
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid red blood cell membrane coated porous silicon nanoparticles functionalized with cancer antigen induce depletion of T cells†
Antti Rahikkala *a,
Flavia Fontanaa,
Tomás Bauleth-Ramosbcd,
Alexandra Correiaa,
Marianna Kemelle,
Jani Seitsonenf,
Ermei Mäkiläg,
Bruno Sarmentoh,
Jarno Saloneng,
Janne Ruokolainenf,
Jouni Hirvonena and
Hélder A. Santos*ai
*a,
Flavia Fontanaa,
Tomás Bauleth-Ramosbcd,
Alexandra Correiaa,
Marianna Kemelle,
Jani Seitsonenf,
Ermei Mäkiläg,
Bruno Sarmentoh,
Jarno Saloneng,
Janne Ruokolainenf,
Jouni Hirvonena and
Hélder A. Santos*ai
aDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00014, Helsinki, Finland. E-mail: antti.rahikkala@helsinki.fi; helder.santos@helsinki.fi
bInstituto de Investigação e Inovação em Saúde (I3S), University of Porto, Rua Alfredo Allen, 208, 4200-135, Porto, Portugal
cInstituto de Engenharia Biomédica (INEB), University of Porto, Rua Alfredo Allen, 208, 4200-135, Porto, Portugal
dInstituto Ciências Biomédicas Abel Salazar (ICBAS), University of Porto, Rua Jorge Viterbo 228, 4150-180, Porto, Portugal
eDepartment of Chemistry, University of Helsinki, FI-00014, Helsinki, Finland
fNanomicroscopy Center, Aalto University, FI-02150, Espoo, Finland
gLaboratory of Industrial Physics, Department of Physics, University of Turku, FI-20014, Turku, Finland
hCESPU, Instituto de Investigação e Formação Avançada em Ciências e Tecnologias da Saúde, Instituto Universitário de Ciências da Saúde, 4585-116 Gandra, Portugal
iHelsinki Institute of Life Science (HiLIFE), University of Helsinki, FI-00014, Helsinki, Finland
First published on 23rd September 2020
Abstract
Erythrocyte-based drug delivery systems have been investigated for their biocompatibility, long circulation time, and capability to transport cargo all around the body, thus presenting enormous potential in medical applications. In this study, we investigated hybrid nanoparticles consisting of nano-sized autologous or allogeneic red blood cell (RBC) membranes encapsulating porous silicon nanoparticles (PSi NPs). These NPs were functionalized with a model cancer antigen TRP2, which was either expressed on the surface of the RBCs by a cell membrane-mimicking block copolymer polydimethylsiloxane-b-poly-2-methyl-2-oxazoline, or attached on the PSi NPs, thus hidden within the encapsulation. When in the presence of peripheral blood immune cells, these NPs resulted in apoptotic cell death of T cells, where the NPs having TRP2 within the encapsulation led to a stronger T cell deletion. The deletion of the T cells did not change the relative proportion of CD4+ and cytotoxic CD8+ T cells. Overall, this work shows the combination of nano-sized RBCs, PSi, and antigenic peptides may have use in the treatment of autoimmune diseases.
Introduction
Recent years have seen an increased interest in investigating biomaterials as carriers of vaccine subunits for immunotherapy.1–5 These carriers include cells and their fragments, providing biocompatibility and natural mechanisms for transport, localization, and responsivity regarding microenvironments in the body.6 Due to their high biocompatibility, red blood cells (RBC)7–12 have potential as delivery systems in therapeutic applications. Indeed, therapeutic agents have already been loaded into carrier RBCs by, for example, electroporation, osmosis, bio-bridge methods, and cell-penetrating peptide methods.13 A key role for carrier purposes is played by the dense glycan coating present in RBC membrane,3 which provides immune evasion and stability in the blood circulation.12,14,15 After a RBC has served its purpose, it is removed from the circulation by macrophages in spleen, liver, or lymph node, hence representing potential targets for designed RBC-based therapies.13Several studies have shown that engineered RBCs may have use in treating autoimmune diseases. Kontos et al. found that RBCs modified with an RBC-targeted ovalbumin (OVA) induced deletion of both CD4+ and CD8+ T cells.16,17 Copp et al. used RBC-cloaked poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) to act as alternative binding sites for anti-RBC polyclonal immunoglobulin G in order to treat anemia.18 In another study, Pishesha et al. covalently attached disease-associated autoantigens to RBCs, which blunted specific B, CD4, and CD8 T cell responses, as well as provided protection against experimental autoimmune encephalomyelitis and type 1 diabetes in separate animal models.19
Previously, we have used cell membrane-derived NPs to study biomimetic nanoreactors20,21 and immunotherapeutic nanoparticles.2,22,23 In the current study, inspired by the benefits of RBCs as carrier cells for antigens and their potential to treat autoimmune diseases, we have developed a hybrid nanoparticle system by encapsulating porous silicon nanoparticles (PSi NP) within nanosized RBC membranes. Our aim was to use RBC membrane cloaked nanoparticles to deliver model antigenic peptides to immune cells and evaluate the immunological properties and antigen-delivery capability of these biohybrid nanocomposite particles. The used model peptide is derived from the tyrosinase-related protein-2 (TRP2).24,25
Two different formulations, with surface-anchored or encapsulated peptides, were fabricated in order to evaluate if the position of the antigen in our nanosystems had any role in triggering an immune response (Fig. 1). Differential light scattering (DLS) and electrophoretic light scattering (ELS) were used to determine the size, polydispersity index (PDI), and ζ-potential of the hybrid nanocomposites. We used transmission and scanning electron microscopy (TEM and SEM, respectively) to characterize the morphology of the nanocomposites, while energy-dispersive X-ray spectroscopy in combination with SEM showed the successful UnPSi encapsulation inside the RBC membranes. Flow cytometry was used to study how the nanocomposites affect CD4+ and CD8+ T cells obtained from peripheral blood mononuclear cells (PBMCs). In these experiments we also compared RBC-encapsulated NPs of allogeneic (alloRBC) and autologous (autoRBC) origins, respectively, depending on whether they were extracted from the same blood donor as the PBMCs (autologous, blood type A+) or from another donor (allogeneic, blood type B+).
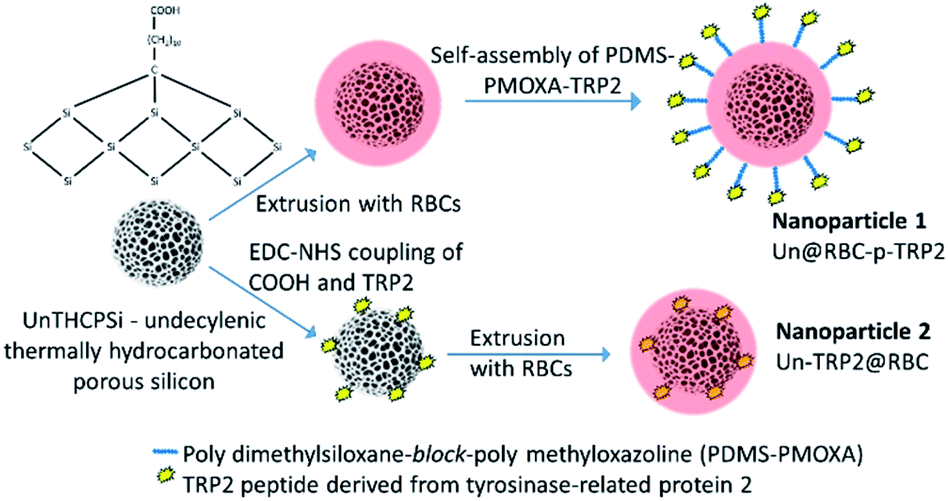 | ||
| Fig. 1 This study features two different types of hybrid nanoparticles. The first type has UnPSi encapsulated within RBC membrane. TRP2 peptides are anchored on the RBC membrane via a cell membrane-mimicking block copolymer PDMS-PMOXA. The second type has UnPSi, surface-functionalized with TRP2 peptides, which are encapsulated within RBC membranes. | ||
Experimental
Preparation of undecylenic thermally hydrocarbonated porous silicon (UnPSi) nanoparticles
UnPSi NPs were prepared as described elsewhere.26,27 In brief, boron doped p+ silicon 〈100〉 wafers were anodized in a hydrofluoric acid (38%)–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) electrolyte mixture. Pulsing low/high etching current created high porosity fracture planes, after which the PSi films were detached from the wafers by an abrupt increase in the etching current and stabilized by thermal hydrocarbonization under steady flow of N2/acetylene (1:1) at 500 °C to generate thermally hydrocarbonated (THC) PSi. The THCPSi films were then thermally treated in undecylenic acid for 16 h at 120 °C, which created a surface functionalization of carboxylic groups (UnPSi). The UnPSi films were wet milled in a 5 vol% undecylenic acid–dodecan solution. The nanoparticles presenting different sizes were separated by centrifugation and stored at 4 °C in ethanol until use.
:1) electrolyte mixture. Pulsing low/high etching current created high porosity fracture planes, after which the PSi films were detached from the wafers by an abrupt increase in the etching current and stabilized by thermal hydrocarbonization under steady flow of N2/acetylene (1:1) at 500 °C to generate thermally hydrocarbonated (THC) PSi. The THCPSi films were then thermally treated in undecylenic acid for 16 h at 120 °C, which created a surface functionalization of carboxylic groups (UnPSi). The UnPSi films were wet milled in a 5 vol% undecylenic acid–dodecan solution. The nanoparticles presenting different sizes were separated by centrifugation and stored at 4 °C in ethanol until use.
Erythrocyte and lymphocytes extraction from blood samples
Blood samples from anonymous donors were provided by the Finnish Red Cross. A buffy coat from an A+ blood type donor was used as the source of PBMCs and the RBCs for autologous samples. The buffy coat, was ca. 70 mL in volume, diluted with phosphate buffered saline (PBS, pH 7.4) in 1:1 ratio. Ficoll-Paquet density gradient medium was applied into six-50 mL centrifuge tubes (15 mL per tube) and 20 mL of the blood–PBS mixture was carefully layered on top. The blood samples were then centrifuged for 40 min at 400g. After the centrifugation, the RBCs were deposited in the bottom of the falcons, Ficoll-Paquet layer on top of the RBCs, and a thin layer of PBMCs between Ficoll-Paquet and plasma, which was the topmost. The buffy coat and the RBCs in the bottom were carefully collected and used immediately in the experiments. The total extracted amount of PBMCs from the buffy coat was ca. 650 M cells. The allogeneic RBCs were obtained from a 6 mL blood sample with EDTA as anticoagulant, provided by the Finnish Red Cross.
Preparation of the RBC ghosts
The RBCs were washed in PBS (pH 7.2) for four times by centrifuging at 4020g, removing the supernatant, and resuspending the cells in PBS. Finally, the RBCs were resuspended in lysis buffer (20 mM of Tris HCl (pH 7.5), 10 mM of KCl, 2 mM of MgCl2 in Milli-Q water). After a brief ultrasonication, the solution was centrifuged at 4020g and the supernatant collected. The pellet was resuspended in lysis buffer, centrifuged again at 4020g, and the supernatant was collected and pooled together with the first supernatant. Then, the supernatant was centrifuged at 20000g for 20 min at 4 °C using a TLA 120.2 rotor in an ultracentrifuge (Optima MAX, Beckmann Coulter, USA). The pellet was discarded and the supernatant was finally centrifuged at 100000g for 5 min at 4 °C. The resulting pellet was redispersed in Milli-Q water.
Conjugating TRP2 with PDMS-b-PMOXA
The carboxylic end-group of TRP2 (SVYDFFVWL, N-terminus conjugated with FITC-Ahx, 1175.4 g mol−1, purity 99.45%, lot number 88297U001, United Peptide, VA, USA) were first activated with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) and N-hydroxysuccinimide (NHS) in 2-(N-morpholino)ethanesulfonic acid (MES) buffered Milli-Q, pH 5.5, for 2 h. Then, an equimolar amount of PDMS-b-PMOXA-NH (6185 g mol−1) was added into the solution and the pH was increased to 7.0. The conjugation proceeded for 24 h under stirring in the dark.Fabrication of UnPSi@RBC-PDMS-PMOXA-TRP2 nanoparticles
500 μL of PBS (pH 7.2) containing 500 μg of UnPSi NPs were combined with 450 μL of RBC ghosts and 50 μL of PDMS-PMOXA-TRP2. The solution was then extruded 21 times through a porous membrane (0.4 μm pore size, Nucleopore Track-Etch Membrane, Whatman, UK) yielding RBC-encapsulated UnPSi nanoparticles with self-assembled PBMS-PMOXA-TRP2 molecules within the RBC membrane.Fabrication of UnPSi-TRP2@RBC
First, 3 mg of UnPSi was dispersed in 500 μL MES at pH 5.5. The carboxylic surface groups of UnPSi were activated by adding 0.11 mmol of EDC and stirring in dark for 1 h. Then, 0.125 mmol of NHS and 0.2 mmol of spermine linker were added to the solution to conjugate the spermine to the activated carboxyl groups, and the pH was adjusted at 7.2. The reaction was allowed to proceed for two hours under stirring in dark. Subsequently, the UnPSi-spermine dispersion was washed twice with Milli-Q water and then dispersed in 500 μL PBS at pH 7.2. In a separate simultaneous activation reaction, 200 μg TRP2 was dissolved in 40 μL ethanol to produce a 0.2 μmol solution. Again, 0.11 mmol of EDC was added into the solution to activate the carboxyl end groups of TRP2. After 1 h under stirring in dark, 0.125 mmol of NHS was added into the solution. Subsequently, the solution was poured under vigorous stirring into a vial containing the washed UnPSi-spermine. The pH was readjusted to 7.2. The coupling of TRP2 to the free amine group of spermine was allowed to take place overnight under stirring in dark. Then, the UnPSi-spermine-TRP2 nanoparticle solution was washed twice with Milli-Q water and dispersed in PBS at pH 7.2 and diluted to 1 mg mL−1. Finally, 500 μL of PBS containing 500 μg UnPSi-spermine-TRP2 was combined with 450 μL of RBC ghosts (ca. 20 × 106 cells per mL) and 50 μL of PBS. The solution was then extruded as described previously to yield UnPSi-spermine-TRP2@RBC, abbreviated as UnPSi-TRP2@RBC.Electron microscopy
Samples for standard TEM and SEM were prepared by dropping a 5 μL droplet of the NP suspension on a formvar/carbon electron microscopy grid (200 mesh; Electron Microscopy Sciences, Hatfield, PA, USA). Samples for cryo-TEM were prepared using an automatic plunge freezer (Vitrobot Mark IV, FEI, Hillsboro, OR, USA). The grids were imaged with a TEM (JEOL JEM-1400, Jeol Ltd. Japan, Tokyo) using 80 kV acceleration voltage and cryo-TEM (JEOL JEM-3200FSC) using 100 kV acceleration voltage on a bright-field mode, or a SEM (S-4800, Hitachi, Tokyo, Japan) using 30 kV acceleration voltage in scanning and backscatter modes. The SEM was also equipped with an energy-dispersive X-ray spectrometer (INCA 350, Oxford Instruments, Abingdon, UK) to perform elemental analysis.Immunostimulation studies
PBMCs were incubated at room temperature in a solution containing 10 μM CellTrace Violet (ThermoFisher Scientific, Waltham, MA, USA) for 20 min, which was used to distinguish cells from debris or aggregated nanoparticles. Subsequently, the PBMCs were seeded in 48-well plates at ca. 500000 cells per well in 400 μL Roswell Park Memorial Institute 1640 medium (RPMI), supplemented with 10% heat-inactivated fetal bovine serum (FBS, ThermoFisher), 1% penicillin streptomycin, 1% minimum essential medium (MEM) non-essential amino acids solution, and 1% L-glutamine. The PBMCs were co-incubated with the samples, each as a triplicate, at 50 μg mL−1 for 72 h at 37 °C, 5% CO2. On day three, each sample was washed once in 100 μL of 1 M of PBS (pH 7.2), and then resuspended in 100 μL of a staining solution. The staining solution for each sample was composed of 2.5 μL anti-human mouse CD3-PE (BioLegend, San Diego, CA, USA), 2 μL anti-human mouse CD4-APC-Vio770 (Miltenyi Biotec, Bergish Gladbach, Germany), 10 μL anti-human mouse CD8a-APC (BD Biosciences, San Jose, CA USA), and 20 μL FcR blocking reagent (Miltenyi Biotec) to prevent non-specific binding, and filled-up to 100 μL with 1 M of PBS. The PBMCs were incubated in the staining solutions for 30 min at 4 °C, after which they were washed once with PBS (pH 7.2) and resuspended in 300 μL of 1 M PBS (pH 7.2). Prior to the flow cytometry analysis, the samples were further stained for 10 min with 2.5 μL 7-Amino-Actinomycin D (7-AAD, BD Biosciences) to distinguish PBMCs going through apoptosis.
Flow cytometry analysis
Cells were analyzed using an LSRFortessa flow cytometer (BD Biosciences, San Jose, CA, USA) using excitation at 405 nm and bandpass filter 450/50 nm for CellTrace Violet, 488 nm excitation with 530/30 bandpass filter for TRP2-FITC, 561 nm excitation with 575/26 bandpass filter for CD3-PE and 670/30 bandpass filter for 7-AAD, and 640 nm excitation with 670/30 nm bandpass filter for CD8-APC and 780/60 nm bandpass filter for CD4-APC-H7. Compensation controls for the used antibodies were prepared using compensation beads. CellTrace Violet-labeled PBMCs were used as compensation controls for CellTrace. Compensations for 7-AAD viability dye were prepared by incubating PBMCs for 10 min in 40% ethanol, after which the cells were washed and 7-AAD was applied to the PBMC sample according to the manufacturer instructions.Ethical statement
All human procedures were performed in accordance with the guidelines of the Finnish Red Cross with proper consent of the anonymous donors, according to the national ethical approvals.Statistics
The values obtained in all the experiments are reported as mean ± standard deviation from at least three independent measurements. A one-way analysis of variance (ANOVA) and the Tukey's test were used to evaluate the statistical significances of the obtained values. The levels of significances were set at probabilities *p < 0.05, **p < 0.01, and ***p < 0.001. The statistical analyses were performed using OriginPro 2018b (OriginLab Corporation, Northampton, MA, USA). The calibration curve of the concentration dependent TRP2 fluorescence was calculated by a least squares fit using Matlab R2018b (Mathworks, Natick, MA, USA).Results and discussion
We prepared two different hybrid nanoparticle systems comprising of RBC membrane, UnPSi NPs, and TRP2, functionalized with isothiocyanate (FITC) in the N-terminus. In the first, we modified the RBC membranes with the self-assembling membrane-mimicking poly(dimethylsiloxane-block-2-methyl-2-oxazoline) (PDMS65-b-PMOXA14-NH, abbreviated PDMS-PMOXA) diblock copolymer,28 which was functionalized with the TRP2 model cancer antigen.29 Previously, PDMS-PMOXA has been used to fabricate polymersomes with the average membrane thickness of 15.4 ± 0.7 nm.30 Subsequently, UnPSi NPs were encapsulated within the modified RBCs to fabricate the UnPSi@RBC-PDMS-PMOXA-TRP2 nanocomposites, abbreviated here as UnPSi@RBC-p-TRP2 in the following sections. In the second type of particle, we conjugated TRP2 on the carboxyl groups presented on the surface on UnPSi NPs, which we subsequently encapsulated within the RBC membranes to generate UnPSi-TRP2@RBC nanocomposites.We isolated PBMCs and RBCs from buffy coat by Ficoll-Paquet gradient density centrifugation. The obtained RBC concentration was ∼40 × 106 cells per mL. Subsequently, we prepared RBC ghosts by hemolysis in a hypotonic solution. Undecylenic thermally hydrocarbonated PSi (UnPSi) was encapsulated within the RBC ghosts as a model nanoparticle. UnPSi allows straightforward surface functionalization of peptides by 1-ethyl-3-(3-dimethylamino)propyl carbodiimide/N-hydroxysuccinimide (EDC/NHS) click chemistry.31 Furthermore, high surface area and porosity32 improve the potential of UnPSi as a delivery vehicle for therapeutic molecules.33,34 Non-functionalized UnPSi induce proliferation of CD3+ T cells; however, they do not activate CD3+CD4+ or CD3+CD8+ cells. They also promote and cause Th2 cells to show increased secretion of regulative cytokines.35
To prepare the UnPSi@RBC-p-TRP2, we first conjugated the TRP2 peptide to the amphiphilic PDMS-b-PMOXA block copolymer's hydrophilic PMOXA block, end-functionalized with piperazine to provide a secondary amine, using 1-ethyl-3-(3-dimethylamino)propyl carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemical reaction.28 The purpose of the PDMS-b-PMOXA was to act as an anchor to immobilize the TRP2 on the surface of the RBC membrane. The conjugation was confirmed with by fluorescence provided by FITC functionalization of the TRP2 (Fig. S1†). We added the polymer–peptide conjugate into a solution containing the lysed RBCs after which we encapsulated UnPSi NPs within the RBC ghosts by extrusion through a semipermeable membrane with 400 nm average pore size.36,37 To prepare the UnPSi-TRP2@RBC, we used again EDC/NHS chemistry to first conjugate spermine as a linker to the carboxylic groups in UnPSi NPs, after which we used EDC/NHS chemistry to conjugate TRP2 to the free ends of the spermine linkers. This was followed by the encapsulation of UnPSi-TRP2 inside RBC ghosts. Using radioactive 21Tb-implanted THCPSi, we have previously determined that the yield of PSi@RBC encapsulation by extrusion is ∼22%.38
Comparing to the size (Table 1) of extruded RBC ghosts (359 ± 12 nm), we determined that PDMS-PMOXA-TRP2 fuses into the RBC membrane, since the nanocomposite size almost doubled (634 ± 210 nm) with unimodal size distribution. The PDMS-PMOXA-TRP2 conjugate self-assembles within the RBC membrane during the extrusion as the lipophilic PDMS block includes itself within the phospholipidic bilayer of the membrane, while the shorter hydrophilic PMOXA block sticks out from the membrane, expressing TRP2 as its outmost component.30 We hypothesized the likelihood of the fusion to increase, also due to the fact that the high manually applied pressure in the syringe drives both the PDMS-PMOXA micelles and RBC ghosts through the 400 nm pores simultaneously. On the contrary, UnPSi-TRP2@RBC showed ca. 135 nm increase compared to non-functionalized UnPSi, while being approximately the same size as extruded RBC ghosts. PDMS-PMOXA increased the ζ-potential and the size of the RBC NPs. This result is in line with a previous study, where PDMS-PMOXA polymersomes were found to have a ζ-potential of +17.0 ± 0.4 mV.28 However, the addition of TRP2 decreased the ζ-potential of both UnPSi-TRP2 and UnPSi@RBC-p-TRP2. The calculated net charge of TRP2 is −1.0 at pH 7.0.39 The PDI of the RBC-encapsulated samples was ca. 0.2 for both the UnPSi-TRP2@RBC and the UnPSi@RBC-PDMS-PMOXA. Both the UnPSi-TRP2@RBC and UnPSi@RBC-PDMS-PMOXA-TRP2 showed good stability by maintaining their size and PDI during a 24 h stability experiment (Fig. S2†).The successful encapsulation of PSi NPs within the RBC-PDMS-PMOXA ghosts was shown using TEM (Fig. 2a and b) and SEM (Fig. 2c and d) with EDX (Fig. 2e). Interestingly, in a previous report on PSi-induced hemotoxicity, Shahbazi et al. found that UnPSi had high hemolytic activity due to its surface hydroxyl groups, which caused increased interaction with the erythrocyte membrane.40 However, in our study, the imaged nanoparticles avoided adhering to the RBC membrane. Our TEM observations showed that UnPSi NPs are not tightly encapsulated with RBC membranes (Fig. 2a–d), which is mostly related to the irregular shape of PSi NPs and negative ζ-potential of both the PSi and RBC membranes. Comparing UnPSi-TRP2@RBC to UnPSi@RBC-p-TRP2, the latter displays clearly a darkened thicker surface than the former. This effect may result from the addition of PDMS-PMOXA-TRP2 and the subsequent drying of the sample during TEM sample preparation increases the density of the RBC membrane in the Un@RBC-p-TRP2 compared to the UnPSi-TRP2@RBC. Micrographs obtained by cryo-TEM showed RBC ghosts with ca. 300 nm diameter (Fig. S3a†) and two fused UnPSi@RBC-p-TRP2 NPs (Fig. S3b†), where the addition of PDMS-PMOXA is apparent through the thicker membrane width. Micrographs obtained by cryo-TEM preserve the membrane morphology of the hybrid NPs, since drying is not involved in the sample preparation. Furthermore, the latter micrograph indicates poor stability of the PDMS-PMOXA modified RBCs, which is evident due to the fusion of two NPs and also mirrors the high PDI in the DLS measurements. The SEM were operated in scanning electron (Fig. 2c) and scanning transmission electron microscopy mode (Fig. 2d). The area-specific EDX confirmed successful encapsulation by showing the presence of elemental silicon (from UnPSi particles) and phosphorus (from the phospholipids of the RBC membrane).
| Sample | Hydrodynamic diameter [nm] | PDI | ζ-Potential [mV] |
|---|---|---|---|
| UnPSi | 209 ± 3 | 0.21 ± 0.01 | −20.6 ± 1.6 |
| UnPSi-TRP2 | 216 ± 1 | 0.11 ± 0.01 | −29.3 ± 0.4 |
| RBC ghost | 359 ± 12 | 0.24 ± 0.12 | −9.5 ± 0.2 |
| PDMS-PMOXA micelles | 242 ± 2 | 0.09 ± 0.02 | 17.7 ± 1.5 |
| UnPSi-TRP2@RBC | 344 ± 5 | 0.20 ± 0.01 | −16.3 ± 0.2 |
| UnPSi@RBC-PDMS-PMOXA | 687 ± 9 | 0.21 ± 0.01 | −10.9 ± 1.1 |
| UnPSi@RBC-p-TRP2 | 634 ± 210 | 0.55 ± 0.03 | −27.5 ± 1.7 |
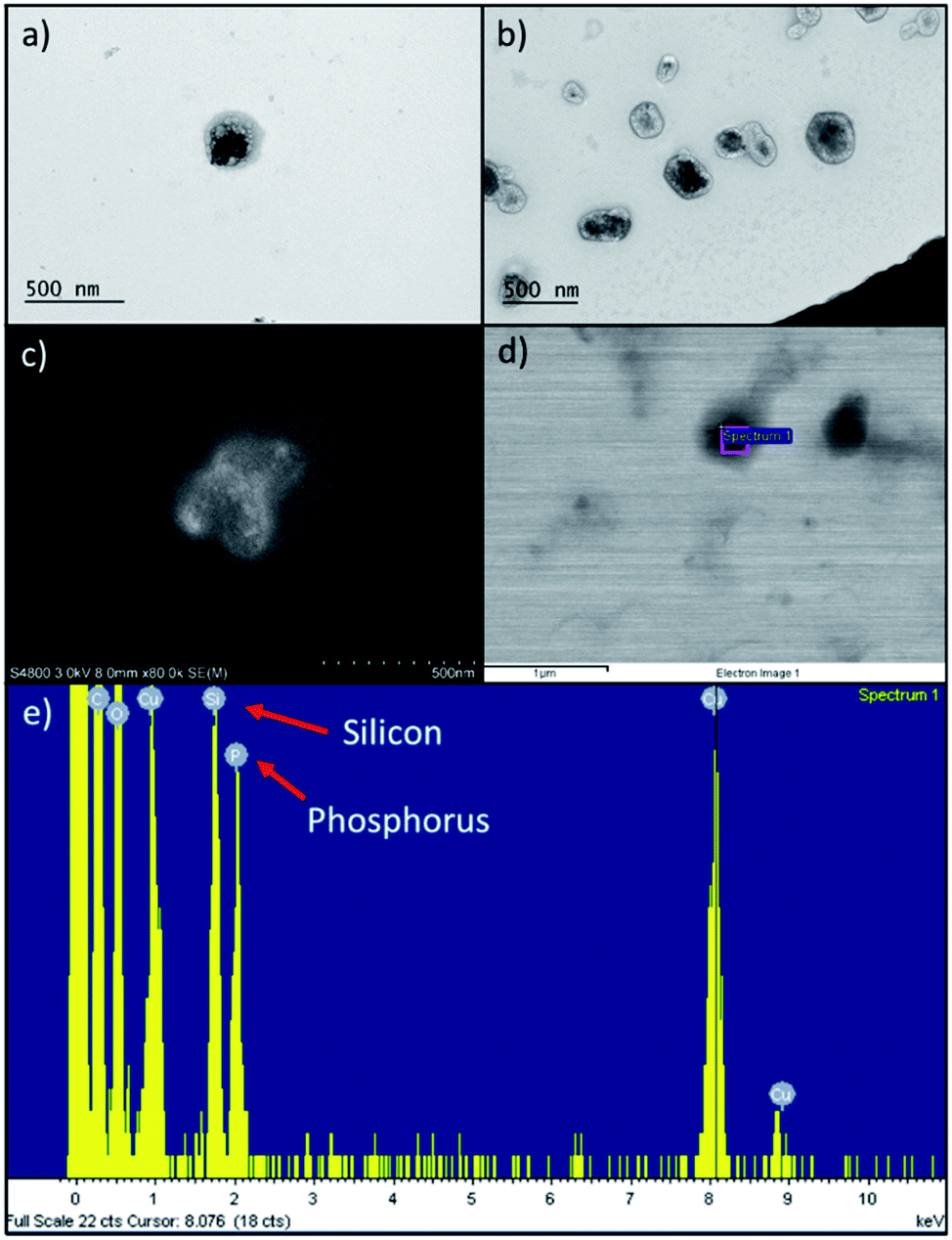 | ||
| Fig. 2 (a) TEM micrograph of Un-TRP2@RBC. Encapsulated UnPSi NP appear as a dark irregular object inside the light dark RBC membrane. (b) TEM micrograph of UnPSi@RBC-p-TRP2. (c) A secondary electron SEM micrograph showing UnPSi nanoparticle enveloped by a RBC membrane. (d) Backscattered electron SEM micrograph of UnPSi-TRP2@RBC. The rectangle inside a particle shows the area where the EDX spectrum in (e) was measured from. (e) EDX spectrum, which indicates elemental Si from the PSi NP and P from the RBC membrane. | ||
We investigated the effects of the NPs on lymphocytes in PBMC populations by evaluating the expression of T-cell markers CD3, CD4, and CD8. The cells were stained with CellTrace™ Violet prior to seeding to distinguish PBMCs from potential NP aggregates or cell debris during the flow cytometry measurements. The samples were incubated in triplicates, at the concentration of 50 μg mL−1, with ca. 0.5 × 106 PBMCs. After 72 h, we washed the PBMCs, blocked unspecific binding with human Fc receptor blocking antibody (CD16/23/32/64/89), and stained them with CD3-PE, CD4-APC-H7 and CD8-APC to investigate the T-cell phenotypes. Furthermore, all the samples were stained with 7-AAD dye prior to flow cytometry analysis to evaluate the cellular viability. The most significant change in T cell populations was the depletion of T cells (Fig. 3), where we gated the cells as Celltrace+, 7-AAD−, CD3+, and lymphocyte gate (Fig. S4†). We chose this gating strategy to first include lymphocytes, then distinguish cells from debris and NPs, remove apoptotic cells, and finally compare the count of CD3+ T cells to PBMC control. The percentage of CD3+ cells in the untreated PBMC sample was 60% ± 1%, which decreased to 44% ± 1% after gating 7-AAD− to remove apoptotic cells. It is notable, that the T cell amounts in the samples with TRP2 and PDMS-PMOXA-TRP2 did not deviate from the PBMC control sample (Fig. S5†). The amount of T cells reduced systemically in all the samples involving the RBC NPs. After removing the apoptotic PBMCs, the UnPSi@autoRBC-p-TRP2 sample had the lowest percentage of CD3+ T cells with 22% ± 8%. This depletion was even more pronounced when we measured the cell counts from the lymphocyte gate (Fig. S5†), where the T cell percentage ranged from 14% ± 4% (autoRBC) to as low as 2% ± 1% (UnPSi@alloRBC-p-TRP2), compared to 35% ± 1% in the untreated PBMC sample. In samples with UnPSi encapsulated within both the allo- and auto-RBCs, the T cell depletion was in the similar levels as with allo- and auto-RBC ghosts. However, when TRP2 was conjugated on the encapsulated UnPSi, the results showed a slightly higher amount of T cells going through apoptosis, especially when comparing autoRBC and UnPSi-TRP2@autoRBC. There was no difference between UnPSi-TRP2 samples encapsulated within allo- or auto-RBCs, which indicates that in this case the TRP2 is a dominant factor that dominates the effect of the RBC phenotype. This indicates that the T cells may be going through apoptosis and they lose their membrane integrity, which causes them to shift from the lymphocyte gate in the side scatter-forward scatter plot.41 Furthermore, the changes in CD3+ CD4+ and CD3+ CD8+ differentiation was insignificant compared to the control (Fig. S6†). The gating was otherwise similar to the depletion analysis, but CD3+ T cells were additionally divided into CD4+ and CD8+.
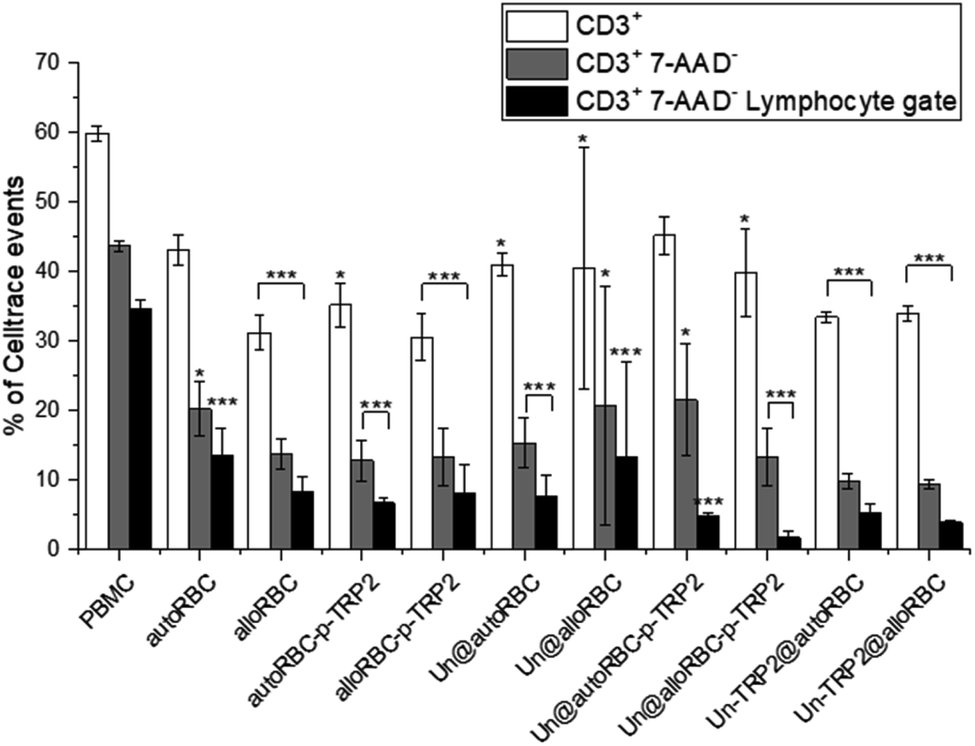 | ||
| Fig. 3 T cell depletion induced by the NP samples after 72 h co-incubation with 700000 PBMCs. N = 3 for all experiments; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005. | ||
A further analysis showed that 27% ± 1% of CD3+ cells were apoptotic in the PBMC control sample. From the UnPSi-TRP2@RBC samples, the allogeneic had the highest apoptosis rate of all samples with 72% ± 1% being 7-AAD positive, while the autologous had 71% ± 3% (Fig. 4). The apoptosis rates for T cells in the Un@autoRBC-p-TRP2 and Un@alloRBC-p-TRP2 were 53% ± 5% and 67% ± 16%, respectively.
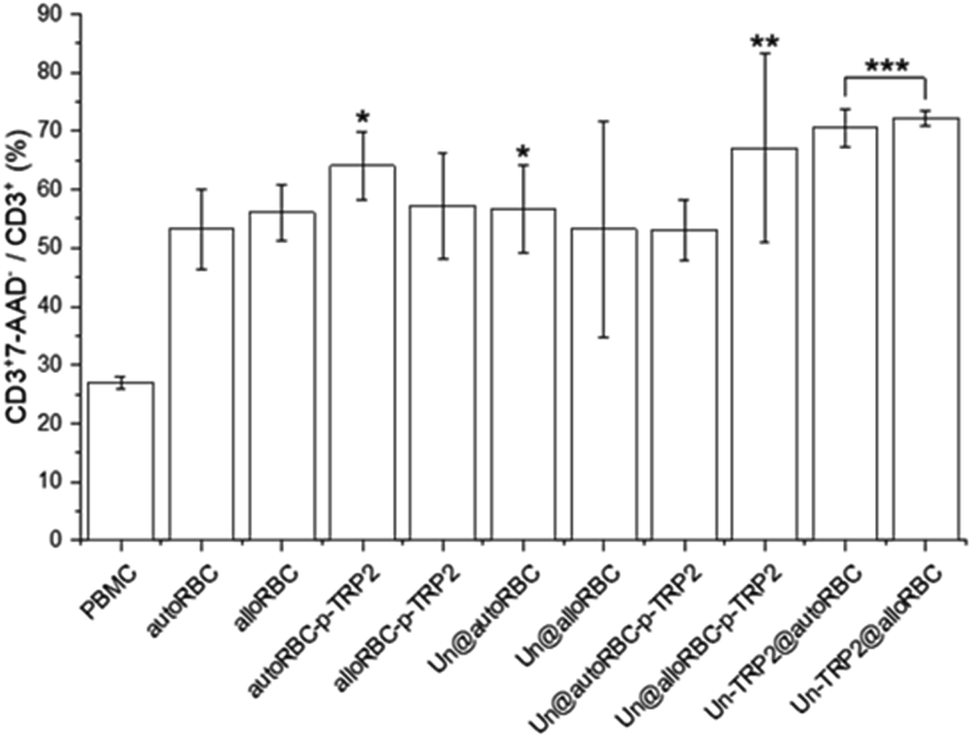 | ||
| Fig. 4 The percentage of apoptotic CD3+ T-cells in the T cell population. N = 3 for all experiments; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005. | ||
The DLS and EM-imaging showed that the encapsulation of UnPSi within the RBCs was successful. The NPs have a spherical morphology and the encapsulation is not tight, which is due to the rigidity of the RBC membrane when forced to nano-size42 and the electrostatic repulsion between the negatively charged RBC membrane and the UnPSi. PDMS-PMOXA has been shown to be able to fuse into the lipid bilayer and induce phase separation in lipid bilayers in both synthetic and biological cell membranes.43–45 Accordingly, PDMS-PMOXA almost doubled the size of the NPs, which was accompanied with reduced stability and high PDI. Furthermore, the addition of TRP2 and PDMS-PMOXA changed the ζ-potential of the NPs, as was expected regarding their charge.
Macrophages and dendritic cells (DCs) clear aged erythrocytes from systemic circulation on a daily basis.19 In normal circumstances, this task is mainly performed by red pulp macrophages; however, in inflammatory conditions also plasmacytoid dendritic cells (pDCs) and monocytes increase erythrocyte consumption, which also initiates production of RBC antibodies.46 Ciana et al. speculated that multiple passes through the spleen reduce the size, leads to losses of portions of the membrane, and consequently result in the rigidity of the membrane.42 This leads to phagocytes removing these aged RBCs from the circulation.
A large number of phagocytosed RBCs may induce a specific T cell activation against the RBCs, which then leads to the apoptotic cell death of the T cells. In liver, where the majority of aged erythrocytes are removed, this process is taking place constantly.47 The resident DC subtype present in liver represent an immature phenotype, and thus, incapable of inducing antigen specific T cell activation.47 Furthermore, Kupffer cells resident to the liver have been shown to act immunosuppressively towards T cell activation. These cells are even capable of suppressing T cell proliferation induced by splenic DCs, which are known to be very capable T cell activators.48 Vitali et al. showed that in peripheral blood the migratory DCs maintain self-tolerance and prevent autoimmunity, which they do by local induction of regulatory T cells.49 They also noted that they could not exclude if DCs, in an environment containing autoreactive T cells, could use other means, such as anergy or TCR down-regulation, to tolerize T cells. Indeed, Grimm et al. found that RBCs modified with OVA induced an antigen-specific deletion of CD4+ and CD8+ T cells, which was caused by antigen-specific regulatory T cells (Treg).17 Their experiments showed an induction of CD25+ FOXP3+ Tregs, both in CD4+ and CD8+ compartments.
Previously, it has been shown that antigens attached on cells via EDC-chemistry induces antigen specific T cell tolerance, where the tolerance results from clonal death of T cells.50 While we cannot exclude the EDC-chemistry from having an influence on our results, the general trend shows that the main driving factor on the clonal death of T cells in this study are the engineered RBC NPs. We speculate that our NPs are phagocytosed by macrophages and migratory DCs, where the latter activate T cells against autoantigens found in RBCs, which are abundant and perceived as aged erythrocytes. TRP2 has been shown to provide a potent immune response when coupled with liposomal carrier and an adjuvant.51 Furthermore, Liu et al. showed that antigens encapsulated within NPs elicit stronger immune response compared to antigens adsorbed on the NP surface.52 This was evident in our study, as the apoptosis of T cells was more pronounced when the RBC NPs were carrying antigenic TRP2, where the encapsulated TRP2 was slightly more potent and resulted in a more efficient T cell apoptosis compared to where the TRP2 was anchored on the surface of the RBCs.
Conclusions
In summary, we prepared hybrid NPs consisting of UnPSi NPs encapsulated within RBC membrane and functionalized them with a model antigen TRP2. The functionalization was achieved either through conjugation of TRP2 to UnPSi, in which the TRP2 was shielded within the RBC encapsulation, or by conjugating the TRP2 to cell membrane mimicking PDMS-b-PMOXA block copolymer, which acts as a surface anchor for the TRP2. Our studies on T cell immunostimulation showed depletion of T cells after 72 h co-incubation with the RBC encapsulated NPs due to programmed cell death of T cells via apoptosis. The NPs did not have influence on the percentage of CD3+ CD4+ or cytotoxic CD3+ CD8+ T cells. Previously, Luk et al. conducted a 24 h in vivo safety experiment on RBC coated PLGA nanoparticles, concluding that their nanoparticles were safe.53 Our results show that these NPs induce apoptosis to T cells, even when they are prepared from autologous RBCs. Furthermore, the ensuing T cell deletion was slightly stronger when TRP2 was within the RBC membrane compared to when it was expressed on the RBC surface via the PDMS-b-PMOXA anchor.Overall, the reported hybrid nanoparticles show potential in inducing autoantigen specific immune tolerance for the treatment of autoimmune diseases. Especially Un-TRP2@RBC NPs, regardless of the phenotype of the RBCs, had a strong influence on the clonal cell death of T cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. acknowledges financial support from the Finnish Cultural Foundation. H. A. S. acknowledges financial support from the University of Helsinki Research Funds, the Sigrid Juselius Foundation, the HiLIFE Research Funds, the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013; Grant No. 310892) and European Research Council Proof-of-Concept Grant (Decision No. 825020). The authors acknowledge the following core facilities funded by Biocenter Finland: Electron Microscopy Unit of the University of Helsinki for providing the facilities for TEM imaging and the Flow Cytometry Unit for providing the facilities for FACS experiments, and Nanomicroscopy Center of Aalto University for providing the facilities for the cryo-TEM imaging.References
- S. Corinti, L. Chiarantini, S. Dominici, M. E. Laguardia, M. Magnani and G. Girolomoni, J. Leukoc. Biol., 2002, 71, 652–658 CAS.
- F. Fontana, M.-A. Shahbazi, D. Liu, H. Zhang, E. Mäkilä, J. Salonen, J. T. Hirvonen and H. A. Santos, Adv. Mater., 2017, 29, 1603239 CrossRef.
- J. Li, S. Huang, Z. Zhou, W. Lin, S. Chen, M. Chen and Y. Ye, Cancer Manage. Res., 2018, 10, 4945–4957 CrossRef CAS.
- Q. Li, D. Zhang, J. Zhang, Y. Jiang, A. Song, Z. Li and Y. Luan, Nano Lett., 2019, 19, 6647–6657 CrossRef CAS.
- H. Zhang, J. Zhang, Q. Li, A. Song, H. Tian, J. Wang, Z. Li and Y. Luan, Biomaterials, 2020, 245, 119983 CrossRef CAS.
- C. H. Villa, A. C. Anselmo, S. Mitragotri and V. Muzykantov, Adv. Drug Delivery Rev., 2016, 106, 88–103 CrossRef CAS.
- N. Doshi, A. S. Zahr, S. Bhaskar, J. Lahann and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21495–21499 CrossRef CAS.
- W. Gao, C.-M. J. Hu, R. H. Fang, B. T. Luk, J. Su and L. Zhang, Adv. Mater., 2013, 25, 3549–3553 CrossRef CAS.
- D. Dehaini, X. Wei, R. H. Fang, S. Masson, P. Angsantikul, B. T. Luk, Y. Zhang, M. Ying, Y. Jiang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2017, 29, 1606209 CrossRef.
- J. Su, G. Liu, Y. Lian, Z. Kamal, X. Que, Y. Qiu and M. Qiu, RSC Adv., 2018, 8, 20068–20076 RSC.
- J. S. Brenner, D. C. Pan, J. W. Myerson, O. A. Marcos-Contreras, C. H. Villa, P. Patel, H. Hekierski, S. Chatterjee, J.-Q. Tao, H. Parhiz, K. Bhamidipati, T. G. Uhler, E. D. Hood, R. Y. Kiseleva, V. S. Shuvaev, T. Shuvaeva, M. Khoshnejad, I. Johnston, J. V. Gregory, J. Lahann, T. Wang, E. Cantu, W. M. Armstead, S. Mitragotri and V. Muzykantov, Nat. Commun., 2018, 9, 2684 CrossRef.
- F. Xiao, J. Fan, C. Tong, C. Xiao, Z. Wang, B. Liu, M. Daniyal and W. Wang, RSC Adv., 2019, 9, 27911–27926 RSC.
- S. Tan, T. Wu, D. Zhang and Z. Zhang, Theranostics, 2015, 5, 863 CrossRef CAS.
- E. H. Eylar, M. A. Madoff, O. Brody and J. Oncley, J. Biol. Chem., 1962, 237, 1992–2000 CAS.
- R. Schauer, Curr. Opin. Struct. Biol., 2009, 19, 507–514 CrossRef CAS.
- S. Kontos, I. C. Kourtis, K. Y. Dane and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E60–E68 CrossRef CAS.
- A. J. Grimm, S. Kontos, G. Diaceri, X. Quaglia-Thermes and J. A. Hubbell, Sci. Rep., 2015, 5, 15907 CrossRef CAS.
- J. A. Copp, R. H. Fang, B. T. Luk, C.-M. J. Hu, W. Gao, K. Zhang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13481–13486 CrossRef CAS.
- N. Pishesha, A. M. Bilate, M. C. Wibowo, N.-J. Huang, Z. Li, R. Deshycka, D. Bousbaine, H. Li, H. C. Patterson, S. K. Dougan, T. Maryama, H. F. Lodish and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3157–3162 CrossRef CAS.
- V. Balasubramanian, A. Correia, H. Zhang, F. Fontana, E. Mäkilä, J. Salonen, J. Hirvonen and H. A. Santos, Adv. Mater., 2017, 29, 1605375 CrossRef.
- V. Balasubramanian, A. Poillucci, A. Correia, H. Zhang, C. Celia and H. A. Santos, ACS Biomater. Sci. Eng., 2018, 4, 1471–1478 CrossRef CAS.
- F. Fontana, S. Albertini, A. Correia, M. Kemell, R. Lindgren, E. Mäkilä, J. Salonen, J. T. Hirvonen, F. Ferrari and H. A. Santos, Adv. Funct. Mater., 2018, 28, 1801355 CrossRef.
- F. Fontana, M. Fusciello, C. Groeneveldt, C. Capasso, J. Chiaro, S. Feola, Z. Liu, E. M. Mäkilä, J. J. Salonen, J. T. Hirvonen, V. Cerullo and H. A. Santos, ACS Nano, 2019, 6477–6490 CrossRef CAS.
- K. Tsukamoto, I. J. Jackson, K. Urabe, P. M. Montague and V. J. Hearing, EMBO J., 1992, 11, 519–526 CrossRef CAS.
- R.-F. Wang, E. Appella, Y. Kawakami, X. Kang and S. A. Rosenberg, J. Exp. Med., 1996, 184, 2207–2216 CrossRef CAS.
- L. M. Bimbo, M. Sarparanta, H. A. Santos, A. J. Airaksinen, E. Mäkilä, T. Laaksonen, L. Peltonen, V.-P. Lehto, J. Hirvonen and J. Salonen, ACS Nano, 2010, 4, 3023–3032 CrossRef CAS.
- M.-A. Shahbazi, P. V. Almeida, E. M. Mäkilä, M. H. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 7488–7500 CrossRef CAS.
- P. Figueiredo, V. Balasubramanian, M.-A. Shahbazi, A. Correia, D. Wu, C. G. Palivan, J. T. Hirvonen and H. A. Santos, Int. J. Pharm., 2016, 511, 794–803 CrossRef CAS.
- G. Liu, Y. Akasaki, H. T. Khong, C. J. Wheeler, A. Das, K. L. Black and J. S. Yu, Oncogene, 2005, 24, 5226–5234 CrossRef CAS.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef CAS.
- M. P. A. Ferreira, S. Ranjan, A. M. R. Correia, E. M. Mäkilä, S. M. Kinnunen, H. Zhang, M.-A. Shahbazi, P. V. Almeida, J. J. Salonen, H. J. Ruskoaho, A. J. Airaksinen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2016, 94, 93–104 CrossRef CAS.
- H. A. Santos, E. Mäkilä, A. J. Airaksinen, L. M. Bimbo and J. Hirvonen, Nanomedicine, 2014, 9, 535–554 CrossRef CAS.
- M. Kovalainen, J. Mönkäre, E. Mäkilä, J. Salonen, V.-P. Lehto, K.-H. Herzig and K. Järvinen, J. Pharmacol. Res., 2012, 29, 837–846 CAS.
- M. P. A. Ferreira, S. Ranjan, S. Kinnunen, A. Correia, V. Talman, E. Mäkilä, B. Barrios-Lopez, M. Kemell, V. Balasubramanian, J. Salonen, J. Hirvonen, H. Ruskoaho, A. J. Airaksinen and H. A. Santos, Small, 2017, 13, 1701276 CrossRef.
- M.-A. Shahbazi, T. D. Fernández, E. M. Mäkilä, X. Le Guével, C. Mayorga, M. H. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 9224–9235 CrossRef CAS.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS.
- B. T. Luk, C.-M. J. Hu, R. H. Fang, D. Dehaini, C. Carpenter, W. Gao and L. Zhang, Nanoscale, 2014, 6, 2730–2737 RSC.
- U. Jakobsson, E. Mäkilä, A. Rahikkala, S. Imlimthan, J. Lampuoti, S. Ranjan, J. Heino, P. Jalkanen, U. Köster, K. Mizohata, H. A. Santos, A. J. Airaksinen, M. Sarparanta and K. Helariutta, Nucl. Med. Biol., 2020, 84–85, 102–110 CrossRef CAS.
- B. AG, Peptide calculator, https://www.bachem.com/service-support/peptide-calculator/, accessed, December, 2019.
- M.-A. Shahbazi, M. Hamidi, E. M. Mäkilä, H. Zhang, P. V. Almeida, M. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2013, 34, 7776–7789 CrossRef CAS.
- W. Swat, L. Ignatowicz and P. Kisielow, J. Immunol. Methods, 1991, 137, 79–87 CrossRef CAS.
- A. Ciana, C. Achilli, A. Gaur and G. Minetti, Cell. Physiol. Biochem., 2017, 42, 1127–1138 CrossRef CAS.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS.
- K. Kita-Tokarczyk, F. Itel, M. Grzelakowski, S. Egli, P. Rossbach and W. Meier, Langmuir, 2009, 25, 9847–9856 CrossRef CAS.
- A. L. Richards, J. E. Hendrickson, J. C. Zimring and K. E. Hudson, Transfusion, 2016, 56, 905–916 CrossRef CAS.
- M. Cremel, N. Guérin, F. Horand, A. Banz and Y. Godfrin, Int. J. Pharm., 2013, 443, 39–49 CrossRef CAS.
- Q. You, L. Cheng, R. M. Kedl and C. Ju, Hepatology, 2008, 48, 978–990 CrossRef CAS.
- C. Vitali, F. Mingozzi, A. Broggi, S. Barresi, F. Zolezzi, J. Bayry, G. Raimondi, I. Zanoni and F. Granucci, Blood, 2012, 120, 1237–1245 CrossRef CAS.
- M. K. Jenkins and R. H. Schwartz, J. Exp. Med., 1987, 165, 302–319 CrossRef CAS.
- V. Jérôme, A. Graser, R. Müller, R. E. Kontermann and A. Konur, J. Immunother., 2006, 29, 294–305 CrossRef.
- L. Liu, P. Ma, H. Wang, C. Zhang, H. Sun, C. Wang, C. Song, X. Leng, D. Kong and G. Ma, J. Controlled Release, 2016, 225, 230–239 CrossRef CAS.
- B. T. Luk, R. H. Fang, C.-M. J. Hu, J. A. Copp, S. Thamphiwatana, D. Dehaini, W. Gao, K. Zhang, S. Li and L. Zhang, Theranostics, 2016, 6, 1004 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05900e |
| This journal is © The Royal Society of Chemistry 2020 |