
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTLC-spectrodensitometric method for simultaneous determination of dapagliflozin and rosuvastatin in rabbit plasma: stability indicating assay and kinetic studies†
Noha S. Abbas *a,
Sayed M. Derayeab,
Mahmoud A. Omarbc and
Gamal A. Salehd
*a,
Sayed M. Derayeab,
Mahmoud A. Omarbc and
Gamal A. Salehd
aMinistry of Health and Population, Assiut, Egypt. E-mail: noha.abbas1987@gmail.com; noha.abbas2008@aun.edu.eg
bDepartment of Analytical Chemistry, Faculty of Pharmacy, Minia University, Egypt
cDepartment of Pharmacognosy and Pharmaceutical Chemistry, College of Pharmacy, Taibah University, Medinah, Saudi Arabia
dDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
First published on 9th November 2020
Abstract
Herein, a sensitive and reliable eco-friendly TLC-spectrodensitometric method has been established for the simultaneous determination of dapagliflozin (DAPA) and rosuvastatin (ROSV) for the first time. TLC separation was carried out on silica gel F254 using ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (5:0.1, v/v) as a mobile phase and UV measurement at 243 nm. The method was fully validated according to ICH guidelines. Acceptable separation was achieved with Rf values of 0.23 and 0.44 for DAPA and ROSV, respectively. Regression plots revealed linear relationships in the concentration range 20–2500 ng per band and 10–2500 ng per band with LODs of 6.60 and 3.57 ng per band for both DAPA and ROSV, respectively. The relative standard deviations (RSDs%) were found to be 1.35 and 0.53 for DAPA and ROSV, respectively. Moreover, kinetic studies were conducted for measurement of degradation rate constant (k) and half life time (t1/2) of DAPA and ROSV via forced photo-degradation.
:methanol (5:0.1, v/v) as a mobile phase and UV measurement at 243 nm. The method was fully validated according to ICH guidelines. Acceptable separation was achieved with Rf values of 0.23 and 0.44 for DAPA and ROSV, respectively. Regression plots revealed linear relationships in the concentration range 20–2500 ng per band and 10–2500 ng per band with LODs of 6.60 and 3.57 ng per band for both DAPA and ROSV, respectively. The relative standard deviations (RSDs%) were found to be 1.35 and 0.53 for DAPA and ROSV, respectively. Moreover, kinetic studies were conducted for measurement of degradation rate constant (k) and half life time (t1/2) of DAPA and ROSV via forced photo-degradation.
1. Introduction
Diabetes mellitus is a complex long-term metabolic disorder, which induces malfunctioning of cholesterol biosynthesis. As per the current American Diabetes Association guidelines, all adults with diabetes should be managed to achieve a low density lipoprotein (LDL) cholesterol less than 100 mg dL−1 employing statins for first-line therapy.1,2 As a result, a combination of oral hypoglycemic drugs and anti-hyperlipidemic agents e.g. statins is effective at decreasing cardiovascular problems. This combination has shown high efficacy and good recovery levels in patients treated with drugs. For examples, atorvastatin mainly used with glimepiride and metformin while simvastatin used with sitagliptin.3–5 Some hypoglycemic drugs (e.g. glibenclamide) when taken with statins have the potential to increase the maximum plasma concentration (Cmax) and the area under the curve (AUC) by up to 20%. Fortunately, a meta-analysis found that SGLT2 inhibitors may mildly increase in LDL and HDL levels, so co-administration with statins is suggested.6 Recently, many studies found that the low dose of rosuvastatin (ROSV) is more effective to reduce LDL than the high dose, and help treatment of low HDL in patients with type II diabetes and dyslipidemia than other statins.7–9 ROSV is preferred to co-administer with dapagliflozin (DAPA) where there is no pharmacokinetic interaction between them so, no need to dose adjustment.10In addition, the high-fat high-fructose diet (HFF) in obesity can favor lipid accumulation in liver and kidney, which related to insulin resistance and lipophilicity induced cellular damage. So, co-administration of dapagliflozin (DAPA) and rosuvastatin (ROSV) could improve the lipid-accumulation induced kidney and liver injury in HFF induced insulin resistance, lipogenesis and lipotoxicity-related renal oxidative stress, inflammation, fibrosis and apoptosis leading to kidney dysfunction recovery. Liver injury-associated inflammation was also improved by these two regimens. Notably, the reduced lipid accumulation in liver and kidney that linked to an improvement of lipid oxidation was prominent in the combination treatment.11
DAPA, a member of sodium-glucose transport protein 2 (SGLT2), has high ability to inhibit SGLT2 receptor leading to reduce glucose reabsorption. It involves in the direct elimination of glucose by the kidney (insulin-independent mechanism).
There are various methods for individual determination of DAPA and ROSV such as RP-HPLC,12,13 LC-MS/MS,14,15 spectrophotometry,16,17 spectrofluorometry,18,19 capillary electrophoresis,20,21 HPTLC22 and RP-UPLC/UV.23 DAPA and ROSV were co-administered in many mixtures such as DAPA–saxagliptin,24 DAPA–metformin,25 DAPA–glimepiride,26 ROSV–atorvastatin,27 ROSV–aspirin28 and ROSV–propranolol.29 To the best of our knowledge there is no analytical method for simultaneous determination of DAPA with ROSV.
Spectrodensitometric methods of analysis have many advantages such as simplicity, rapidity, sensitivity, and inexpensive tools. In comparison to HPLC method, TLC methods need small volumes of mobile phases that is considered economic point of view.30
Inspired by these reports, simple and reliable TLC method was developed for analysis of DAPA–ROSV binary mixture in standards, dosage forms and spiked rabbit plasma. Moreover, stability indicating and kinetic studies were applied using photo-degradation at different time intervals, and to calculate degradation rate constant (k) and half lifetime (t1/2).
2. Experimental
2.1. Instrumentation
The sample application was done in the form of bands using Camag 100 μL sample syringe (Hamilton, Bonaduz, Switzerland) applied by Camag Linomat V (Muttenz, Switzerland) sample automatic applicator and CAMAG TLC Scanner III operated using UV lamp short-wavelength 254 nm (Vilber Lournate 220 V 50 Hz, Marne-la-Vallee, Cedex, France). The absorption spectra were recorded over the wavelength range from 200 to 400 nm with slit dimensions of 3 × 0.45 mm and scanning speed of 20 mm s−1. The spectrophotometric measurements were carried out using a double beam UV-visible spectrophotometer (Shimadzu, Kyoto, Japan) model UV-1601 PC connected to an IBM compatible computer, with UV PC personal spectroscopy software version 3.7.2.2. Chemicals and reagents
DAPA and ROSV with purity of 99% were obtained as a gift from NODCAR, El-Dokki, Giza, Egypt. Methanol (HPLC grade) was purchased from Fisher, Loughborough, UK. Ethyl acetate was purchased from El-Nasr pharmaceutical, Chemical Co, Abu-Zabaal, Cairo, Egypt. All reagents and chemicals were of analytical grade. TLC plates (20 × 20 cm) pre-coated with silica gel F254 were purchased from Merck, Darmstadt, Germany. The standard solutions of DAPA and ROSV were prepared in methanol. FORXIGA® tablets were purchased from AstraZeneca for Pharmaceutical, Cairo-Egypt, and labeled to contain 10 mg of DAPA per tablet. ROSVVAST® tablets were purchased from Chemipharm for Pharmaceutical Industries, Cairo-Egypt, and labeled to contain 10 mg of ROSV per tablet.2.3. Preparation of standard solution and working solution
Stock solutions of 1.0 mg mL−1 of DAPA and ROSV were prepared in methanol. Different aliquots were diluted with methanol in 10.0 mL standard flasks to prepare concentration ranges of 20–500 μg mL−1 for DAPA and ROSV. A stock standard solution for forced degradation was prepared by dissolving 10 mg of DAPA and ROSV in 10 mL flask in methanol then further dilution with water to give the concentration of 200 μg mL−1 for DAPA and ROSV.2.4. Chromatographic conditions
Firstly, the silica gel G 60 F254 TLC plates were cut into 20 cm × 7 cm, 0.2 mm thickness was used as a stationary phase and prewashed with methanol.Then, the samples were spotted in the form of bands of width 4 mm with a Camag 100 μL sample syringe by sample automatic applicator. A constant application rate of 150 nL s−1 was used, and spots were spaced 10 mm from the bottom edge of the plate. For the application of samples by a spray-on technique, the application volume was optimized, and the actual amount used was 5.0 μL. The slit dimension was kept at 3 × 0.45 mm and 20 mm s−1 scanning speed were used. Ethyl acetate:methanol (5:0.1, v/v) as a mobile phase was developed over a distance of 65 cm of the plate in a closed container saturated with mobile phase for 30 min; the plates were completely dried. Spectrodensitometric scanning was carried out using Camag TLC scanner III in the reflectance/absorbance mode at 243 nm (the optimum wavelength for both). Data were integrated using WIN CATS Software. The development of mobile phase was carried out at room temperature (25 ± 2 °C).
2.5. Applications
2.6. Method validation
The proposed method was validated according to (ICH) guidelines31 including linearity, selectivity, precision, accuracy, and robustness.2.7. Forced degradation study
The stability indicating properties and specificity of the proposed method was assessed by applying forced degradation studies. In order to quantify drugs in the presence of their possible degrades; the test solution of DAPA and ROSV were exposed to stress conditions to generate their degradation products. Intentional degradation was performed according to the recommendation of regulatory guidelines (ICH Q1A, Q2A) for the necessity of developing and validating stability-indicating methods for the drugs.31,32 These guidelines were used to select the type of the stress conditions depending on the nature of individual analytes present. Forced degradation conditions of the drugs were based on the above ICH guidelines in addition to different previously reported researches.33–37The light effect is considered an important factor in the stability of drugs. Great problems of stability arise by the light of the shorter and longer wavelength, in particular from ultraviolet and visible.
Degradation of samples were prepared by dissolving 10 mg of DAPA and ROSV in methanol in 10 mL standard flasks and successive dilutions with water to prepare 200 μg mL−1 of DAPA and ROSV and then subjected to UV light at 365 nm. After exposing to UV light at the 6 time interval (30, 60, 90, 120, 150, 180 min), and take a sample after each stress time. After that, the solutions were allowed to cool at room temperature prior to application of the proposed spectrodensitometric method.
3. Results and discussion
3.1. Spectral analysis
The UV absorption spectra of the DAPA and ROSV demonstrate that the two drugs have maximum absorbance at 225 nm and 243 nm, respectively. To detect DAPA and ROSV simultaneously, λmax = 243 nm was used to measure the binary mixture (Fig. 1). Fig. 2 shows the comparison between 2D and 3D TLC spectrodensitograms of binary mixture at 225 nm, 243 nm and 286 nm. Moreover, the quantitative parameters for analysis of the binary mixture at different wavelengths were introduced in Table 1. It is seen that the lowest LODs for DAPA and ROSV were obtained at λ = 243 nm. | ||
| Fig. 1 UV spectrum of DAPA (500 ng per band) and ROSV (500 ng per band) in methanol as a solvent. | ||
 | ||
| Fig. 2 3D and 2D spectrodensitograms of 500 ng per band of DAPA and ROSV at 225 nm (A), 286 nm (B) and 243 nm (C), respectively in methanol. | ||
| Parameters | 243 nm | 225 nm | 286 nm | |||
|---|---|---|---|---|---|---|
| DAPA | ROSV | DAPA | ROSV | DAPA | ROSV | |
| a Average of three replicates. | ||||||
| Linearity range, (ng per band) | 20–2500 | 10–2500 | 200–2500 | 33–2500 | 130–2500 | 430–2500 |
| Limit of detection (LOD), (ng per band) | 6.60 | 3.57 | 66.44 | 11.09 | 390.76 | 142.70 |
| Regression equation | Y = 3.7457x + 892.94 | Y = 4.4748x + 1028.7 | y = 3.7319x + 1019 | y = 4.4637x + 598.14 | y = 3.7126x + 974.93 | y = 4.4547x + 1864.3 |
| Intercept (a) ± SDa | 892.94 ± 7.50 | 1028.66 ± 4.84 | 1018.97 ± 75.14 | 727.20 ± 14.75 | 974.92 ± 145.07 | 1670.89 ± 192.63 |
| Slope (b) ± SDa | 3.75 ± 0.05 | 4.47 ± 0.02 | 3.73 ± 0.08 | 4.38 ± 0.04 | 3.71 ± 0.10 | 4.45 ± 0.27 |
| Correlation coefficient (r) | 0.9828 | 0.9819 | 0.9729 | 0.9817 | 0.9790 | 0.9810 |
3.2. Optimization of variables
Different mobile systems of different compositions and ratios, and different saturation times were tried for separation of binary mixture. The selection of mobile phase based on well separated peaks, symmetric bands and reasonable Rf values (Table S1†). The optimum mobile phase composition was found to be ethyl acetate:methanol with ratio of 5:0.1 v/v (Fig. 3). Different saturation times were tested (15–45 min.) and it was found that the best saturation time with low % RSD for Rf values at 30 min. After this time there is no significant change in the Rf obtained as indicated in Table S2.† The choice of the best saturation time was based on the least time gave the best separation between two bands and the most robust results of Rf as represented by % RSD.
 | ||
| Fig. 3 Representative 2D spectrodensitogram for separation of DAPA and ROSV using ethyl acetate:methanol with ratio of 5:0.1 v/v using saturation time of 30 min. | ||
3.3. Validation
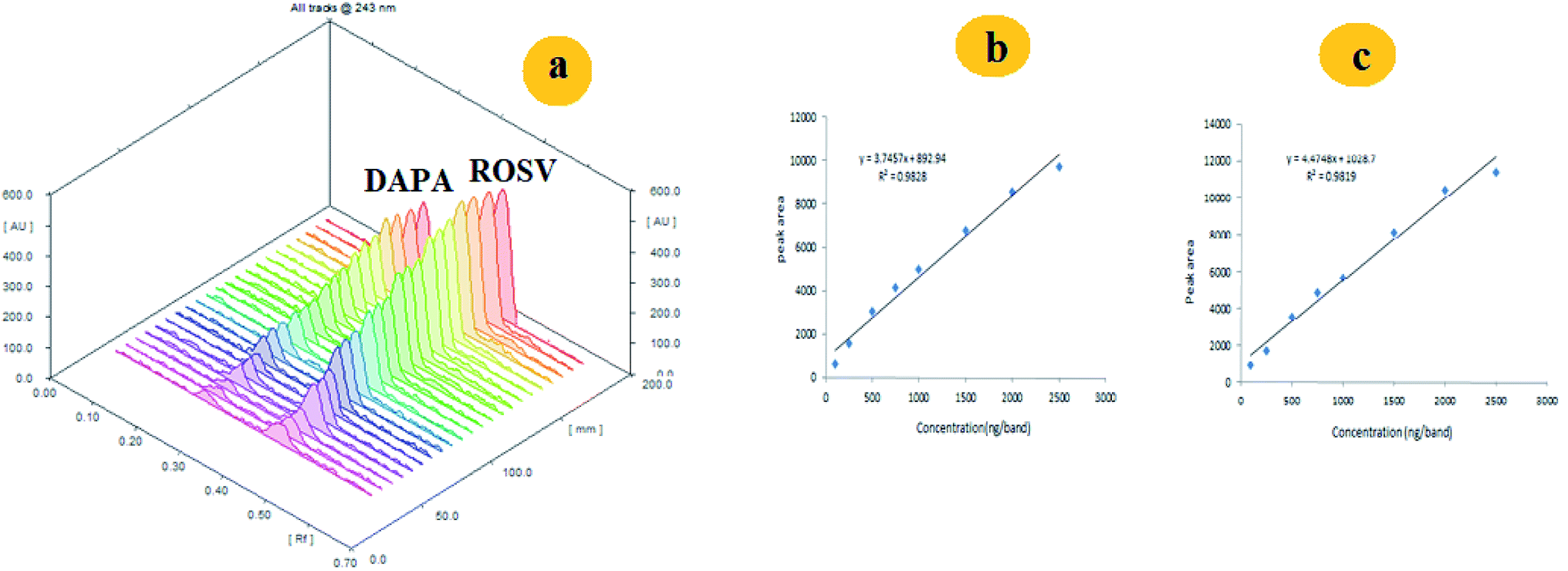 | ||
| Fig. 4 (a) Representative 3D spectrodensitogram, (b) and (c) are calibration plots for separation of DAPA and ROSV, respectively using ethyl acetate:methanol with ratio of 5:0.1 v/v using saturation time of 30 min. | ||
3.4. Applications of the TLC method
The proposed method was applied for analysis of DAPA and ROSV in their tablet dosage forms. Three replicates were made in each one at three concentration levels and the recovery% was found to be in the range of 98.2–100.0%. The results were compared with those obtained by the reported methods38,39 as shown in Table 2. After applying t- and F-tests at a 95% confidence level indicating (p = 0.05), there are no significant differences between the investigated and reported methods, indicating the accuracy and precision of the method. The standard addition method was also applied by using three different concentration levels of the standard at 50%, 100% and 150% of the target concentration 1000 ng per band of DAPA and 500 ng per band of ROSV in triplicate and calculating the recoveries% (Table S5†).| Pharmaceutical tablets | Amount of sample taken | Proposed method | Reported method38,39 | F- and t- valuesb |
|---|---|---|---|---|
| Recovery (%) ± SDa | Recovery (%) ± SDa | |||
| a Average of three determinations.b Theoretical values for t- and F-test at 95% confidence limit (n = 3) were 3.18 and 19, respectively. | ||||
| FORXIGA® | 250 | 98.5 ± 4.58 | 98.6 ± 3.28 | F = 1.80 |
| 750 | 100.4 ± 4.46 | 100.1 ± 2.16 | ||
| 1500 | 98.2 ± 2.12 | 98.4 ± 1.22 | t = 1.50 | |
| ROSUVAST® | 250 | 99.9 ± 4.80 | 99.5 ± 3.25 | F = 2.78 |
| 750 | 100.1 ± 4.65 | 99.9 ± 2.24 | ||
| 1500 | 98.3 ± 2.23 | 98.5 ± 1.56 | t = 1.58 | |
The method is applied also for the determination of DAPA and ROSV in rabbit plasma, extraction recovery of DAPA and ROSV was calculated and it ranged from 98.3 to 100.1% (for DAPA) and from 98.5 to 100.2% (for ROSV). These results indicate that the effect of the plasma matrix on the studied drug extraction was (Table 3). It is worth mentioning that due to low sensitivity of UV detection (in μg level), this do not allow the determination of DAPA and ROSV metabolites (identified in ng levels). Fig. 5 shows the plasma and urine spectrodensitograms after oral administration of binary mixture where no metabolites peaks were observed.
| Drug | Amount added (ng per band) | Amount found (ng per band) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| DAPA | 100 | 98.33 | 98.3 | 2.52 |
| 500 | 499.81 | 99.9 | 1.92 | |
| 1500 | 1489.30 | 99.2 | 2.11 | |
| 2000 | 2003.12 | 100.1 | 0.95 | |
| 2500 | 1476.56 | 98.4 | 3.21 | |
| ROSV | 100 | 99.54 | 99.5 | 3.63 |
| 500 | 492.89 | 98.5 | 1.67 | |
| 1500 | 1487.23 | 99.1 | 1.20 | |
| 2000 | 1998.03 | 99.9 | 2.32 | |
| 2500 | 2505.21 | 100.2 | 2.54 |
 | ||
| Fig. 5 Representative 2D spectrodensitogram of (a) plasma and (b) urine samples after oral administration of DAPA and ROSV. | ||
3.5. Specificity
The comparative spectra of standards and samples for DAPA and ROSV at the peak start (s), peak apex (m) and peak-end (e) positions of the band were investigated, the values were found to be very close to 1, which indicate the closeness in these positions between dosage forms and standards., as shown in Table S6,† which indicates that all peaks from different peak sites were attributable to the investigated drugs. The recovery obtained upon dividing the response of samples by that of standards using the same concentration level 1000 ng per band (DAPA) and 500 ng per band (ROSV) for both sample and standard by applying standard addition method, were in the ranges of 99.95 ± 0.25% to 100.01 ± 0.16%, which indicates suitable peak purity and selectivity. Moreover, the appearance of DAPA and ROSV spots at specific Rf different from each other and from their degrades indicates the specificity of the proposed method. Developed spectrodensitograms were observed for resolution of degraded products and spots were assessed for peak purity (Fig. 6). | ||
| Fig. 6 Specificity of DAPA and ROSV at concentration (1000 ng per band) for DAPA and (500 ng per band) for ROSV. Mobile phase is ethyl acetate:methanol with ratio of 5:0.1 v/v and saturation time of 30 min. | ||
3.6. Kinetic study
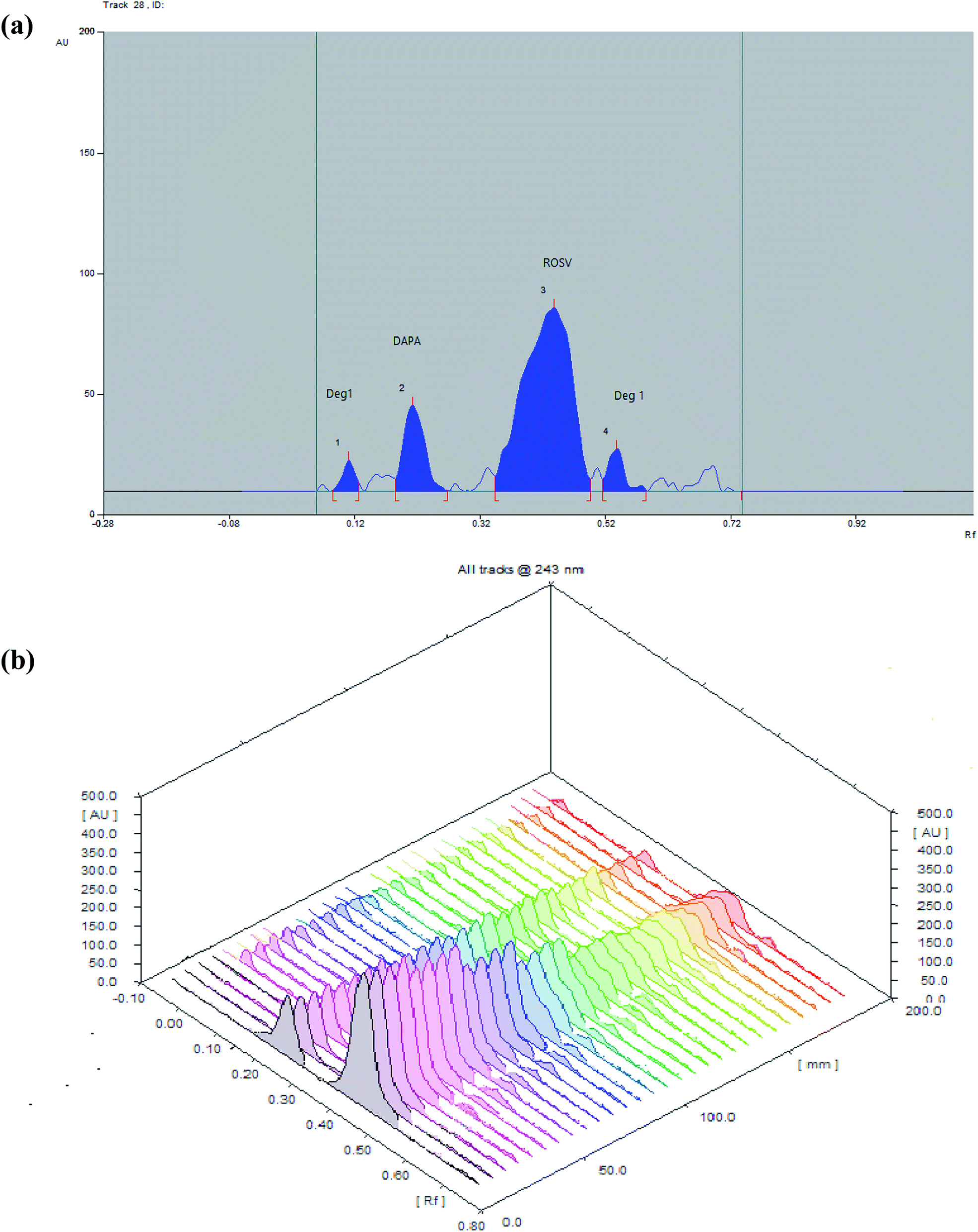 | ||
| Fig. 7 (a) 2D and (b) 3D spectrodensitograms for photodegradation of DAPA and ROSV (1000 ng per band) at (0, 30, 60, 90, 120, 150, 180 min). | ||
| Time (min) | DAPA | ROSV | ||
|---|---|---|---|---|
| Content remained (ng per band) ± SDa | % recovery | Content remained (ng per band) ± SDa | % recovery | |
| a Average of three determinations. | ||||
| 0 | 1000.00 ± 1.06 | 100.0 | 1000.00 ± 0.36 | 100.0 |
| 30 | 840.77 ± 1.31 | 84.1 | 802.16 ± 0.33 | 80.2 |
| 60 | 700.11 ± 1.24 | 70.0 | 520.43 ± 0.45 | 52.0 |
| 90 | 620.70 ± 0.52 | 62.1 | 400.37 ± 1.72 | 40.0 |
| 120 | 534.18 ± 1.66 | 53.4 | 275.42 ± 1.65 | 27.5 |
| 150 | 240.77 ± 0.61 | 24.1 | 153.98 ± 1.54 | 15.4 |
| 180 | 190.03 ± 0.74 | 19.0 | 113.94 ± 0.55 | 11.4 |
During stressed degradation experiments, it was noticed that DAPA and ROSV were more sensitive toward photolysis in water solution than in methanol as, the DAPA is relatively stable in pure methanol,40,41 and a mixture of a buffer with organic solvent42 while ROSV is more degradable in pure water than other organic solvent43,44 at 365 nm, and found that degradation of DAPA depends on the time, but ROSV depends on initial concentration at the same conditions.
The degradation rate constant (k) and the regression coefficient (r2) of the two drugs calculated from Table 4. From this data can conclude that there is a linear relationship between remained concentration and time in case of DAPA which suggests zero-order reaction mechanism for degradation of DAPA and linear relationship between logarithm of concentration and time in case of ROSV which suggests first-order reaction mechanism for the degradation of ROSV which is similar to the previous reports, as the regression coefficient values were 0.9899, 0.9882 for DAPA and ROSV, respectively.
The zero-order rate constant (Kobs) for the degradation of DAPA was obtained from the slopes of the linear plots of concentration vs. time in min (Kobs = −slope). It was determined at 4.52 min−1 and the t1/2 obtained from rate constant (Kobs) value equal (t1/2 = a/(2 × Kobs)) was determined at 110.6 min, while the first order rate constant for the degradation of ROSV obtained from the slope of log concentration versus time in min (K = −slope × 2.303). It was determined at 5.4 min−1 and t1/2 obtained from rate constant K(t1/2 = 0.693/k) was determined at 128.3 min.
3.7. Mechanism of degradation
Devrukhakar and Shankar have reported degradation pathways for DAPA under different stressed conditions. These conditions included acid, alkaline and neutral degradation using 2 N HCl, 2 N NaOH and H2O, respectively at 60 °C reflux for one day.45 Moreover, they identified the degrades of DAPA after exposure to heat at 50 °C for one day, and 3% H2O2 for 5 days at ambient condition. Many studies proved the stability of DAPA in methanol under UV light at (254 nm).40,41 In this paper, our study was based on the exposure of DAPA to UV light at (365 nm) for 180 min in presence of neutral conditions. Exposure of DAPA to UV light and water resulted in one degradation product, probably of low polarity than DAPA, by certain pathway (Fig. 7), which needs specific tools for its identification such as 1H-NMR, 13C-NMR and MS. Our future studies will be focused on the elucidation of DAPA degradation pathways under UV light and/or in presence of neutral conditions.Scheme 1 shows the photo-degradation of ROSV where stains are very sensitive to light and easily decomposed to form different types of degradation products.46,47 Photo-degradation of ROSV yields diastereomeric dihydrophenanthrenes, which are more polar than ROSV so that they were eluted after ROSV peak (Fig. 5a). In a first step ROSV is cyclized by the action of irradiation to yield unstable diastereomers 8a,9-dihydrophenanthrenes 1a and 1b, characterized with two novel chiral centers. Due to loss of aromaticity, these intermediates are unstable and undergo thermal intramolecular sigmatropic 1,5-hydrogen shift to yield stable dihydrophenanthrenes (R,S,R)-2 and (S,S,R)-2.
 | ||
| Scheme 1 The proposed mechanism of photo-degradation of ROSV in aqueous medium at 365 nm. | ||
3.8. Stability
The time may affect the stability of samples in solution when they are left in the solvent before chromatographic development. For this purpose, solutions of DAPA and ROSV were prepared and stored in dark at room temperature for 24 h. Then, the spectrodensitograms were analyzed for the appearance of any additional bands or changes in peak areas or positions. The average of the % recovery obtained for methanolic solutions and aqueous solutions was 98.7% and 94.5% for DAPA and 99.3% and 93.2% for ROSV, respectively. This test proves the stability of DAPA and ROSV in methanolic solution rather than the aqueous solutions. Apparently, this may be the cause of solid formulations (tablets) are preferred over the aqueous formulations.4. Conclusion(s)
TLC spectrodensitometric method was developed using ethyl acetate:methanol (5:0.1 v/v) as mobile phase for simultaneous separation and determination of DAPA and ROSV in bulk, dosage forms and rabbit plasma for the first time. The method offers high selectivity and sensitivity with acceptable reproducibility. Regression plots revealed linear relationships in the concentration range 20–2500 ng per band and 10–2500 ng per band with LODs of 6.60 and 3.57 ng per band for both DAPA and ROSV, respectively. The forced degradation was carried out according to ICH guidelines which indicate that DAPA and ROSV undergo degradation to a different extent under UV light. The investigated TLC spectrodensitometric method was capable of resolving analytes in the presence of their degradation products. From the peak purity profile studies, it was found that the peaks of the analytes were not interfered with the degradation products.
Conflicts of interest
The author declares that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- A. D. Association, Diabetes Care, 2003, 26(Suppl 1), S33–S50 Search PubMed.
- D. Sangeetha and M. Vadlamudi, Indian J. Pharm. Sci., 2019, 81, 365–372 CAS.
- S. R. Polagani, N. R. Pilli, R. Gajula and V. Gandu, J. Pharm. Anal., 2013, 3, 9–19 CrossRef CAS.
- M. Rahman and M. Begum, Am. J. Pharmacol. Toxicol., 2016, 10, 1–7 Search PubMed.
- H. M. Lotfy, M. A. M. Hegazy and S. A. N. Abdel-Gawad, Eur. J. Chem., 2013, 4, 414–421 CrossRef CAS.
- H. E. Bays, P. Sartipy, J. Xu, C. D. Sjöström and J. A. Underberg, J. Clin. Lipidol., 2017, 11, 450–458 e451 CrossRef.
- T. Katabami, M. Murakami, S. Kobayashi, T. Matsui, M. Ujihara, S. Takagi, M. Higa, T. Ichijo, A. Ohta and Y. Tanaka, J. Int. Med. Res., 2014, 42, 457–467 CrossRef.
- J. Tuomilehto, L. Leiter and D. Kallend, Int. J. Clin. Pract. Symp. Suppl., 2004, 58, 30–40 CrossRef.
- J. Tuomilehto, L. Leiter and D. Kallend, Int. J. Clin. Pract., Suppl., 2004, 58, 30–40 CrossRef.
- S. Kasichayanula, M. Chang, X. Liu, W. C. Shyu, S. C. Griffen, F. P. LaCreta and D. W. Boulton, Adv. Ther., 2012, 29, 163–177 CrossRef CAS.
- L. Thongnak, V. Chatsudthipong, A. Kongkaew and A. Lungkaphin, Toxicol. Appl. Pharmacol., 2020, 396, 114997 CrossRef CAS.
- S. Manasa, K. Dhanalakshmi, G. N. Reddy and S. Sreenivasa, Int. J. Pharm. Sci. Drug Res., 2014, 6, 250–252 CAS.
- T. R. Kumar, N. R. Shitut, P. K. Kumar, M. C. Vinu, V. V. Kumar, R. Mullangi and N. R. Srinivas, Biomed. Chromatogr., 2006, 20, 881–887 CrossRef CAS.
- Q. C. Ji, X. Xu, E. Ma, J. Liu, S. Basdeo, G. Liu, W. Mylott, D. W. Boulton, J. X. Shen, B. Stouffer, A. F. Aubry and M. E. Arnold, Anal. Chem., 2015, 87, 3247–3254 CrossRef CAS.
- D. Zhang, J. Zhang, X. Liu, C. Wei, R. Zhang, H. Song, H. Yao, G. Yuan, B. Wang and R. Guo, J. Pharm. Pharmacol., 2011, 2, 341–346 CrossRef CAS.
- G. V. Mante, K. R. Gupta and A. T. Hemke, Pharm. Methods, 2017, 8, 102–107 CrossRef CAS.
- R. Abdul Aziz, M. Hasna and A.-a. Rafif, Int. J. Pharmacol. Pharm. Sci., 2015, 7, 191–198 Search PubMed.
- M. A. Omar, H. M. Ahmed, M. A. Abdel Hamid and H. A. Batakoushy, J. Lumin., 2019, 1–9 Search PubMed.
- V. Braga, T. Mancilha, R. Cassella and W. Pacheco, J. Fluoresc., 2012, 23, 49–55 CrossRef.
- H. M. Maher, A. E. Abdelrahman, N. Z. Alzoman and H. I. Aljohar, J. Liq. Chromatogr. Relat. Technol., 2019, 42, 161–171 CrossRef CAS.
- İ. Süslü, M. Celebier and S. Altınöz, Chromatographia, 2007, 66, 65–72 CrossRef.
- S. S. K. R. T. Sane, S. N. Menon, S. R. Inamdar and M. R. Mote, J. Phys. Chem. C, 2005, 18, 194–198 Search PubMed.
- H. Trivedi and M. Patel, Sci. Pharm., 2012, 80, 393–406 CrossRef CAS.
- C. P. Desai, A. B. Chaudhary and B. D. Patel, World J. Pharm. Sci., 2018, 7, 803–812 CAS.
- T. Deepan, M. V. B. Rao and M. D. Dhanaraju, IDOS, 2017, 9, 189–199 CAS.
- A. Patel and D. D. maheshwari, EJPMR, 2017, 4, 416–434 Search PubMed.
- Y. Shah, Z. Iqbal, L. Ahmad, A. Khan, M. I. Khan, S. Nazir and F. Nasir, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 557–563 CrossRef CAS.
- J. Gawad, S. Bari and G. Sugandhi, Int. J. Pharm. Biol. Sci., 2013, 3, 173–184 CAS.
- N. M. El-Abasawi, K. A. M. Attia, A. A. M. Abo-Serie, S. Morshedy and A. Abdel-Fattah, Spectrochim. Acta, Part A, 2018, 198, 322–330 CrossRef CAS.
- L. Cai, Current protocols, 2014, 2014, 6.3.1–6.3.18 Search PubMed.
- I. H. T. GUIDELINE, International Conference on Harmonization, 2005, pp. 1–17 Search PubMed.
- I. C. H. GUIDELINES, International Conference on Harmonization, 2003, pp. 1–16 Search PubMed.
- G. Chakarvarty, N. Seth and V. Sharma, Int. Res. J. Pharm., 2013, 4, 36–39 CrossRef.
- I. C. H. GUIDELINE, International Conference on Harmonization, 1996, vol. 4, pp. 1–15 Search PubMed.
- B. Prathap, G. H. S. Rao, G. Devdass, A. Dey and N. Harikrishnan, Int. J. Innovative Pharm. Sci. Res., 2012, 3, 229–237 Search PubMed.
- Y. Rini, S. Iyan and M. Muchtaridi, Int. J. Appl. Pharm., 2018, 10, 38–42 CrossRef.
- B. A. Moussa, M. A. Mahrouse and M. G. Fawzy, Spectrochim. Acta, Part A, 2018, 1, 15–29 Search PubMed.
- K. Priya Chitra, M. C. Eswaraiah and M. V. B. Rao, J. Chem. Pharm. Res., 2015, 7, 45–49 Search PubMed.
- B. Uyar, M. Celebier and S. Altinoz, Pharmazie, 2007, 62, 411–414 CAS.
- H. M. Ahmed, M. A. Omar, H. A. Batakoushy and M. A. Abdel Hamid, Microchem. J., 2019, 1–21 Search PubMed.
- S. Sura, R. R. Modalavalasa and C. B. Kothapalli, Der Pharma Chemica, 2018, 10, 93–102 CAS.
- N. Singh, P. Bansal, M. Maithani and Y. Chauhan, NewJ.Chem., 2018, 42, 2459–2466 RSC.
- F. Belal, F. Ibrahim, A. Khedr and T. Elawady, J. Liq. Chromatogr. Relat. Technol., 2014, 37, 1114–1132 CrossRef CAS.
- T. C. Machado, T. M. Pizzolato, A. Arenzon, J. Segalin and M. A. Lansarin, Sci. Total Environ., 2015, 502, 571–577 CrossRef CAS.
- P. S. Devrukhakar and M. S. Shankar, Chromatographia, 2020, 83, 1233–1245 CrossRef CAS.
- M. Litvić, K. Smic, V. Vinković and M. Filipan-Litvić, J. Photochem. Photobiol., A, 2013, 252, 84–92 CrossRef.
- A. Astarita, M. DellaGreca, M. R. Iesce, S. Montanaro, L. Previtera and F. Temussi, J. Photochem. Photobiol., A, 2007, 187, 263–268 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05628f |
| This journal is © The Royal Society of Chemistry 2020 |