
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,4-Dithiolbenzene, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl molecular systems: electronic devices with possible applications in molecular electronics
J. H. Ojeda *ab,
Lina K. Piracón Muñozb,
Julian A. Guerra Pinzónb and
Jovanny A. Gómez Castañob
*ab,
Lina K. Piracón Muñozb,
Julian A. Guerra Pinzónb and
Jovanny A. Gómez Castañob
aGrupo de Física de Materiales, Universidad Pedagógica y Tecnológica de Colombia, Tunja, Colombia. E-mail: judith.ojeda@uptc.edu.co
bLaboratorio de Química Teórica y Computacional, Grupo de Investigación Química-Física Molecular y Modelamiento Computacional (QUIMOL), Facultad de Ciencias, Universidad Pedagógica y Tecnológica de Colombia, Tunja, Boyacá, Colombia
First published on 28th August 2020
Abstract
A theoretical study of the electronic transport properties of the 1,4-dithiolbenzene, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl molecules coupled to two metal contacts is carried out. The tight binding Hamiltonian approximation is applied to describe each of the molecular systems using the real space renormalization analytical method. Using Green's functions with the Landauer formalism, the transmission probability, current, shot noise and Fano factor of these three systems are calculated and analyzed in order to identify their behavior as insulators, semiconductors, or conductors, and their possible applications, such as quantum wires. The theoretical results are compared with experimental results that have been reported in the literature. The results indicate a high concordance between the results obtained by the proposed method and the experimental results.
1 Introduction
Molecular electronics has had an enormous impact on the development of industry and technology during the last decades, establishing itself as an interdisciplinary branch that involves sciences such as chemistry, physics, materials science and other areas of the molecular domain, which are directed to the use of individual molecules or small groups of molecules (molecular aggregates) as fundamental units.1–4With these fundamental units, colloids, polymers, supramolecular systems, quantum dots, quantum wires, and nanotubes, among others, can be built for use in the design and development of molecular systems with specific structural and electronic properties that fulfill morphological, self-assembling and electronic transport in a controlled way, especially when connected to electrodes, allowing new devices (switches, capacitors, rectifiers, storage devices, to name a few) to become possible candidates for the application of molecular electronics.2,5–10 These unions combine the fundamental processes of intramolecular and intermolecular transfer of electrons through molecular and continuous levels with the electrodes and processes outside the equilibrium of the currents generated by a voltage.11
The aromatic molecules that behave like quantum wires are of great interest since they contain a high degree of conjugation from the delocalization of electrons through their π bonds, which provides them with a high electron transfer.12,13 In particular, the thiol groups (R–SH) have high electronic density and high affinity to bond with metal electrodes. In addition, these groups have the ability to self-assemble when they are in contact with metal surfaces, forming stable covalent bonds.14 According to theoretical and experimental studies, the addition of thiol groups to aromatic molecules causes a decrease in the width of the prohibited band, which generates a better electron transfer.15,16 The HOMO–LUMO gap in this type of sulfur aromatic compounds ranges from 2 eV to 4 eV, similar to the width of the gap of an ordinary semiconductor.17
In terms of the molecular orbital (MO) theory, the aromatic molecules replaced by –SH groups have bonding and anti-bonding states, formed by the p orbitals of carbon and sulfur perpendicular to the plane of the benzene ring (which are responsible for the majority of the electric current that passes through these molecules), and σ bonds due to the presence of a nodal plane between these two atoms.17,18 Complementary to this, the sulfur atom generates high polarizability in conjugated molecular systems stabilizing the cationic species in various oxidation states and therefore, providing greater ease of transport of electric charge through such systems.
Self-assembled monolayers (SAM) of aromatic dithiols have received special attention due to the possibility of using them as connectors between metal or semiconductors contacts.19 For this reason, it is of particular interest to study their electronic behavior, employing three systems of molecules with these characteristics, which are shown in Fig. 1.
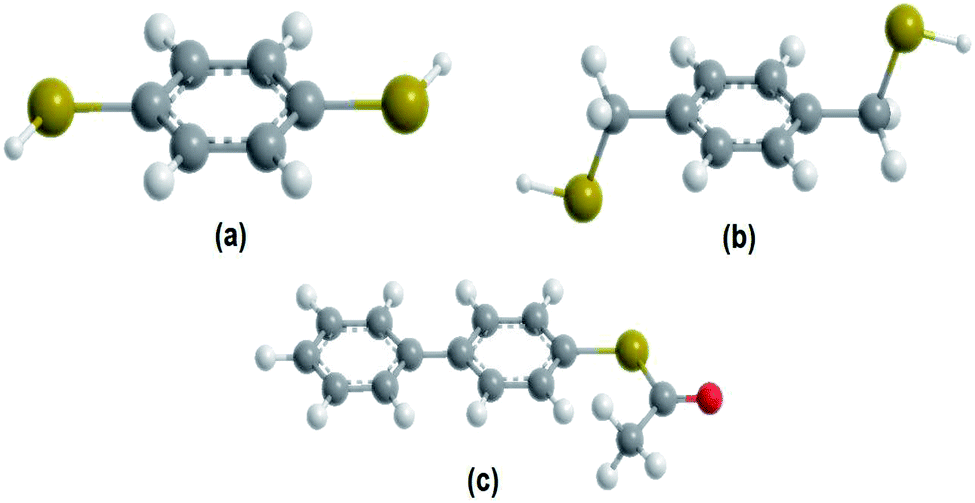 | ||
| Fig. 1 Molecular representation (a) 1,4-dithiolbenzene, (b) 1,4-dimethanethiolbenzene and (c) 4-thioacetylbiphenyl. | ||
In particular, fused aromatic systems and π-polyconjugated systems as the 1,4-dithiolbenzene, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl molecular systems, which we will study here as molecular wires are prototypes that attract attention due to their useful electrical properties, which not only give stability to the structural skeleton (generated by the assembly of the macromolecule), but which could also be the molecular basis for intra and inter-molecular transport of electrons. In this sense, a large number of experiments have been carried out with the main objective of extracting conclusive information about the transfer properties of electrical charge of this category of molecules.
To investigate the electrical effects on the joints of the molecular systems, we first derive an exact expression of the probability of transmission  , using this result we calculate the electric current
, using this result we calculate the electric current  depending of the molecule-contact coupling (Γ). Finally we calculate the shot noise
depending of the molecule-contact coupling (Γ). Finally we calculate the shot noise  and Fano factor
and Fano factor  .
.
2 Model
The 1,4-dithiolbenzene molecule (Fig. 1a) has at its ends two thiol groups which can be strongly bound to each electrode, generating an efficient electrical conductance.20 The current and conductance for this system was measured experimentally by Reed et al.,21 were able to restrict the number of active molecules between two statically stable gold electrodes through the mechanically controlled break-junction (MCBJ) technique.The second molecular system studied here corresponds to 1,4-dimethanethiolbenzene (Fig. 1b), from which monolayers can be prepared experimentally through self-assembling processes.22 Dorogi et al. measured the current and conductance for self-assembled monolayers of this molecule using gold clusters and deposition in vacuum at room temperature. Using a scanning tunneling microscope (STM), it was possible to obtain images of the clusters, with which the I/V profiles of the system were obtained.
The 4-thioacetylbiphenyl molecule (Fig. 1c) is the third molecular system studied in the present work and differs from the other molecules in that it has a geometric asymmetry caused by a single thioacetyl group located at carbon atom with respect to the C–C bond of the biphenyl. This asymmetry makes the profile of the transmission probability different when the charge carriers go from the left lead to the right lead than it is when the charge carriers go from the right lead to the left lead.23
The behavior of this molecule was studied experimentally, generating highly organized self-assembled monolayers by nanodeposition and lithography, where the acetyl group was removed from the molecule taking into account that the interface of Au–S is much more stable than the junction with another atom of high electron density (such as oxygen from the acetyl group).21,24
The three molecular systems analyzed in this work are modeled through the tight binding (TB) Hamiltonian, which is characterized by enabling a special approach to calculating the structure of the electronic bands in systems of low dimensionality.25 The TB Hamiltonian uses a basis for the wave functions, allowing studying and analyzing the electronic properties that have or lack translational symmetry. This Hamiltonian assumes that the valence electrons of the atoms of the molecular system remain strongly linked to its atomic nucleus, generating an atomic component plus a small perturbation due to the interaction of the electronic cloud.
Thus, the three systems are modeled through the TB Hamiltonian given by the equation:
| H = Ham + Hml + Hι, | (1) |
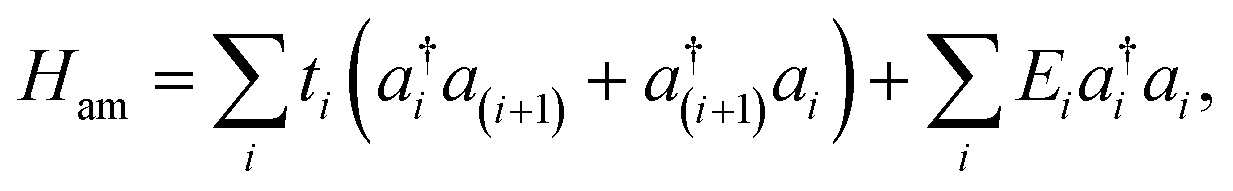 | (2) |
On the other hand, the Hamiltonian of the metal leads and the molecule–metal lead interaction Hml (left–right) and Hι, are given respectively by
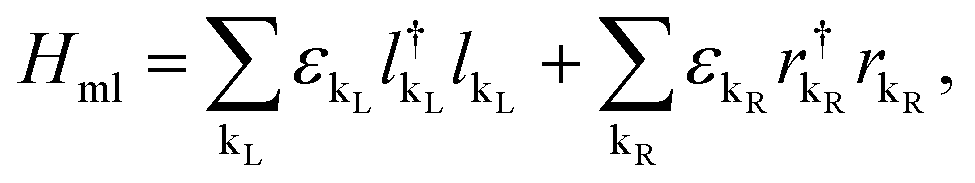 | (3) |
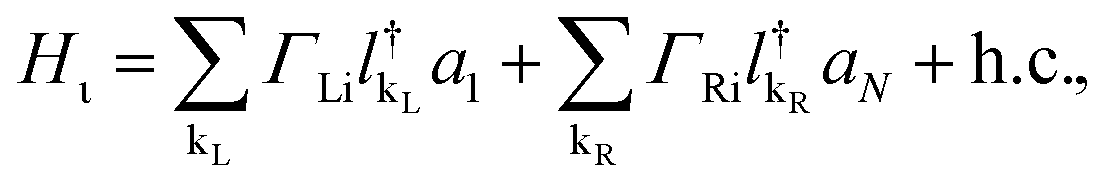 | (4) |
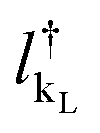 and
and 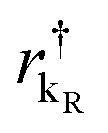 are the creation operators of an electron in the state kL,R with energy εkL,R, while ΓL(R)i represents the coupling between each metallic contact and the molecular system, and h.c. is the conjugate complex Hamiltonian.
are the creation operators of an electron in the state kL,R with energy εkL,R, while ΓL(R)i represents the coupling between each metallic contact and the molecular system, and h.c. is the conjugate complex Hamiltonian.
3 Method
3.1 Green's functions and dynamic equation
Starting from the time-independent Schrödinger equation for a single particle| [E − H(r)]Ψ(r) = 0, | (5) |
| [E − H(r)]G(r,r′;E) = δ(r − r′), | (6) |
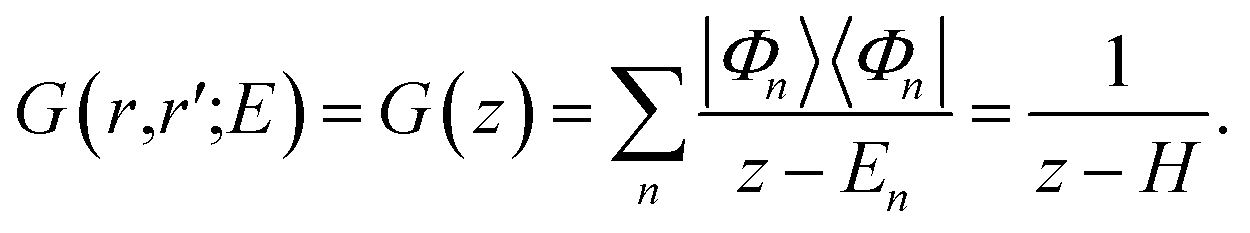 | (7) |
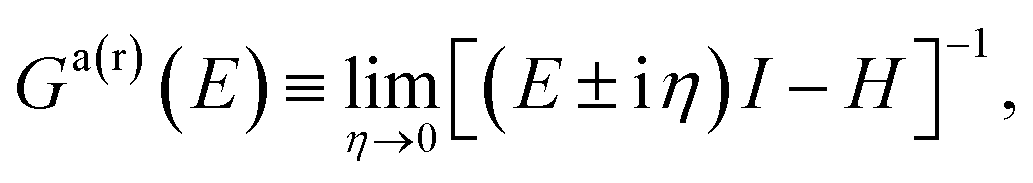 | (8) |
The Hamiltonian H of the given system given in eqn (7) and (8) can be expressed as
| H = H0 + Σ(E), | (9) |
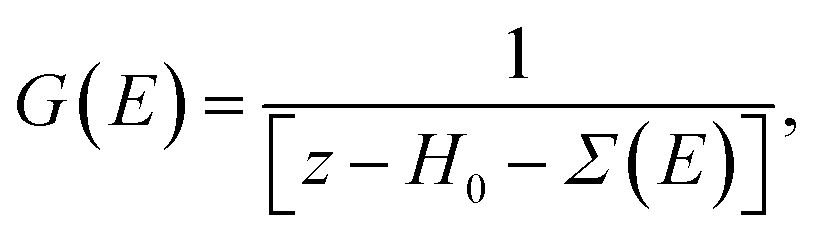 | (10) |
| G(E) = g(E) + g(E)Σ(E)G(E) | (11) |
| G = G0 + G0(ΣL + ΣR)G | (12) |
3.2 Electronic transport properties
The current that passes through a molecule coupled to two electrodes will be deduced by taking into account the transmission probability and its relation with the conductance, employing the Landauer formalism, which depends on the availability of states around the chemical potential of the electrodes μS (source) and μD (drain), where the potential difference μS − μD reduces the energy levels in the drain contact with respect to the source contact, resulting in two different Fermi's functions, given by
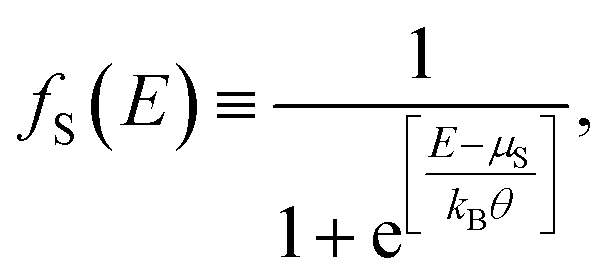 | (13) |
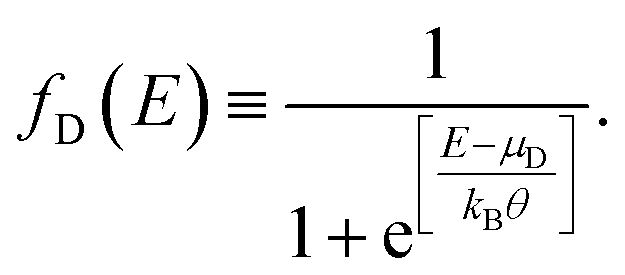 | (14) |
Each contact seeks to balance the molecular system with itself, while the source continues sending electrons to the channel (molecular aromatic system), with the purpose of establishing an equilibrium, which is not achieved because the drain continues receiving electrons. Taking into account the above, the electric current is given by
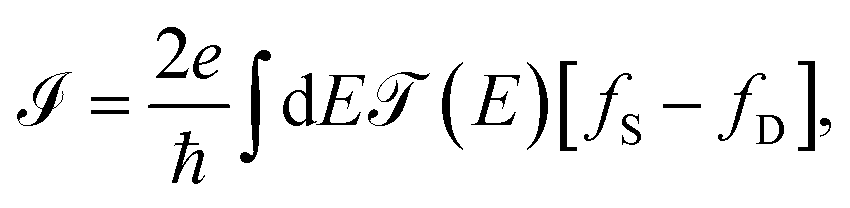 | (15) |
On the other hand, the function 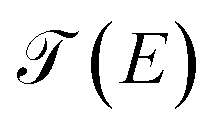 is the transmission probability which describes both flows, always supposing that the transport is coherent.30 The transmission probability T(E) given in eqn (15) can be defined as:
is the transmission probability which describes both flows, always supposing that the transport is coherent.30 The transmission probability T(E) given in eqn (15) can be defined as:
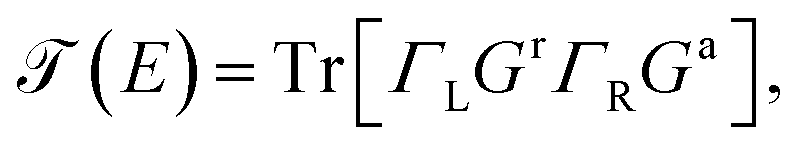 | (16) |
Within the analysis of the electronic properties, complementary to the above is also the shot noise 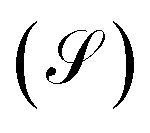 , which describes the current's fluctuations due to the electron's discrete charge. In a low dimensional conductor system, these fluctuations have a quantum origin, arising from the probability that electrons can be transmitted or reflected through the molecular system.32 Quantum noise is caused by the random movement of electric charge carriers across the molecule-contact interface.33 For low dimensional systems, such as quantum wells or resonant tunnels, this type of random fluctuations can be modeled by means of a Poisson process, using the expression
, which describes the current's fluctuations due to the electron's discrete charge. In a low dimensional conductor system, these fluctuations have a quantum origin, arising from the probability that electrons can be transmitted or reflected through the molecular system.32 Quantum noise is caused by the random movement of electric charge carriers across the molecule-contact interface.33 For low dimensional systems, such as quantum wells or resonant tunnels, this type of random fluctuations can be modeled by means of a Poisson process, using the expression
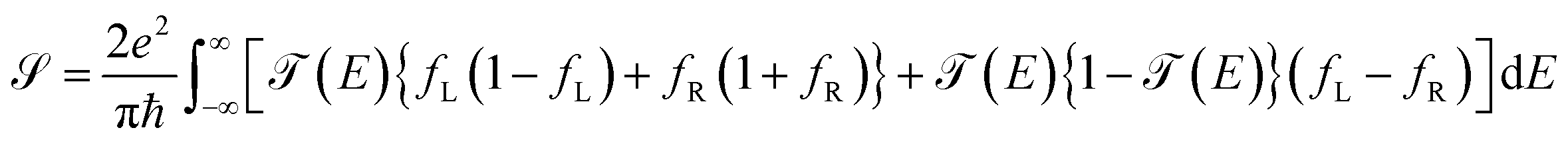 | (17) |
Finally, the power of the noise of the current fluctuations provides additional important information about the electronic correlation by calculating the Fano factor  , which is the ratio between the quantum noise and the current, directly giving information about whether the magnitude of the quantum noise reaches the Poisson limit
, which is the ratio between the quantum noise and the current, directly giving information about whether the magnitude of the quantum noise reaches the Poisson limit 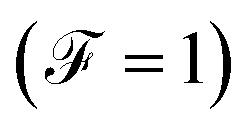 , where there is no correlation between the charge carriers, or a limit at which
, where there is no correlation between the charge carriers, or a limit at which 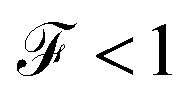 , where the quantum noise reaches a sub-Poisson limit, generating an electron correlation. So, the Fano factor is then given by the relationship:34,35
, where the quantum noise reaches a sub-Poisson limit, generating an electron correlation. So, the Fano factor is then given by the relationship:34,35
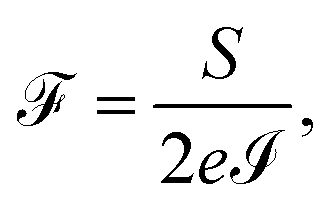 | (18) |
3.3 Renormalization process and calculation of Green's functions
We make use of the renormalization or decimation process in order to obtain the Green's functions in eqn (16) for the three molecular systems (1,4-dithiolbenzene, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl). This process involves transforming them into effective one-dimensional systems, taking into account a geometric rearrangement (illustrated in Fig. 2).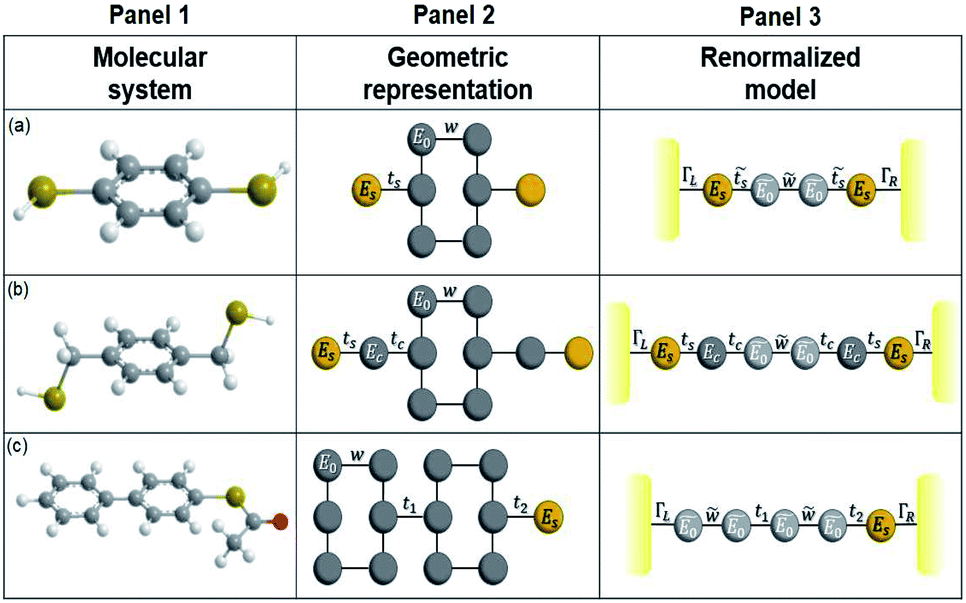 | ||
| Fig. 2 Molecular systems, geometric representation and renormalized models of (a) 1,4-dithiolbenzene, (b) 1,4-dimethanethiolbenzene and (c) 4-thioacetylbiphenyl. | ||
Applying the Dyson equation (eqn (12)), the effective energies of each site and their corresponding couplings are obtained. This allows obtaining new Green's functions, which contain all the information of the flat molecules. Thus, the transmission probability for the new molecular and effective one-dimensional systems can be calculated using the relation
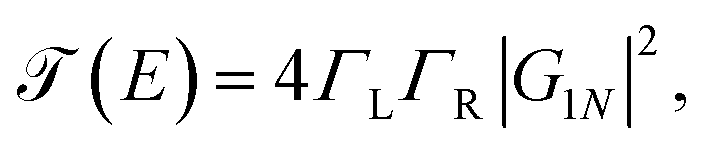 | (19) |
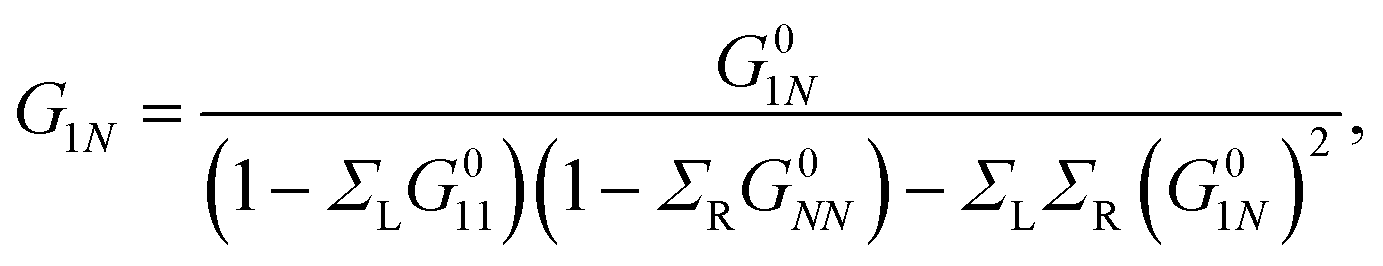 | (20) |
Taking into account that G011 = G0NN and ΣL = ΣR = iΓ/2, we substitute eqn (20) in eqn (19) to get36
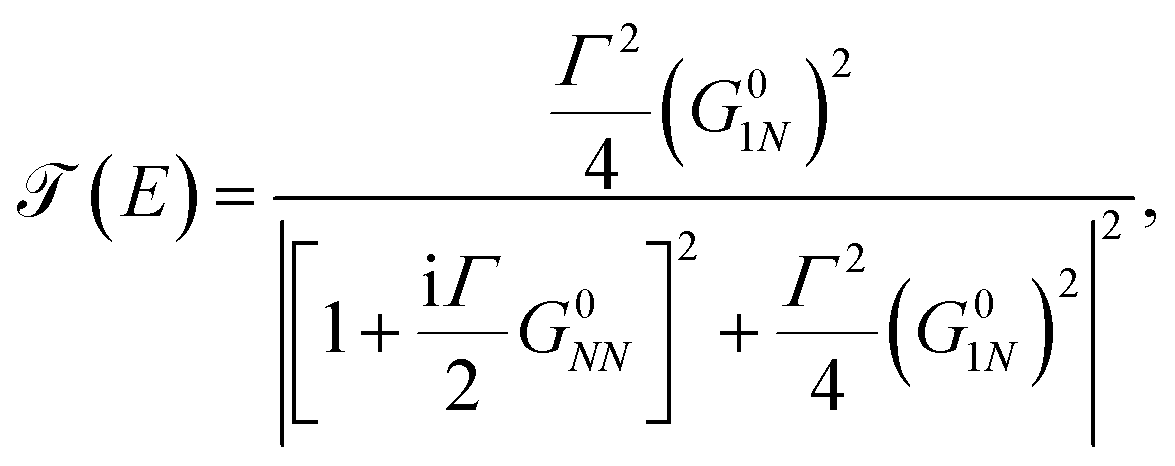 | (21) |
The renormalization method is a method that transforms the real space into an effective space, where the degrees of freedom are reduced and the set of linear equations that characterize the system is converted into a set of nonlinear equations with energies and couplings effective. This method is characterized by its low computational cost (compared to the density functional theory.37,38
It is important to mention that in this renormalization process, the hydrogen atom is neglected in each model, due to the experimental fact that it is absorbed by the deposited surface, and therefore the sulfur atoms will be linked strongly to the metal electrode.39
4 Results and discussion
The energy values of each site and couplings are given by approximations derived from experimental data or predicted by theoretical calculations taken from other authors.28In the first instance and in order to validate the renormalization method, the transmission probability 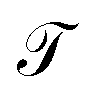 as a function the energy of the injection of the electron is calculated for the 1,4-dithiolbenzene molecular system (Fig. 2a), through two regimes of molecule-contact coupling: (a) a weak regime (Γ < ts, W) where Γ = 0.1 eV and (b) a strong regime (Γ > ts, W) where Γ = 2.0 eV. Then
as a function the energy of the injection of the electron is calculated for the 1,4-dithiolbenzene molecular system (Fig. 2a), through two regimes of molecule-contact coupling: (a) a weak regime (Γ < ts, W) where Γ = 0.1 eV and (b) a strong regime (Γ > ts, W) where Γ = 2.0 eV. Then 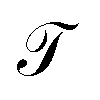 is determined taking into account a set of test values given by Ec = Es = 0 eV and ts = W = 1 eV, where Ec is the energy of site for the carbon atom, Es is the energy of site for the sulfur atom, ts is the value corresponding to the carbon–sulfur bond (C–S) and W is the carbon–carbon resonant bond (C–C).
is determined taking into account a set of test values given by Ec = Es = 0 eV and ts = W = 1 eV, where Ec is the energy of site for the carbon atom, Es is the energy of site for the sulfur atom, ts is the value corresponding to the carbon–sulfur bond (C–S) and W is the carbon–carbon resonant bond (C–C).
In Fig. 3, the transmission probability is displayed in the weak (or resonant) coupling regime (ts, W ≥ Γ, black line) and the strong regime (ts, W ≤ Γ, dashed red line), where the width of the bands in the transmission has increased due to hybridization, which is a consequence of the strong interaction between the free electron states of each sulfur atom with the electronic states of the electrodes, showing that with an increase of the electrode–molecule bond energy the width of the bands in the transmission profile increases. The quantum effect of the contacts is one of the main characteristics that regulate the transport properties in this class of low dimensional systems, where a system can pass from a conductor to an insulator or vice versa.
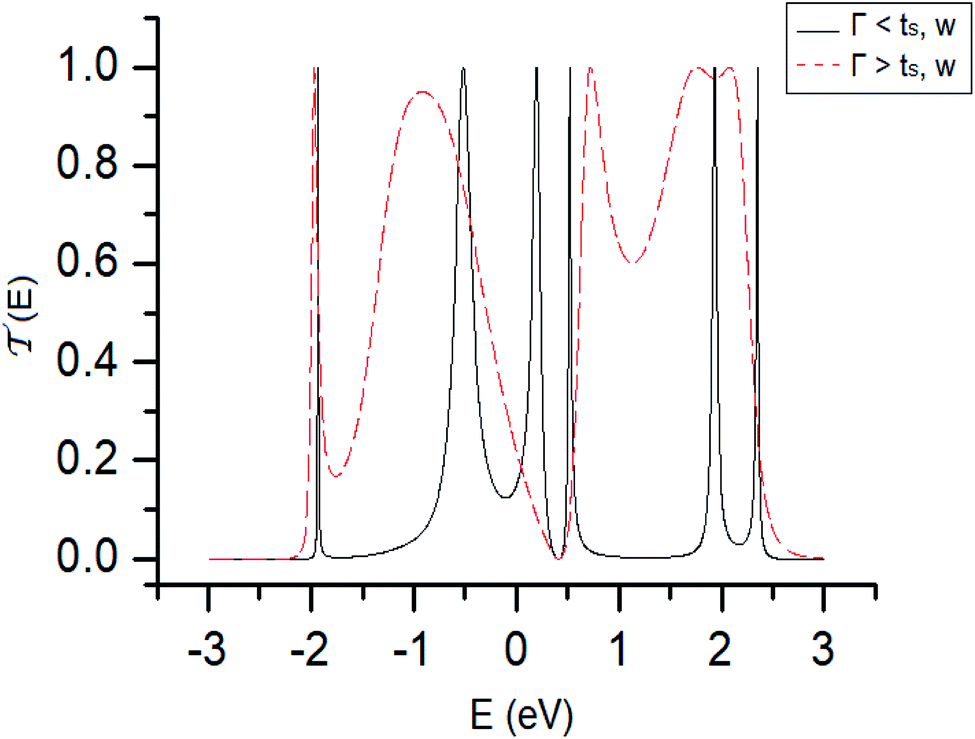 | ||
Fig. 3 Transmission probability  as a function of the energy of the injection of the electron for the 1,4-dithiolbenzene molecular system. Weak regime (Γ < ts, W, black line) and strong regime (Γ > ts, W, dashed red line). as a function of the energy of the injection of the electron for the 1,4-dithiolbenzene molecular system. Weak regime (Γ < ts, W, black line) and strong regime (Γ > ts, W, dashed red line). | ||
It is important to note that the N resonances that appear in the profile of the transmission (especially when we have a weak regime) can be associated with the eigenvalues of each atomic site of the molecule around the equilibrium Fermi energy Ef = 0.0 eV. These values were calculated through the tight binding Hamiltonian given by the eqn (2), and we found that the eigenvalues are −1.93, 0.37, 1.93, 0.56, −0.56 and 2.47 eV. As we can see, these values coincide with the values of the energy where there are resonances in the probability of transmission (Fig. 3, black line).
Therefore, we validated the renormalization method used in our calculations.
Accordingly, we calculate the transmission probability  for the molecular systems 1,4-dithiolbenzene, 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl, using the values of the energy within the Landauer formalism given by Ec = 0.1 eV for the site energy of the carbon atom, tc = 0.52 eV for the non-resonant carbon–carbon bond,40 Es = −0.98 eV for the site energy of the sulfur atom,41 ts = 3.86 eV the carbon–sulfur bond energy, and W = 3.0 eV the resonant C–C bond energy.42 It is important to clarify that 4-thioacetylbiphenyl is an asymmetric molecular system all its transport properties are determined taking into account the injection of electrons in both directions: from the left lead to the right lead and from the right lead to the left lead.
for the molecular systems 1,4-dithiolbenzene, 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl, using the values of the energy within the Landauer formalism given by Ec = 0.1 eV for the site energy of the carbon atom, tc = 0.52 eV for the non-resonant carbon–carbon bond,40 Es = −0.98 eV for the site energy of the sulfur atom,41 ts = 3.86 eV the carbon–sulfur bond energy, and W = 3.0 eV the resonant C–C bond energy.42 It is important to clarify that 4-thioacetylbiphenyl is an asymmetric molecular system all its transport properties are determined taking into account the injection of electrons in both directions: from the left lead to the right lead and from the right lead to the left lead.
The transmission probability is calculated as function of the electron's energy and molecule-contact bonding energy Γ within an energy range of 0.1 eV (weak regime) to 3.0 eV (strong regime) (Fig. 4).
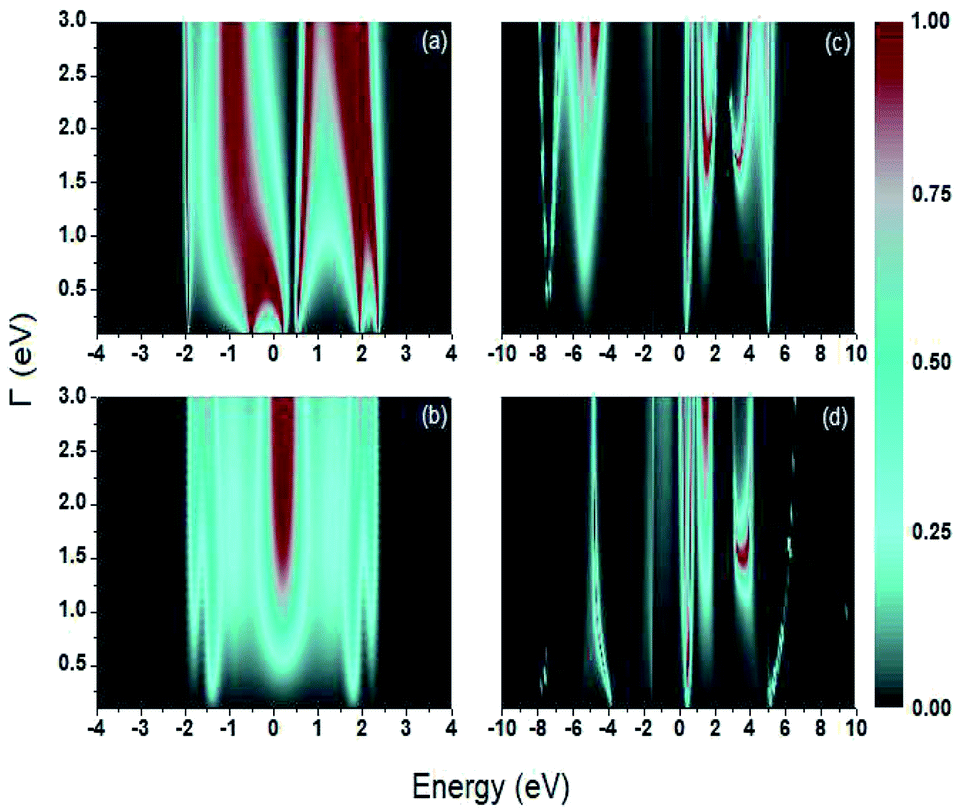 | ||
| Fig. 4 Contour plot. Transmission probability as function of the electron's energy and molecule-contact bonding energy (Γ) for (a) 1,4-dithiolbenzene molecule, (b) 1,4-dimethanethiolbenzene molecule, (c) 4-thioacetylbiphenyl molecule, where the electric charge enters from the left contact to the right contact and (d) 4-thioacetylbiphenyl molecule, where the electric charge enters from the right contact to the left contact. | ||
For all molecular systems there is a change in the profile of  , presenting semiconductor behavior (Fig. 4a), where we can observe that when E = 0 eV and Γ ∼ 0.1 eV, the transmission is zero
, presenting semiconductor behavior (Fig. 4a), where we can observe that when E = 0 eV and Γ ∼ 0.1 eV, the transmission is zero 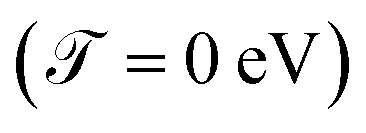 and when E ∼ 0.57 eV the transmission probability is a maximum, generating a gap of ∼0.7 eV, which coincides with the gap obtained experimentally by Reed et al.21 As well, the behavior of an insulator is found (Fig. 4b–d), in this same weak coupling regime (Γ = 0.1 eV). On the other hand, we can see conductive behavior for all systems within the strong coupling regime (Γ ≥ 2.0 eV). These behaviors as insulator, semiconductor or conductor occur with a decrease or increase in the HOMO–LUMO (gap) bandwidth, within which the Fermi level is located (Ef = 0 eV).
and when E ∼ 0.57 eV the transmission probability is a maximum, generating a gap of ∼0.7 eV, which coincides with the gap obtained experimentally by Reed et al.21 As well, the behavior of an insulator is found (Fig. 4b–d), in this same weak coupling regime (Γ = 0.1 eV). On the other hand, we can see conductive behavior for all systems within the strong coupling regime (Γ ≥ 2.0 eV). These behaviors as insulator, semiconductor or conductor occur with a decrease or increase in the HOMO–LUMO (gap) bandwidth, within which the Fermi level is located (Ef = 0 eV).
In the same way, in Fig. 4c and d, slightly different behaviors are observed in the profile of  , due to the fact that when the electron enters through the left electrode and goes to the right electrode, the electron's state enters in resonance with the electronic cloud of the aromatic ring of biphenyl, unlike when the electron enters through the right electrode to the left electrode, where it interacts with the free electronic pairs of the sulfur atom. However, noting the transmission around E = 0 eV as Γ increases, the behavior of
, due to the fact that when the electron enters through the left electrode and goes to the right electrode, the electron's state enters in resonance with the electronic cloud of the aromatic ring of biphenyl, unlike when the electron enters through the right electrode to the left electrode, where it interacts with the free electronic pairs of the sulfur atom. However, noting the transmission around E = 0 eV as Γ increases, the behavior of  presenting a maximum value is very similar for the two cases. This therefore, allows us to consider the system as a conductive system within the strong regime.
presenting a maximum value is very similar for the two cases. This therefore, allows us to consider the system as a conductive system within the strong regime.
In Fig. 5, the transmission probability is evaluated as a function of the electron's energy and ts, the coupling between the sulfur and carbon atoms (S–C), taking a range of energy values between 2 eV to 5 eV, for a value of Γ = 2.0 eV. This range of values for ts was used in this paper since it includes the value of the coupling energy ts = 3.86 eV taken by Sanderson.43
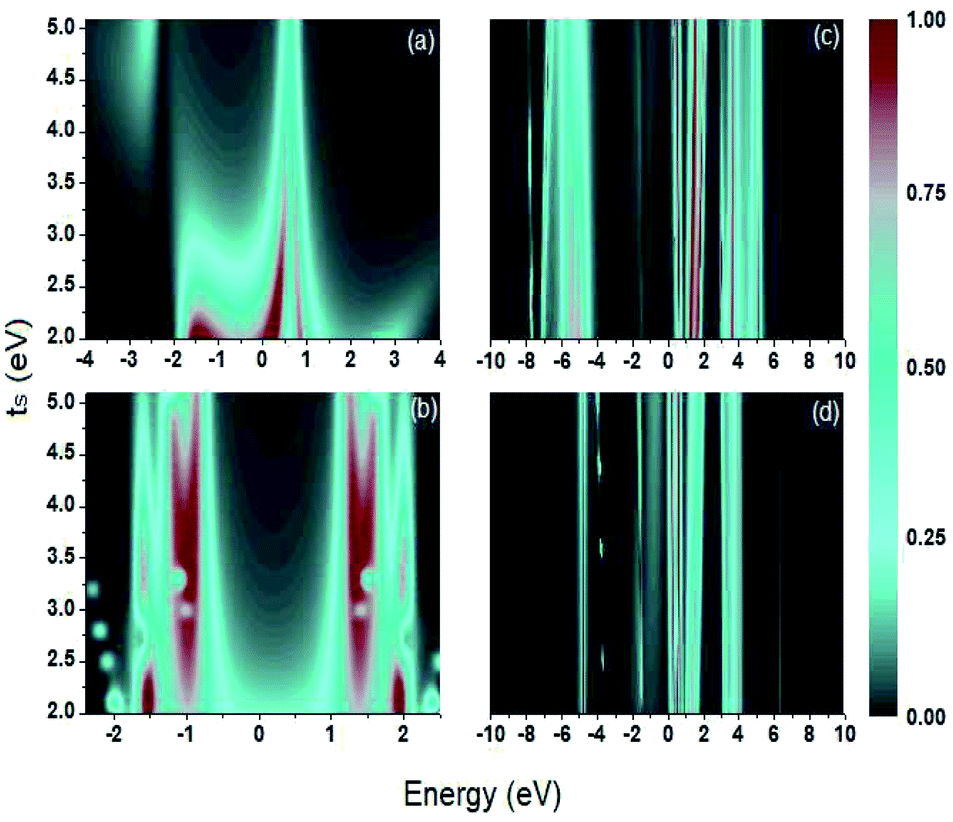 | ||
| Fig. 5 Contour plot. Transmission probability as a function of the electron's energy and bonding energy ts for (a) 1,4-dithiolbenzene, (b) 1,4-dimethanediolbenzene and (c) 4-thioacetylbiphenyl with left–right electron input and (d) 4-thioacetylbiphenyl with right–left electron input. | ||
For each system, the transmission probability has maximum values around certain values of the energy, but the width of the bands does not vary markedly as ts increases, which indicates that the variation of this bonding does not contribute significantly to the behavior of the transport properties through these molecular systems. An important observation can be made for the sweep of this coupling energy: the profile of  reveals a single representative signal in Fig. 5a for an electron injection energy between 0 eV and 1 eV, which indicates that this system can behave like a quantum dot, with an effective energy value approximately equal to 0.5 eV.
reveals a single representative signal in Fig. 5a for an electron injection energy between 0 eV and 1 eV, which indicates that this system can behave like a quantum dot, with an effective energy value approximately equal to 0.5 eV.
The graphs shown in Fig. 6 show the current as a function of the voltage and the energy of the molecule–electrode coupling (Γ) for 1,4-dithiolbenzene systems (Fig. 6a), 1,4-dimethanethiolbenzene (Fig. 6b), 4-thioacetylbiphenyl with left–right electron input (Fig. 6c) and 4-thioacetylbiphenyl with right–left electron input (Fig. 6d).
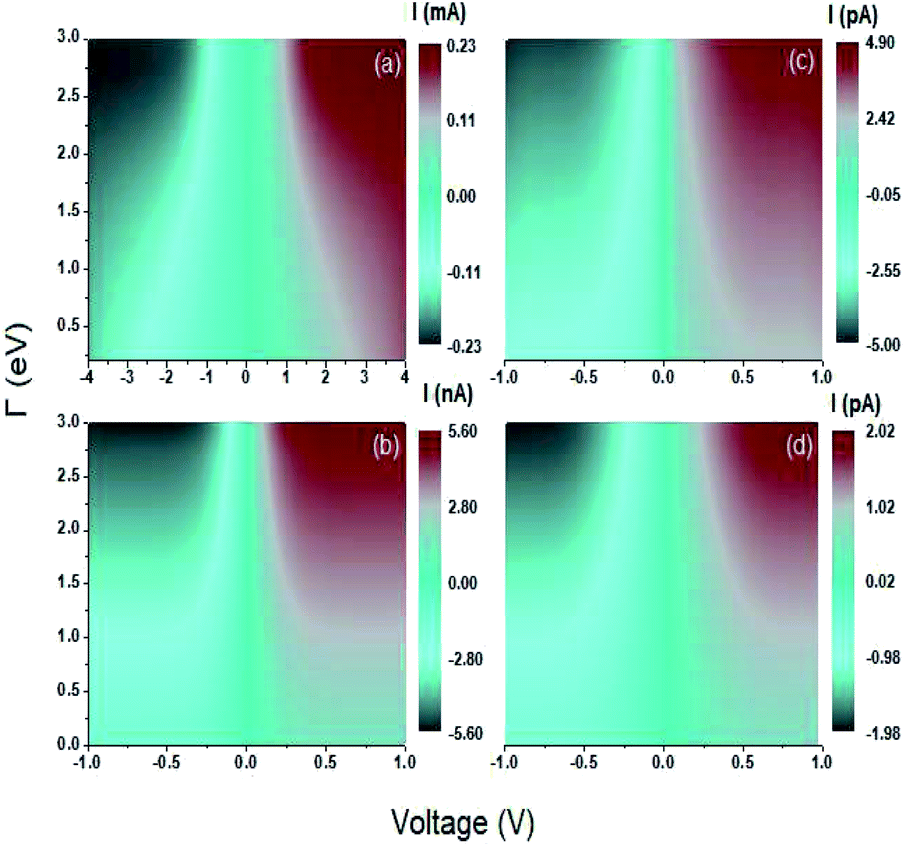 | ||
| Fig. 6 Contour plot. Current as a function of voltage and molecule-contact bonding energy (Γ) for (a) 1,4-dithiolbenzene, (b) 1,4-dimethanediolbenzene and (c) 4-thioacetylbiphenyl with left–right electron input and (d) 4-thioacetylbiphenyl with right–left electron input. | ||
As can be seen in Fig. 6a there is a voltage gap that decreases as the molecule–electrode coupling increases. This gap indicates that the system is not a conductor and can be characterized as an almost insulating molecular device within the weak coupling regime (Γ ∼ 0.1 eV) and has a voltage gap ∼3 V and can pass to semiconductor behavior within the strong coupling regime (Γ ∼ 3 eV) with a voltage gap of ∼0.7 V, where we find that the HOMO and LUMO bands are very close.
For the other systems, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl (Fig. 6b–d) the gap in each of the characteristic curves of 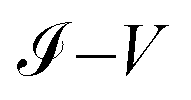 are very small for the strong coupling regime (Γ ∼ 3 eV) and can be considered approximately equal to 0 V, leading to the characterization of each of these molecular systems as conducting molecular electronic devices. However, it is noteworthy that for these systems, the electronic behavior is that of an insulating device for weak couplings.
are very small for the strong coupling regime (Γ ∼ 3 eV) and can be considered approximately equal to 0 V, leading to the characterization of each of these molecular systems as conducting molecular electronic devices. However, it is noteworthy that for these systems, the electronic behavior is that of an insulating device for weak couplings.
On the other hand, the behavior of the C–V characteristic curves shows that as the number of atoms increases in each molecular system, the amplitude of the threshold current and the width of the gap decrease, showing maximum values in the current amplitude of the order of μA for the 1,4-dithiolbenzene molecule, nA for the 1,4-dimethanethiolbenzene molecule, and pA for the 4-thioacetylbiphenyl molecule.
Another transport property as stated above are the quantum current fluctuations 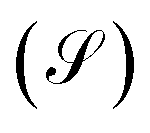 . Fig. 7 shows the normalized current fluctuations
. Fig. 7 shows the normalized current fluctuations 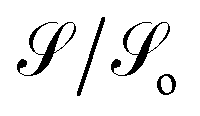 as a function of the input voltage and the molecule–electrode coupling (Γ), as was done for the current characteristic curves.
as a function of the input voltage and the molecule–electrode coupling (Γ), as was done for the current characteristic curves.
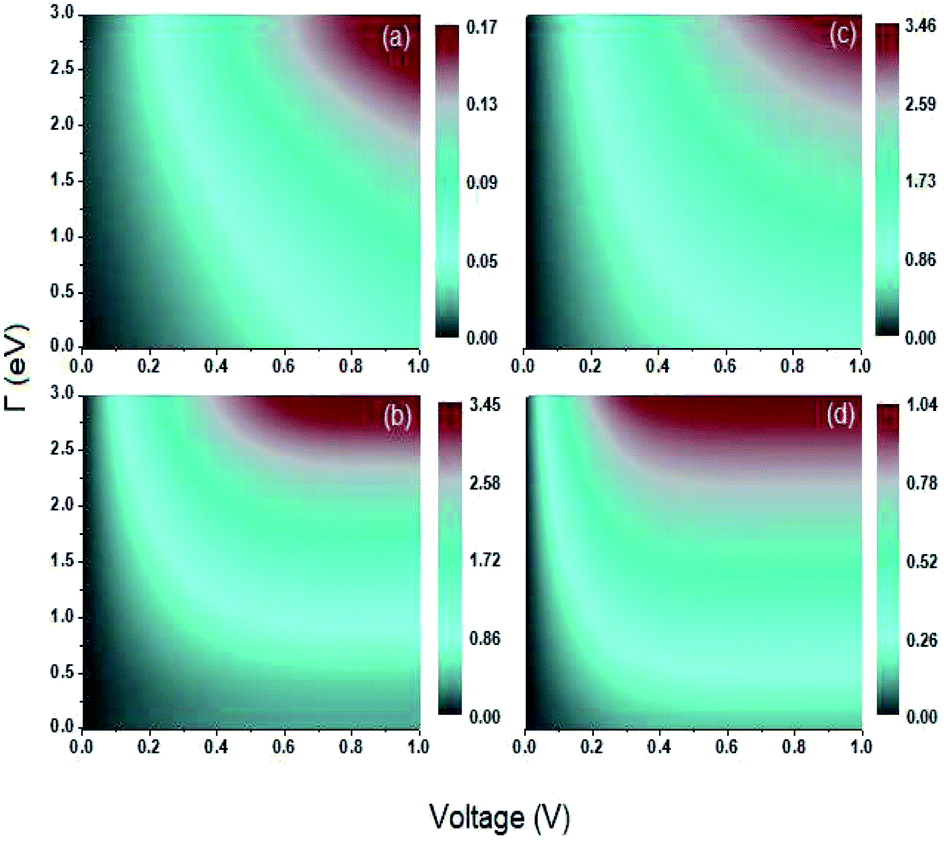 | ||
| Fig. 7 Contour plot. Shot noise as a function of the voltage and molecule-contact bonding energy (Γ) for (a) 1,4-dithiolbenzene, (b) 1,4-dimethanediolbenzene and (c) 4-thioacetylbiphenyl with left–right electron input and (d) 4-thioacetylbiphenyl with right–left electron input. | ||
As we can see in Fig. 7, these fluctuations are shown for all molecular systems, where there are certain maximum values of the fluctuations of current or quantum noise  for which they occur at an average value of the transmission probability. The maximum values of
for which they occur at an average value of the transmission probability. The maximum values of  correspond to a large value of Γ compared to the coupling between the atoms of each molecule, these values being in a regime corresponding to a strong coupling and match with the values of the same transmission amplitude at
correspond to a large value of Γ compared to the coupling between the atoms of each molecule, these values being in a regime corresponding to a strong coupling and match with the values of the same transmission amplitude at 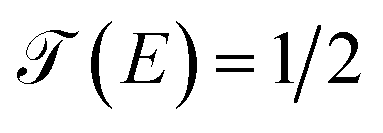 .
.
Fig. 8 show the Fano factor 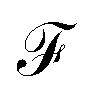 as a function of the voltage and the energy of the molecule–electrode coupling (Γ) for 1,4-dithiolbenzene systems (Fig. 8a), 1,4-dimethanethiolbenzene (Fig. 8b), 4-thioacetylbiphenyl with left–right electron input (Fig. 8c) and 4-thioacetylbiphenyl with right–left electron input (Fig. 8d).
as a function of the voltage and the energy of the molecule–electrode coupling (Γ) for 1,4-dithiolbenzene systems (Fig. 8a), 1,4-dimethanethiolbenzene (Fig. 8b), 4-thioacetylbiphenyl with left–right electron input (Fig. 8c) and 4-thioacetylbiphenyl with right–left electron input (Fig. 8d).
 | ||
| Fig. 8 Contour plot. Fano factor as a function of the voltage and the molecule-contact bonding energy (Γ) for (a) 1,4-dithiolbenzene, (b) 1,4-dimethanediolbenzene and (c) 4-thioacetylbiphenyl with left–right electron input and (d) 4-thioacetylbiphenyl with right–left electron input. | ||
The Fano factor predicts the correlation between the electrons in the system, which only occurs in the sense that one electron is perturbed by the presence of the other charges according to the Pauli exclusion principle (since all other electron–electron interactions have been neglected in this formalism). Taking this into account, in Fig. 8 we observe that for small voltages, the electronic correlation of the system is null 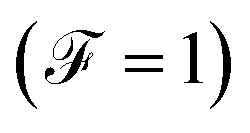 , but it grows proportionally with the increase of the coupling Γ,
, but it grows proportionally with the increase of the coupling Γ, 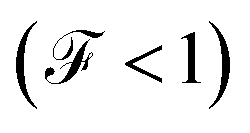 .
.
When the quantum noise ranges from a Poissonian limit 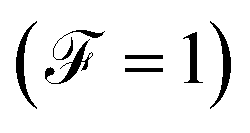 to a sub-Poissonian limit
to a sub-Poissonian limit 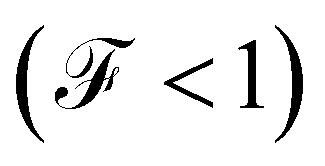 as both Γ and the voltage increase, a tunneling process will be observed. It is notable that, for the weak coupling regime, although the threshold voltage is exceeded, the electronic correlation is quite low
as both Γ and the voltage increase, a tunneling process will be observed. It is notable that, for the weak coupling regime, although the threshold voltage is exceeded, the electronic correlation is quite low 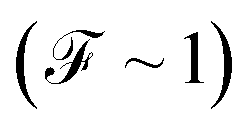 and decreases with an increase in the number of atoms or the length of each molecular system; which is also reflected in the decreased amplitude of both quantum noise and current.
and decreases with an increase in the number of atoms or the length of each molecular system; which is also reflected in the decreased amplitude of both quantum noise and current.
4.1 Comparison of theoretical and experimental results comparison of the 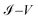 characteristic curves
characteristic curves
Fig. 9a–c show the 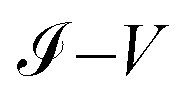
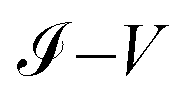 characteristic curves for the 1,4-dithiolbenzene, 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl systems, comparing the calculations performed analytically in this work by the method of Green's functions (blue line) with the experimental results reported in Reed et al.21 (Fig. 9a), Dorogi et al.22 (Fig. 9b), and Dameron et al.42 (Fig. 9c), respectively.
characteristic curves for the 1,4-dithiolbenzene, 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl systems, comparing the calculations performed analytically in this work by the method of Green's functions (blue line) with the experimental results reported in Reed et al.21 (Fig. 9a), Dorogi et al.22 (Fig. 9b), and Dameron et al.42 (Fig. 9c), respectively.
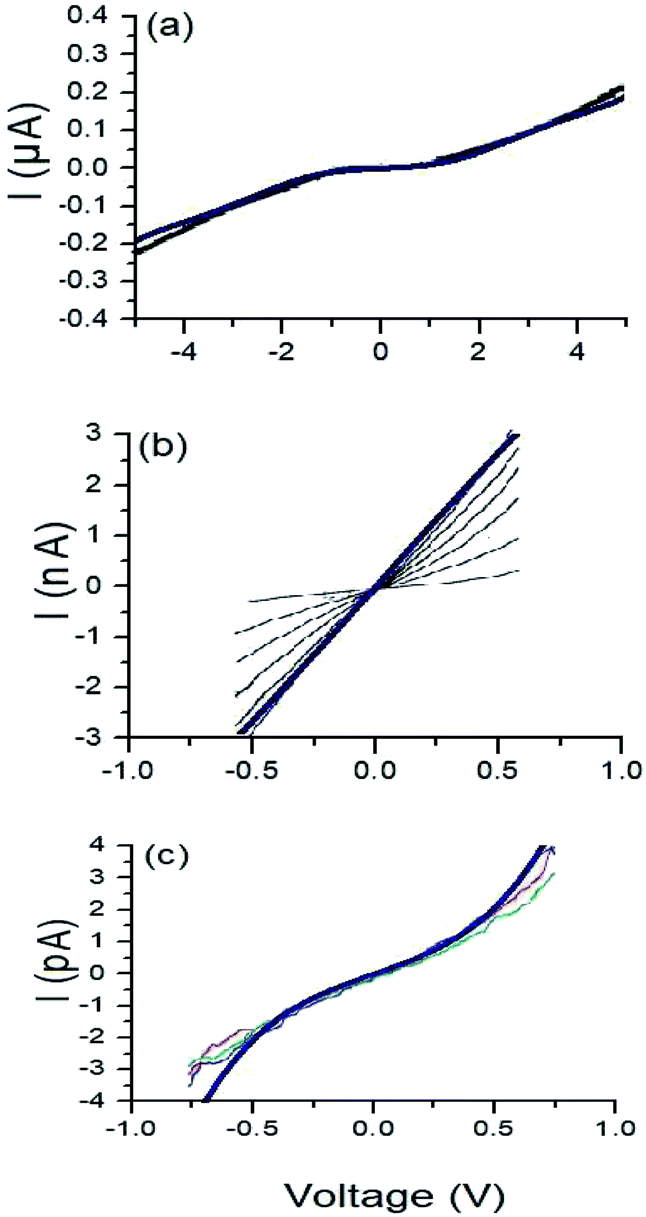 | ||
Fig. 9 Characteristic curves 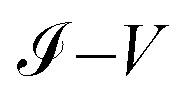 for the (a) 1,4-dithiolbenzene molecule, (b) 1,4-dimethanethiolbenzene molecule and (c) 4-thioacetylbiphenyl molecule: theoretical (blue) and experimental (other colors). for the (a) 1,4-dithiolbenzene molecule, (b) 1,4-dimethanethiolbenzene molecule and (c) 4-thioacetylbiphenyl molecule: theoretical (blue) and experimental (other colors). | ||
As we can see, both the experimental and theoretical 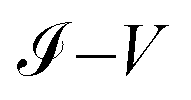 characteristic curves are similar for all molecular systems. Therefore, this validates the Green's functions method used in this work.
characteristic curves are similar for all molecular systems. Therefore, this validates the Green's functions method used in this work.
Is noteworthy that the theoretical calculations displayed in Fig. 9, were made within a single strong coupling regime given a value of Γ = 2.0 eV, coinciding with one of the experimental measurements made in each of the cases shown, specifically in the 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl molecular systems.
In the particular case of the molecular system 1,4-dimethanethiolbenzene (Fig. 9b), if the coupling Γ decreases from 2.0 eV to 0.5 eV, the amplitude of the current decreases and we get curves similar to those found by Dorogi et al., reaching amplitudes in the current of the order of 0.30 nA, as can be verified in Fig. 6b.
Finally, the 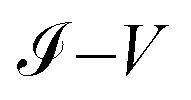 characteristic curves of all systems show a semiconductor behavior for the 1,4-dithiolbenzene and a conductive behavior for the 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl systems. In addition, we can see that as the size of the molecular wire increases (i.e., the number of molecular atomic sites increases), the amplitude of the current decreases. This behavior represents physically a decrease in the overlap between the charge density of the border states (sulfur states) and the other molecular sites of all molecules. With weak hybridization of the states of contacts with the states of the molecular wire, the change in the energy gap HOMO–LUMO of the molecular systems 1,4-dithiolbenzene, 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl (which is proportional to the inverse of the molecular length), increases and therefore a decrease in the electronic conductance is presented. This increase in the gap was seen in Fig. 4.
characteristic curves of all systems show a semiconductor behavior for the 1,4-dithiolbenzene and a conductive behavior for the 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl systems. In addition, we can see that as the size of the molecular wire increases (i.e., the number of molecular atomic sites increases), the amplitude of the current decreases. This behavior represents physically a decrease in the overlap between the charge density of the border states (sulfur states) and the other molecular sites of all molecules. With weak hybridization of the states of contacts with the states of the molecular wire, the change in the energy gap HOMO–LUMO of the molecular systems 1,4-dithiolbenzene, 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl (which is proportional to the inverse of the molecular length), increases and therefore a decrease in the electronic conductance is presented. This increase in the gap was seen in Fig. 4.
5 Conclusions
The electronic transport properties of the sulfurized aromatic molecules of 1,4-dithiolbenzene, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl were determined using a tight binding Hamiltonian. For this purpose, the decimation or renormalization method of the real space was used through the Green's functions technique, which was validated with the test energy values in the transmission profile of 1,4-dithiolbenzene.The transmission probability, current, quantum noise and, Fano factor were calculated for different values of the strength of the bonding between the molecule and the electrode, from small values corresponding to a weak coupling (Γ = 0.1 eV) to a strong coupling (Γ = 3.0 eV), observing different behaviors, where the 1,4-dithiolbenzene molecule behaves as a semiconductor–conductor and the 1,4-dimethanethiolbenzene and 4-thioacetylbiphenyl systems behave as insulators–conductors. Varying the S–C coupling energy did not influence the transmission profiles and therefore neither did it influence the transport properties of the molecular systems studied.
The current as a function of the bias voltage was theoretically determined for the three molecular systems with a strong coupling of 2.0 eV affording a comparison with the experimental results obtained by Reed et al.,21 Dorogi et al.,22 and Dameron et al.42 Excellent agreement was observed between the theoretical and experimental results, validating the analytical method employed in this work, which can thus be applied to study similar molecular systems to be used as possible devices in molecular electronics.
A Appendix-renormalization process
A.1 System 1: 1,4-dithiolbenzene
The molecule is constituted by an aromatic ring with two thiol groups in para position, i.e., the two substituents occupy the opposite positions (Fig. 10A). This molecule contains six carbon, two sulfur, and two hydrogen atoms. The system is geometrically reorganized, where each atomic site is associated with a local Green's function given by gi = 1/(E − Ei), where i represents the carbon or sulfur atom. In this case E0 is the carbon atom's energy of the molecule and Es the sulfur atom's energy.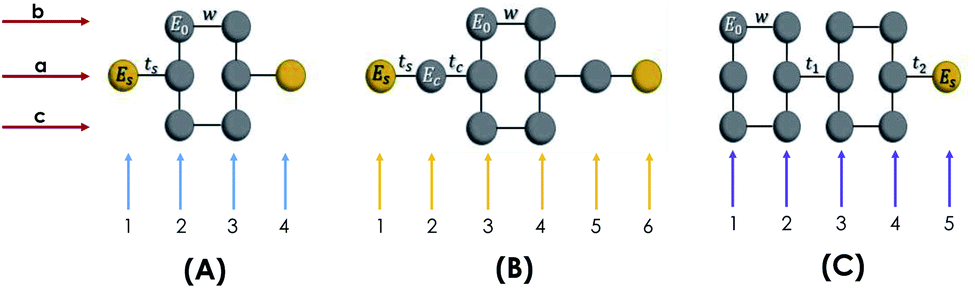 | ||
| Fig. 10 Geometric representation (A) 1,4-dithiolbenzene molecule, (B) 1,4-dimethanethiolbenzene molecule and (C) 4-thioacetylbiphenyl molecule. | ||
In this scheme, w represents each of the resonant bonds of the aromatic ring, ts represents the carbon–sulfur bond and Γ the energy of the bond between the molecule and the electrode. The geometric structure of the molecule is labeled vertically by sites 1, 2, 3 and 4 and horizontally by rows a, b and c. Our purpose is to reduce the number of degrees of freedom of the molecule to one dimension, so it will only be necessary to renormalize sites 2 and 3 towards row a.
Starting from the Dyson equation, we rewrite the dynamic equation for site 2 in the form
| Ga22 = g0 + g0wGb22 + g0wGc22 + g0tsGa12, | (22) |
| Gc22 = g0 + g0wGa22 + g0wGc23, | (23) |
| Gb22 = g0 + g0wGa22 + g0wGb23. | (24) |
The resulting eqn (22)–(24) are linearly independent and therefore they can be expressed in terms of the central zone labeled with row a, where
| Gc23 = g0wGc22 + g0wGa23, | (25) |
| Gb23 = g0wGb22 + g0wGa23. | (26) |
Solving this system of equations we have
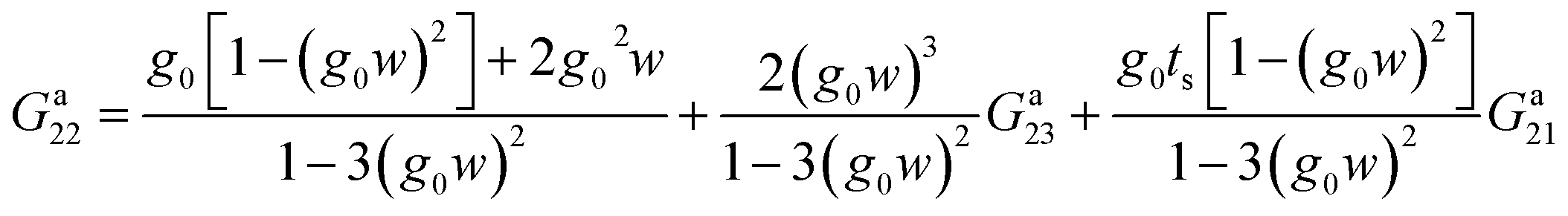 | (27) |
For the fourth site labeled 3, we proceed in the same way:
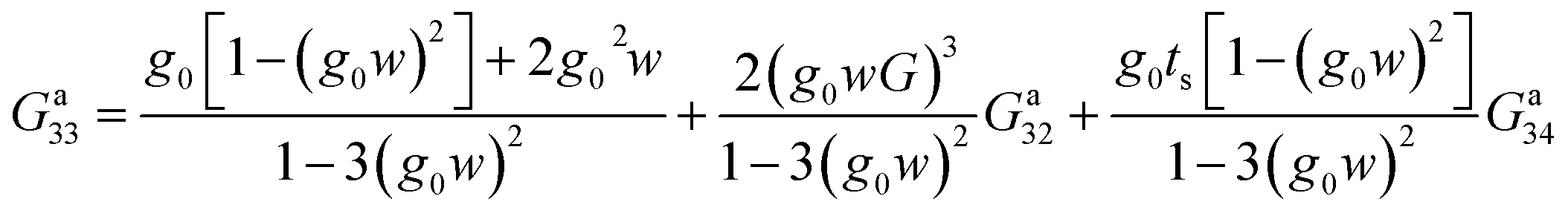 | (28) |
Eqn (27) and (28) represent Dyson's effective dynamic equations for the two-site and three-site molecules, respectively, so that there results
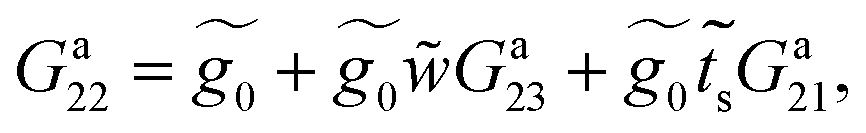 | (29) |
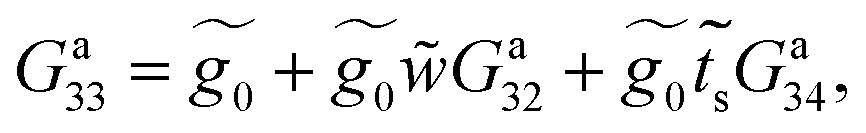 | (30) |
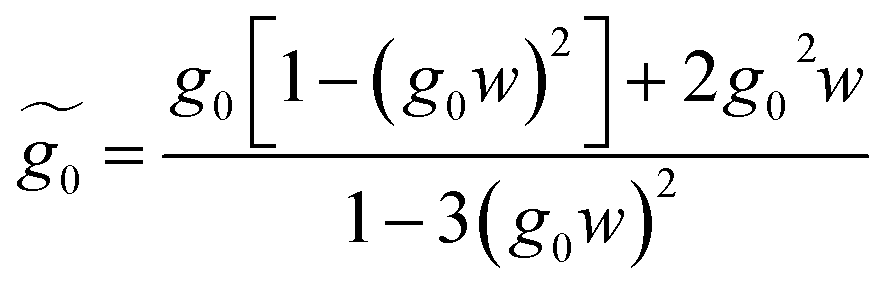 | (31) |
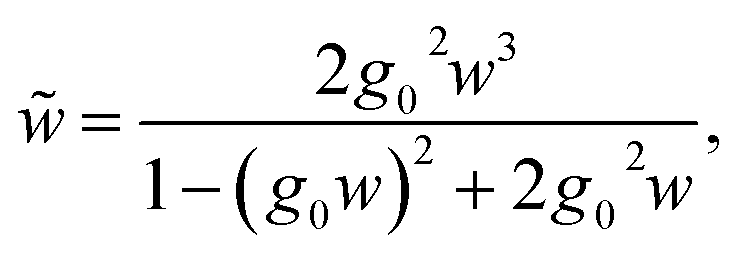 | (32) |
In the same way we get 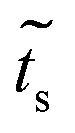 , which is the coupling between sites 1–2 or 3–4:
, which is the coupling between sites 1–2 or 3–4:
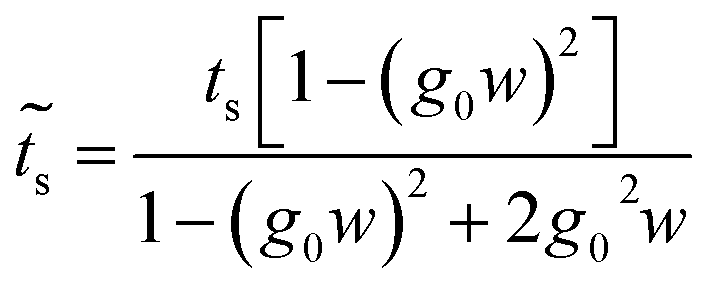 | (33) |
Once the molecule has been decimated (towards the a row), it is necessary to find the Green's function of the electron that travels through the chain from site n = 1 to n = 4. Taking the resulting one-dimensional chain (illustrated in Fig. 2, panel 3) and retaking the dynamic equation, we obtain
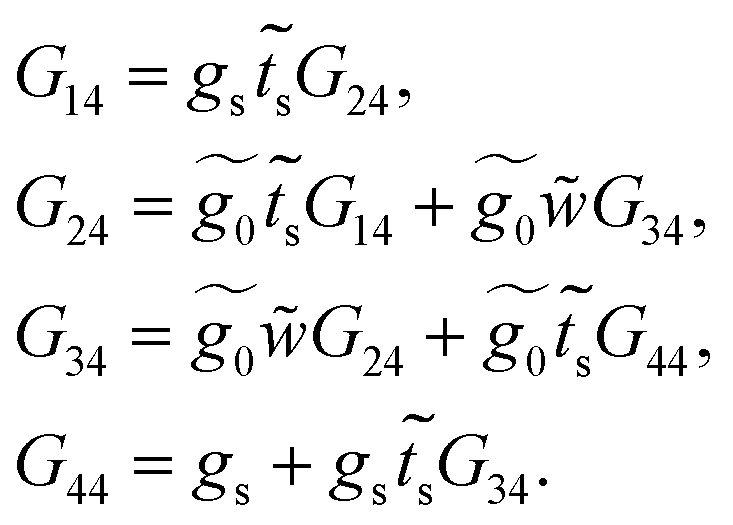 | (34) |
Finally, with the solution of eqn (34), the Green's functions G14, G11 and G44 are found to be
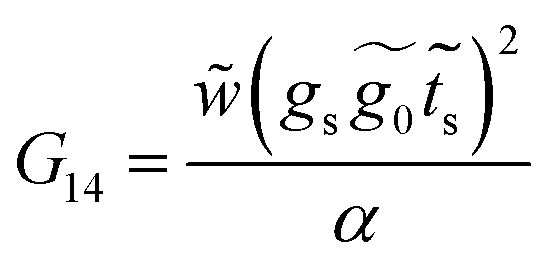 | (35) |
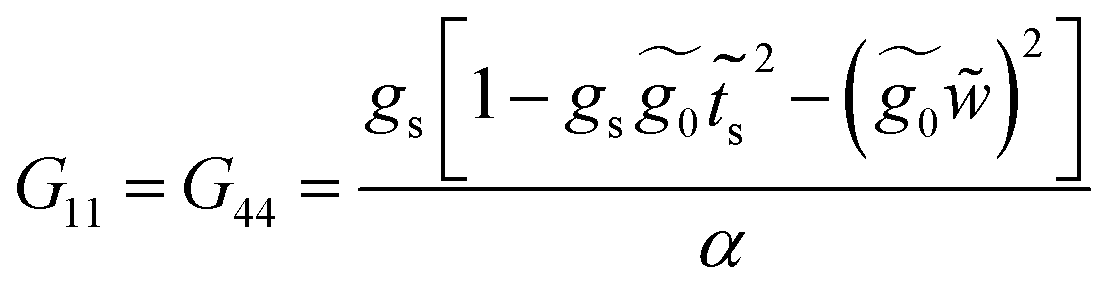 | (36) |
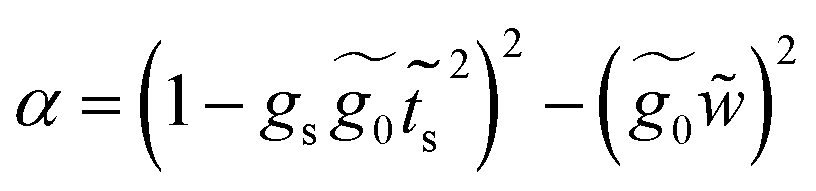
A.2 System 2: 1,4-dimethanediolbenzene
This molecule is composed of a benzene ring substituted with two methanothiols in para position, in all it contains eight carbon atoms, two sulfur atoms, and four hydrogen atoms. Fig. 10B shows the geometry taken for this molecule.The effective Green's function 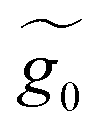 of this system is equal to that obtained for system 1, as are the effective couplings
of this system is equal to that obtained for system 1, as are the effective couplings ![[w with combining tilde]](https://www.rsc.org/images/entities/i_char_0077_0303.gif) and
and 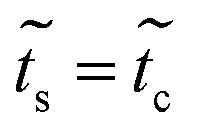 ; therefore a one-dimensional effective linear chain has been obtained (Fig. 2b – panel 3).
; therefore a one-dimensional effective linear chain has been obtained (Fig. 2b – panel 3).
Applying the same solution method to the resulting linear system with six effective sites for this molecule, the Green's functions G11, G16 and G66 are given by
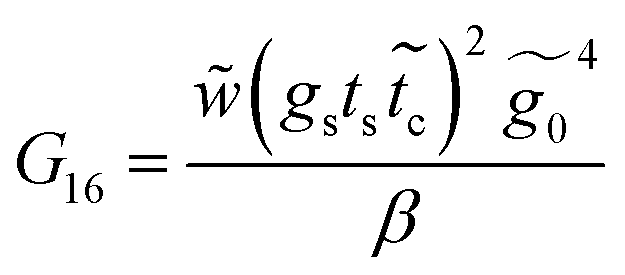 | (37) |
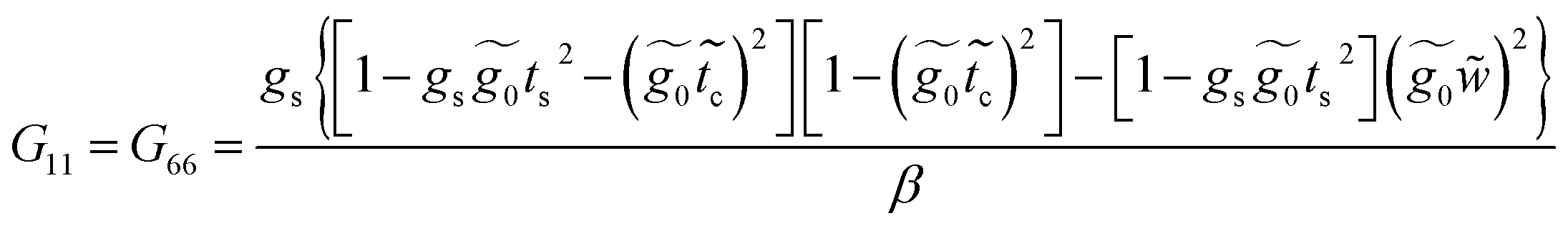 | (38) |
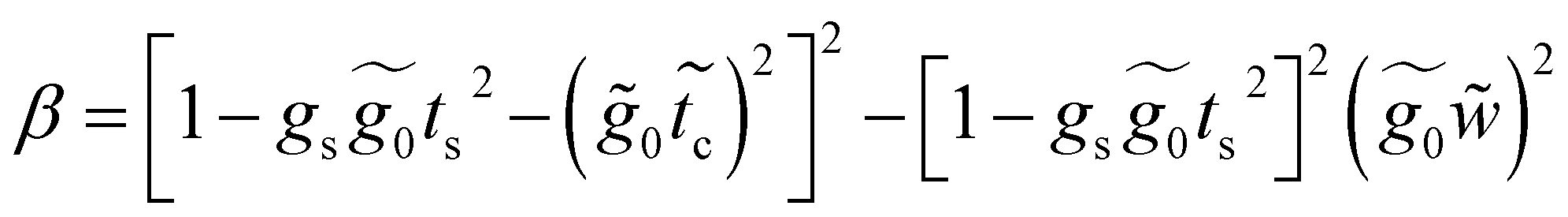
A.3 Renormalization of 4-thioacetylbiphenyl
This molecule contains two benzene rings, where one is substituted by a thioacetyl group, the OCCH3 group is removed by binding the monolayer to the metal surface. Fig. 10C shows the geometry taken for this molecule.Applying the same method of solution to the resulting linear system with five (Fig. 2c – panel 3) effective sites for this molecule, the Green's functions G11, G15 and G55 are given by
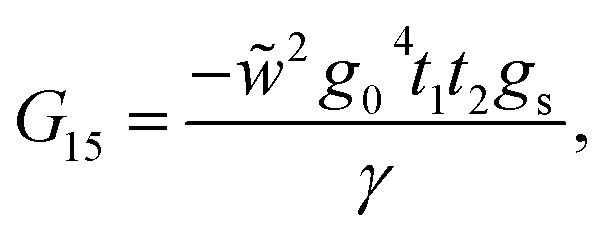 | (39) |
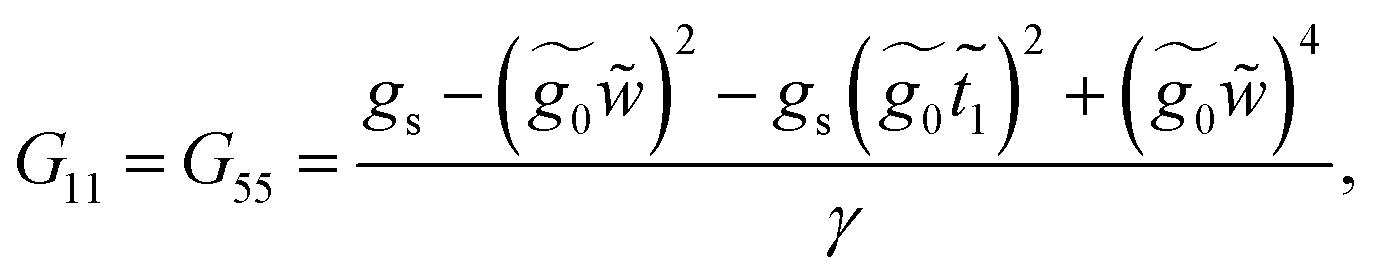 | (40) |
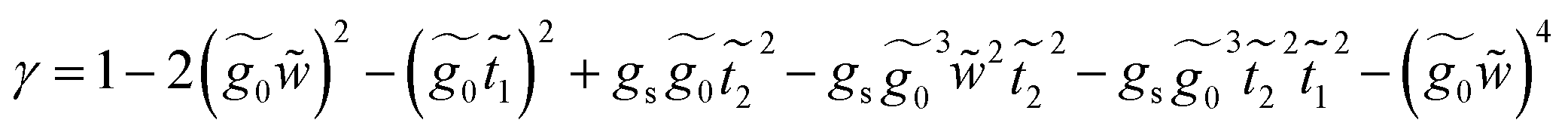 .
.
Conflicts of interest
The authors declare not to have any financial and/or personal relationships with other people or organizations that could inappropriately influence in the work entitled “1,4-dithiolbenzene, 1,4-dimethanediolbenzene and 4-thioacetylbiphenyl molecular systems: electronic devices with possible applications in molecular electronics” or state.Acknowledgements
JHO acknowledges the financial support from El Patrimonio Autónomo Fondo Nacional de Financiamiento para la Ciencia, la Tecnología y la Innovación Francisco José de Caldas (project: CD 111580863338, CT FP80740-173-2019).Notes and references
- H. Choi and C. C. M. Mody, Soc. Stud. Sci., 2009, 39, 11 CrossRef.
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541 CrossRef CAS PubMed.
- A brief history of some landmark papers, Nat. Nanotechnol., 2010, 5, 237, DOI:10.1038/nnano.2010.80.
- N. Invernizzi and G. Foladori, El despegue de las nanotecnologías, CIENCIA ergo-sum, Revista Científica Multidisciplinaria de Prospectiva [Internet], 2005, vol. 12, pp. 321–327, https://www.redalyc.org/articulo.oa?id=10412314 Search PubMed.
- E. Coronado Miralles, An. Quim., 2011, 107, 21 Search PubMed.
- G. M. Whitesides, Small, 2005, 1, 172 CrossRef CAS PubMed.
- J. M. Tour, Molecular Electronics: Commercial Insights, Chemistry, Devices, Architecture and Programming, World Scientific, 2003 Search PubMed.
- J. R. Heath and M. A. Ratner, Phys. Today, 2003, 56, 43 CrossRef CAS.
- A. Bendounan, 2010, arXiv:1001.1692v1.
- Y. Ie, M. Endou, A. Han, R. Yamada, H. Tada and Y. Aso, Pure Appl. Chem., 2012, 84, 931 CAS.
- G. Cuniberti, G. Fagas, and K. Richter, Introducing Molecular Electronics, Springer-Verlag, 2005 Search PubMed.
- H. Sellers, A. Ulman, Y. Shnidman and J. E. Eilers, J. Am. Chem. Soc., 1993, 115, 9389 CrossRef CAS.
- C. Majumder, H. Mizuseki and Y. Kawazoe, J. Mol. Struct.: THEOCHEM, 2004, 681, 65 CrossRef CAS.
- R. Brasca and P. G. Bolcatto, Ann. Funct. Anal., 2005, 17, 56 Search PubMed.
- M. Di Ventra, S. T. Pantelides and N. D. Lang, Phys. Rev. Lett., 2000, 84, 979 CrossRef CAS PubMed.
- M. P. Samanta, W. Tian, S. Datta, J. I. Henderson and C. P. Kubiak, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 7626 CrossRef PubMed.
- W. Tian, S. Datta, S. Hong, R. G. Reifenberger, J. I. Henderson and C. P. Kubiak, Phys. E, 1997, 1, 304 CrossRef CAS.
- S. Hong, R. Reifenberger, W. Tian, S. Datta, J. Henderson and C. P. Kubiak, Superlattices Microstruct., 2000, 28, 289 CrossRef CAS.
- M. A. Daza Millone, Monocapas autoensambladas de alcanotioles y α, ω-alcanoditioles sobre oro: adsorción no específica de moléculas bioactivas, biomoléculas y vesículas, Doctoral thesis, Universidad Nacional de La Plata-Argentina, 2011.
- J. H. Tian, B. Liu, X. Li, Z. L. Yang, B. Ren, S. T. Wu, N. Tao and Z. Q. Tian, J. Am. Chem. Soc., 2006, 128, 14748 CrossRef CAS PubMed.
- M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour, Science, 1997, 278, 252 CrossRef CAS.
- M. Dorogi, J. Gómez, R. Osifchin, R. P. Andres and R. Reifenberger, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 9071 CrossRef CAS PubMed.
- J. H. Ojeda, R. Rey-González and D. Laroze, J. Appl. Phys., 2013, 114, 213702 CrossRef.
- J. M. Tour, L. Jones, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh and S. V. Atre, J. Am. Chem. Soc., 1995, 117, 9529 CrossRef CAS.
- P. E. A. Turchi, A. Qonis and L. Colombo, Tight-binding approach to computational materials science, Materials Research Society Symposia Proceedings, Massachusetts, U.S.A, 1997 Search PubMed.
- D. K. Ferry and S. M. Goodnick, Transport in Nanostructures, Cambridge University Press, Cambridge UK, 1999 Search PubMed.
- C. A. Plazas Riaño, Transporte electrónico a través de alambres moleculares: una aplicación al ADN, Master thesis, Universidad Nacional de Colombia, 2011.
- J. H. Ojeda, P. Orellana and D. Laroze, J. Chem. Phys., 2014, 140, 104308 CrossRef CAS PubMed.
- M. Di Ventra, Electrical Transport in Nanoscale Systems, Cambridge University Press, Cambridge UK, 2008 Search PubMed.
- S. Datta, Quantum Transport: Atom to Transistor, Cambridge University Press, 2005 Search PubMed.
- E. N. Economou, Green's Functions in Quantum Physics, Springer, Berlin, Heidelberg, 2006 Search PubMed.
- Y. M. Blanter and M. Büttiker, Phys. Rep., 2000, 336, 1 CrossRef CAS.
- S. Wolf, R. F. M. Smith and V. González Pozo, Guía para mediciones electrónicas y prácticas de laboratorio, Prentice-Hall Hispanoamericana, 1992 Search PubMed.
- S. K. Maiti, Int. J. Quantum Chem., 2008, 108, 135 CrossRef CAS.
- J. H. Ojeda, C. A. Duque and D. Laroze, Phys. E, 2014, 62, 15 CrossRef CAS.
- J. C. Cuevas and E. Scheer, Molecular electronics: an introduction to theory and experiment, World Scientific, 2010 Search PubMed.
- M. A. Rivera Mateus, J. H. Ojeda and D. Gallego, AIP Adv., 2020, 10, 065021 CrossRef CAS.
- J. H. Ojeda Silva, J. S. Paez Barbosa and C. A. Duque Echeverri, Molecules, 2020, 25(14), 3215 CrossRef PubMed.
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152 CrossRef CAS.
- M. Szwarc, J. Chem. Phys., 1949, 17, 431 CrossRef CAS.
- J. Jia, A. Kara, L. Pasquali, A. Bendounan, F. Sirotti and V. A. Esaulov, J. Chem. Phys., 2015, 143, 104702 CrossRef PubMed.
- A. A. Dameron, J. W. Ciszek, J. M. Tour and P. S. Weiss, J. Phys. Chem. B, 2004, 108, 16761 CrossRef CAS.
- R. Sanderson, Polar Covalence, Elsevier Science, 1983 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |