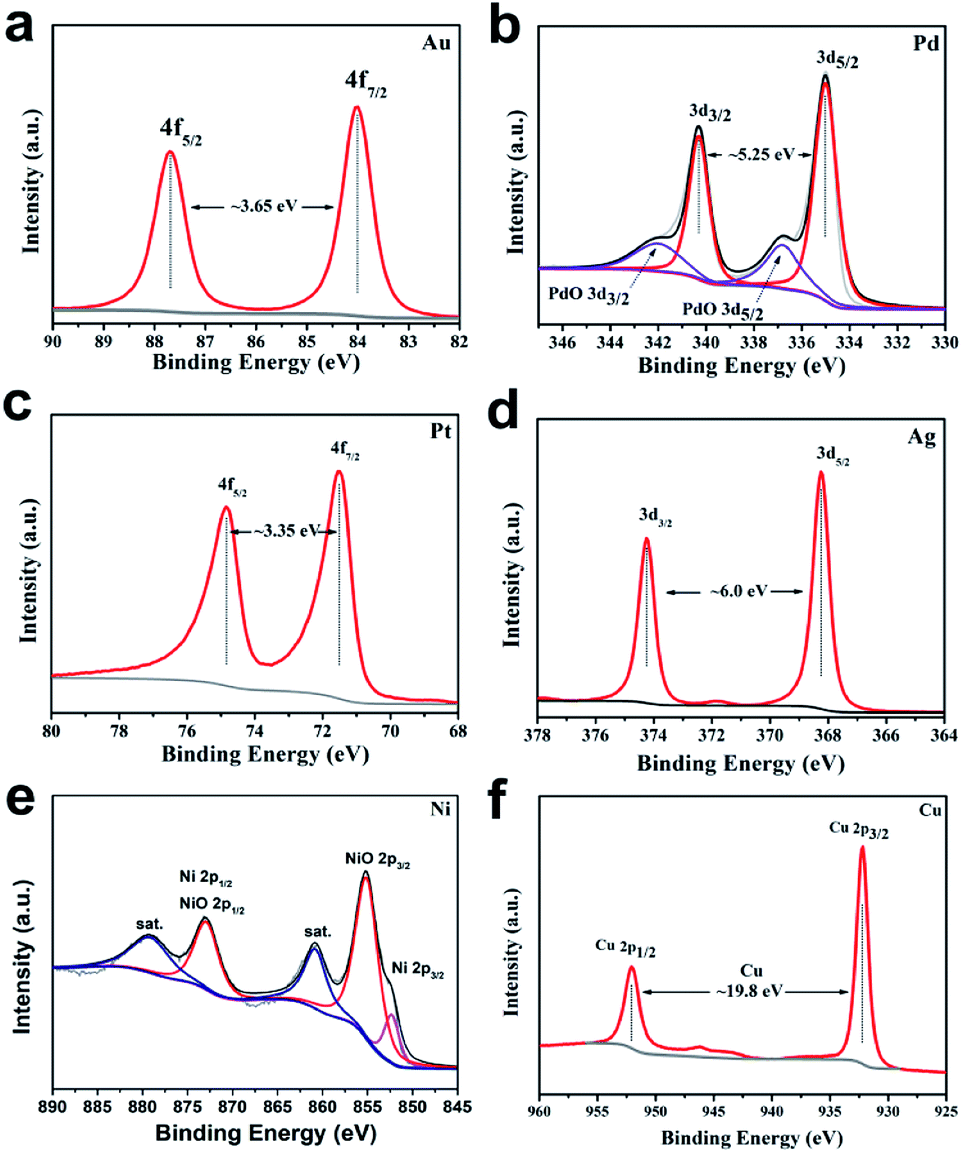 | ||
| Fig. 2 XPS results of metal materials prepared with Cs[B6H7] and NaBH4 as reducing agents. (a) Converted Au3+; (b) converted Pd2+; (c) converted Pt4+; (d) converted Ag+; (e) converted Ni2+; and (f) converted Cu2+. | ||
In NaBH4, H exists in the form of −1 valence, so it has a strong reducing ability for transition metal ions, and itself is converted into sodium borate in aqueous solution. In closo-[B6H7]−, due to the strong polarization of B, each H atom carries an excess of electrons (0.00345e),21 so it exhibits properties similar to NaBH4 (the consistency from the PXRD results has been verified). The difference is that the reducing ability of NaBH4 is too strong, and the metal ions that are instantly converted (or reduced) tend to agglomerate, which seriously hinders the uniformity and nanometerization of zero-valent metals. Closo-[B6H7]− has a mild reducing ability, and Au3+, Pd2+, Pt4+, and Ag+ are converted into sols at room temperature (Fig. 3a–d). On the other hand, the color of these sols is lighter than that of NaBH4, indicating better dispersibility and smaller particle size (generally, the stronger the aggregation of nanomaterials, the more the color they show tends to blue shift). FE-SEM and TEM can also show that Au3+ (Fig. 3e and f), Pd2+ (Fig. 3g and h), Pd2+ (Fig. 3i and j) and Ag+ (Fig. 3k and l) evolved into nano-scale particles under the action of closo-[B6H7]− (due to the chemical adsorption between Pt and closed cage borane, so Pt nanoparticles appear to be encapsulated20). For non-noble metal ions Ni2+ and Cu2+, the reduction ability of closo-[B6H7]− is gentler, and the reduction reaction needs to reach about 60 °C before it can start. As we all know, NaBH4 in aqueous solution preferentially reduces the metal ions that are arranged behind the metal activity sequence. Therefore, when there are metal ions stronger than H+, NaBH4 preferentially reduces H+. Since closo-[B6H7]− is reductively similar to NaBH4, it can almost convert the noble metal ions Au3+, Pd2+, Pt4+ and Ag+ into the corresponding zero-valent state. For Cu2+ and Ni2+, one part evolves into an oxide, and the other part is in a zero-valent state (where only a small part of Ni2+ is converted to a zero-valent state). In order to compare the similarities and differences between Cs[B6H7] and NaBH4 reducing metal ions, we have performed the following experiments. The feed ratio between the metal ions (Au3+) and Cs[B6H7] (or NaBH4) was increased to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to consume all Cs[B6H7] (or NaBH4). Then, the supernatant after the reaction was collected and the solvent was distilled off under reduced pressure, and the Fourier transform infrared spectroscopy (FT-IR, Fig. 4a) signal of the solid was collected. The results showed that after Cs[B6H7] and NaBH4 reduced metal ions, the FT-IR signals of the products were almost the same (BO2− species were generated), indicating that Cs[B6H7] and NaBH4 had the same conversion pathway after reducing metal ions (Fig. 4b). In addition, the B–H stretching vibration at 2480 cm−1 and the cage-effect signal at 1000–1300 cm−1 disappeared, which proved that the cage structure of Cs[B6H7] was dissolved and the B–H bond became a B–O bond.
:1 to consume all Cs[B6H7] (or NaBH4). Then, the supernatant after the reaction was collected and the solvent was distilled off under reduced pressure, and the Fourier transform infrared spectroscopy (FT-IR, Fig. 4a) signal of the solid was collected. The results showed that after Cs[B6H7] and NaBH4 reduced metal ions, the FT-IR signals of the products were almost the same (BO2− species were generated), indicating that Cs[B6H7] and NaBH4 had the same conversion pathway after reducing metal ions (Fig. 4b). In addition, the B–H stretching vibration at 2480 cm−1 and the cage-effect signal at 1000–1300 cm−1 disappeared, which proved that the cage structure of Cs[B6H7] was dissolved and the B–H bond became a B–O bond.
 | ||
| Fig. 3 The phenomenon when sodium borohydride and Cs[B6H7] act on Au3+, Pd2+, Pt4+, and Ag+, respectively. (a) Converted Au3+; (b) converted Pd2+; (c) converted Pt4+; and (d) converted Ag+. Among them, (i) and (ii) marked in red represent the situation when NaBH4 and Cs[B6H7] are used as reducing agents. The FE-SEM images of Cs[B6H7] acting on Au3+, Pd2+, Pt4+ and Ag+ are shown in (e, g, i and k), respectively. The TEM images of Cs[B6H7] acting on Au3+, Pd2+, Pt4+ and Ag+ are shown in (f, h, j and l), respectively. | ||
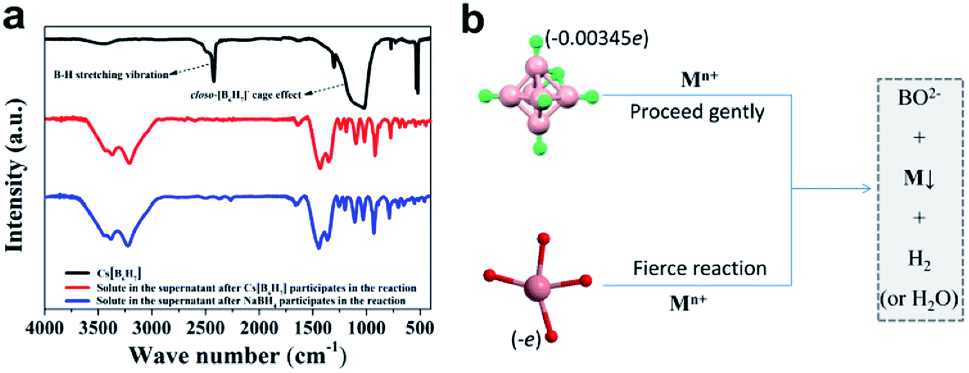 | ||
| Fig. 4 (a) FT-IR results. The black line is the FT-IR signal of Cs[B6H7], the red line is the FT-IR signal after Cs[B6H7] acts on Au3+, and the blue line is the FT-IR signal after NaBH4 acts on Au3+. (b) Closo-[B6H7]− and BH4− have similar conversion paths when reducing metal ions. | ||
In order to evaluate the pros and cons of metal-based materials obtained by the closo-[B6H7]− reduction method in catalytic reactions, Pt and Cu were elected as representatives of precious metals and non-precious metals, respectively, to explore their catalytic properties. In the electrochemical hydrogen evolution reaction (E-HER) of water, simulation 20 wt% Pt/C showed excellent performance. Compared to commercial 20 wt% Pt/C, simulation 20 wt% Pt/C only needs to apply an overpotential of 71 mV to achieve a current density of 10 mA cm−2 (Fig. 5a), and the corresponding Tafel slope is as low as 43 mV dec−1 (Fig. 5b). Then, the catalytic performance of Cu NPs obtained by closo-[B6H7]− was tested by the model reaction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP), which is usually catalyzed by metal nanomaterials. Initially, in the UV-vis spectrum of the mixed aqueous solution of 4-NP and NaBH4, a strong absorption peak at 400 nm was shown and attributed to the formation of 4-nitrophenolate anions. Even after a long period of time, this peak remained unchanged, indicating that the reduction reaction did not occur in the absence of catalyst. Once the Cu-based nanomaterials were added into the system, the reduction reaction proceeded immediately, accompanied by a distinct color change from dark to yellow to colorless in a short time. The reaction progress was monitored through time-dependent UV-vis spectroscopy (Fig. 5c), which showed a rapid decrease of the absorbance value at 400 nm with time. The corresponding conversion over time was obtained and presented in Fig. 5c. It clearly showed that Cu-based materials can completely convert 4-NP to 4-AP in less than 10 min. The pseudo-first-order kinetic equations shown in the inset were further obtained (Fig. 5d). Under the same conditions, the S. Sun team's report needs about 90 min to complete.22 These results indicate the superiority of the metal-based materials prepared with closo-[B6H7]− as a reducing agent.
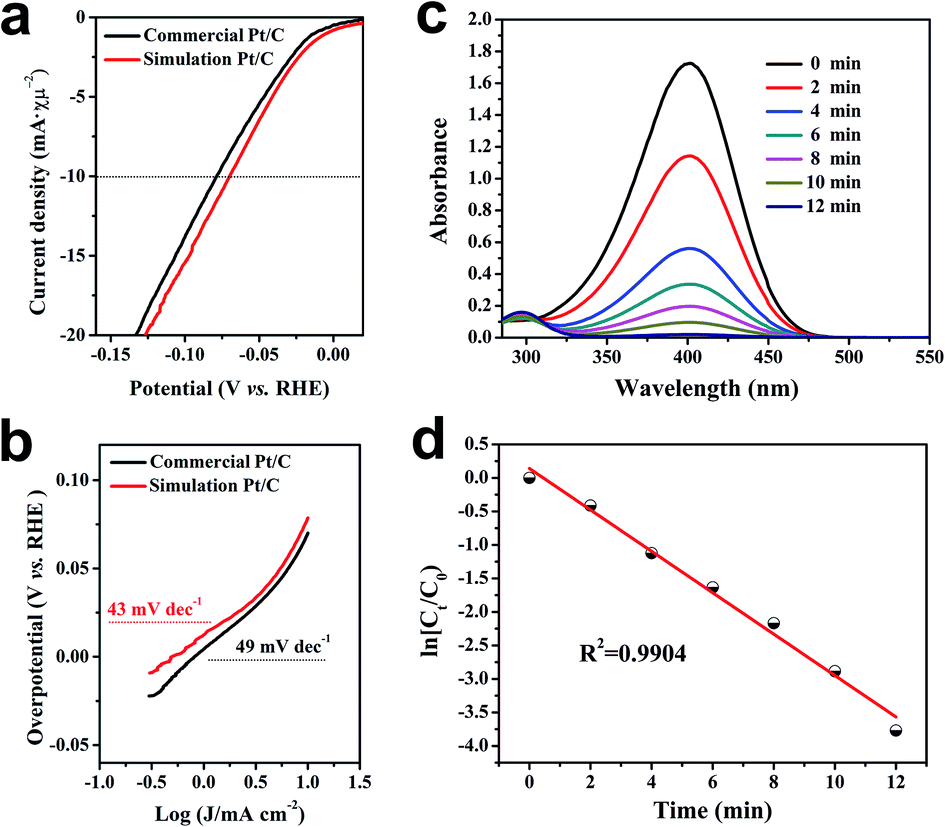 | ||
| Fig. 5 LSV curves (a) and corresponding Tafel slopes (b) when commercial 20 wt% Pt/C and simulated 20 wt% Pt/C are used as the working electrodes, respectively. (c and d) UV-vis spectra and kinetic fitting results of the conversion of 4-nitrophenol to 4-aminophenol catalyzed by Cu-based materials prepared by Cs[B6H7]. | ||
Conclusions
In summary, this research revealed for the first time the potential of closo-[B6H7]− in the closed borane family in the preparation of metal-based materials, and developed a completely new route for the preparation of metal-based catalysts. Closo-[B6H7]− has a milder reduction capacity than NaBH4, which can effectively avoid the aggregation (or evolution into bulk) of metal-based materials. Compared with closo-[B12H12]2−, closo-[B6H7]− not only realizes the conversion of Pt4+ to Pt0 at room temperature, but also expands the reduction range of the closed borane family from noble metals to non-noble metals. In the E-HER of water and reduction reaction of 4-nitrophenol, the Pt-based materials and Cu-based materials prepared by this strategy showed excellent performance. With the continuous upgrading of catalyst preparation technology, this research provides researchers with an alternative way to choose a suitable preparation strategy, which is of great significance for the development of nanomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (no. 2042016 HF1054) and Wuhan University Experiment Technology Project Funding (no. WHU-2016-SYJS-06).References
- A. Kirkeminde, S. Spurlin, L. Draxler-Sixta, J. Cooper and S. Ren, Angew. Chem., Int. Ed., 2015, 54, 4203–4207 CrossRef.
- Q. M. Kainz and O. Reiser, Acc. Chem. Res., 2014, 47, 667–677 CrossRef.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef.
- X. Xu, X. Zhang, H. Sun, Y. Yang, X. Dai, J. Gao, X. Li, P. Zhang, H.-H. Wang, N.-F. Yu and S.-G. Sun, Angew. Chem., Int. Ed., 2014, 53, 12522–12527 Search PubMed.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef.
- K. B. Tang, Y. T. Qian, J. H. Zeng and X. G. Yang, Adv. Mater., 2003, 15, 448–450 CrossRef.
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef.
- R. M. Penner, Acc. Chem. Res., 2017, 50, 1902–1910 CrossRef.
- A. M. A. Mohamed and T. D. Golden, ITexLi, 2016, 283 pages, ISBN: 9535122703 9535122709 Search PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef.
- B. Qi, C. Wu, L. Xu, W. Wang, J. Cao, J. Liu, S. Zhang, D. Gabel, H. Zhang and X. Zhou, Chem. Commun., 2017, 53, 11790–11793 RSC.
- B. Qi, C. Wu, X. Li, D. Wang, L. Sun, B. Chen, W. Liu, H. Zhang and X. Zhou, ChemCatChem, 2018, 10, 2285–2290 CrossRef CAS.
- B. Qi, X. Li, L. Sun, B. Chen, H. Chen, C. Wu, H. Zhang and X. Zhou, Nanoscale, 2018, 10, 19846–19853 RSC.
- X. Zhao, Y. Fu, C. Yao, S. Xu, Y. Shen, Q. Ding, W. Liu, H. Zhang and X. Zhou, ChemCatChem, 2019, 11, 2362–2369 CrossRef.
- X. Zhao, Z. Yang, W. Wang, Y. Li, X. Zhou and H. Zhang, J. Mater. Chem. A, 2020, 8, 7171–7176 RSC.
- X. Zhao, C. Yao, H. Chen, Y. Fu, C. Xiang, S. He, X. Zhou and H. Zhang, J. Mater. Chem. A, 2019, 7, 20945–20951 RSC.
- X. Zhao, Z. Yang, A. V. Kuklin, G. V. Baryshnikov, H. Ågren, W. Wang, X. Zhou and H. Zhang, J. Mater. Chem. A, 2020, 8, 13086–13094 RSC.
- X. Zhao, Z. Yang, A. V. Kuklin, G. V. Baryshnikov, H. Agren, W. Liu, H. Zhang and X. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 31419–31430 CrossRef CAS.
- Z. Wang, Y. Liu, H. Zhang and X. Zhou, J. Colloid Interface Sci., 2020, 566, 135–142 CrossRef CAS.
- N. Jiao, Y. Zhang, L. Liu, J. n. M. Shreeve and S. Zhang, J. Mater. Chem. A, 2017, 5, 13341–13346 RSC.
- M. Shen, H. Liu, C. Yu, Z. Yin, M. Muzzio, J. Li, Z. Xi, Y. Yu and S. Sun, J. Am. Chem. Soc., 2018, 140, 16460–16463 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |