
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the rate of radiation energy on the charge-carrier kinetics application of all-inorganic CsPbBr3 perovskite nanocrystals†
Virendra Kumara,
Vandana Nagalb,
Rahul Kumara,
Shubhda Srivastavacd,
Bipin Kumar Gupta d,
Mahesh Kumard,
Aurangzeb Khurram Hafizb and
Kedar Singh*a
d,
Mahesh Kumard,
Aurangzeb Khurram Hafizb and
Kedar Singh*a
aNanotechnology Lab, School of Physical Sciences, Jawaharlal Nehru University (JNU), New Delhi-110067, India. E-mail: kedarbhu08@gmail.com; virendra2181@gmail.com
bQuantum and Nanophotonics Research Laboratory, Centre for Nanoscience and Nanotechnology, Jamia Millia Islamia (A Central University), New Delhi-110025, India
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
dCSIR – National Physical Laboratory, Dr. K. S. Krishnan Road, New Delhi-110012, India
First published on 24th September 2020
Abstract
In the field of optoelectronics, all-inorganic CsPbBr3 perovskite nanocrystals (PNCs) have gained significant interest on account of their superb processability and ultra-high stability among all the counterparts. In this study, we conducted an in-depth analysis of CsPbBr3 PNCs using joint transient optical spectroscopies (time-resolved photoluminescence and ultrafast transient absorption) in a very comprehensive manner. In order to understand the in-depth analysis of excited-state kinetics, the transient absorption spectroscopy has been performed. The structure of interest of CsPbBr3 PNCs was subjected to the rates of the radiation energy of 0.10 mW (κr/κnr = ∼0.62) and 0.30 mW (κr/κnr = ∼0.64). With the rate of radiation energy 0.30 mW, it was observed that there was a significant increase in hot carrier relaxation together with high radiative recombination, resulting in a decrease in charge trappings. Herein, we demonstrate that the tuning of the rate of radiation energies helps to understand the charge-carrier kinetics of CsPbBr3 PNCs, which would thus improve the manufacturing of efficient photovoltaic devices.
1. Introduction
All-inorganic colloidal CsPbX3 (X = Cl, Br, and I) PNCs1 have emerged as competitive candidates in the domain of illumination, display and optoelectronics applications,2 for instance, in solar cells,3 light-emitting diodes,4 photo-detectors,5 lasers,6 photo-catalysts,7 luminescent solar concentrators,8 and backlight displays.1,9 These novel optical and electrical attributes such as the large coefficient of absorption,10 high carrier mobility,11 low recombination rate1,12 together with high photoluminescence quantum yield (PLQY)1 and superior stability13 are the wide range attributes of CsPbX3 (X = Cl, Br, and I)1,13 in comparison to their hybrid counterparts MAPbX3 (MA = CH3NH3, X = Cl, Br, and I).14 From the above-mentioned features, one can easily comprehend that the kinetics of the photo-generated electron–hole15,16 pairs inside PNCs plays a decisive role in their optoelectronic properties.17 The time-resolved photoluminescence (TRPL)18–29 and transient absorption (TA)30–37 spectroscopies used to study the ultrafast photo-physical operation of charge carrier kinetics33 in the context of the generation and recombination,30,34 relaxation of the hot carriers,30 properties of the interfacial charge transport30,38–40 and pathways of negligible electron–hole confirmed with the treatment of these spectroscopic techniques, which accounts for a high PLQY1 in the CsPbX3 (X = Cl, Br, and I) PNC and its composites.These key findings provided a broad perspective on the optimization of halide-based perovskites for optoelectronic device applications.41,42 Substantial defect reductions and the corresponding increase in the radiative recombination rates have been reported.43 Kamat et al. reported a pump fluence-dependent charge carrier kinetics study in a thin film of CH3NH3PbI3.44 With the variation in the halide composition from CsPbBr3 to CsPbI3, Kim et al. reported a comparative study of hot carrier cooling.45 The pump fluence-based study of the mechanism behind hot carrier cooling in MAPbI3 NCs was reported by Fu et al.46 The intensity-dependent investigation of the hot carrier cooling process in CsPbI3 quantum dots was examined by Feng Liu et al.47 Similarly, the wavelength-dependent mechanism of the hot-carrier cooling phenomenon in CsPbBr3 NC/MO2 (M = Si, Ti, and Sn) composites was investigated by Kuang et al.30 Herein, all-inclusive charge-carrier kinetics was looked into the CsPbBr3 PNCs using the TRPL and the rate of radiation energy-dependent TA spectroscopy. To the best of our knowledge, we have also reported the kinetic Burstein–Moss shift attribute of the band edge carrier aggregation in the CsPbBr3 PNC material in the context of the rate of radiation dependence. As a result, the application of a 0.30 mW rate of radiation energy helps more to improve hot-carrier relaxation and significantly promotes radiative recombination in order to reduce charge trappings. In addition, the ratio obtained from the TRPL (κr/κnr = ∼0.63) is consistent with the result obtained by TA (κr/κnr = ∼0.64). The systematic use of different high-power laser sources for the study of charge-carrier kinetics into CsPbBr3 PNCs opens up a new way of minimizing charge trapping states to make this inorganic perovskite more stable, and the observation of these findings allows for critical insights into the relatively undiscovered excited-state feature of PNCs.
2. Experimental section
2.1 Materials
Cesium carbonate (Cs2CO3, 99.995%), lead bromide (PbBr2, 99.999%), hexane (laboratory reagent, ≥95%), oleic acid (OA, 90%), 1-octadecene (ODE, 90%) and oleylamine (OLA, 80–90%) were used in the preparation of CsPbBr3 PNCs.2.2 Formulation of cesium oleate
A cesium oleate precursor was formulated by adding 2.50 mmol of Cs2CO3, 8.85 mmol of oleic acid, and 158.4 mmol of 1-octadecene in a three-neck 100 mL flask, followed by vacuum desiccation for 1 h at 120 °C, and then heating the flask at 130 °C under a nitrogen atmosphere for the stark solubilization of Cs2CO3. The obtained solution stored in a freeze and reheated to 100 °C before using it for the preparation of all inorganic CsPbBr3 PNC.2.3 Preparation of all inorganic CsPbBr3 PNC
Colloidal all-inorganic CsPbBr3 PNCs were prepared using a more general modified hot-injection route. For this purpose, 1-octadecene (19.8 mmol) and lead bromide (0.188 mmol) were taken in a three-neck 25 mL flask, followed by vacuum desiccation at 120 °C for 1 h. Desiccated oleylamine (1.87 mmol) and oleic acid (1.77 mmol) were fed into the above mixture at 120 °C under a nitrogen environment. After stark solubilization of the PbBr2 salt for approximately 1 h under the nitrogen environment, the temperature of the reaction was increased to 180 °C and maintained for approximately 30 min (for adjusting the size of NC); and the mixture was quenched by dipping into the ice-water bath just after the Cs-oleate (0.4 mL) solution was injected.12.4 Separation and purgation of all-inorganic CsPbBr3 PNC
After cooling the mixture in an ice-water bath, the mixture was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min in order to isolate the agglomerated PNCs. In order to prepare the long-term colloidal stable solution, we centrifuged the suspended part that has been disposed and the particles in small amounts of hexane (49.9 mmol) were re-dispersed.
000 rpm for 10 min in order to isolate the agglomerated PNCs. In order to prepare the long-term colloidal stable solution, we centrifuged the suspended part that has been disposed and the particles in small amounts of hexane (49.9 mmol) were re-dispersed.
2.5 Characterizations
Rigaku MiniFlex 600 with a Cu Kα radiation (λ = 1.540593 Å) source was used to obtain the powder X-ray diffraction pattern. A Cary 100 spectrometer in an absorption mode was used for the acquisition of UV-Vis spectra in the range of 400–700 nm for colloidal solutions. A Cary Eclipse (MY17060005) was used to collect the spectra in the range of 460–700 nm for the steady-state photoluminescence (PL, excitation at 450 nm). Fluorescein1 dye was used as a reference fluorophore to calculate the photoluminescence quantum yield (PLQY). A photoluminescence lifetime spectrometer (EPL470, Edinburg Instruments Ltd with a picosecond pulsed diode laser as an exciting source having a pulse duration 93.2 ps and the peak wavelength of 466.2 nm with a maximum average power of 5 mW) was used to perform the PL lifetime measurements in a time-correlated single-photon counting mode. Transmission electron microscopy (TEM), selected area electron diffraction (SAED) and high-resolution TEM (high-angle annular dark-field scanning transmission electron microscopy JEOL 2100F) studies were carried out to understand the nanostructure in the material. An Omicron electron spectrometer of Oxford Instrument Germany equipped with a monochromatic Al Kα X-ray source (hν = 1486.7 eV) operating at 150 W was used for the elemental analysis (i.e., X-ray photoelectron spectroscopy). Binding energies were in accordance with the sp3 hybridization (C–C) carbon from OLA and OA and studied in comparison with the C1s peak (fixed at around 284.46 eV) used as a reference. An IRAffinity-1S SHIMADZU 00841 was used to record the Fourier transform-infrared spectra between 3500 cm−1 and 584 cm−1. The TA measurements were performed using the TA spectroscopy system consisting of Ti:sapphire oscillator, regenerative amplifier coupled with an optical parametric amplifier (OPA). The oscillator is a Verdi Pumped Micra system whose output (∼350 mW power with 800 nm pulses) is seeded to the regenerative amplifier, which produces 4 mJ energy output in the form of 800 nm pulses with the pulse width of 35 fs at 1 kHz repetition rate. This output was further divided into 2 portions: one is seeded into OPA (TOPAS) giving rise 290 nm to 2600 nm thus addressing from the UV to NIR region with an output pulse duration of 80 fs, while the other is introduced into spectrometer (HELIOS) moving through a delay stage of 0–8 ns, which further descends onto the sapphire crystal giving rise to a continuum of white light (420–780 nm) as a probe pulse. The former pulse of 410 nm (pump pulse) stimulates the sample under study and transfers the information of excited-state dynamics to the probe pulse. Then, the probe pulse renders the information to the detector (CMOS), and the acquired carrier dynamics have been studied using the Surface Explorer Software.3. Results and discussion
The all-inorganic colloidal CsPbBr3 PNC was prepared using a chemical route of modified hot-injection.30 A yellowish color precipitate was obtained by centrifugation and was dispersed in hexane, a non-polar solvent.3.1 X-ray diffraction
The powder X-ray diffraction (PXRD) pattern of the as-prepared all-inorganic colloidal CsPbBr3 PNC depicted in Fig. S1† asserts an orthorhombic structure of the as-formed material, similar likewise to the earlier reports.13.2 Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) revealed that the as-synthesized CsPbBr3 PNCs were of the cubic morphology and exhibited an average size of 11.58 ± 0.21 nm, as shown in Fig. 1a.3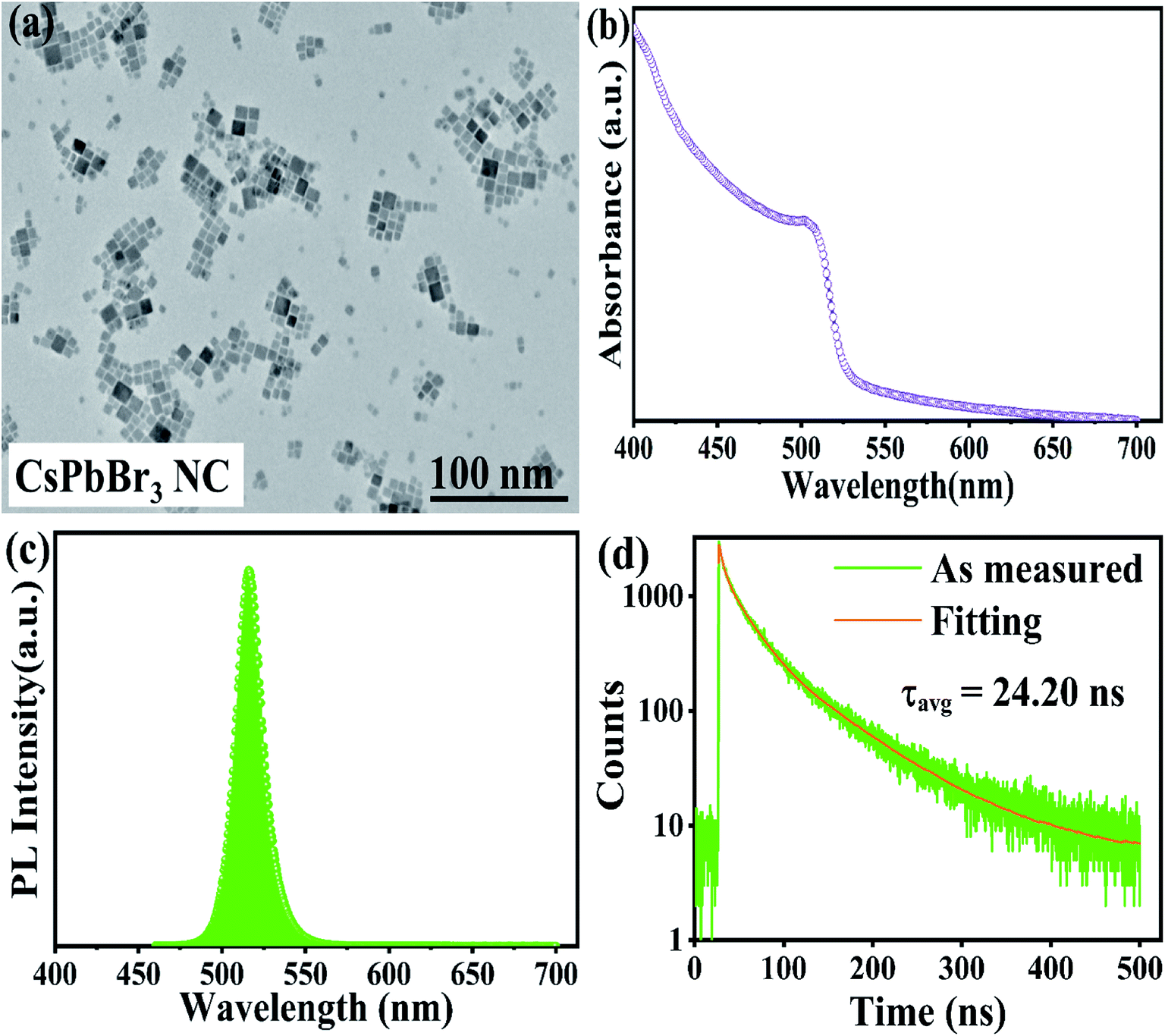 | ||
| Fig. 1 (a) A TEM image; (b) ultraviolet-visible spectra; (c) PL; (d) TRPL spectra of all-inorganic CsPbBr3 PNC. | ||
3.3 Ultraviolet-visible spectroscopy and photoluminescence
Ultraviolet-visible absorption spectroscopy and photoluminescence spectroscopy were used to study the optical attributes of the all-inorganic CsPbBr3 PNCs. The measured wavelength at the sharp absorption band edge was 501 nm, as displayed in Fig. 1b30 and the emission peak was seen at 516 nm, derived from the photoluminescence (PL) emission spectrum, as depicted in Fig. 1c,30 and PLQY was calculated to be 38.7%, as listed in Table S1.†3.4 Time-resolved photoluminescence (TRPL) spectroscopy
The TRPL spectroscopy was further carried out in order to get further insight into the carrier kinetics in a quantitative way.30,48–50 Using tri-exponential functions,22 the PL fitted the decay curve pictured in Fig. 1d, and the computed values are listed in Table S2.† Although the PL spectra in Fig. 1c shows a single peak at 516 nm, the UV-Vis results in Fig. 1b indicates that the transitions of the direct bandgap of PNCs are dominated by other radiative transitions, which are further confirmed by TRPL spectroscopy as a presence of three exponential components. Moreover, the fluorescence decay lifetime (τ1) and its contribution (β1) corroborates with the intrinsic exciton relaxation.50–53Similarly, an intermediate fluorescence decay lifetime (τ2) and its contribution (β2) supports interaction between excitons and phonons50–53 and similarly ultralong-lived fluorescence decay lifetime (τ3) and its contribution (β3) corresponds to the interactions between excitons and defects.52–54 The long-lived lifetime τ3 maybe a result of less non-radiative energy transfer to the trap states.51–54 Moreover, we have also computed the radiative (κr) and non-radiative (κnr) recombination decay rates,22,55–59 as listed in Table S3.† The computed values of (κr) and (κnr) are 15.95 × 106 s−1 and 25.37 × 106 s−1, respectively, and (κr/κnr) is about 0.63, which suggests that the non-radiative process here is relatively fast.22,55–59
From smaller decay lifetime component (τ1), longer τavg, and larger (κr/κnr), one can infer that the as-synthesized sample features fewer trap states.22,55,57
3.5 Ultrafast transient absorption spectroscopy
The TA spectroscopy was used to overcome the time resolution limitations of the TRPL technique to probe into the hot carrier kinetics.30 The fundamental physical processes of TA have three contributions namely (i) ground state bleach (GSB), (ii) stimulated emission (SE), and (iii) photo-induced absorption (PIA). In order to produce a TA spectrum, the probe pulse is transmitted through the sample at several time delays concerning with or without the pump pulse. Therefore, a differential transmission spectrum ΔT(λ,τ) (T = (I/I0), where I stands for transmitting intensity and I0 the incident intensity of light), is produced that gives the difference of transmission measured with and without the pump pulse. If the differential transmission spectrum ΔT(λ,τ) has a positive sign, then it refers to photo-induced transmission (PIT) owing to the reduced population of the ground state (i.e., GSB)37 or SE, and the negative sign stands for the photo-induced absorption (PIA)35–37 as result of a higher population of the photo-excited states.31,37 Similarly, if we take into consideration the differential absorption ΔA(λ,τ) spectrum that quantifies the difference of absorption with and without the pump pulse. The relationship between absorption (A) and transmission (T),60| A(λ,τ) = 2 − log10[T(λ,τ)] |
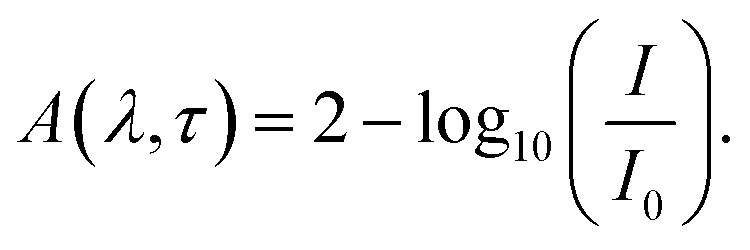 | (1) |
On adding the differential change to the above equation,60 we get
| A(λ,τ) + ΔA(λ,τ) = 2 − log10[T(λ,τ) + ΔT(λ,τ)], | (2) |
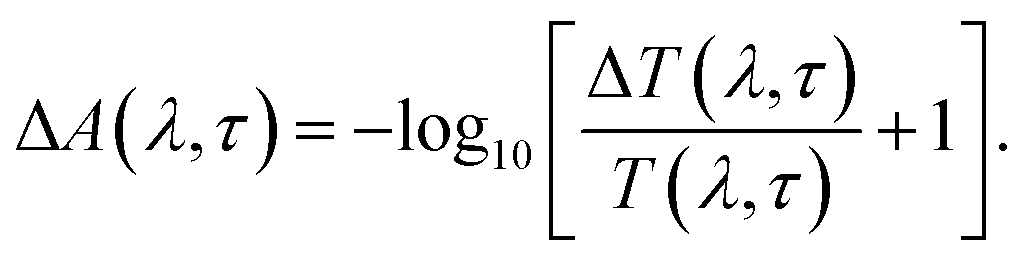 | (3) |
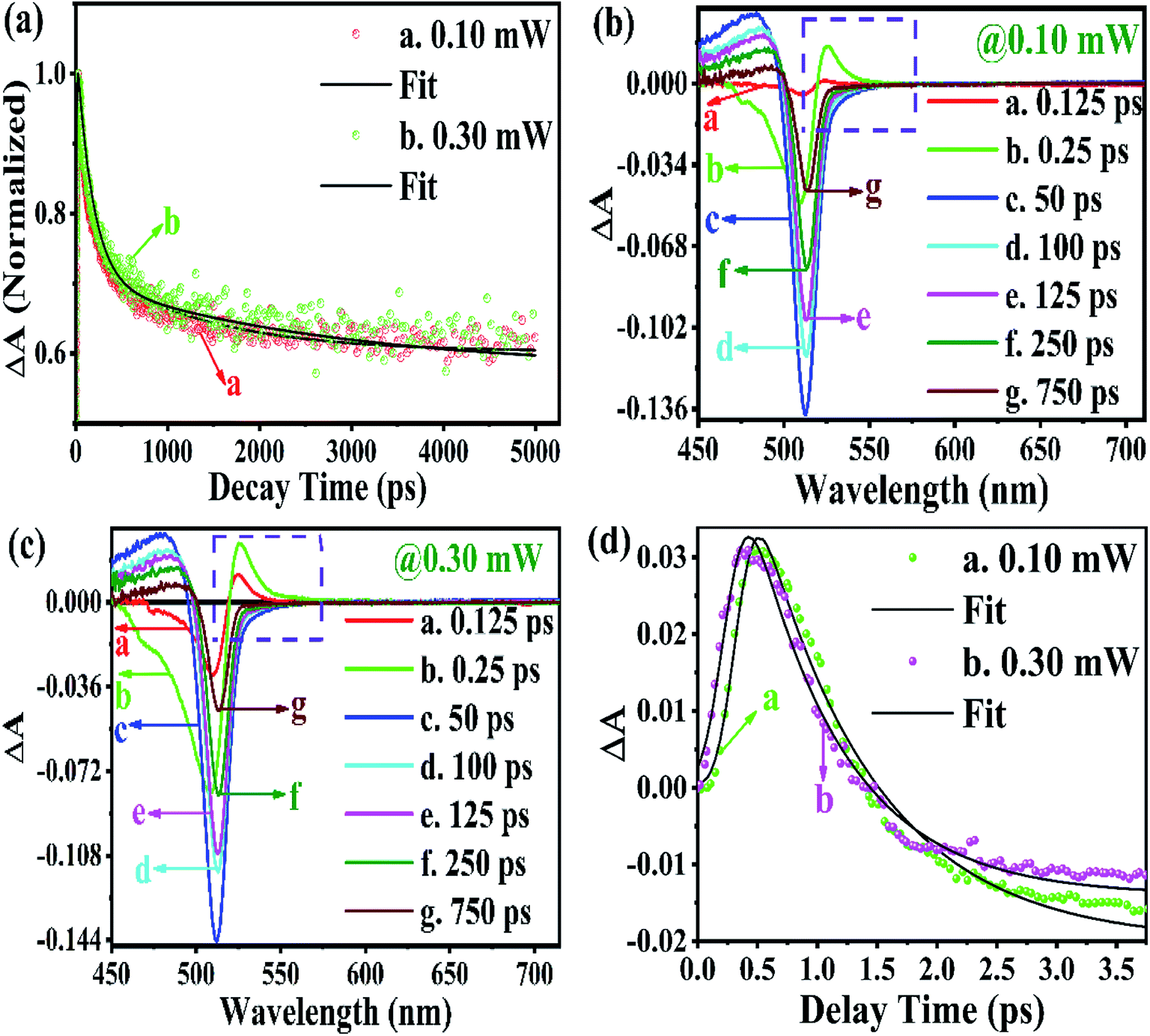
From the above equations, it can be easily observed that ΔA and ΔT will have opposite signs. For example, in the case of photo-induced transmission (PIT), ΔT has a positive value, while the corresponding negative value is observed for ΔA.60 On the other hand, for photo-induced absorption (PIA), we have a positive value for ΔA, which is a negative value for ΔT.31,60 In this study, a pump pulse with an operating wavelength 410 nm was used to excite CsPbBr3 PNCs, while, a white light probe was used to study the variations in induced absorption as a function of wavelength (λ) and delay time (τ). The plotted curves of TA spectra of the as-synthesized CsPbBr3 PNCs for various time delays as well as at different pump powers (0.10 mW, 0.30 mW) are shown in Fig. 2. For small delay times, TA spectra with a dominant peak at 516 nm show positive PIA,36 which steadily declines due to an increase in the delay time. The signal of GSB localized at 511 nm indicates the population occupancy of different energy states of the PNCs, as shown in Fig. 2c.30,61–65 Fig. 2d represents the intraband state of the surface trap of PNC.30,35,37 The ΔA intensity of the GSB signal helps to evaluate the carrier density in terms of the bandgap.35,37,63 Fig. 3a shows that the peak intensity of GSB increases at a higher pump power of 0.30 mW as compared to that at lower power of 0.10 mW at the same delay time. As a result, the rate of recombination of charge carriers increases with increasing pump power, as shown in Fig. 3a.30,44 The three-exponential parameters model is used to fit the decay kinetics of GSB, and the computed values are listed in Table S4.†63
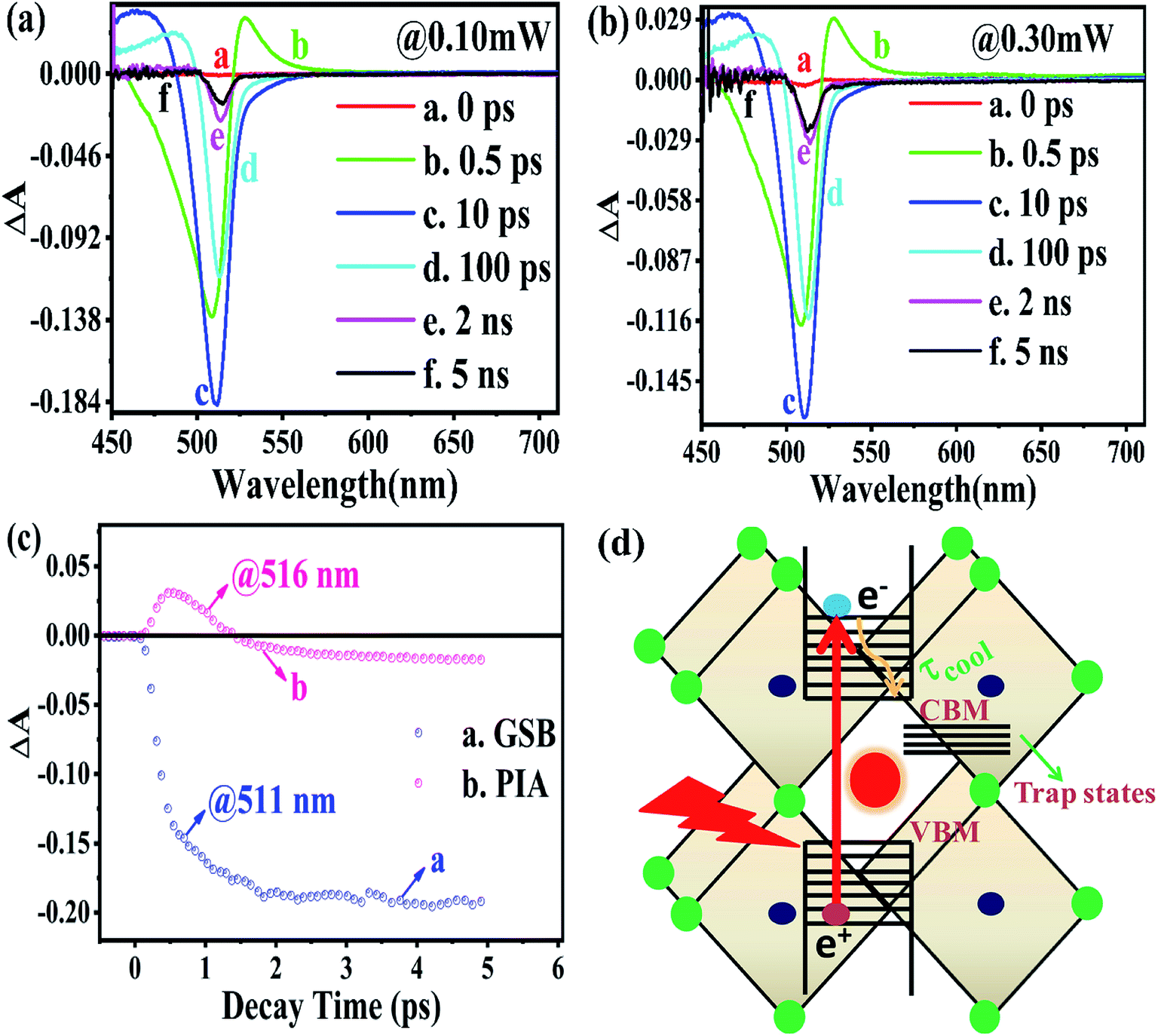 | ||
| Fig. 2 Transient absorption spectroscopy under a 410 nm pump pulse of the rate of radiation energy transported by a laser source (a) at 0.10 mW; (b) at 0.30 mW; (c) the positions of PIA and GSB features explored by exploiting the transient absorption kinetics; (d) schematic depiction of the hot carrier cooling process in the CsPbBr3 PNCs. | ||
 | ||
| Fig. 3 (a) GSB; temporal evolutions after photo-excitation at the sort of rate of radiated energies transported by the laser source for instance (0.10 mW (b), 0.30 mW (c)) are depicted in the transient absorption (TA) spectrums of CsPbBr3 PNC; (d) PIA, respectively. | ||
Similarly, from the results derived from the TRPL quantitative analysis, τavg of CsPbBr3 PNC for 0.10 mW is 455.37 ps, which is much lower than 884.05 ps obtained with a pump power of 0.30 mW. Similarly, as mentioned in TRPL calculations, the values of κr and κnr using the TA spectroscopy are 0.84 × 109 s−1 and 1.35 × 109 s−1, respectively, and κr/κnr is about 0.62 for CsPbBr3 PNCs for the pump power of 0.10 mW.
For the pump power of 0.30 mW, κr and κnr are 0.44 × 109 s−1 and 0.69 × 109 s−1, respectively, and κr/κnr is about 0.64 (similar to the TRPL value, as listed in Table S3†), as listed in Table S5.† The values for κr/κnr are suggesting a fact that the non-radiative processes get suppressed in the presence of a high-power radiation source. It can be easily inferred that at a high rate of radiation energy transported (for instance 0.30 mW) to the PNCs, the values for smaller decay lifetime components, longer τavg and larger κr/κnr, lead to a reduction in the intermediate energy states or traps both at the surface as well as inside the PNCs resulting in the improvement of PL and PLQY of the PNCs. The PIA signal intensity is less for the lower pump power, as observed in Fig. 3b and c, and the PIA decay time was almost indistinguishable to that build-up on the GSB band-edge, as shown in Fig. 2c.34–37,64,65 From the experimental results, we assert that when radiation energy is incident at a high rate on CsPbBr3 PNCs, GSB shows a charge carrier-density involving a blue-shift of the peak wavelength together with the broadening of spectral width resulting from the aggregation of charge carriers at the band edge and can be explicated by the kinetics of Burstein–Moss shift.35 From Fig. 3d, one can understand that the cooling time of hot carriers is observed for CsPbBr3 PNCs for the 0.10 mW pump power is 526.65 fs, while it is 417.47 fs for the pump power of 0.30 mW. Once again it can be easily understood that there is a larger decrease in the defect states as more power of radiation energy is delivered to PNCs, which can be easily observed in the improvement of PL and PLQY of the PNCs. However, such evidences were not observed during PL and TRPL measurements due to the use of the xenon lamp as a source of excitation, which was less capable of releasing the trapped energy from the defect states as observed in Fig. 3d, where we used a laser as a source of excitation with different power tuneability. Hence, the laser-induced ultrafast study is legitimate as a probe to study the kinetics of influence of the rate of radiation energy on charge carrier kinetics of CsPbBr3 PNC.
4. Conclusions
In this study, we have presented the charge-carrier kinetics of all-inorganic CsPbBr3 PNCs. The hot-carrier cooling physical process got augmented in the case of radiation power of 0.30 mW delivered to the CsPbBr3 PNCs. The reduction in the charge trapping states by a factor of 1.03 was observed compared to the radiation power of 0.10 mW incident on the CsPbBr3 PNCs, which was analysed by the ultrafast transient absorption spectroscopy. In our work, we observe that with higher radiation energy (0.30 mW) in the context of transient absorption spectroscopy, it is easier to suppress the intermediate energy states or charge trapping states, resulting in the enhancement of PL and PLQY of the PNCs. The current findings make perovskite NCs highly promising and more competitive applicant for future device applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work supported financially by the UGC-JRF as well as AIRF JNU for allowing all the required characterization facilities. A grant furnished by the SERB-DST project (EEQ/2016/000652), funded by Govt. of India, as well appreciatively acknowledged.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- Y. Chang, Y. J. Yoon, G. Li, E. Xu, S. Yu, C.-H. Lu, Z. Wang, Y. He, C. H. Lin and B. K. Wagner, ACS Appl. Mater. Interfaces, 2018, 10, 37267–37276 CrossRef CAS.
- H. Liu, Z. Liu, W. Xu, L. Yang, Y. Liu, D. Yao, D. Zhang, H. Zhang and B. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 14256–14265 CrossRef CAS.
- B. Xin, Y. Pak, S. Mitra, D. Almalawi, N. Alwadai, Y. Zhang and I. S. Roqan, ACS Appl. Mater. Interfaces, 2019, 11, 5223–5231 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS.
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee and J. H. Heo, Science, 2015, 350, 1222–1225 CrossRef CAS.
- N. Wang, L. Cheng, R. Ge, S. Zhang, Y. Miao, W. Zou, C. Yi, Y. Sun, Y. Cao and R. Yang, Nat. Photonics, 2016, 10, 699–704 CrossRef CAS.
- B. R. Sutherland and E. H. Sargent, Nat. Photonics, 2016, 10, 295–302 CrossRef CAS.
- S. Chen, C. Teng, M. Zhang, Y. Li, D. Xie and G. Shi, Adv. Mater., 2016, 28, 5969–5974 CrossRef CAS.
- D. Li, D. Zhou, W. Xu, X. Chen, G. Pan, X. Zhou, N. Ding and H. Song, Adv. Funct. Mater., 2018, 28, 1804429 CrossRef.
- J. Luo, S. Li, H. Wu, Y. Zhou, Y. Li, J. Liu, J. Li, K. Li, F. Yi and G. Niu, ACS Photonics, 2017, 5, 398–405 CrossRef.
- X. Wu, B. Zhou, J. Zhou, Y. Chen, Y. Chu and J. Huang, Small, 2018, 14, 1800527 CrossRef.
- J. Yang, K. Liu, Z. Cheng, P. Jing, Q. Ai, X. Chen, B. Li, Z. Zhang, L. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 34744–34750 CrossRef CAS.
- S. W. Eaton, M. Lai, N. A. Gibson, A. B. Wong, L. Dou, J. Ma, L.-W. Wang, S. R. Leone and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1993–1998 CrossRef CAS.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS.
- C. H. Lin, Q. Zeng, E. Lafalce, M. J. Smith, S. T. Malak, J. Jung, Y. J. Yoon, Z. Lin, Z. V. Vardeny and V. V. Tsukruk, Adv. Opt. Mater., 2017, 5, 1700011 CrossRef.
- C. H. Lin, Q. Zeng, E. Lafalce, S. Yu, M. J. Smith, Y. J. Yoon, Y. Chang, Y. Jiang, Z. Lin and Z. V. Vardeny, Adv. Opt. Mater., 2018, 6, 1800474 CrossRef.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem.–Eur. J., 2017, 23, 9481–9485 CrossRef CAS.
- F. Meinardi, Q. A. Akkerman, F. Bruni, S. Park, M. Mauri, Z. Dang, L. Manna and S. Brovelli, ACS Energy Lett., 2017, 2, 2368–2377 CrossRef CAS.
- J. Tong, J. Wu, W. Shen, Y. Zhang, Y. Liu, T. Zhang, S. Nie and Z. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 9317–9325 CrossRef CAS.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS.
- P. Ramasamy, D.-H. Lim, B. Kim, S.-H. Lee, M.-S. Lee and J.-S. Lee, Chem. Commun., 2016, 52, 2067–2070 RSC.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko and M. I. Bodnarchuk, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS.
- M. A. Prelas, C. L. Weaver, M. L. Watermann, E. D. Lukosi, R. J. Schott and D. A. Wisniewski, Prog. Nucl. Energy, 2014, 75, 117–148 CrossRef CAS.
- M. Sychov, A. Kavetsky, G. Yakubova, G. Walter, S. Yousaf, Q. Lin, D. Chan, H. Socarras and K. Bower, Appl. Radiat. Isot., 2008, 66, 173–177 CrossRef CAS.
- S. Kumar, 2015, arXiv preprint arXiv:1511.07427.
- J.-F. Liao, Y.-F. Xu, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, ACS Appl. Mater. Interfaces, 2018, 10, 42301–42309 CrossRef CAS.
- R. Berera, R. van Grondelle and J. T. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS.
- Y. Yamada, T. Nakamura, M. Endo, A. Wakamiya and Y. Kanemitsu, J. Am. Chem. Soc., 2014, 136, 11610–11613 CrossRef CAS.
- M. T. Trinh, X. Wu, D. Niesner and X.-Y. Zhu, J. Mater. Chem. A, 2015, 3, 9285–9290 RSC.
- N. S. Makarov, S. Guo, O. Isaienko, W. Liu, I. Robel and V. I. Klimov, Nano Lett., 2016, 16, 2349–2362 CrossRef CAS.
- M. B. Price, J. Butkus, T. C. Jellicoe, A. Sadhanala, A. Briane, J. E. Halpert, K. Broch, J. M. Hodgkiss, R. H. Friend and F. Deschler, Nat. Commun., 2015, 6, 8420 CrossRef CAS.
- Y. Zhai, C. X. Sheng, C. Zhang and Z. V. Vardeny, Adv. Funct. Mater., 2016, 26, 1617–1627 CrossRef CAS.
- Y. Yang, D. P. Ostrowski, R. M. France, K. Zhu, J. Van De Lagemaat, J. M. Luther and M. C. Beard, Nat. Photonics, 2016, 10, 53–59 CrossRef CAS.
- Y. Fang, C. Bi, D. Wang and J. Huang, ACS Energy Lett., 2017, 2, 782–794 CrossRef CAS.
- H. S. Rao, W. G. Li, B. X. Chen, D. B. Kuang and C. Y. Su, Adv. Mater., 2017, 29, 1602639 CrossRef.
- M. He, F. Qiu and Z. Lin, J. Phys. Chem. Lett., 2013, 4, 1788–1796 CrossRef CAS.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS.
- J.-S. Yao, J. Ge, B.-N. Han, K.-H. Wang, H.-B. Yao, H.-L. Yu, J.-H. Li, B.-S. Zhu, J.-Z. Song and C. Chen, J. Am. Chem. Soc., 2018, 140, 3626–3634 CrossRef CAS.
- M. Zhang, H. Yu, M. Lyu, Q. Wang, J.-H. Yun and L. Wang, Chem. Commun., 2014, 50, 11727–11730 RSC.
- J. S. Manser and P. V. Kamat, Nat. Photonics, 2014, 8, 737–743 CrossRef CAS.
- H. Chung, S. I. Jung, H. J. Kim, W. Cha, E. Sim, D. Kim, W.-K. Koh and J. Kim, Angew. Chem., 2017, 56, 4160–4164 CrossRef CAS.
- J. Fu, Q. Xu, G. Han, B. Wu, C. H. A. Huan, M. L. Leek and T. C. Sum, Nat. Commun., 2017, 8, 1–9 CrossRef CAS.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase and T. Minemoto, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- Z. Hu, Z. Liu, Y. Bian, S. Li, X. Tang, J. Du and Z. Zang, et al., Adv. Opt. Mater., 2018, 6, 1700997 CrossRef.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS.
- C. Bi, S. Wang, Wen Wen, J. Yuan, G. Cao and J. Tian, J. Phys. Chem. C, 2018, 122, 5151–5160 CrossRef CAS.
- Z. Liang, S. Zhao, Z. Xu, B. Qiao, P. Song, D. Gao and X. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 28824–28830 CrossRef CAS.
- H. Shi, X. Zhang, X. Sun, R. Chen and X. Zhang, J. Phys. Chem. C, 2019, 123, 19844–19850 CrossRef CAS.
- G. Li, J. Huang, H. Zhu, Y. Li, J.-X. Tang and Y. Jiang, Chem. Mater., 2018, 30, 6099–6107 CrossRef CAS.
- Z. Liu, Y. Zhang, Y. Fan, Z. Chen, Z. Tang, J. Zhao and Y. Lv, et al., ACS Appl. Mater. Interfaces, 2018, 10, 13053–13061 CrossRef CAS.
- N. Kawano, M. Koshimizu, Y. Sun, N. Yahaba, Y. Fujimoto, T. Yanagida and K. Asai, J. Phys. Chem. C, 2014, 118, 9101–9106 CrossRef CAS.
- Z. Yuan, S. Yu, Y. Tian, Y. Xin and B. Ma, Chem. Commun., 2015, 51, 16385–16388 RSC.
- Z. Shi, S. Li, Y. Li, H. Ji, X. Li, D. Wu and T. Xu, et al., ACS Nano, 2018, 12, 1462–1472 CrossRef CAS.
- K. E. Knowles, M. D. Koch and J. L. Shelton, J. Mater. Chem. C, 2018, 6, 11853–11867 RSC.
- P. Maity, J. Dana and H. N. Ghosh, J. Phys. Chem. C, 2016, 120, 18348–18354 CrossRef CAS.
- S. Sarkar, V. K. Ravi, S. Banerjee, G. R. Yettapu, G. B. Markad, A. Nag and P. Mandal, Nano Lett., 2017, 17, 5402–5407 CrossRef CAS.
- X. Luo, G. Liang, Y. Han, Y. Li, T. Ding, S. He, X. Liu and K. Wu, J. Am. Chem. Soc., 2020, 142, 11270–11278 CrossRef CAS.
- J. Aneesh, A. Swarnkar, V. K. Ravi, R. Sharma, A. Nag and K. V. Adarsh, J. Phys. Chem. C, 2017, 121, 4734–4739 CrossRef CAS.
- A. Mondal, J. Aneesh, V. K. Ravi, R. Sharma, W. J. Mir, M. C. Beard, A. Nag and K. V. Adarsh, Phys. Rev. B, 2018, 98, 115418 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05766e |
| This journal is © The Royal Society of Chemistry 2020 |