
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperation of Fe(II) and peroxymonosulfate for enhancement of sulfamethoxazole photodegradation: mechanism study and toxicity elimination†
Han Gonga,
Wei Chu*b,
He Gongc,
Airu Huanga,
Jingjun Lina and
Muting Yan *a
*a
aJoint Laboratory of Guangdong Province and Hong Kong Region on Marine Bioresource Conservation and Exploitation, College of Marine Sciences, South China Agricultural University, Guangzhou, China. E-mail: marineymt@scau.edu.cn
bDepartment of Civil and Environmental Engineering, Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. E-mail: wei.chu@polyu.edu.hk
cSchool of Chemical Engineering, ShengLi College, China University of Petroleum, Dongying, Shandong, China
First published on 28th September 2020
Abstract
This study aims at systematically examining the potential of removing the emerging pollutant sulfamethoxazole (SMX) from aqueous solution under photo-assisted peroxymonosulfate (PMS) activation by Fe(II). The residual SMX was determined by HPLC analysis. The concentration of Fe(II) ([Fe(II)]) was monitored during SMX degradation. Fe(II) and PMS cooperated with each other for faster SMX photodegradation; a relatively lower or higher molar ratio between Fe(II) and PMS led to lower SMX removal efficiency due to the insufficient radicals or scavenging effect. A fixed reaction ratio of [Fe(II)]Δ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PMS]0 with 1.6:1 at the first 5 min was detected for reactions with [Fe(II)]0 ≥ 0.5 mM or [PMS]0 ≤ 0.25 mM. The pH level of around 6.0 was recommended for optimal SMX removal under the treatment process UVA + Fe(II) + PMS. Six transformation products were detected through UPLC/ESI-MS analysis, and four of the proposed intermediates were newly reported. Concentrations of the intermediates were proposed based on the isoxazole-ring balance and the Beer–Lambert law. Total Organic Carbon (TOC) reduction was mainly attributed to the loss of benzene ring, N–S cleavage, and isoxazole ring opening during SMX degradation. The contributions of reactive species OH˙ and SO4˙− were determined based on quench tests. The acute toxicity of SMX to the rotifers was eliminated after the proposed treatment, demonstrating that the process was effective for SMX treatment and safe to the environment.
:[PMS]0 with 1.6:1 at the first 5 min was detected for reactions with [Fe(II)]0 ≥ 0.5 mM or [PMS]0 ≤ 0.25 mM. The pH level of around 6.0 was recommended for optimal SMX removal under the treatment process UVA + Fe(II) + PMS. Six transformation products were detected through UPLC/ESI-MS analysis, and four of the proposed intermediates were newly reported. Concentrations of the intermediates were proposed based on the isoxazole-ring balance and the Beer–Lambert law. Total Organic Carbon (TOC) reduction was mainly attributed to the loss of benzene ring, N–S cleavage, and isoxazole ring opening during SMX degradation. The contributions of reactive species OH˙ and SO4˙− were determined based on quench tests. The acute toxicity of SMX to the rotifers was eliminated after the proposed treatment, demonstrating that the process was effective for SMX treatment and safe to the environment.
1. Introduction
Pharmaceuticals and personal care products, PPCPs, being detected in natural waters, have raised global concern for their potential environmental and health effects.1 Sulfamethoxazole (SMX) is a pharmaceutical that is largely used for the treatment of bacterial infections such as urinary tract infections, bronchitis, and prostatitis. A previous study showed that SMX has low biodegradability and may result in direct or indirect toxicological effects on the environment and human health.2 Especially, as an antibiotic, residues of SMX may pose a potential risk for developing antibiotic resistant bacteria.3Common water/wastewater treatments processes cannot effectively remove antibiotics including SMX because of its antibacterial nature. Owing to the low removal efficiency of SMX in wastewater by activated sludge treatment process,4 a large amount of SMX enters the environment annually. SMX has been detected in the effluents of sewage treatment plants as well as surface water, groundwater, and even tap water.5 The concentration of SMX was detected as high as 2000 ng L−1 in many municipal sewage treatment plants and ranging from 30 to 480 ng L−1 in surface water.6,7 Governments of different countries and researchers are getting aware of the discharged antibiotics and their by-products in nature water. Therefore, effective treatment techniques for enhancement of the SMX removal efficiency are urgently in need.
Advanced Oxidation Processes (AOPs) provide prospective procedures in degrading the refractory pollutants including POPs (persistent organic pollutants) and antibiotics.8–17 In terms of the treatment of SMX, photolysis, photocatalysis, Fenton, ozonation, sulfate radicals-based processes, and ultrasonic process have been widely explored.18–22 Among the treatment processes, sulfate radicals-based processes raised great interests in recent years because of its high efficiency, and less pH sensitivity.23
Peroxymonosulfate (PMS) was widely used as the oxidant to generate powerful sulfate radicals (SO4˙−).23–26 It is comparatively more stable and easier to handle because of its solid state at ambient temperature.27 Though using PMS directly to react with organics gives a slow reaction rate, PMS can be activated into highly reactive radicals by heat or transition metals e.g. Fe(II), the environmental friendly metal.28 The limitation of coupling Fe(II) and PMS is its slow regeneration rate of Fe(II) from Fe(III), and UV irradiation may solve this drawback.29
Previous studies prove the high efficiency in SMX removal by processes based on sulfate radicals including UV + Persulfate, Thermo + Persulfate, Fe(II) + Persulfate, Benzoquinone + PMS, and Hydroxylamine + Fe(II) + PMS.20,30–33 Despite the high efficiency, the treated SMX was reported to be more toxic for heat activated persulfate process.34 Therefore, in case the degradation by-products of SMX possess toxicity to the aquatic environment, it is also essential to monitor the toxicity of the treated solution.
In this study, the process based on photo-assisted Fe(II) + PMS process was studied for SMX degradation. The effects of the parameters were investigated by evaluating the SMX removal under different light source, Fe(II) concentration, PMS concentration, and pH level. Prediction of degradation rate by varying SMX concentration was conducted. Besides, the reaction mechanism including mineralization, degradation products and pathways were determined and the toxicity of the treated solution was assessed.
2. Methodology
2.1 Chemicals and reagents
All chemicals are of analytic reagent grade and all solvents as mobile phases are of HPLC or LC/MS grade. SMX (C10H11N3O3S: 99.0%), PMS (KHSO5·0.5KHSO4·0.5K2SO4: 95%), and ferrous sulfate (FeSO4·7H2O: 99.0%) were purchased from Sigma-Aldrich Inc. (USA). Sulfuric acid (H2SO4) and sodium hydroxide (NaOH) were used to adjust the initial pH of the solutions; monopotassium phosphate (KH2PO4), phosphate acid (H3PO4), and sodium nitrite (NaNO2) were obtained from British Drug Houses (BDH, England). Acetonitrile (ACN, C2H3N), tertbutyl alcohol (TBA, C4H10O) and methanol (MeOH, CH4O) were purchased from Tedia Company, USA. 1,10-Phenanthroline (C12H10N2O) was obtained from International Laboratory (IL, USA). Ultrapure water generated from a Barnstead NANO pure water treatment system (Thermo Fisher Scientific Inc., USA) was employed for preparation of the solutions.2.2 Experimental procedures
For SMX degradation, all experiments were performed in cylindrical borosilicate glass quartz beakers at 23 ± 2 °C. The beakers were placed on a magnetic stirrer at the center of a CCP-4V computer controlled photochemical reactor (Luzchem Research Inc.). Two phosphor-coated low-pressure mercury lamps emitting monochromatic light were symmetrically arranged on the top center within the reactor as the irradiation source. Four different types of lamps at 254, 300, 350 and 420 nm as UVC, UVB, UVA and VIS were investigated for the effect of light source. The power of the UV lamps was approximately 25 watts. The absorbance spectrum of relative energy of the UV lamps provided by the producer was shown in Fig. S1.†The lamps were turned on 10 minutes before the start of the experiment for warming up and to ensure stable light irradiation. A cooling fan was installed in the reactor for temperature control. The fan was internally installed by the producer of the reactor. Therefore, the stable temperature (23 ± 2 °C) in the reactor can be ensured during the reaction and circulating cooling water was not needed. The reaction was initiated by the addition of predetermined amount of PMS and Fe(II) solution. The initial volume of each reaction solution was fixed at 400 mL. The beakers were stirred for a complete mixture and homogeneity throughout the reactions. Samples were taken at the preset time intervals and quenched with methanol (v/v = 1:1) for determination of the remaining SMX and transformation products. For TOC measurement, concentrated NaNO2 was used as the quencher. For determination of the remaining Fe(II), samples of 1.5 mL were taken and mixed with 1.5 mL 1,10-phenanthroline. To identify the possible species in the reaction system, quenchers including TBA and MeOH were employed. The mole ratio of the quencher to PMS is 1000:1.
2.3 Analytical methods
The residual SMX was quantified by High Performance Liquid Chromatography (HPLC) which consists of a Waters 515 HPLC pump and a Waters 2487 UV detector. An isocratic flow running at a flow rate 1 mL min−1 was used with the mobile phase of ACN/10 mM monopotassium phosphate (50/50, pH adjustment to 3.0 with H3PO4). The injection volume was 10 μL. The UV detector wavelength was set at single wavelength at 258 nm. The retention time of SMX in this mobile phase was around 4.70 min. The peak area of detected SMX was recorded. Linear correlation was detected between varied SMX concentrations and the corresponding peak areas. SMX removal was presented in C/C0, representing the normalized concentration with reference to the initial SMX concentration.The identification of the transformation products was performed by the Ultra-High Performance Liquid Chromatography/Electrospray Ionization-Mass Spectrometry (UPLC/ESI-MS) system. The UPLC (Dionex UltiMate 3000) comprises a degasser, a pump, an autosampler, a diode array detector (DAD), a column compartment and a column (Thermo Hypersil GOLD, 1.9 μm, 50 × 2.1 mm). A gradient flow of the mobile phase containing ACN (A) and 0.1% formic acid (B) at 0.15 mL min−1 was employed. The injection volume was 5 μL. The ratio of A was kept at 10% during the first 2 min, then increased to 60% linearly during 2–15 min and held at 60% for 3 min. Finally, the ratio of mobile phase was returned to the initial composition until the end of the run. For mass analysis, alternative ion mode was employed. A Bruker amaZon SL ion trap mass analyzer coupled with an ion trap mass spectrometer detector (MSD) was performed.
TOC of the solution was measured with a TOC analyzer (Shimadzu, TOC-L). The concentration of Fe(II) was determined with a UV-vis spectrophotometer (MRC, Spectro UV-11) at 510 nm.
2.4 Toxicity test
The rotifer (Brachionus calyciflorus), an important aquatic species widely used in the toxicity assessment of pollutants, was employed for acute toxicity in this study. Toxicity testing was conducted using a 24-well polystyrene plate with the test solution adjusted to pH 7.0 ± 0.5. Ultrapure water was used as the culture medium for the control group and starve group. Ten healthy juveniles with age 0–2 h were selected into each well and exposed to the test sample of 1 mL for 24 h at 20 ± 0.5 °C in the dark. The green algae Chlorella pyrenoidesa of 50 μL was introduced as the food of rotifers to all groups except for the starve group. The survival of the rotifers was measured as the test parameter. Three replicates were performed for each group.3. Results and discussion
3.1 Degradation of SMX in different systems
Fig. 1a presents the decay curves of SMX in different reaction systems. Only about 3% of SMX was degraded by UVA + PMS system, indicating that PMS could be slightly activated by UVA (eqn (1)).35
 | (1) |
 | ||
| Fig. 1 SMX degradation in different reaction systems. (a) SMX decay curves. (b) Concentration change of Fe(II). Experimental conditions: [SMX]0 = 0.05 mM, [Fe(II)]0 = 0.25 mM, [PMS]0 = 0.25 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
In the system of UVA + Fe(II), around 15% of SMX was removed within 25 min. As SMX degradation was negligible in the presence of sole UVA or sole Fe(II) (data not shown), the SMX degradation in this system was mainly due to the generation of OH˙ from [Fe(OH)]2+, a common species resulting from the hydrolysis of Fe(III) (eqn (2)–(4)).36 From Fig. 1b, the concentration of Fe(II) kept unchanged throughout the whole reaction because of the Fe(II)/Fe(III) cycle in the presence of UVA.
 | (2) |
| Fe3+ + H2O → [Fe(OH)]2+ + H+ | (3) |
 | (4) |
Compared to UVA, Fe(II) is more effective in activating PMS, with a SMX removal of 35% achieved within 25 min in the system of PMS + Fe(II). Besides, the possible involvement of Fe(IV)O2+ in the reaction system PMS + Fe(II) may also contribute to the relatively higher removal efficiency.37 The SMX degradation is much more significant in the first 5 minutes (with approximately 30% removal efficiency) than that in the following 20 minutes (with only about 5% additional removal). From Fig. 1b, the concentration of Fe(II) was decreased during the process because of the activation of PMS (to form sulfate radicals) and the transformation to Fe(III), simultaneously. Fe(II) was used up during the first 5 minutes, and cannot be regenerated from the beneficial Fe(II)/Fe(III) cycle without the assistant of UVA. Because of the approaching of complete consumption of both Fe(II) and PMS, the reaction was retarded afterwards.
| Fe2+ + HSO5− → SO4˙− + Fe3+ + OH− | (5) |
The removal efficiency of SMX in the UVA + Fe(II) + PMS system is substantially higher than those in the above systems. It is shown in Fig. 1a that the degradation process possesses a relatively stable curve during the 25 minute reaction time, which corresponds to the pseudo first-order kinetics. In this system, SO4˙− can be formed through the activation of PMS by either Fe(II) or UVA (eqn (1) and (2)). And OH˙ can be formed through the activation of PMS or Fe(II) by UVA (eqn (1), (3)–(5)). Though Fe(II) can be transformed to Fe(III) by both UVA and PMS, the remaining of [Fe(II)] at the 5th min (0.025 mM) is apparently high enough to maintain a healthy continuity of the process compared to that of the depletion of Fe(II) in the dark system. The regeneration of Fe(II) from Fe(III) due to the presence of UVA is apparently the key (Fig. 1b).
3.2 SMX degradation in Fe(II) + PMS system activated with different light sources
The contribution of hydrolysis to the SMX degradation was evaluated and proven to be insignificant in this study (data not shown). However, SMX removal efficiency via direct photolysis under UVC and UVB was found to be 97% and 55%, respectively (Fig. 2a). Direct photolysis was not helpful for TOC removal21 and the shortwave UV is not available in the sunlight, thus UVC and UVB was not recommended for SMX degradation.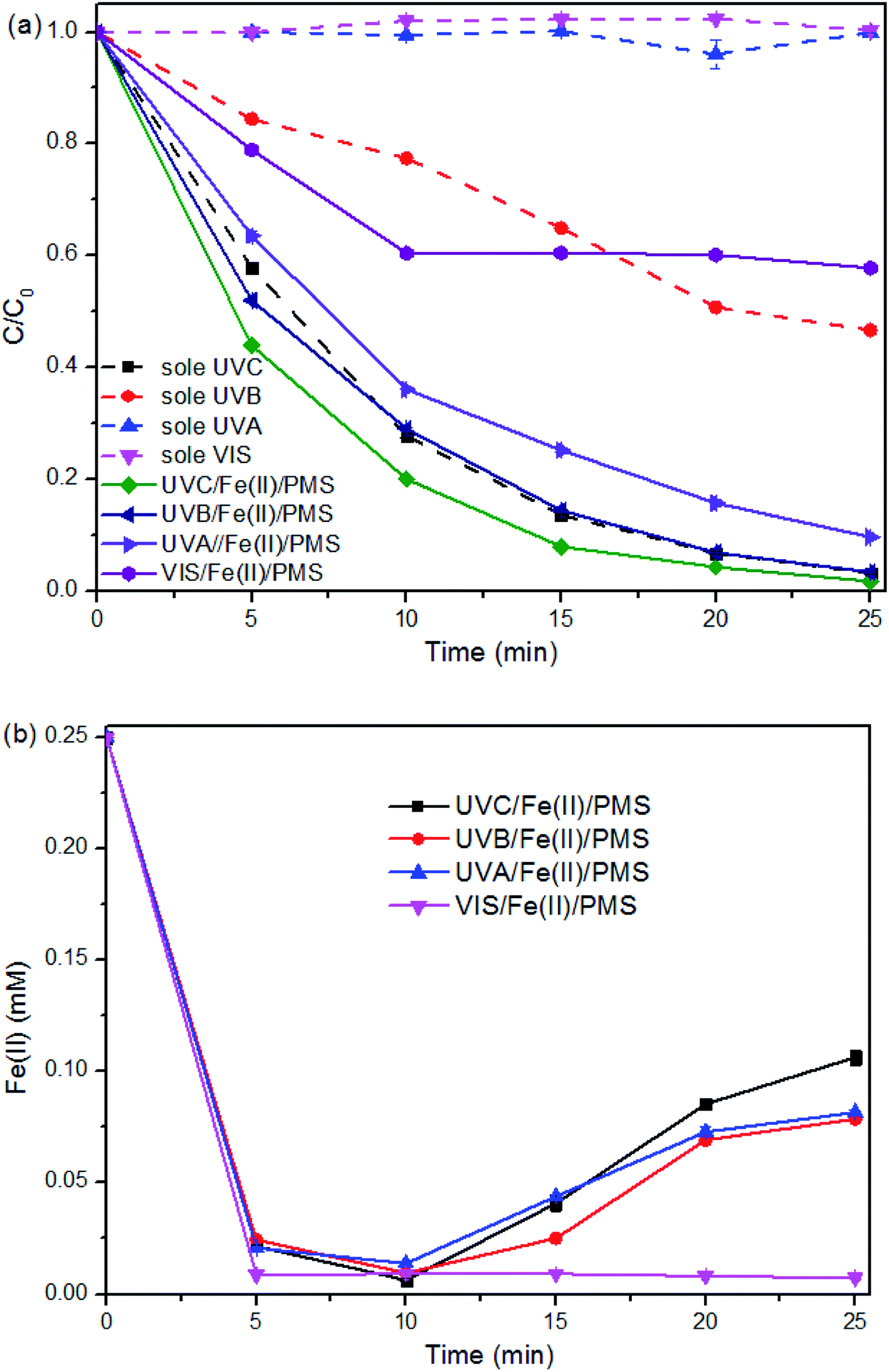 | ||
| Fig. 2 SMX degradation in the Fe(II) + PMS system with varied light source. (a) SMX decay curves. (b) Concentration change of Fe(II). Experimental conditions: [SMX]0 = 0.05 mM, [PMS]0 = 0.25 mM, [Fe(II)]0 = 0.25 mM, [pH]0 = 6.09, two lamps were employed. | ||
The direct photolysis of SMX under UVA and VIS was insignificant due to their low energy and the lower absorption of SMX at higher range of the light wavelength. However, the degradation performance under UVA + Fe(II) + PMS process is much better than that under VIS + Fe(II) + PMS. For the treatment process VIS + Fe(II) + PMS, SMX degradation was limited to the initial 10 min and being retarded afterward, indicating the limited ability of VIS to activate Fe(II) + PMS.
The limited ability was also illustrated from Fig. 2b, where the [Fe(II)] evolution in the VIS + Fe(II) + PMS was similar to that in the Dark + Fe(II) + PMS. While the [Fe(II)] evolution in the UV + Fe(II) + PMS systems, despite the wavelength, shows similar patterns. Therefore, the regeneration of Fe(II) can be realized under the irradiation of UV. As UVA also has the potential for using solar light as the free energy source, UVA + Fe(II) + PMS was further investigated for the SMX degradation in this study.
3.3 SMX degradation performance in the Fe(II) + PMS + UVA system
 | ||
| Fig. 3 Effect of Fe(II) concentration on SMX degradation in the Fe(II) + PMS + UVA system. (a) SMX degradation. (b) Concentration change of Fe(II). Experimental conditions: [SMX]0 = 0.05 mM, [PMS]0 = 0.25 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
As [Fe(II)]0 was overdosed at or above 0.50 mM, it is interesting to note that, around 0.40 mM of Fe(II) ([Fe(II)]Δ) was consumed with a fixed [PMS]0 at 0.25 mM in 5 min of the reaction regardless of the [Fe(II)]0 (i.e., [Fe(II)]Δ:[PMS]0 = 1.6:1). In this case, a large amount of free Fe(II) was left in the solution and resulted in additional scavenging effect (eqn (7) and (8)).29,39 The sulfate radicals and hydroxyl radicals may be transformed by the overdosed Fe(II) to SO42− and OH−, respectively.27
| HSO5− + SO4˙−→ SO5˙−+ SO42− + H+ | (6) |
| Fe2+ + SO4˙− → Fe3+ + SO42− | (7) |
| Fe2+ + OH˙ → Fe3+ + OH− | (8) |
Similarly, the SMX removal was enhanced within the [PMS]0 from 0.0625 to 0.25 mM and then reduced with the continue increase of [PMS]0 (Fig. 4a). As can be seen from Fig. 4b, for the reactions with [PMS]0 lower than 0.25 mM, the reaction ratio of [Fe(II)]Δ:[PMS]0 at the first 5 min was also 1.6:1. However, higher [PMS]0 at 0.25 mM or above was unable to react with [Fe(II)] effectively, with around 0.20 mM Fe(II) consumed. Low dosage of PMS means insufficient radicals, as well as surplus Fe(II) leading to scavenging effect (eqn (6) and (7)). While overdosed PMS increases the chance to quench the radicals (eqn (5)). Therefore, the properly adjusted [Fe(II)]0 and [PMS]0 played a critical role in SMX degradation. Relatively lower or higher Fe(II)/PMS would retard the reaction, and the optimal ratio of [Fe(II)]0:[PMS]0 for SMX degradation in this study was determined at 1:1.
 | ||
| Fig. 4 Effect of PMS concentration on SMX degradation in the Fe(II) + PMS + UVA system. (a) SMX degradation. (b) Concentration change of Fe(II). Experimental conditions: [SMX]0 = 0.05 mM, [Fe(II)]0 = 0.25 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
 | ||
| Fig. 5 Effect of pH on SMX degradation in the Fe(II) + PMS + UVA system. Experimental conditions: [SMX]0 = 0.05 mM, [Fe(II)]0 = 0.25 mM, [PMS]0 = 0.25 mM, two UVA lamps were employed. | ||
The removal efficiency of SMX was enhanced significantly with the increase of the initial pH from 2.07 to 6.09. Previous studies stated that the free Fe(II) was reduced in extremely acidic condition or at high pH levels due to the formation of [Fe·H2O]2+ or Fe(OH)2, respectively.40 Besides, large amount of H+ could plays the role of SO4˙− scavenger,41 extremely low pH was not favorable for SMX removal in this process. With further increase of pH to 9.27, SMX decay was significantly retarded, with final removals around 40%. In alkaline conditions, the self-dissociation may occur to PMS, and the active oxygen of PMS starts to decompose.42,43 This would lead to less sulfate radicals available for SMX degradation, and thus inhibited SMX degradation at high pH levels. Therefore, the pH level of around 6.0 is recommended for SMX degradation, which is also a great advantage of the proposed process for real application.
| k = 0.0089/[SMX]0 (R2 = 0.9708) | (9) |
 | ||
| Fig. 6 Prediction of kinetics with varied SMX concentration in the Fe(II) + PMS + UVA system. Experimental conditions: [Fe(II)]0 = 0.25 mM, [PMS]0 = 0.25 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
| 2Cl− → Cl2 + 2e− | (10) |
| Cl2 + H2O → HOCl + Cl− + H+ | (11) |
 | ||
| Fig. 7 Effect of inorganic ions on SMX degradation by Fe(II) + PMS + UVA process. Experimental conditions: [Fe(II)]0 = 0.25 mM, [PMS]0 = 0.25 mM, [Ion]0 = 0.10 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
3.4 Reaction mechanism of SMX degradation
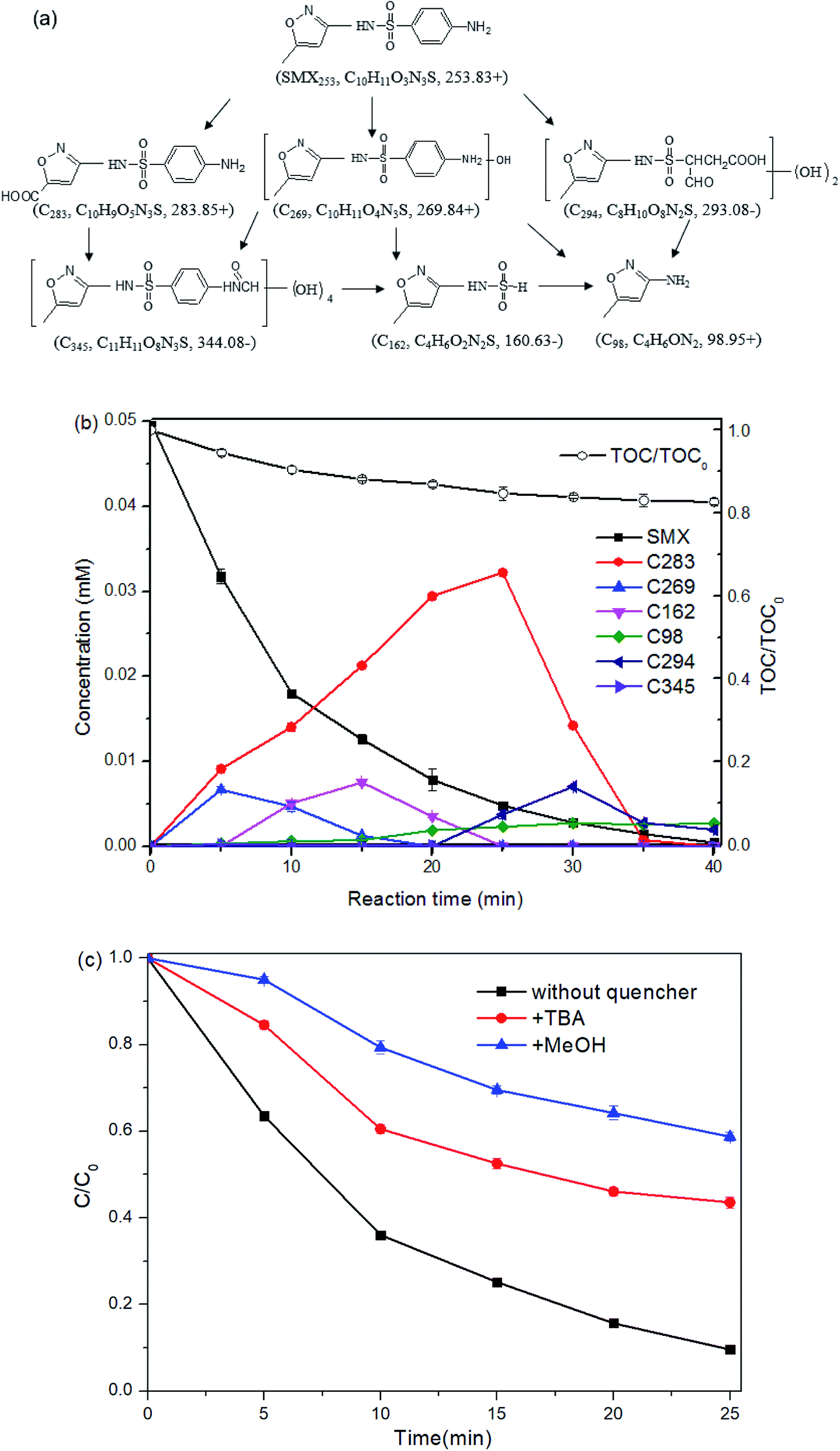 | ||
| Fig. 8 Reaction mechanism of SMX degradation in the Fe(II) + PMS + UVA system. (a) Proposed degradation pathways. (b) TOC reduction and proposed concentration of the degradation products. (c) SMX degradation with quenchers. Experimental conditions: [SMX]0 = 0.05 mM, [Fe(II)]0 = 0.25 mM, [PMS]0 = 0.25 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
The product C269 (m/z = 269.82 for [M + H]+), increasing by 16 mass units from the parent compound SMX, suggests the addition of one hydroxyl group to SMX. The location of the hydroxyl group cannot be confirmed due to the insufficient information provided in the MS/MS2 spectrum. The presence of C269 was also detected during SMX degradation by ultrasound + ozone oxidation,22 thermo + persulfate oxidation,32 ferrate oxidation,46 ferrate + US process,47 and Fe(II) + montmorillonite catalyzed ozonation.48
C283, at m/z 283.83 in positive ion mode, was supposed to result from the carboxylation of the methyl group on the isoxazole ring. The variation of 30 mass units from the parent compound SMX, was proposed to come from the introduction of two oxygen molecular, as well as the loss of two hydrogen molecular. The amino group on the benzene ring of C283 was confirmed by the mass difference of 91 units between the product ion at m/z 283.82 and that at m/z 192.70.
The intermediate C162 detected in negative ion mode was proposed to result from the loss of the benzene ring (together with the amino group) from the parent compound.
The intermediate C98 was proposed to result from the bond cleavage of S–N between sulfonyl and benzene ring. It was also detected in SMX degradation by treatment processes including Fe(II) + persulfate49 and UV + H2O2.18
The species C294, with m/z 293.08 in negative ion mode, differing by 40 mass units from the SMX molecular, was attributed to the addition of two hydroxyl groups, as well as the oxidation of the benzene ring. The product ion at m/z 201.68, corresponding to loss of 92 units from the product ion at m/z 293.08, was obtained from the loss of the isoxazole and one amino group. The product ions at m/z 235.87 in the MS2 spectra, with variation of around 57 mass units from m/z 293.08, was proposed to be –CH2COOH group formed from the oxidation of the benzene ring.
The MS data which shows the fragment ion at m/z 344.08 in negative ion mode suggests the introduction of groups with mass 92 based on the parent compound (m/z 253.83). The fragment at m/z 313.04, with difference value of 64 units m/z from the fragment ion at m/z 247.79 shown from MS2 data, indicates four hydroxyl groups. And the remaining mass difference of 28 indicates the addition of –CO group. Therefore, the intermediate C345 comes from the introduction of four hydroxyl groups, as well as the transformation of –NH2 to –NH2CO on the benzene ring. The latter one may be obtained from the combination of the –NH2 group with the –HCOOH group from the formic acid, a common product formed during AOPs.
According to the proposed intermediates, carboxylation, hydroxylation, isoxazole ring opening, loss of benzene ring, N–S cleavage and dehydration condensation were illustrated as SMX degradation pathways under the process UVA + Fe(II) + PMS (Fig. 8a).
The peak areas of the detected intermediates from the chromatography were recorded in Fig. S3.† As the standards of these generated intermediates were not available from the market, it is impractical to provide the exact concentrations of each intermediates. According to the structures of the detected intermediates and the low TOC reduction, it was assumed that no isoxazole-ring was opened at the initial stage and hence the isoxazole-ring balance was followed during the reaction. Besides, the concentrations of the intermediates are assumed to be linear to the corresponding areas from the chromatographic peaks based on the Beer–Lambert law. Then the concentration of the detected intermediates may be proposed with the following equation.
 | (12) |
Six formulas were built up as the experiment data (ASMX and Aint at 5, 10, 15, 20, 25, and 30 min) were put into the above eqn (12). The unknown values of §SMX/§int for C283, C269, C162, C98, C294 and C345 are solved to be 0.67, 1.05, 1.52, 3.23, 1.32 and 3.47, respectively. Therefore, the concentration of the intermediate can be determined and the result was shown in Fig. 8b.
3.5 Toxicity of the treated solution to rotifers
The survival of the rotifers was threatened by SMX solution, only around 30% rotifers survived as exposed to 0.05 mM SMX. After treatment for 40 min, the parent compound was removed completely (at least the concentration was lower than the detection limitation of LC). Though only around 17% TOC reduction was reached, only two intermediates (C98 and C294 at around 0.002 mM) were detected at the 40th min of the treatment. Though higher toxicity of C98 was predicted by ECOSAR in previous studies by Yang et al., and Milh et al.,20,51 no adverse effect of C98 at 5.6 mM was detected by testing growth inhibition of Vibrio fischeri by Majewsky et al.52 In this study, the solution after treatment for 40 min showed no significant difference compared to that in control group (Fig. 9). Though the toxicity of C294 was unknown, the result indicates the mixture of C98 and C294 at around 0.002 mM was safe to the survival of rotifers. Taking into consideration of the residual 87% TOC, it can be predicted that the undetected low-molecular organic substances have no significant adverse effect on the survival of rotifers. Therefore, it is believed that the treatment process was efficient to transform the toxic SMX to harmfulless substances. | ||
| Fig. 9 The effect of SMX solution to rotifers before and after treatment. Treatment conditions: [SMX]0 = 0.05 mM, [Fe(II)]0 = 0.25 mM, [PMS]0 = 0.25 mM, [pH]0 = 6.09, two UVA lamps were employed. | ||
4. Conclusion
In this study, SMX was removed efficiently under the treatment process UVA + Fe(II) + PMS. The performance of the treatment process was affected by operating parameters such as the concentration of [Fe(II)], [PMS], and the pH level of solution. The optimum SMX degradation efficiency was observed in the pH level of around 6.0, with [Fe(II)]:[PMS] = 1:1. A model was established to predict the removal efficiency or reaction rate in terms of the initial concentration of SMX. Six transformation products and the corresponding concentrations were proposed. Around 17% TOC reduction was reached as SMX was removed completely. Carboxylation, hydroxylation, isoxazole ring opening, loss of benzene ring, N–S cleavage and dehydration condensation were involved as the degradation pathways. The radicals OH˙ and SO4˙− were involved and OH˙ contributed more to SMX degradation. The treated SMX showed no acute toxicity to the tested rotifers. This study provided an efficient method to remove SMX.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the National Natural Science Foundation of China (41807476), Science and Technology Program of Guangdong, China (2019A141405034) and Hong Kong Polytechnic University (1-ZVH6).References
- H. Xie, H. Hao, N. Xu, X. Liang, D. Gao, Y. Xu, Y. Gao, H. Tao and M. Wong, Sci. Total Environ., 2019, 659, 230–239 CrossRef CAS.
- M. Isidori, M. Lavorgna, A. Nardelli, L. Pascarella and A. Parrella, Sci. Total Environ., 2005, 346, 87–98 CrossRef CAS.
- G. G. Ying, L. Y. He, A. J. Ying, Q. Q. Zhang, Y. S. Liu and J. L. Zhao, Environ. Sci. Technol., 2017, 51, 1072–1073 CrossRef CAS.
- P. Verlicchi, M. Al Aukidy and E. Zambello, Sci. Total Environ., 2012, 429, 123–155 CrossRef CAS.
- A. Puckowski, K. Mioduszewska, P. Lukaszewicz, M. Borecka, M. Caban, J. Maszkowska and P. Stepnowski, J. Pharm. Biomed. Anal., 2016, 127, 232–255 CrossRef CAS.
- T. B. Minh, H. W. Leung, I. H. Loi, W. H. Chan, M. K. So, J. Q. Mao, D. Choi, J. C. Lam, G. Zheng, M. Martin, J. H. Lee, P. K. Lam and B. J. Richardson, Mar. Pollut. Bull., 2009, 58, 1052–1062 CrossRef CAS.
- A. L. Spongberg, J. D. Witter, J. Acuna, J. Vargas, M. Murillo, G. Umana, E. Gomez and G. Perez, Water Res., 2011, 45, 6709–6717 CrossRef CAS.
- M. J. Chen, Y. Huang and W. Chu, Chin. J. Catal., 2019, 40, 673–680 CrossRef CAS.
- L. Xu, Y. Sun, L. Gan, J. Han, P. Wang, L. Yu, X. Mei, W. Li, B. Lyu, C. Pei and W. Chu, Appl. Catal., B, 2019, 259, 117958 CrossRef CAS.
- M. J. Chen and W. Chu, Chem. Eng. J., 2016, 296, 310–318 CrossRef CAS.
- A. Hassani, A. Khataee, S. Karaca and M. Fathinia, RSC Adv., 2016, 6, 87569–87583 RSC.
- F. Ghanbari, M. Riahi, B. Kakavandi, X. Hong and K.-Y. A. Lin, J. Water Process. Eng., 2020, 36, 101321 CrossRef.
- F. Ghanbari, F. Zirrahi, K.-Y. A. Lin, B. Kakavandi and A. Hassani, J. Environ. Chem. Eng., 2020, 8, 104167 CrossRef CAS.
- F. Ghanbari, F. Zirrahi, D. Olfati, F. Gohari and A. Hassani, J. Mol. Liq., 2020, 310, 113217 CrossRef CAS.
- A. Hassani, M. Faraji and P. Eghbali, J. Photochem. Photobiol., A, 2020, 400, 112665 CrossRef CAS.
- A. Hassani, A. Khataee, M. Fathinia and S. Karaca, Process Saf. Environ. Prot., 2018, 116, 365–376 CrossRef CAS.
- P. Y. Motlagh, A. Khataee, A. Hassani and T. Sadeghi Rad, J. Mol. Liq., 2020, 302, 112532 CrossRef CAS.
- R. Zhang, Y. Yang, C. H. Huang, N. Li, H. Liu, L. Zhao and P. Sun, Environ. Sci. Technol., 2016, 50, 2573–2583 CrossRef CAS.
- H. W. Yu, T. Anumol, M. Park, I. Pepper, J. Scheideler and S. A. Snyder, Water Res., 2015, 81, 250–260 CrossRef CAS.
- Y. Yang, X. Lu, J. Jiang, J. Ma, G. Liu, Y. Cao, W. Liu, J. Li, S. Pang, X. Kong and C. Luo, Water Res., 2017, 118, 196–207 CrossRef CAS.
- H. Y. Kim, T. H. Kim and S. Yu, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50, 292–300 CrossRef CAS.
- W. Q. Guo, R. L. Yin, X. J. Zhou, J. S. Du, H. O. Cao, S. S. Yang and N. Q. Ren, Ultrason. Sonochem., 2015, 22, 182–187 CrossRef CAS.
- L. J. Xu, W. Chu and L. Gan, Chem. Eng. J., 2015, 263, 435–443 CrossRef CAS.
- L. Gan, Q. Zhong, A. Geng, L. Wang, C. Song, S. Han, J. Cui and L. Xu, Sci. Total Environ., 2019, 694, 133705 CrossRef CAS.
- L. Xu, X. Wang, Y. Sun, H. Gong, M. Guo, X. Zhang, L. Meng and L. Gan, Ultrason. Sonochem., 2019, 60, 104749 CrossRef.
- Y. Rao, F. Han, Q. Chen, D. Wang, D. Xue, H. Wang and S. Pu, Chemosphere, 2019, 218, 299–307 CrossRef CAS.
- Y. R. Wang and W. Chu, Chem. Eng. J., 2013, 215–216, 643–650 CrossRef CAS.
- L. Xu, L. Meng, X. Zhang, X. Mei, X. Guo, W. Li, P. Wang and L. Gan, J. Hazard. Mater., 2019, 379, 120795 CrossRef CAS.
- Y. R. Wang and W. Chu, Appl. Catal., B, 2012, 123–124, 151–161 CrossRef CAS.
- Y. Zhou, J. Jiang, Y. Gao, J. Ma, S. Y. Pang, J. Li, X. T. Lu and L. P. Yuan, Environ. Sci. Technol., 2015, 49, 12941–12950 CrossRef CAS.
- G. Liu, X. Li, B. Han, L. Chen, L. Zhu and L. C. Campos, J. Hazard. Mater., 2017, 322, 461–468 CrossRef CAS.
- Y. Ji, Y. Fan, K. Liu, D. Kong and J. Lu, Water Res., 2015, 87, 1–9 CrossRef CAS.
- Y. Ji, C. Ferronato, A. Salvador, X. Yang and J. M. Chovelon, Sci. Total Environ., 2014, 472, 800–808 CrossRef CAS.
- Y. Q. Gao, N. Y. Gao, Y. Deng, D. Q. Yin, Y. S. Zhang, W. L. Rong and S. D. Zhou, Desalin. Water Treat., 2014, 56, 2225–2233 CrossRef.
- E. Hayon, A. Treinin and J. Wilf, J. Am. Chem. Soc., 1972, 94, 47–57 CrossRef CAS.
- J. J. Pignatello, Environ. Sci. Technol., 1992, 26, 944–951 CrossRef CAS.
- Z. Wang, W. Qiu, S. Y. Pang, Y. Zhou, Y. Gao, C. Guan and J. Jiang, Chem. Eng. J., 2019, 371, 842–847 CrossRef CAS.
- J. Madhavan, P. Maruthamuthu, S. Murugesan and M. Ashokkumar, Appl. Catal., A, 2009, 35–39 CrossRef CAS.
- I. Sires, E. Guivarch, N. Oturan and M. A. Oturan, Chemosphere, 2008, 72, 592–600 CrossRef CAS.
- H. L. So, W. Chu and Y. H. Wang, Chemosphere, 2019, 218, 110–118 CrossRef CAS.
- J. W. T. Spinks and R. J. Woods, Introduction to radiation chemistry, Wiley Press, New York, 1990 Search PubMed.
- L. B. Donald and O. E. John, J. Am. Chem. Soc., 1956, 78, 1125–1129 CrossRef.
- A. Rastogi, S. R. Ai-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 85, 171–179 CrossRef CAS.
- V. Diaz, R. Ibanez, P. Gomez, A. M. Urtiaga and I. Ortiz, Water Res., 2011, 45, 125–134 CrossRef CAS.
- J. Gaffney Vde, V. V. Cardoso, M. J. Benoliel and C. M. Almeida, J Environ. Manag., 2016, 166, 466–477 CrossRef.
- Z. Zhou and J. Q. Jiang, J. Pharm. Biomed. Anal., 2015, 106, 37–45 CrossRef CAS.
- K. Zhang, Z. Luo, T. Zhang, N. Gao and Y. Ma, BioMed Res. Int., 2015, 2015, 169215 Search PubMed.
- D. Shahidi, A. Moheb, R. Abbas, S. Larouk, R. Roy and A. Azzouz, J. Hazard. Mater., 2015, 298, 338–350 CrossRef CAS.
- S. Wang and J. Wang, RSC Adv., 2017, 7, 48670–48677 RSC.
- J. M. Monteagudo, A. Durán, I. San Martín and P. Carrillo, Chem. Eng. J., 2019, 364, 257–268 CrossRef CAS.
- H. Milh, B. Schoenaers, A. Stesmans, D. Cabooter and R. Dewil, Chem. Eng. J., 2020, 379, 122234 CrossRef CAS.
- M. Majewsky, D. Wagner, M. Delay, S. Brase, V. Yargeau and H. Horn, Chem. Res. Toxicol., 2014, 27, 1821–1828 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05704e |
| This journal is © The Royal Society of Chemistry 2020 |