
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the reactivity of neutral water-soluble Ru(II)–PTA carbonyls towards the model imine ligands pyridine and 2,2′-bipyridine†
Federica Battistin‡
 *,
Alessio Vidal,
Gabriele Balducci and
Enzo Alessio*
*,
Alessio Vidal,
Gabriele Balducci and
Enzo Alessio*
Department of Chemical and Pharmaceutical Sciences, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy. E-mail: alessi@units.it
First published on 16th July 2020
Abstract
As a continuation of our strategy for preparing new Ru(II) precursors to be exploited as building blocks in the construction of metal-mediated supramolecular assemblies with improved solubility in water, here we describe the reactivity of selected neutral Ru(II)–PTA carbonyls (PTA = 1,3,5-triaza-7-phosphaadamantane) towards the model imine ligands pyridine (py) and 2,2′-bipyridine (bpy) and the preparation and characterization of several neutral and cationic water-soluble derivatives: trans,trans,trans-[RuCl2(CO)(py)(PTA)2] (7), cis,cis,trans-[RuCl2(CO)2(py)(PTA)] (9), cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10), mer-[Ru(bpy)(CO)(PTA)3](Cl)2 (12), cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13), cis,trans-[Ru(bpy)(CO)2(PTA)2](NO3)2 (14NO3). In addition, we found that light-induced isomerization in some bpy compounds could be induced. The following species, either side-products isolated in low yield or compounds obtained exclusively in solution, were also unambiguously identified: cis,cis,trans-[RuCl2(CO)(py)(PTA)2] (8), trans-[RuCl2(bpy)(CO)(PTA)] (11), cis,cis-[Ru(bpy)Cl(CO)(PTA)2]Cl (15) and cis,cis-[Ru(bpy)(CO)2Cl(PTA)]Cl (16). The X-ray structures of 7, 11·H2O, and 12·7H2O are also reported. All compounds are new and – with few exceptions – show a good solubility in water.
Introduction
The cage-like monodentate phosphane 1,3,5-triaza-7-phosphaadamantane (PTA, Fig. 1)1 is an amphiphilic, air-stable, neutral ligand that – besides dissolving in several organic solvents – is characterized by a high solubility in water (ca. 235 g L−1) by virtue of H-bonding to the tertiary amine nitrogens. The coordination chemistry of PTA has been thoroughly addressed by Peruzzini and co-workers in a series of review papers.2 PTA typically makes strong bonds with most transition metal ions through the soft P atom (PTA-κP). P-bound PTA has moderate steric demand (cone angle 103°), good σ- and π-bonding abilities and, above all, it typically imparts excellent water solubility to its complexes. For this reason PTA metal compounds have been investigated as potential anticancer drugs,3–7 and as homogeneous catalysts in aqueous solution or in biphasic aqueous–organic conditions.2,8–10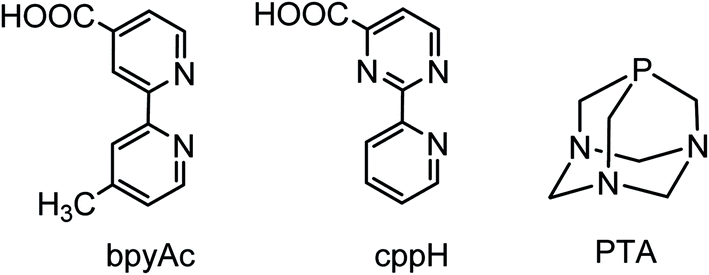 | ||
| Fig. 1 Schematic structures of the chelating diimine linkers 4′-methyl-2,2′-bipyridine-4-carboxylic acid (bpyAc) and 2-(2′-pyridyl)pyrimidine-4-carboxylic acid (cppH) and 1,3,5-triaza-7-phosphaadamantane (PTA). | ||
In addition, PTA-κP can bind also to other metal ions through the three hard N atoms, thus acting as a bridging ligand,2,11 and undergo N-protonation at relatively mild pH (in Ru(II) complexes PTA-κP has a pKa value of ca. 3),12,13 thus altering the charge and solubility of its complexes.
In recent years we have actively investigated the chemistry of Ru(II)–PTA compounds, mainly with the aim of obtaining new precursors to be exploited as building blocks in the construction of metal-mediated conjugates and supramolecular assemblies with improved solubility in water. Ideally, such water-soluble fragments might contribute to the solubilization in aqueous media of neutral assemblies containing hydrophobic linkers (e.g. synthetic porphyrins). In particular, we focused on Ru–PTA–dmso and Ru–PTA–CO compounds, describing several new derivatives and finding systematic trends.14,15 Here we report an explorative investigation on the reactivity of a series of Ru–PTA carbonyls towards the imine ligands pyridine (py) and 2,2′-bipyridine (bpy), used as models for monodentate pyridyl-linkers (e.g. 4,4′-bpy or pyridylporphyrins) and for chelating diimine linkers, such as 4′-methyl-2,2′-bipyridine-4-carboxylic acid (bpyAc) and 2-(2′-pyridyl)pyrimidine-4-carboxylic acid (cppH) (Fig. 1),16 respectively.
In particular we investigated the six neutral and water-soluble Ru(II)–PTA–CO precursors shown in Fig. 2 that have at least one or two potentially labile ligands, i.e. H2O, dmso-S, Cl.15,17
 | ||
| Fig. 2 Schematic structures of the neutral Ru(II)–PTA–CO precursors investigated in this paper. Top row: the monocarbonyls trans,trans,trans-[RuCl2(CO)(OH2)(PTA)2] (1), cis,cis,trans-[RuCl2(CO)(dmso-S)(PTA)2] (2), cis,mer-[RuCl2(CO)(PTA)3] (3), trans-[RuCl2(CO)(PTA)3] (4); bottom row: the dicarbonyls [RuCl(μ-Cl)(CO)2(PTA)]2 (5) and cis,cis,trans-[RuCl2(CO)2(PTA)2] (6). The potentially leaving ligands are colored. | ||
We describe here the preparation and characterization of a series of new derivatives. The compounds trans,trans,trans-[RuCl2(CO)(py)(PTA)2] (7), cis,cis,trans-[RuCl2(CO)2(py)(PTA)] (9), cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10), mer-[Ru(bpy)(CO)(PTA)3](Cl)2 (12), cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13), cis,trans-[Ru(bpy)(CO)2(PTA)2](NO3)2 (14NO3), were obtained in good yield and fully characterized, whereas cis,cis,trans-[RuCl2(CO)(py)(PTA)2] (8), trans-[RuCl2(bpy)(CO)(PTA)] (11), cis,cis-[Ru(bpy)Cl(CO)(PTA)2]Cl (15), and cis,cis-[Ru(bpy)(CO)2Cl(PTA)]Cl (16) were either identified in solution only (8, 15, 16) or isolated in low amounts (11). The X-ray molecular structures of 7, 11·H2O, and 12·7H2O are also reported, together with a comprehensive collection of the typical 31P NMR chemical shift in Ru(II)–PTA complexes as a function of the trans ligand.
Finally, it is interesting to note that water-soluble Ru–PTA–CO complexes and their derivatives are potentially of interest as homogeneous catalysts in aqueous medium or in biphasic systems for reactions that involve CO or CO2,2,8b,9,10,13,18–24 and as controlled CO-releasing and photo-releasing systems (CORMs and photoCORMs) for medicinal application.25–35
Results and discussion
Reactions with pyridine (py)
To the best of our knowledge, there are very few examples of Ru(II)–PTA coordination compounds with monodentate pyridine or azole ligands, i.e. not included in polydentate ligands.5b,36–40 Only recently the group of Romerosa described a series of neutral Ru(II)–PTA half-sandwich compounds that bear a natural purine bound through the N7 or N9 of the imidazolic ring.41When trans,trans,trans-[RuCl2(CO)(OH2)(PTA)2] (1) was treated with a slight excess of pyridine in water at room temperature, the yellow complex trans,trans,trans-[RuCl2(CO)(py)(PTA)2] (7) was selectively obtained upon evaporation of the solvent (Scheme 1) and fully characterized by ESI-MS, NMR, and IR spectroscopies (ESI†). Thus, pyridine easily and selectively replaced the labile aqua ligand with retention of the geometry. The 31P{1H} NMR spectrum of 7 (D2O) presents a singlet at −50.0 ppm, in the typical region of mutually trans PTAs. The IR spectrum shows a carbonyl stretching band at 1946 cm−1.
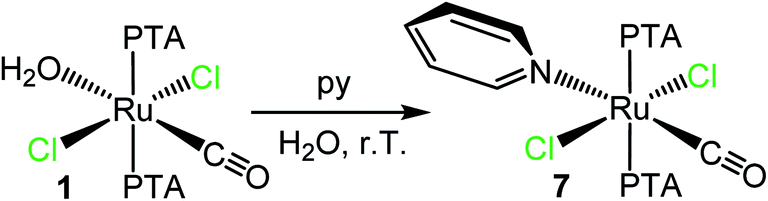 | ||
| Scheme 1 Preparation of trans,trans,trans-[RuCl2(CO)(py)(PTA)2] (7) upon treatment of trans,trans,trans-[RuCl2(CO)(OH2)(PTA)2] (1) with py in water. | ||
The geometry of complex 7 was confirmed by single crystal X-ray diffraction analysis (Fig. 3). In the structure, the trans CO/py ligands were found to be disordered by exchanging their mutual positions, with a major population of 74.7%.
 | ||
| Fig. 3 X-ray molecular structure (50% probability ellipsoids) of trans,trans,trans-[RuCl2(CO)(py)(PTA)2] (7) (major population: 74.7%). Coordination distances (Å): Ru1–Cl1 = 2.409(1), Ru1–Cl2 = 2.419(1), Ru1–N3 = 2.207(6), Ru1–C36 = 1.861(7), Ru1–P1 = 2.349(1), Ru1–P2 = 2.344(1). | ||
Conversely, treatment of cis,cis,trans-[RuCl2(CO)(dmso-S)(PTA)2] (2) with a slight excess of pyridine in water afforded mixtures of products, comprising 7, 8 (see below) and other unidentified minor species with coordinated py. However, when the reaction was performed in chloroform under microwave assisted conditions (100 °C) it afforded a mixture of 7 (see above) and of a minor component (ca. 20%) that was identified as the stereoisomer cis,cis,trans-[RuCl2(CO)(py)(PTA)2] (8) (that formally derives from 2 upon replacement of the dmso without geometrical rearrangement) (Scheme 2). In fact, the main NMR resonances of 8 (i.e. the py signals in the proton spectrum and the singlet for the two PTAs in the phosphorous spectrum) are only slightly shifted compared to those of 7, and integration confirmed the stoichiometry (ESI†). The IR spectrum of the mixture shows a broad carbonyl stretching band at 1977 cm−1.
 | ||
| Scheme 2 Reactivity of cis,cis,trans-[RuCl2(CO)(dmso-S)(PTA)2] (2) with py in chloroform under MW assisted conditions (100 °C) affording a mixture of the stereoisomers trans,trans,trans-[RuCl2(CO)(py)(PTA)2] (7) and cis,cis,trans-[RuCl2(CO)(py)(PTA)2] (8). | ||
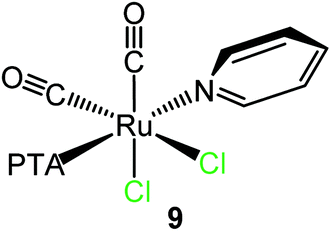
As previously observed by us, in aqueous solution the dinuclear species [RuCl(μ-Cl)(CO)2(PTA)]2 (5) behaves as a monomer with a formally vacant position trans to PTA after the selective and rapid cleavage of the chloride bridges leading to the formation of the neutral aqua species cis,cis,trans-[RuCl2(CO)2(OH2)(PTA)].15 Consistently, treatment of 5 with a slight excess of py in D2O at ambient temperature afforded crystals of cis,cis,trans-[RuCl2(CO)2(py)(PTA)] (9). Even though the crystals were of low quality, the X-ray structure (ESI†) unambiguously confirmed the geometry of the complex, with the pyridine trans to PTA (first example in Ru(II) species), i.e. in the place of the cleaved bridging chloride (Scheme 3).
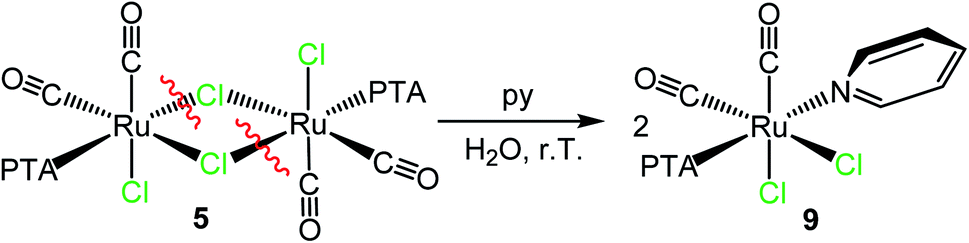 | ||
| Scheme 3 Reactivity of the dinuclear species [RuCl(μ-Cl)(CO)2(PTA)]2 (5) with pyridine in water affording cis,cis,trans-[RuCl2(CO)2(py)(PTA)] (9). | ||
The ESI-MS and NMR spectra of 9 in CDCl3 (ESI;† the solubility of this species was much higher in chloroform than in water) are fully consistent with this formulation. The IR spectrum presents two bands (2058 and 1994 cm−1) at frequencies very similar to those found in the precursor 5,15 in agreement with the presence of two adjacent CO ligands that, in both compounds, are trans to chlorides. The 31P{1H} NMR spectrum shows a singlet at −28.7 ppm.
Reactions with 2,2′-bipyridine (bpy)
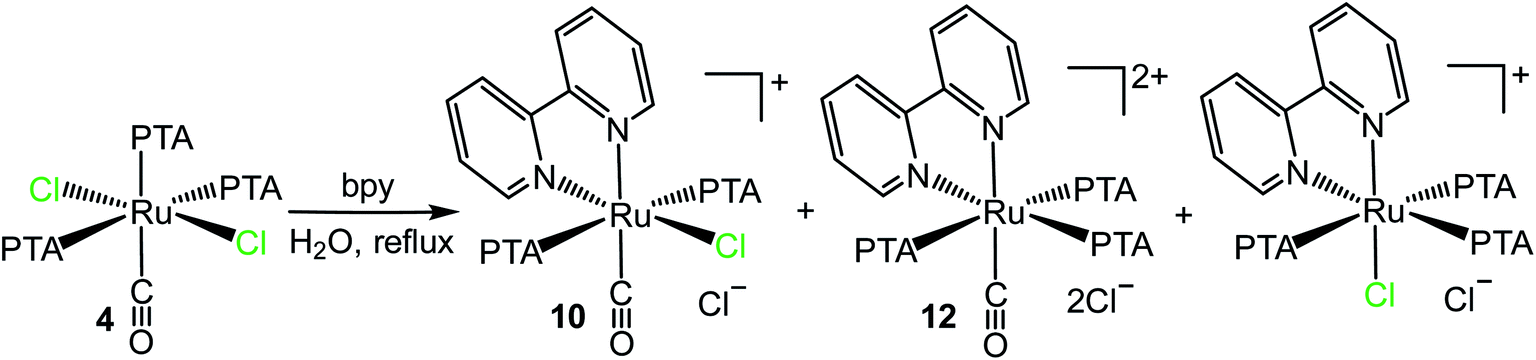
Examples of neutral and cationic Ru(II)–PTA complexes, containing one (cis,cis- and cis,trans-[Ru(bpy)Cl2(PTA)2], fac- and mer-[Ru(bpy)Cl(PTA)3](PF6)) or two (cis- and trans-[Ru(bpy)2(PTA)2](PF6)2, and cis-[Ru(bpy)2(OH2)(PTA)](CF3SO3)2) bpy ligands have been described by us and other groups.7,14,42 They were obtained either by treatment of Ru–PTA precursors with bpy, or through a complementary approach (i.e. by reaction of Ru–bpy complexes with PTA).In this work, we found that trans,trans,trans-[RuCl2(CO)(OH2)(PTA)2] (1) reacts with bpy in refluxing water by replacing the water molecule and one of the adjacent chlorides affording the orange cationic complex cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10) (Scheme 4).
 | ||
| Scheme 4 Reactivity of trans,trans,trans-[RuCl2(CO)(OH2)(PTA)2] (1) and cis,cis,trans-[RuCl2(CO)(dmso-S)(PTA)2] (2) with bpy in water affording cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10). | ||
Compound 10 was fully characterized by NMR spectroscopy and mass spectrometry. The 1H NMR spectrum in D2O shows eight equally intense resonances in the aromatic region, consistent with the asymmetric environment of the bpy ligand (ESI†). In agreement with previous findings,16a,43 the highest frequency doublet was assigned to H6, i.e. the proton with a partial positive charge that points towards the adjacent chloride (Scheme 4). The 31P{1H} NMR spectrum presents a singlet in the region of mutually trans PTAs (δ = −50.6 ppm). The CO stretching frequency in the IR spectrum (1984 cm−1) is shifted to higher wavenumbers compared to the precursor 1 (1941 cm−1), in agreement with a lower π back-bonding contribution from ruthenium because of the positive charge. With time, a very small amount of colorless crystals grew spontaneously from the D2O NMR solution of the raw product.44 They were found to belong to the unexpected neutral Ru(II) complex trans-[RuCl2(bpy)(CO)(PTA)] (11), whose formation implies replacement of one PTA. The full spectroscopic characterization and X-ray molecular structure of this minor species are reported in the ESI.†
Compound 10 was obtained also by treatment of cis,cis,trans-[RuCl2(CO)(dmso-S)(PTA)2] (2) with bpy in water (either 16 h at reflux or 30 min at 150 °C in a microwave reactor, Scheme 4). In this case bpy replaces the molecule of dmso and the adjacent chloride.
The reaction of cis,mer-[RuCl2(CO)(PTA)3] (3) with bpy in refluxing water led to the replacement of both chlorides by bpy, affording the orange dicationic complex mer-[Ru(bpy)(CO)(PTA)3](Cl)2 (12) (Scheme 5).
 | ||
| Scheme 5 Preparation of mer-[Ru(bpy)(CO)(PTA)3](Cl)2 (12) upon treatment of cis,mer-[RuCl2(CO)(PTA)3] (3) with bpy in water at reflux. | ||
Compound 12 was fully characterized by NMR and IR spectroscopy, mass spectrometry, and its single-crystal X-ray structure was also determined (Fig. 4 and ESI†). The PTA region of the 1H NMR spectrum in D2O consists of two pairs of signals, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. The two low-frequency multiplets (12H each) centered at 4.41 and 3.83 ppm and correlated in the H–H COSY spectrum, were attributed to the two equivalent trans PTA ligands whose protons fall in the shielding cone of the adjacent bpy. The other two signals (6H each), centered at 4.81 ppm (partially overlapped with the water signal) and 4.65 ppm, respectively, belonged to the PTA trans to N. The 1H–13C HSQC spectrum established that, in each set, the high-frequency quartet belongs to the NCH2N protons and the other signal to the NCH2P protons. The downfield region of the spectrum (eight resonances, three of which overlapping in an unresolved multiplet centered at 8.42 ppm) is consistent with the asymmetric coordination environment of bpy. The aromatic protons were assigned through a 1D NOESY spectrum: saturation of the singlet of the NCH2P protons of the PTA trans to bpy gave an NOE effect with the multiplet at 8.42 ppm thus implying that it contains the resonance of proton H6′, i.e. the one closest to the adjacent PTA. The other bpy resonances were then assigned through an H–H COSY spectrum. The 31P{1H} NMR spectrum presents an AX2 system. The triplet that belongs to the PTA trans to bpy (−49.6 ppm) is upfield shifted of ca. 10 ppm compared to most of the known monocationic and dicationic Ru(II) complexes featuring one or two PTAs trans to bpy, i.e. mer-[Ru(bpy)Cl(PTA)3](PF6) (−30.2 ppm), fac-[Ru(bpy)Cl(PTA)3](PF6) (−43.5 ppm),14 cis-[Ru(bpy)2(PTA)2](Cl)2 (−38.8 ppm) and cis-[Ru(bpy)2(PTA)(H2O)](CF3SO3)2 (−31.6 ppm).42b The difference might be attributed to the presence of the CO ligand in 12.
:2 ratio. The two low-frequency multiplets (12H each) centered at 4.41 and 3.83 ppm and correlated in the H–H COSY spectrum, were attributed to the two equivalent trans PTA ligands whose protons fall in the shielding cone of the adjacent bpy. The other two signals (6H each), centered at 4.81 ppm (partially overlapped with the water signal) and 4.65 ppm, respectively, belonged to the PTA trans to N. The 1H–13C HSQC spectrum established that, in each set, the high-frequency quartet belongs to the NCH2N protons and the other signal to the NCH2P protons. The downfield region of the spectrum (eight resonances, three of which overlapping in an unresolved multiplet centered at 8.42 ppm) is consistent with the asymmetric coordination environment of bpy. The aromatic protons were assigned through a 1D NOESY spectrum: saturation of the singlet of the NCH2P protons of the PTA trans to bpy gave an NOE effect with the multiplet at 8.42 ppm thus implying that it contains the resonance of proton H6′, i.e. the one closest to the adjacent PTA. The other bpy resonances were then assigned through an H–H COSY spectrum. The 31P{1H} NMR spectrum presents an AX2 system. The triplet that belongs to the PTA trans to bpy (−49.6 ppm) is upfield shifted of ca. 10 ppm compared to most of the known monocationic and dicationic Ru(II) complexes featuring one or two PTAs trans to bpy, i.e. mer-[Ru(bpy)Cl(PTA)3](PF6) (−30.2 ppm), fac-[Ru(bpy)Cl(PTA)3](PF6) (−43.5 ppm),14 cis-[Ru(bpy)2(PTA)2](Cl)2 (−38.8 ppm) and cis-[Ru(bpy)2(PTA)(H2O)](CF3SO3)2 (−31.6 ppm).42b The difference might be attributed to the presence of the CO ligand in 12.
 | ||
| Fig. 4 X-ray molecular structure (50% probability ellipsoids) of mer-[Ru(bpy)(CO)(PTA)3](Cl)2·7H2O (12·7H2O) obtained by recrystallization of the raw product from water/acetone. Coordination distances (Å): Ru1–C1 = 1.8746(14), Ru1–N51 = 2.119(1), Ru1–N52 = 2.139(1), Ru1–P2 = 2.3650(4), Ru1–P3 = 2.3391(4), Ru1–P4 = 2.3631(4). | ||
The IR spectrum shows a carbonyl stretching band at 2010 cm−1, i.e. at higher wavenumbers than in the precursor (1942 cm−1), in agreement with the 2+ charge of 12. For comparison, the CO stretching band in the dicationic monocarbonyl Ru(II) complex fac-[Ru(CO)(dmso-O)3(dmso-S)2](PF6)2 falls at 2012 cm−1,45 suggesting that this parameter is not particularly affected by the nature of the ligands, but rather by the net charge of the complex.
X-ray quality crystals of 12 were obtained upon recrystallization of the raw product from water/acetone (Fig. 4). The coordination distances are in general agreement with the known trans influence of the ligands: thus, the Ru–P bond lengths of the two trans PTA ligands are longer than the Ru–P distance trans to N. Similarly, the Ru–N bond length trans to CO is longer than that trans to P, consistent with the previous finding that CO has a stronger trans influence than PTA.14
The reaction of trans-[RuCl2(CO)(PTA)3] (4) – a stereoisomer of 3 – with bpy led to a mixture of compounds (Scheme 6), identified through the 31P{1H} NMR spectrum as 10, 11, and mer-[Ru(bpy)Cl(PTA)3]+ (previously described by us as PF6 salt),14 in ca. 1:7.5:3 ratio. This finding suggests that, even though bpy preferentially replaces the two chlorides, it can also replace either a PTA/Cl− or a CO/Cl− pair.
 | ||
| Scheme 6 Reactivity of trans,mer-[RuCl2(CO)(PTA)3] (4) towards bpy in refluxing water. | ||
Treatment of the dinuclear species [RuCl(μ-Cl)(CO)2(PTA)]2 (5) with bpy in refluxing water led to the isolation of the cationic dicarbonyl red-orange compound cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13), formally derived from the replacement in each half of the precursor of the two chlorides trans to CO, one bridging and the other terminal (Scheme 7). In fact, the 31P{1H} NMR spectrum in D2O presents a singlet at −25.4 ppm, i.e. in the typical region of PTA trans to Cl. The 1H NMR spectrum shows in the aromatic region four equally intense signals (2H each), in agreement with bpy coordinated in a symmetrical environment. The integration ratio of bpy and PTA resonances is consistent with the stoichiometry of the complex. No hydrolysis of the chloride was observed in D2O solution (i.e. no new set of proton resonances attributable to [Ru(bpy)(CO)2(OH2)(PTA)]2+ grew with time), most likely because of the positive charge of the complex. The IR spectrum of 13 presents two CO stretching bands at 2085 and 2034 cm−1, in agreement with the presence of two cis carbonyls. For comparison, the CO stretching bands in the cationic dicarbonyl Ru(II) complex cis,fac-[Ru(CO)2Cl(dmso)3](PF6) fall at 2092 and 2030 cm−1.45
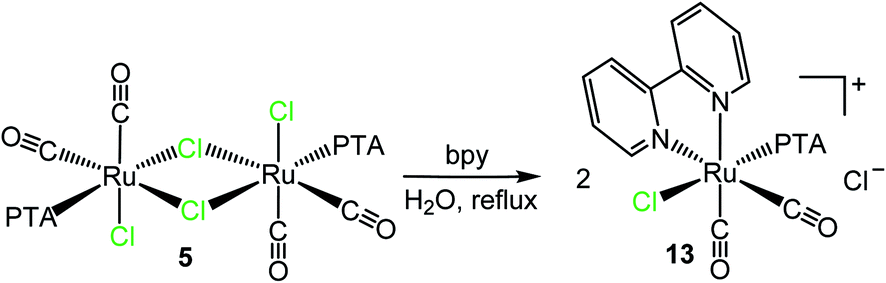 | ||
| Scheme 7 Reactivity of [RuCl(μ-Cl)(CO)2(PTA)]2 (5) towards bpy in refluxing water affording cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13). | ||
The reaction between cis,cis,trans-[RuCl2(CO)2(PTA)2] (6) and bpy yielded a mixture of 13 with a new product 14 (in ca. 1/2 ratio) (Scheme 8). The new species is characterized by a set of four equally intense bpy signals in the aromatic region of the 1H NMR spectrum, and by a singlet at −50.9 ppm in the 31P{1H} NMR spectrum, typical of mutually trans PTAs. Based on this spectroscopic evidence, 14 was formulated as the dicationic complex with two mutually trans PTAs, cis,trans-[Ru(bpy)(CO)2(PTA)2](Cl)2 (14Cl), derived from 6 upon replacement of both chlorides. To be noted that the formation of 13 from 6 involves isomerization: Cl, that was trans to CO in the precursor, is trans to PTA in 13.
 | ||
| Scheme 8 Reactivity of cis,cis,trans-[RuCl2(CO)2(PTA)2] (6) towards bpy in refluxing water yielding a mixture of cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13) and cis,trans-[Ru(bpy)(CO)2(PTA)2](Cl)2 (14Cl). | ||
Compound 14 was selectively obtained, as the orange nitrate salt (14NO3), when 6 was reacted with bpy in refluxing water after the addition of 2.1 eq. of AgNO3 (Scheme 9). The 1H, 31P{1H} NMR, and ESI-MS spectra of 14NO3, are coincident with those of 14Cl (Scheme 8). Consistent with the cis geometry of the CO ligands, the complex has two CO stretching bands (2086 and 2038 cm−1) in the IR spectrum. Compared with the precursor 6, the CO stretching frequencies of both 13 and 14 are shifted to higher wavenumbers, in accordance with the positive charge of the two products; nevertheless, it is remarkable that in 14 they are very similar to those found in 13, despite the higher positive charge. Compound 14 is the first dicationic dicarbonyl Ru(II) compound that we have isolated thus far; the previously prepared dicationic species have a single CO (e.g. [Ru(CO)(dmso)5](PF6)2), whereas the dicarbonyl compounds were monocationic (e.g. 13).
 | ||
| Scheme 9 Preparation of cis,trans-[Ru(bpy)(CO)2(PTA)2](NO3)2 (14NO3). | ||
Light-induced rearrangements in Ru(II)–PTA–bpy carbonyls
Both Ru(II)–PTA and RuX2(CO)n(P)4−n (n = 1–3) complexes have shown to be photoreactive.14,15,24,46–49 The group of Romerosa has recently reported a study of the effects of visible light on a series of water-soluble Ru(II) complexes with PTA.24 For this reason we decided to perform a preliminary investigation of the effect of visible light on selected Ru(II)–PTA–bpy carbonyls, that are characterized by the presence of an absorption band in the range of 400–525 nm. In general, D2O solutions of the complexes were directly irradiated in the NMR tube and the products were characterized spectroscopically, without being isolated.We found that 24 h irradiation with blue light (λ = 470 nm) of an orange D2O solution of cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10) induces its clean and complete transformation to the new cis-isomer cis,cis-[Ru(bpy)Cl(CO)(PTA)2]Cl (15) (Scheme 10). In fact, the 31P{1H} NMR spectrum of the product (ESI†) presents an AX system of two doublets, one in the region of PTA trans to Cl (−25.1 ppm, 2JP–P = 24.7 Hz) and the second in that of PTA trans to bpy (−42.3 ppm). The PTA region of the 1H NMR spectrum shows a broad singlet at 3.93 ppm and three partially overlapped AB systems centered respectively at 4.41, 4.58, and 4.71 ppm (4H each), consistent with the non-equivalence of the two PTAs. The two most upfield signals, correlated in the 1H–1H COSY spectrum (ESI†), were attributed to the PTA trans to Cl that falls into the shielding cone of the adjacent bpy. As above for 12, the aromatic protons were assigned through a 1D NOESY spectrum, followed by an 1H–1H COSY spectrum (ESI†). Finally, the IR spectrum of compound 15 shows a single carbonyl stretching band at 1992 cm−1, i.e. a frequency similar to that of the trans-isomer 10 (1984 cm−1).
 | ||
| Scheme 10 Light-induced isomerization (λ = 470 nm, 24 h) of cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10) to cis,cis-[Ru(bpy)Cl(CO)(PTA)2]Cl (15). | ||
A similar light-induced isomerization was observed with cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13) (Scheme 11). In this case the process was slower (and less selective) and complete transformation required the irradiation of an aqueous solution of 13 with blue light for 3 days. The major product of this reaction, 16, shows a singlet in the region of PTA trans to bpy (−39.6 ppm) in the 31P{1H} NMR spectrum, and eight resonances in the aromatic region of the 1H NMR spectrum for an asymmetrically bound bpy (with two multiplets of overlapping resonances). Consistent with these spectroscopic evidence, the new compound 16 was formulated as cis,cis-[Ru(bpy)(CO)2Cl(PTA)]Cl. The IR spectrum presents two carbonyl stretching bands at 2006 and 1979 cm−1 in agreement with the presence of two cis CO ligands. Compared to the starting trans isomer 13 (2085 and 2034 cm−1), the two bands of 16 are shifted to lower wavenumbers, possibly because one CO is trans to the good π-donor Cl.
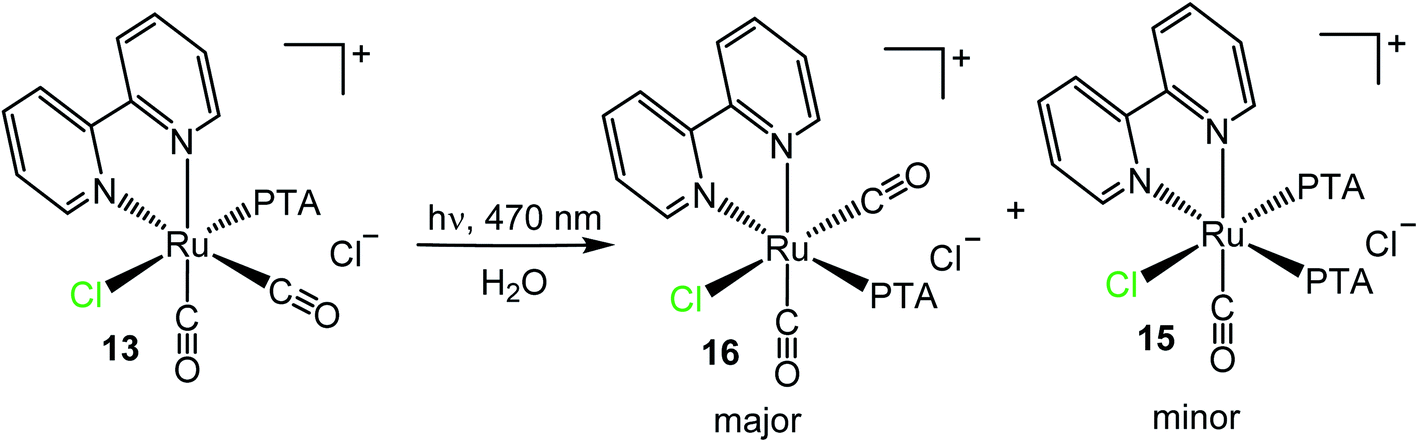 | ||
| Scheme 11 Light-induced isomerization (λ = 470 nm, 72 h) of cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13) to cis,cis-[Ru(bpy)(CO)2Cl(PTA)]Cl (16) and cis,cis-[Ru(bpy)Cl(CO)(PTA)2]Cl (15). | ||
To our surprise, the 31P{1H} NMR spectrum showed that formation of 16 is accompanied by that of 15 (ca. 6/1 ratio), in addition to other uncharacterized minor species (ESI†). Since there is no external source of PTA, the formation of 15 from 13 implies the occurrence of a decarbonylation process followed by disproportionation.
Conversely, compound cis,trans-[Ru(bpy)(CO)2(PTA)2](NO3)2 (14NO3) was light-stable. The 31P NMR spectrum of a D2O solution of 14NO3 remained unchanged even after one week of irradiation either with blue or white light.
Conclusions
We have investigated the reactivity of selected neutral Ru(II)–PTA carbonyls with potentially labile ligands (i.e. H2O, dmso and/or Cl) towards the model imine ligands pyridine (py) and 2,2′-bipyridine (bpy). The results are graphically summarized in Tables 1 and 2 that collect the main products with their respective precursors. To be noted that 5 is the only precursor that affords derivatives with a single PTA bound to ruthenium. Whereas the monodentate pyridine replaces exclusively neutral ligands, i.e. H2O or dmso-S, bpy is capable of replacing also the chlorides, thus affording mono- and dicationic products. PTA and supporting CO ligands are typically not replaced. Consistent with the expected lability order, the aqua ligand is easily replaced by pyridine at room temperature (e.g. 7), whereas replacement of dmso-S required more forcing conditions (refluxing chloroform or ethanol) that induced partial isomerization. Replacement of chlorides by bpy was typically performed in refluxing water, and was occasionally silver-assisted. Most of the complexes are well soluble in water and alcohols (methanol and ethanol). Exceptions are the neutral carbonyls 9 and 11, both containing a single PTA ligand.| Precursor | 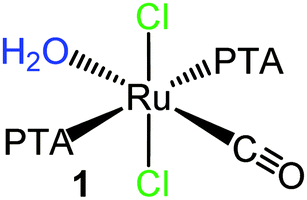 |
 |
| Product |  |
 |
| Precursor |  |
 |
 |
 |
| Product |  |
 |
 |
 |
Coordination of bpy is often accompanied by stereochemical changes. We notice that bpy does not like being bound trans to two PTA ligands. In fact, in all the Ru(II)–PTA–bpy complexes described in the literature that feature two or three PTA ligands, the coplanar {Ru(bpy)(PTA)2} fragment is found only in fac-[Ru(bpy)Cl(PTA)3](PF6), that however is a kinetic product that thermally isomerizes to the thermodynamic meridional stereoisomer.14 We believe that this is due to both steric and electronic factors (π back-bonding competition).
We found that in some bpy compounds light-induced isomerization could be induced. Thus, irradiation with blue light selectively transformed cis,trans-[Ru(bpy)Cl(CO)(PTA)2]Cl (10) into cis,cis-[Ru(bpy)Cl(CO)(PTA)2]Cl (15) (Scheme 10), whereas the isomerization of cis,trans-[Ru(bpy)(CO)2Cl(PTA)]Cl (13) to cis,cis-[Ru(bpy)(CO)2Cl(PTA)]Cl (16) was accompanied by formation of 15 (Scheme 11).
We have already observed that is possible to use 31P{1H} NMR chemical shifts as a diagnostic tool for establishing the geometry of octahedral Ru–PTA complexes.14,15 We report here an updated list of the typical 31P{1H} NMR chemical shift intervals (Δ) for PTA bound to Ru(II) as a function of the nature of the trans ligand, that contains the data from the new py and bpy derivatives (Table 3). We observe that when PTA is trans to an imine nitrogen (i.e. py, bpy), the 31P{1H} NMR chemical shift is remarkably affected by the nature of the ligand: when trans to py it falls at −28 ppm (we have only one example: complex 9), whereas when trans to bpy it falls in a rather broad range which is shifted to lower frequencies compared to py: −30 ÷ −50 ppm (but most often between −30 a −40 ppm, see below). Pyridine is known to be a weaker field ligand compared to bpy, being both a worse σ-donor and π-acceptor. In addition, the orientation of the pyridyl rings are different: for bpy they are in the plane containing PTA, whereas pyridine is typically canted with respect to such plane, implying a different involvement of their π orbitals. Indeed, in the complex trans-[RuCl2{(R,R)-iPr-pybox}(PTA)] where pyridine is part of a tridentate chelating ligand, and is forced to lay in the equatorial plane that contains the trans PTA, the 31P chemical shift falls at −41.5 ppm, i.e. in the range typical for bpy.5b
| Ligand trans to PTA | Δ 31P{1H}a (ppm) | Δ Ru–P distance (Å) | Ref.b |
|---|---|---|---|
| a For comparison, the singlet of free PTA falls at δ = −98.2 ppm in D2O and at −102.3 ppm in CDCl3.b p.w. = present work.c Single hit.d From [9]aneS3 = 1,4,7-trithiacyclononane.e When PTA is trans to 2,2′-bipyridine-6,6′-dicarboxylato (bda), the 31P{1H} chemical shift falls at −51.2 ppm (not included in the table).54f Concerns bpy complexes exclusively. In the single phen complex, mer-[Ru(phen)Cl(PTA)3]Cl (two independent cations in the asymmetric unit), the Ru–P distances are slightly shorter, 2.269(3) and 2.279(3) Å.42bg Single hit, from cyclometallated 2-phenylpyridine. | |||
| OH2/OH | −5 ÷ −16 | — | 14, 15, 24 and 50 |
| Cl | −14 ÷ −30 | 2.232 ÷ 2.283 | p.w. and 8b, 14 and 15 |
| N(azole) | −17 ÷ −49 | 2.2605 ÷ 2.2971 | 36–40 |
| Br | −24 ÷ −27 | 2.266 ÷ 2.281 | 51 |
| H | −26.6c | 2.299 ÷ 2.300 | 48 |
| N(py)c | −28.8 | p.w. | |
| Sd | −30 ÷ −45 | 2.280 ÷ 2.318 | 12, 16b and 43 |
| N(bpy, phen) | −30 ÷ −50e | 2.2849 ÷ 2.3391f | p.w. and 7, 14 and 42b |
| P(PTA) | −42 ÷ −60 | 2.290 ÷ 2.400 | p.w. and, 7, 8b, 13–15, 24, 42, 48, 50 and 52–54 |
| CO | −55 ÷ −70 | 2.3821 ÷ 2.3828 | 15 |
| Cg | −68.4 | 2.395(1) | 55 |
As already observed by us,14,15 Table 3 provides also a trans-influence ranking for the ligands trans to PTA; in fact there is a rough correlation between the Ru–P bond length and the chemical shift of the 31P{1H} NMR resonance: as the Ru–P distance increases, the phosphorous resonance shifts progressively to lower frequencies (i.e. closer to that of free PTA). To this regard we notice that there are only two examples of PTA trans to bpy in which the 31P chemical shift falls at frequencies lower than −40 ppm: fac-[Ru(bpy)Cl(PTA)3](PF6) (−43.5 ppm) and mer-[Ru(bpy)(CO)(PTA)3](Cl)2 (12, −49.6 ppm). In both complexes, due to the overall charge and competing ligands, the π-back donation to such PTAs is expected to be lower than for the other cases, leading to weaker and longer Ru–P bonds.
Experimental section
Materials
All chemicals were purchased from Sigma-Aldrich and used as received. Solvents were of reagent grade. The Ru(II)–PTA carbonyls 1–6 were synthesized and purified as previously reported by us.15Instrumental methods
Mono- and bi-dimensional (1H–1H COSY, 1H–13C HSQC) NMR spectra were recorded at room temperature on a Varian 400 or 500 spectrometer (1H: 400 or 500 MHz, 13C{1H}: 100.5 or 125.7 MHz, 31P{1H}: 161 or 202 MHz). 1H and 13C{1H} chemical shifts were referenced to the peak of residual non-deuterated solvent (δ = 7.26 and 77.16 for CDCl3, 2.50 and 39.52 for DMSO-d6) or were measured relative to the internal standard DSS (δ = 0.00) for D2O. Carbon resonances were assigned through the HSQC spectra. 31P chemical shifts were referenced to external 85% H3PO4 at 0.00 ppm. ESI mass spectra were collected in the positive and negative mode on a PerkinElmer APII spectrometer at 5600 eV. The UV-vis spectra were obtained on an Agilent Cary 60 spectrophotometer, using 1.0 cm path-length quartz cuvettes (3.0 mL). Solid state infrared spectra were obtained as Nujol mulls between NaCl plates and recorded on a PerkinElmer Fourier-transform IR/Raman 2000 instrument in the transmission mode. Solution IR spectra (in chloroform, ethanol or methanol) in the CO stretching region were recorded between CaF2 windows (0.5 mm spacer). A thermostatted Berghof stainless steel vessel (autoclave), equipped with a 100 mL Teflon liner, was used for the reactions with CO under pressure. A CEM Discover microwave reactor was used for the microwave-assisted reactions performed in 10 mL vessels. Elemental analyses were performed in the Department of Chemistry of the University of Bologna (Italy). A homemade LED apparatus, described in detail elsewhere,56 was used for performing the photochemical reactions in NMR tubes.X-ray diffraction
Data collections were performed at the X-ray diffraction beamline (XRD1) of the Elettra Synchrotron of Trieste (Italy) equipped with a Pilatus 2 M image plate detector.Collection temperature was 100 K (nitrogen stream supplied through an Oxford Cryostream 700); the wavelength of the monochromatic X-ray beam was 0.700 Å and the diffractograms were obtained with the rotating crystal method. The crystals were dipped in N-paratone and mounted on the goniometer head with a nylon loop. The diffraction data were indexed, integrated and scaled using the XDS code.57 The structures were solved by the dual space algorithm implemented in the SHELXT code.58 Fourier analysis and refinement were performed by the full-matrix least-squares methods based on F2 implemented in SHELXL.59 The Coot program was used for modeling.60 Anisotropic thermal motion was allowed for all non-hydrogen atoms. Hydrogen atoms were placed at calculated positions with isotropic factors U = 1.2 × Ueq., Ueq. being the equivalent isotropic thermal factor of the bonded non hydrogen atom. Crystal data and details of refinements are in the ESI.†
Synthesis of the complexes
All the synthetic procedures with Ru–PTA carbonyls precursors were performed under dimmed light and in light-protected glassware.Compound 10 was also obtained, in lower yield, by treatment of an aqueous solution of cis,cis,trans-[RuCl2CO(dmso-S)(PTA)2] (2) (40.0 mg, 0.067 mmol) with bpy (10.5 mg, 0.067 mmol) for 16 h at reflux or in a microwave reactor at 150 °C for 30 min. The solvent was removed by rotary evaporation affording an orange solid that was crushed with acetone (2 × 2 mL), then washed with diethyl ether, and dried in vacuo (yield 25.6 mg, 57%).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the University of Trieste (FRA2018), and Fondazione Beneficentia Stiftung, is gratefully acknowledged. We wish to thank BASF Italia Srl for a donation of hydrated ruthenium chloride.References
- (a) D. J. Daigle, A. B. Pepperman Jr and S. L. Vail, J. Heterocycl. Chem., 1974, 11, 407–408 CrossRef CAS; (b) D. J. Daigle, T. J. Decuir, J. B. Robertson and D. J. Darensbourg, Inorg. Synth., 1998, 32, 40–45 CAS.
- (a) A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955–993 CrossRef CAS; (b) J. Bravo, S. Bolaño, L. Gonsalvi and M. Peruzzini, Coord. Chem. Rev., 2010, 254, 555–607 CrossRef CAS; (c) A. Guerriero, M. Peruzzini and L. Gonsalvi, Coord. Chem. Rev., 2018, 355, 328–361 CrossRef CAS.
- D. N. Akbayeva, L. Gonsalvi, W. Oberhauser, M. Peruzzini, F. Vizza, P. Brueggeller, A. Romerosa, G. Sava and A. Bergamo, Chem. Commun., 2003, 264–265 RSC.
- (a) C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC; (b) C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed; (c) P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC; (d) P. J. Dyson, Chimia, 2007, 61, 698–703 CrossRef CAS; (e) A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Internet J. Oncol., 2008, 33, 1281–1289 CAS; (f) W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS; (g) C. M. Clavel, E. Paunescu, P. Nowak-Sliwinska, A. W. Griffioen, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2015, 58, 3356–3365 CrossRef CAS PubMed; (h) M. V. Babak, S. M. Meier, K. V. M. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, W. Berger, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2015, 6, 2449–2456 RSC; (i) A. Weiss, X. Ding, J. R. van Beijnum, I. Wong, T. J. Wong, R. H. Berndsen, O. Dormond, M. Dallinga, L. Shen, R. O. Schlingemann, R. Pili, C.-M. Ho, P. J. Dyson, H. van den Bergh, A. W. Griffioen and P. Nowak-Sliwinska, Angiogenesis, 2015, 18, 233–244 CrossRef CAS PubMed; (j) B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- (a) A. García-Fernández, J. Díez, Á. Manteca, J. Sánchez, R. García-Navas, B. G. Sierra, F. Mollinedo, M. Pilar Gamasa and E. Lastra, Dalton Trans., 2010, 39, 10186–10196 RSC; (b) E. Menéndez-Pedregal, J. Díez, Á. Manteca, J. Sánchez, A. C. Bento, R. García-Navas, F. Mollinedo, M. Pilar Gamasa and E. Lastra, Dalton Trans., 2013, 42, 13955–13967 RSC.
- R. Pettinari, F. Marchetti, F. Condello, C. Pettinari, G. Lupidi, R. Scopelliti, S. Mukhopadhyay, T. Riedel and P. J. Dyson, Organometallics, 2014, 33, 3709–3715 CrossRef CAS.
- A. Wołoszyn, C. Pettinari, R. Pettinari, G. V. Badillo Patzmay, A. Kwiecień, G. Lupidi, M. Nabissi, G. Santoni and P. Smoleński, Dalton Trans., 2017, 46, 10073–10081 RSC.
- (a) D. J. Darensbourg, F. Joó, M. Kannisto, Á. Kathó and J. H. Reibenspies, Organometallics, 1992, 11, 1990–1993 CrossRef CAS; (b) D. J. Darensbourg, F. Joó, M. Kannisto, A. Kath, J. H. Reibenspies and D. J. Daigle, Inorg. Chem., 1994, 13, 200–208 CrossRef; (c) F. Joó, L. Nádasdi, J. Elek and G. Laurenczy, Chem. Commun., 1999, 971–972 RSC; (d) G. Laurenczy, F. Joó and L. Nádasdi, Inorg. Chem., 2000, 39, 5083–5088 CrossRef CAS PubMed; (e) G. Laurenczy, F. Joó and L. Nádasdi, High Pressure Res., 2000, 18, 251–255 CrossRef; (f) G. Kovács, L. Nádasdi, G. Laurenczy and F. Joó, Green Chem., 2003, 5, 213–217 RSC.
- Aqueous-phase Organometallic Catalysis: Concepts and Applications, ed. B. Cornils and W. A. Hermann, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- W.-C. Lee and B. J. Frost, Green Chem., 2012, 14, 62–66 RSC.
- F. Battistin, A. Vidal, P. Cavigli, G. Balducci, E. Iengo and E. Alessio, Inorg. Chem., 2020, 59, 4068–4079 CrossRef CAS PubMed and references therein.
- B. Serli, E. Zangrando, T. Gianferrara, C. Scolaro, P. J. Dyson, A. Bergamo and E. Alessio, Eur. J. Inorg. Chem., 2005, 3423–3434 CrossRef CAS.
- A. Udvardy, A. C. Bényei and Á. Kathó, J. Organomet. Chem., 2012, 717, 116–122 CrossRef CAS.
- F. Battistin, G. Balducci, E. Iengo, N. Demitri and E. Alessio, Eur. J. Inorg. Chem., 2016, 2850–2860 CrossRef CAS.
- F. Battistin, G. Balducci, B. Milani and E. Alessio, Inorg. Chem., 2018, 57, 6991–7005 CrossRef CAS PubMed.
- (a) F. Battistin, G. Balducci, N. Demitri, E. Iengo, B. Milani and E. Alessio, Dalton Trans., 2015, 44, 15671–15682 RSC; (b) F. Battistin, D. Siegmund, G. Balducci, E. Alessio and N. Metzler-Nolte, Dalton Trans., 2019, 48, 400–414 RSC.
- In all the Ru(II) compounds described in this paper PTA is always bonded exclusively through the P atom , thus κP is omitted in the formulas.
- G. Laurenczy, S. Jedner, E. Alessio and P. J. Dyson, Inorg. Chem. Commun., 2007, 10, 558–562 CrossRef CAS.
- P. Crochet and V. Cadierno, Dalton Trans., 2014, 43, 12447–12462 RSC.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017–4024 CrossRef CAS PubMed.
- A. Guerriero, H. Bricout, K. Sordakis, M. Peruzzini, E. Monflier, F. Hapiot, G. Laurenczy and L. Gonsalvi, ACS Catal., 2014, 4, 3002–3012 CrossRef CAS.
- A. Guerriero, H. Bricout, M. Peruzzini, P. J. Dyson, E. Monflier, F. Hapiot, L. Gonsalvi and G. Laurenczy, Inorg. Chim. Acta, 2015, 431, 132–138 CrossRef.
- F. Scalambra, M. Serrano-Ruiz and A. Romerosa, Dalton Trans., 2017, 46, 5864–5871 RSC.
- A. Udvardy, M. Serrano-Ruiz, V. Passarelli, E. Bolyog-Nagy, F. Joó, Á. Kathó and A. Romerosa, Inorg. Chim. Acta, 2018, 470, 82–92 CrossRef CAS.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. J. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722–3729 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS.
- C. C. Romao, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014, 50, 3644–3660 RSC.
- S. Garcia-Gallego and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 53, 9712–9721 CrossRef CAS PubMed.
- I. Chakraborty, S. J. Carrington and P. K. Mascharak, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed.
- U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS PubMed.
- J. Marhenke, L. Trevino and C. Works, Coord. Chem. Rev., 2016, 306, 533–543 CrossRef CAS.
- M. A. Wright and J. A. Wright, Dalton Trans., 2016, 45, 6801–6811 RSC.
- M. Kubeil, R. R. Vernooij, C. Kubeil, B. R. Wood, B. Graham, H. Stephan and L. Spiccia, Inorg. Chem., 2017, 56, 5941–5952 CrossRef CAS PubMed.
- S. Bolaño, J. Bravo, J. Castro, M. M. Rodríguez-Rocha, M. F. C. Guedes da Silva, A. J. L. Pombeiro, L. Gonsalvi and M. Peruzzini, Eur. J. Inorg. Chem., 2007, 5523–5532 CrossRef.
- A. García-Fernández, J. Díez, A. Manteca, J. Sánchez, M. P. Gamasa and E. Lastra, Polyhedron, 2008, 27, 1214–1228 CrossRef.
- S. Bolaño, M. M. Rodriguez-Rocha, J. Bravo, J. Castro, E. Oñate and M. Peruzzini, Organometallics, 2009, 28, 6020–6030 CrossRef.
- S. Miguel, J. Díez, M. P. Gamasa and M. E. Lastra, Eur. J. Inorg. Chem., 2011, 951, 4745–4755 CrossRef.
- A. García-Fernández, J. Díez, M. Pilar Gamasa and E. Lastra, Eur. J. Inorg. Chem., 2014, 5, 917–924 CrossRef.
- L. Hajji, C. Saraiba-Bello, G. Segovia-Torrente, F. Scalambra and A. Romerosa, Eur. J. Inorg. Chem., 2019, 4078–4086 CrossRef CAS.
- (a) F. Scalambra, M. Serrano-Ruiz, S. Nahim-Granados and A. Romerosa, Eur. J. Inorg. Chem., 2016, 1528–1540 CrossRef CAS; (b) F. Scalambra, N. Holzmann, L. Bernasconi, S. Imberti and A. Romerosa, Eur. J. Inorg. Chem., 2019, 1162–1170 CrossRef CAS.
- E. Iengo, N. Demitri, G. Balducci and E. Alessio, Dalton Trans., 2014, 43, 12160–12163 RSC.
- In the proton NMR spectrum of raw 10 a set of very small resonances, partially overlapped with those of 10, presumably belonging to 11 (or perhaps to the corresponding complex with protonated PTA and thus more soluble) is barely visible. 11 could also be the product of a slow transformation of 10 (perhaps light mediated), since it forms in few weeks ESI.†.
- I. Bratsos, S. Calmo, E. Zangrando, G. Balducci and E. Alessio, Inorg. Chem., 2013, 52, 12120–12130 CrossRef CAS PubMed.
- C. F. J. Barnard, J. A. Daniels, J. Jeffery and R. J. Mawby, Dalton Trans., 1976, 953–961 RSC.
- (a) L. M. Wilkes, J. H. Nelson, J. P. Mitchener, M. W. Babich, W. C. Riley, B. J. Helland, R. A. Jacobson, M. Y. Cheng, L. Seff and L. B. McCusker, Inorg. Chem., 1982, 21, 1376–1382 CrossRef CAS; (b) D. W. Krassowski, J. H. Nelson, K. R. Brower, D. Hauenstein and R. A. Jacobson, Inorg. Chem., 1988, 27, 4294–4301 CrossRef CAS.
- C. A. Mebi and B. J. Frost, Inorg. Chem., 2007, 46, 7115–7120 CrossRef CAS PubMed.
- R. Girotti, A. Romerosa, S. Mañas, M. Serrano-Rui and R. N. Perutz, Inorg. Chem., 2009, 48, 3692–3698 CrossRef CAS PubMed.
- J. Kovács, F. Joó, A. Bényei and G. Laurenzy, Dalton Trans., 2004, 2336–2340 RSC.
- F. Battistin, F. Scaletti, G. Balducci, S. Pillozzi, A. Arcangeli, L. Messori and E. Alessio, J. Inorg. Biochem., 2016, 160, 180–188 CrossRef CAS PubMed.
- D. N. Akbayeva, S. Moneti, M. Peruzzini, L. Gonsalvi, A. Ienco and F. Vizza, C. R. Chim., 2005, 8, 1491–1496 CrossRef CAS.
- S. Grguric-Sipka, C. R. Kowol, S.-M. Valiahdi, R. Eichinger, M. A. Jakupec, A. Roller, S. Shova, V. B. Arion and B. K. Keppler, Eur. J. Inorg. Chem., 2007, 2870–2878 CrossRef CAS.
- S. Yazdani, B. E. Silva, T. C. Cao, A. L. Rheingold and D. B. Grotjahn, Polyhedron, 2018, 161, 63–70 CrossRef.
- L. Leyva, C. Sirlin, L. Rubio, C. Franco, R. Le Lagadec, J. Spencer, P. Bischoff, C. Gaiddon, J.-P. Loeffler and M. Pfeffer, Eur. J. Inorg. Chem., 2007, 3055–3066 CrossRef CAS.
- F. Battistin, G. Balducci, J. Wei, A. K. Renfrew and E. Alessio, Eur. J. Inorg. Chem., 2018, 2018, 1469–1480 CrossRef CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 72, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Carbonyl stretching bands of complexes 7, 9, 10, 12–16: crystal and refinement data, and selected coordination distances and angles for compounds 7, 11·H2O, and 12·7H2O (Tables S1–S5); detailed characterization of 11; additional 1D and 2D NMR spectra for compounds 7–16. CCDC 1829778 (7), 1829779 (11) and 2007937 (12). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05898j |
| ‡ Now at IMDEA Nanociencia, Ciudad Universitaria de Cantoblanco, Faraday 9, 28049 Madrid, Spain. Email: E-mail: federica.battistin@imdea.org |
| This journal is © The Royal Society of Chemistry 2020 |