DOI:
10.1039/D0RA05228K
(Paper)
RSC Adv., 2020,
10, 32241-32248
Theoretical studies on carbon dioxide adsorption in cation-exchanged molecular sieves
Received
14th June 2020
, Accepted 17th August 2020
First published on 1st September 2020
Abstract
The capture and storage of the greenhouse gas, CO2, has attracted much interest from scientists in recent years. In this work, density functional theory (DFT) was used to study the adsorption of CO2 in different cation-exchanged molecular sieves. The results show that for the monovalent metal (Li, Na, K, Cu) ion-exchanged molecular sieves (zeolite Y, ZSM-5, CHA and A), the adsorption capacities for CO2 decrease in the order of Li+ > Na+ > K+ > Cu+. Cu+-exchanged zeolites are not suitable as adsorbents for CO2. For the CO2 adsorption capacities in different zeolites with the same exchanged cation, the adsorption energy decreases in the order of Y > A > ZSM-5 ≈ CHA for Li-exchanged zeolites, and ZSM-5 still has the lowest CO2 adsorption energy for both Na- and K-exchanged zeolites. In the cation-exchanged Y zeolites with divalent metals (Be, Mg, Ca and Zn), the CO2 adsorption performance increases in the order of Zn2+ < Be2+ < Ca2+ < Mg2+. Thus, Zn2+-exchanged zeolites are not suitable as adsorbents for CO2.
1. Introduction
The development of fossil fuels has brought huge economic benefits to people, but it has also brought serious environmental pollution, such as acid rain, haze, and global warming. Among them, the emission of large amounts of carbon dioxide is the main factor causing the greenhouse effect. On the other hand, carbon dioxide is also an important industrial resource. Meanwhile, carbon capture and storage is an important step in many processes, such as natural gas sweetening, biogas upgrading, landfill gas purification, and post-combustion processes. If we can effectively capture carbon dioxide, it will greatly slow down the pace of global warming, solve the environmental crisis, and also bring considerable economic benefits. So, the collection and reuse of CO2 have attracted a great deal of interest from scientists in recent years.
Commonly used methods for the separation and capture of CO2 include absorption, membrane separation, and adsorption.1,2 The chemical absorption method using amine as a solvent is relatively mature, but it has disadvantages of high corrosion, high energy consumption, and degradation of amines.3 The adsorption method has broad prospects due to its simplicity of operation, variety of adsorbents, low cost, high product purity, and large temperature or pressure operating ranges. Microporous and mesoporous molecular sieves, such as metal–organic frameworks, zeolites, and porous carbons, are one class of materials that scientists are focused on for CO2 capture and separation.4,5 Among them, cation-exchanged zeolites are of particular interest because of their low cost and relatively high stability. Furthermore, the gas–solid interaction energy can be tuned by changing the metal cation,6–8 cation concentration,9–11 or the size or topology of the zeolite micropores.12,13 The adsorption of CO2 in alkali ion-exchanged zeolites has been extensively investigated experimentally and by computational methods.6–18 It was found that the cation-CO2 geometry is related to the nature of alkali cations on the Y zeolite, where the adsorption enthalpies show a decrease from Li+ to Cs7+. Yang et al. found that the samples of Cs@RWY, Rb@RWY, and K@RWY exhibited excellent CO2 adsorption properties.14 Hedin et al. found that zeolite NaKA was highly selective towards CO2 over N2 adsorption by tuning the size of the pore window apertures.15 In addition, the CO2 adsorption rate on zeolite NaKA of different sizes were fast and relevant for time scales required for adsorption-based carbon capture and storage.16 Goj et al. studied the adsorption of CO2 and N2 in three siliceous zeolites with identical chemical composition, but different pore structures (silicalite, ITQ-3 and ITQ-7). They found that the CO2/N2 selectivities varied strongly as the zeolite structure changed, and the selectivity in ITQ-3 was the highest.17 A strong relationship between the CO2 adsorption energy and the volume of the cavities of the zeolitic imidazolate framework materials was also found.18
However, there is a scarcity of systematic study on interactions between molecular sieves and CO2, such as the effect of the zeolite topology and alkali-metal cation type on CO2 adsorption. In this work, the adsorption structure and energies of carbon dioxide on different metal cation-exchanged zeolites were well investigated, aiming to provide a theoretical understanding of the CO2 adsorption, and help the optimal design of suitable molecular sieves in the experiment. Four zeolites (Y, ZSM-5, CHA and A) with different frameworks and six mono- and di-valent charge-balancing cations (Li, Na, K, Be, Mg and Ca), which are all widely used in experiment and industry, were chosen to investigate the influence of zeolite structures and cation types on the performance of CO2 adsorption.4,19 The transition metal ions with fully occupied d-orbitals, Cu+ and Zn2+, were also selected for comparison with the main group elements without d electrons to increase the diversity and interest.
2. Computational methods
The zeolite cluster models were taken from the crystallographic data of Y, CHA, ZSM-5 and A,20 which are shown in Fig. 1. The Y zeolite (FAU) has a three-dimensional twelve-membered ring pore system with only one T site. Site II is considered a site of interest to study adsorption due to its accessibility by adsorbates.21,22 One Al atom replaces one Si atom in the six-membered ring. The cation Li, Na, K or Cu is put in the center of the ring to counterbalance the negative charge of AlO4−. After fully considering the framework atoms that may interact with CO2, a 30T cluster model of the local environment around this site was used in this work (Fig. 1a). During the structural optimization process, the atoms on the six-membered ring and its connected peripheral O atoms, metal ions, and CO2 molecules were relaxed with the remaining atoms fixed. As shown in Fig. 1, the atoms represented in ball and stick was relaxed, and the others shown in tube were held fixed. The CHA zeolite has a unique eight-membered ring structure, and also has only one T site. An Al atom in the eight-membered ring substituted a Si atom to form the center, and the cluster was extended around it. Finally, a 36T model covering the whole cage framework structure and the Al active site center with at least an Al–(O–Si–O)4–Si– structure was selected to mimic the CHA zeolite framework (Fig. 1b). The ZSM-5 zeolite has a two-dimensional ten-membered ring pore channel. According to the literature, Al is easily exchanged at the T12 position.23,24 To simulate ZSM-5, a 29T model with a channel containing a ten-membered ring with an T12 Al site as the center of an Al–(O–Si–O)4–Si– structure was selected (Fig. 1c). Zeolite A has an eight-membered cyclic structure with only one T site. A 41T model containing the framework structure of the eight-membered ring channel was selected (Fig. 1d), in which the Al has an Al(–O–Si–O)4–Si– structure. The sizes of these selected zeolite models allowed us to take full account of the possible interactions between CO2 and the skeletal structures. The dangling bonds at the edge of the cluster model were saturated with hydrogen atoms. The Si–H bond length was fixed at 1.47 Å, and the direction was consistent with the direction of the previous Si–O bond. During the structural optimization process of the latter three zeolites, the Al–(O–Si)4 structure around the central Al site, metal ions and adsorbed molecules were relaxed and the remaining atoms were fixed. A CO2 molecule was put on each optimized zeolite cluster with various configurations, followed by further optimizations of the adsorption structure. Then, the converged and stable one was presented.
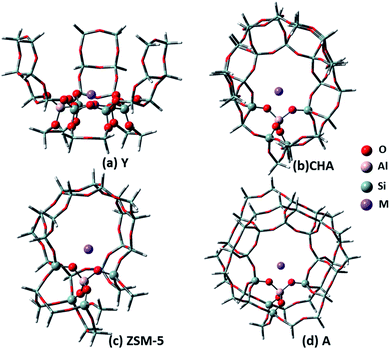 |
| | Fig. 1 Cluster models used to present the zeolite framework: (a) 30T Y, (b) 36T CHA, (c) 29T ZSM-5, and (d) 41T A. Atoms that were allowed to relax are shown in ball and stick representation, others that were held fixed are shown in tube representation. | |
The dispersion interactions between CO2 and the zeolite frameworks are rather large and must be considered in the computational model.11 Hence, the energies were calculated at the level of the B97D Grimme's functional including dispersive interactions,25,26 which has been widely used in the study of weak interactions in zeolites.23,27,28 The 6-311G (2d, p) basis set was used for the Li, Na, K, Be, Mg and Ca cations. Stuttgart RSC 1997 basis set with an effective nuclear potential (ECP)29,30 was used for Cu and Zn cations, and has been widely used for the transition metals.22,31–34 The 6-31G (d,p) basis set was used for other atoms, such as Si, O, Al and C. The adsorption energy of each adsorbate was calculated from the electronic energies of the optimized structures:
| ΔE = ESystem − EAdsorbate − ECluster |
where
ESystem is the energy of the optimized geometries including the adsorbed molecule and the cluster,
EAdsorbate is the energy of the free CO
2 molecule, and
ECluster is the energy of the initial zeolite cluster. The calculated adsorption energy of CO
2 adsorbed on NaY and NaZSM-5 were about −9.56 kcal mol
−1 in our work, which was consistent with the experimental data (−7.05 to −10.28 kcal mol
−1) obtained by calorimetry measurements.
7,35,36 Our calculation indicates that it is hard for Cu
+ to adsorb CO
2, and is also in line with the experimental adsorption isothermal studies.
37,38 All of these can demonstrate that the models, functionals and basis set adopted here are reasonable and suitable. Basis set superposition error (BSSE) corrections evaluated by the counterpoise methods were taken into account.
39,40 It was used to correct the final structure obtained from the normal optimization. All of the above calculations were performed using Gaussian 09 (
ref. 41) on the National Supercomputing Center in Shenzhen.
In order to visualize the interactions between the adsorbed CO2 and the cation-exchanged zeolites, a noncovalent interaction index approach developed by Yang et al.42 was adopted. Here, the reduced density gradient (RDG), defined as RDG(r) = (1/(2(3π2)1/3))((|Δρ(r)|)/(ρ(r)4/3)), together with the electron density ρ, was used to distinguish the covalent and noncovalent interactions. The noncovalent interactions are located in regions with low density and low RDG. The sign of the second largest eigenvalue (λ2) of the electron density Hessian can distinguish between bonded (λ2 < 0) and non-bonded (λ2 > 0) interactions. It can also discern different types of noncovalent interactions: sign(λ2) ρ < 0, H-bonding interaction; sign(λ2) ρ ≈ 0, weak van der Waals (vdW) interaction; and sign(λ2)ρ > 0, strong repulsive interaction. The functions RDG and sign(λ2) ρ were calculated with the Multiwfn software.43
3. Results and discussion
3.1 Adsorption of CO2 on monovalent metal cation-exchanged zeolites
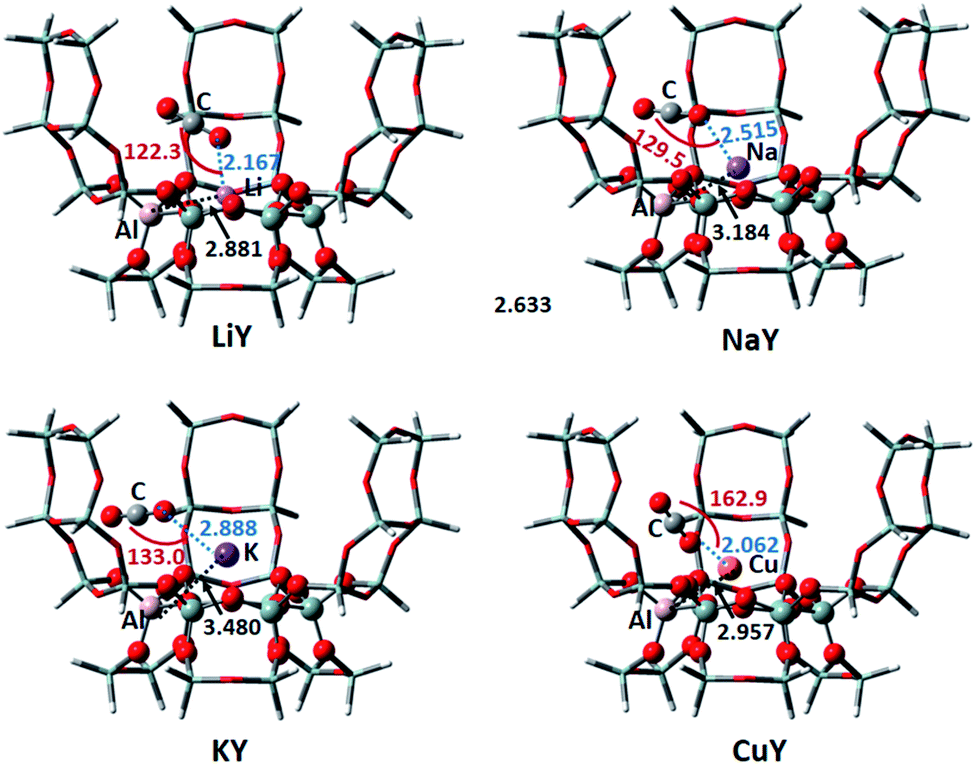
3.1.1 Y zeolite. The 30T model was used to simulate the Y zeolite, and monovalent Li, Na, K, Cu metal cations were selected to neutralize the negative charge of the framework. According to the characteristics of the model structure, several possible adsorption configurations of CO2 were designed. The optimum CO2 adsorption structure determined by comparing the optimized energy and some of the structural parameters are shown in Fig. 2. The distance between the Al atom in the six-membered ring of the framework and the metal cation increases from 2.881 Å to 3.480 Å with the increase of the radius of Li, Na and K, which means that the metal cation is getting farther away from the zeolite framework. The three adsorbed CO2 on Li, Na and K have nearly the same stable configurations, where the CO2 molecule interacts with the metal ion through its O atom. The geometry of CO2 remains almost linear, which is consistent with the result found by Grajciar et al.6 Typically, we can observe an increase of the length of the C–O bond adjacent to the cation, and a decrease of the other bond by approximately the same amount. This result is in good agreement with previous computational results using ab initio calculations.44 The distances between O–Li, O–Na and O–K are 2.167, 2.515 and 2.888 Å, respectively; thus, the distance between the O atom and metal cation is getting farther and farther. This is in line with the DFT study by Pillai et al. that showed the adsorbate-cation distance increasing in the order of Li, Na and K.45 Furthermore, the angle of the cation and CO2 (M⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) is non-linear as shown in Fig. 2, which is in good agreement with the GCMC simulations performed on the NaY system.46 The adsorption energies of CO2 adsorbed on LiY, NaY and KY are −8.85, −8.90 and −7.92 kcal mol−1, respectively. It means that the CO2 adsorption on the Li and Na atoms are similar, and slightly stronger than that for the K atom. The adsorption energy on Li, NaY is consistent with the experimental heat of adsorption of CO2 of −7.05 to −10.28 kcal mol−1 in Li, Na-FAU obtained by differential scanning calorimetry and microcalorimetry measurments.7,36 From the isosurfaces of the reduced density gradient as shown in Fig. 3, it can be seen that all of the isosurfaces of the Li, Na and KY zeolites display some green and brown regions. This is indicative of a strong vdW interaction and a little repulsion between the cation and adsorbed CO2. Previous research studies have found that both electrostatic and van der Waals interactions play crucial roles in the interactions among the metal cation, zeolite and CO2.6,47–49 Based on the analyses of adsorption energy and isosurfaces of the reduced density gradient, we also demonstrate that the vdW interactions make important contributions to the adsorption of CO2 in metal cation-exchanged zeolites. The adsorption capacity of CuY on CO2 is much weaker and the adsorption energy is 31.17 kcal mol−1. This means that the adsorbed complex increased the energy, and the CuY zeolite is not suitable to adsorb CO2. After analyzing the adsorption structure of CO2 on CuY, it can be seen that CO2 points to the zeolite framework, with the O–O distance between CO2 and the zeolite at just 2.489 Å. The distance between the O in CO2 and the Cu+ ion is also very short (2.062 Å). There is a strong repulsive effect between CO2 and the zeolite framework, as well as the Cu atom. It can be proved by its RDG result shown in Fig. 3. The isosurface of CO2 adsorbed on CuY displays a small region in red between the CO2 molecule and Cu+ ion, as well as the oxygen atom in the framework. This indicates a repulsive interaction here.
CO) is non-linear as shown in Fig. 2, which is in good agreement with the GCMC simulations performed on the NaY system.46 The adsorption energies of CO2 adsorbed on LiY, NaY and KY are −8.85, −8.90 and −7.92 kcal mol−1, respectively. It means that the CO2 adsorption on the Li and Na atoms are similar, and slightly stronger than that for the K atom. The adsorption energy on Li, NaY is consistent with the experimental heat of adsorption of CO2 of −7.05 to −10.28 kcal mol−1 in Li, Na-FAU obtained by differential scanning calorimetry and microcalorimetry measurments.7,36 From the isosurfaces of the reduced density gradient as shown in Fig. 3, it can be seen that all of the isosurfaces of the Li, Na and KY zeolites display some green and brown regions. This is indicative of a strong vdW interaction and a little repulsion between the cation and adsorbed CO2. Previous research studies have found that both electrostatic and van der Waals interactions play crucial roles in the interactions among the metal cation, zeolite and CO2.6,47–49 Based on the analyses of adsorption energy and isosurfaces of the reduced density gradient, we also demonstrate that the vdW interactions make important contributions to the adsorption of CO2 in metal cation-exchanged zeolites. The adsorption capacity of CuY on CO2 is much weaker and the adsorption energy is 31.17 kcal mol−1. This means that the adsorbed complex increased the energy, and the CuY zeolite is not suitable to adsorb CO2. After analyzing the adsorption structure of CO2 on CuY, it can be seen that CO2 points to the zeolite framework, with the O–O distance between CO2 and the zeolite at just 2.489 Å. The distance between the O in CO2 and the Cu+ ion is also very short (2.062 Å). There is a strong repulsive effect between CO2 and the zeolite framework, as well as the Cu atom. It can be proved by its RDG result shown in Fig. 3. The isosurface of CO2 adsorbed on CuY displays a small region in red between the CO2 molecule and Cu+ ion, as well as the oxygen atom in the framework. This indicates a repulsive interaction here.
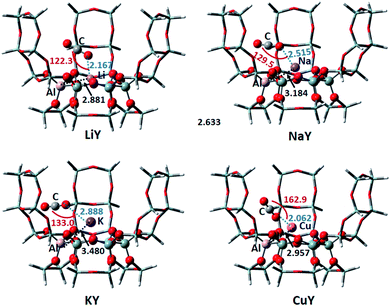 |
| | Fig. 2 The optimized CO2 adsorption structures on the M-Y zeolites. The distances are in Å, and the angles are between the metal ions and CO2. | |
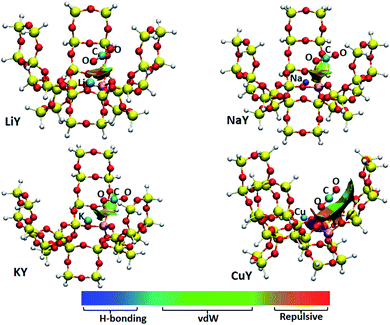 |
| | Fig. 3 Isosurface plots of the reduced density gradient for CO2 adsorbed on various metal cation-Y zeolite structures. The isosurfaces of the reduced density gradient are colored according to the values of the quantity sign(λ2)ρ, and the RGB scale is indicated. vdW represents the van der Waals interaction. | |
3.1.2 CHA zeolite. The stable configurations of the adsorbed CO2 on metal cation-exchanged CHA zeolites, as well as some structural parameters, are shown in Fig. 4. In this adsorption process, the metal cation-CHA zeolite showed many similar characteristics to the metal cation-Y. The distance between the metal cation (Li, Na and K) and Al in the zeolite increases from 2.632 Å to 3.456 Å. The O atom in CO2 interacts with the metal cation, and the distances of O–Li, O–Na and O–K increase from 2.078, 2.478 to 2.912 Å. It shows that the interaction between CO2 and the metal ions would reduce. This is consistent with their CO2 adsorption energy order. The adsorption energies of CO2 adsorbed on LiCHA, NaCHA and KCHA decrease, which are −12.32, −9.62 and −6.26 kcal mol−1, respectively. It indicates that the Li cation has the strongest adsorption effect on CO2. For the CuCHA zeolites, the Cu cation is surrounded by Al and three zeolite oxygen atoms. The distances between Cu and these three framework oxygen atoms are about 2.06 Å, and the Cu–Al atom spacing is 2.323 Å. After adsorption of CO2, Cu interacts with O at a distance of 2.600 Å. Its CO2 adsorption energy is 5.19 kcal mol−1. The positive value indicates that the system has higher energy after CO2 adsorption, so the Cu cation does not adsorb the CO2 molecule easily. This may be related to the structure of the CuCHA zeolite. When CO2 is adsorbed on CuCHA, it is very close to the zeolite framework. In general, the adsorption capacity of CO2 on the metal cation-exchanged CHA zeolites is Li > Na > K > Cu.
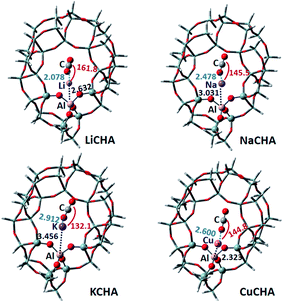 |
| | Fig. 4 The optimized CO2 adsorption structures on the M-CHA zeolites. The distances are in Å, and the angles are between metal ions and CO2. | |
3.1.3 ZSM-5 zeolite. The stable adsorption structures of CO2 on the metal cation-exchanged ZSM-5 zeolite, together with some structural parameters, are shown in Fig. 5. The structural change trends on different metal cations are similar to those of the M-Y and M-CHA zeolites. The adsorption energies of CO2 adsorbed on LiZSM-5, NaZSM-5 and KZSM-5 are −12.25, −10.30 and −10.05 kcal mol−1, respectively. This means that the Li cation and CO2 have the strongest effect. The adsorption energy of CO2 adsorbed on NaZSM-5 is in line with the experimental value of −6.93 to −11.95 kcal mol−1 obtained by calorimetry measurements.36 Unlike the interaction of other metal cations with two backbone oxygen atoms, Cu+ tends to migrate into the ZSM-5 framework and interacts with the multiple oxygen atoms of the zeolite. So, CuZSM-5 may be unfavorable for the adsorption of CO2, which can be further verified through its calculated adsorption energy of 25.66 kcal mol−1. The large positive value shows that CuZSM-5 does not easily adsorb CO2.
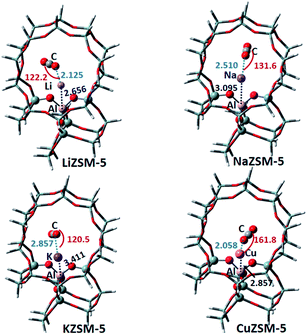 |
| | Fig. 5 The optimized CO2 adsorption structures on the M-ZSM-5 zeolites. The distances are in Å, and the angles are between the metal ions and CO2. | |
3.1.4 Zeolite A. The stable CO2 adsorption structures and some structural parameters on the M-A zeolite are shown in Fig. 6. The structural changes on different metal cation-exchanged zeolites are similar to those of the three zeolites above. The adsorption energies of CO2 adsorbed on LiA, NaA, KA and CuA are −11.29, −8.28, −5.63 and 31.16 kcal mol−1, respectively. It can be seen that the adsorption capacity for CO2 is Li > Na > K. CuA should not be used as a CO2 adsorbent.
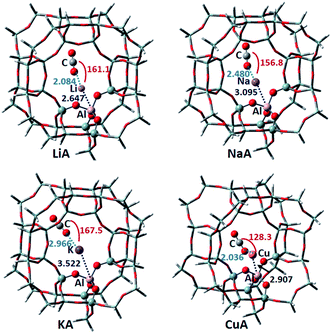 |
| | Fig. 6 The optimized CO2 adsorption structures on the M-A zeolites. The distances are in Å, and the angles are between the metal ions and CO2. | |
As reported in the isosteric heat of adsorption experiment, the adsorption enthalpies of CO2 on zeolite strongly depend on the zeolite topology, the composition (Si/Al and extra-framework cations), the adsorbed CO2 amount and temperature. At low coverage, they vary from −4.78 to −14.34 kcal mol−1.50 The calculated adsorption energies of CO2 on the four molecular sieves studied in this work are all within this range. Moreover, the adsorption energy of CO2 in the same zeolite exchanged by different metal cations is basically reduced in the order of Li, Na, K and Cu. Thus, the Li-exchanged zeolite has the strongest CO2 adsorption capacity, which is consistent with the experimental results.19,51–54 However, the Cu-exchanged zeolite is not suitable as a CO2 adsorbent, which is also in line with the experimental adsorption isotherm studies.37,38 Cu+ binds strongly with CO, rather than CO2, and can be used to separate CO from gas mixtures containing CO2. In order to further explain why the Cu-exchanged zeolites do not easily adsorb CO2 molecules, the electrostatic potential of each metal cation-exchanged zeolite was calculated, and the result is shown in Fig. 7. It was found that the electrostatic potentials of the exchanged Li, Na and K cations on the Y zeolite are positive values. This means that they are more likely to interact with the lone pair electrons of the oxygen atom in CO2. Thus, the abovementioned three metal cation-Y zeolites adsorb CO2 molecules more readily. However, the electrostatic potential of the Cu atoms on CuY is almost zero, and it is not likely to attract electrons. The electrostatic potential results of the other three kinds of zeolites are consistent with that of the Y zeolite. Therefore, the repulsive effect of the geometry and electronic structure may be the reason why the Cu-zeolite does not easily absorb CO2.
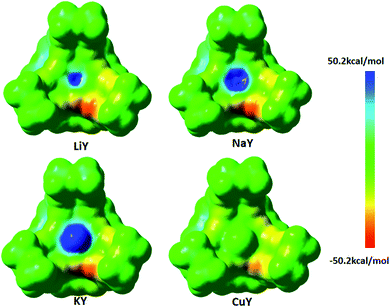 |
| | Fig. 7 The view of the electrostatics potential for various M-Y zeolites. | |
3.2 Comparison of the CO2 absorption capability of different zeolites with the same exchanged metal cation
Comparing the CO2 adsorption capacities of the different zeolites with the same exchanged metal cation (see Fig. 8), we can see that the adsorption energy decreases in the order of Y > A > ZSM-5 ≈ CHA for the Li-exchanged zeolites. It means that LiCHA and LiZSM-5 have the highest adsorption capacities for CO2. In the Na-exchanged zeolite, the adsorption energy decreases in the order of A > Y > CHA > ZSM-5. NaZSM-5 and NaCHA have strong adsorption capacities for CO2. In the K-exchanged zeolites, the adsorption energy decreases in the order of A > CHA > Y > ZSM-5, and the KZSM-5 zeolite has the highest adsorption capacity for CO2. In the Cu-exchanged zeolites, all of them have positive CO2 adsorption energies. This means that the Cu+-exchanged zeolite is not suitable for CO2 adsorption, so they are not shown in this figure. Of the four zeolites, Li, Na-exchanged ZSM-5 and CHA zeolites and KZSM-5 zeolite have strong adsorption capacity for CO2. So, the channel in the ZSM-5 zeolite should be an ideal structure for CO2 adsorption.
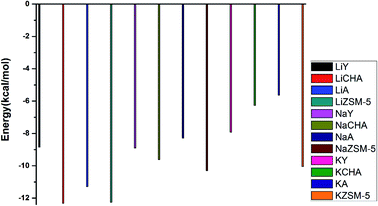 |
| | Fig. 8 The CO2 adsorption capacities of the same metal cation on different zeolites. | |
3.3 Adsorption of CO2 on the divalent cation-exchanged Y zeolites
According to the above calculations, it is known that the orders of the adsorption capacity of CO2 for each cation on different zeolites are the same. Thus, we chose the Y zeolite, which is often used in experiments, as the model to study the effects of divalent cations on the adsorption of CO2 in the zeolite. In this section, the 30T Y zeolite cluster model was selected for the study, where the two Al atoms replaced two Si atoms in the six-membered ring framework. It has two different structures: Al–Si–Al and Al–Si–Si–Al, which are labeled as Y(I) and Y(II), respectively. Different divalent metal cations M (M = Be, Mg, Ca, Zn) were used to neutralize the negative charge of the framework.
3.3.1 Adsorption of CO2 on the Y(I) model. The adsorption structures of CO2 on the Y(I) zeolites exchanged by Be2+, Mg2+, Ca2+ and Zn2+, as well as some structural parameters, are shown in Fig. 9. It can be seen that on each of the four metal cation-exchanged zeolites, the CO2 molecule is directed to the zeolite framework, and the O atom of CO2 interacts with the metal cation. The distances between O–Be, O–Mg and O–Ca are 1.917 Å, 2.137 Å and 2.522 Å, respectively; as the distance increases the interaction between the metal cation and CO2 becomes weaker. This is consistent with the order of the CO2 adsorption energies calculated on the models, which are −6.78, −14.71 and −11.71 kcal mol−1 for BeY(I), MgY(I) and CaY(I), respectively. On the Be2+, Mg2+ and Ca2+ exchanged zeolites, the metal cation is located in the center of the six-membered ring. However, on ZnY(I), Zn is located near one Al atom, and its distance from the two surrounding framework oxygen atoms is about 1.95 Å. In addition, Zn is close to the adsorbed CO2, where the O–Zn distance is just 2.010 Å. This may lead to a large rejection between them. Thus, ZnY may not be a suitable CO2 adsorbent, which is consistent with its high adsorption energy of 23.21 kcal mol−1. The adsorption of CO2 in ZnY is very similar to that on the Cu-exchanged zeolites, where the geometric structure and electron repulsion should be the main reason to reduce its CO2 adsorption ability. Comparing the adsorption energies, it can be seen that the adsorption capacity of CO2 on the M2+Y(I) cluster is ZnY < BeY < CaY < MgY.
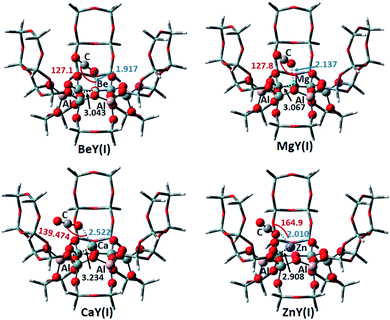 |
| | Fig. 9 The optimized CO2 adsorption structures on the M2+-Y(I) zeolites. The distances are in Å, and the angles are between the metal ions and CO2. | |
3.3.2 Adsorption of CO2 on the Y(II) model. The structures of CO2 adsorbed on the M2+Y(II) cluster models are shown in Fig. 10. The positions of metal cations on the zeolite are similar to that on the Y(I) models. Be, Mg and Ca are basically located in the center of the six-membered ring, and Zn is located near one Al atom. On BeY(II) and ZnY(II), CO2 is directed to the zeolite framework, whereas CO2 on MgY(II) and CaY(II) points to the pore channel. The adsorption energies of CO2 on BeY(II), MgY(II) and CaY(II) are −6.67, −13.83 and −11.47 kcal mol−1, respectively. However, the CO2 adsorption energy on ZnY(II) is 18.91 kcal mol−1, and the distance between Zn and CO2 is short (2.047 Å). It can be seen that the CO2 adsorption capacity here is Zn < Be < Ca < Mg. This is similar to the result of CO2 adsorbed on the Y(I) model, and it can be found that the electron transfer has the similar trend too. So, the different structures of Y(I) and Y(II) have little effect on the CO2 adsorption process. They have similar adsorption structures, adsorption energies and charge transfer trends.
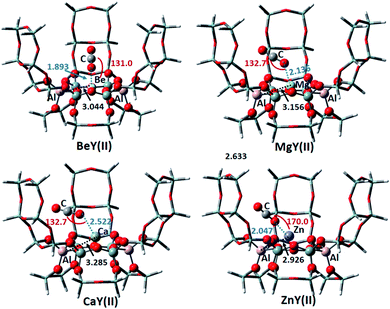 |
| | Fig. 10 The optimized CO2 adsorption structures on the M2+-Y(II) zeolites. The distances are in Å, and the angles are between the metal ions and CO2. | |
4. Conclusion
In this work, DFT theory was used to simulate the adsorption of CO2 on different metal cation M (M = Li, Na, K, Cu, Be, Mg, Ca and Zn) exchanged zeolites (Y, CHA, ZSM-5 and A). It was found that the adsorption capacity of CO2 is basically reduced in the order of Li > Na > K > Cu for the monovalent metal cation exchanged molecular sieves. Compared to the alkali metal cations, the Cu+-exchanged zeolites are not suitable as adsorbents for CO2. For the same metal cation, the CO2 adsorption capacities in different zeolites are also different. The Li-exchanged CHA and ZSM-5 zeolites have similar adsorption energies. For the Na and K-exchanged zeolites, ZSM-5 has the strongest CO2 adsorption capacity. Taken together, ZSM-5 has a good pore structure for CO2 adsorption. In the divalent metal cation-exchanged Y zeolites, the adsorption of CO2 was increased by ZnY < BeY < CaY < MgY. The Ca2+ and Mg2+-exchanged Y zeolites have strong adsorption effects on CO2, and are suitable to be used as CO2 adsorbents.
Funding information
This work was supported by the National Natural Science Foundation of China (21773056 and 21703056), and the Young Backbone Teacher Program of Henan University of Technology (0503/21420046).
Conflicts of interest
There are no conflicts to declare.
References
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 CrossRef CAS.
- J. T. Yeh, K. P. Resnik, K. Rygle and H. W. Pennline, Fuel Process. Technol., 2005, 86, 1533–1546 CrossRef CAS.
- N. Gargiulo, F. Pepe and D. Caputo, J. Nanosci. Nanotechnol., 2014, 14, 1811–1822 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- G. D. Pirngruber, P. Raybaud, Y. Belmabkhout, J. Čejka and A. Zukal, Phys. Chem. Chem. Phys., 2010, 12, 13534–13546 RSC.
- D. F. Plant, G. Maurin, I. Deroche, L. Gaberova and P. L. Llewellyn, Chem. Phys. Lett., 2006, 426, 387–392 CrossRef CAS.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478–487 CrossRef CAS.
- J. Pawlesa, A. Zukal and J. Čejka, Adsorption, 2007, 13, 257–265 CrossRef CAS.
- M. Palomino, A. Corma, F. Rey and S. Valencia, Langmuir, 2010, 26, 1910–1917 CrossRef CAS PubMed.
- A. Zukal, A. Pulido, B. Gil, P. Nachtigall, O. Bludský, M. Rubeš and J. Čejka, Phys. Chem. Chem. Phys., 2010, 12, 6413–6422 RSC.
- T. D. Pham and R. F. Lobo, Microporous Mesoporous Mater., 2016, 236, 100–108 CrossRef CAS.
- A. Henrique, M. Karimi, J. A. C. Silva and A. E. Rodrigues, Chem. Eng. Technol., 2019, 42, 327–342 CrossRef CAS.
- H. Yang, M. Luo, X. Chen, X. Zhao, J. Lin, D. Hu, D. Li, X. Bu, P. Feng and T. Wu, Inorg. Chem., 2017, 56, 14999–15005 CrossRef CAS PubMed.
- Q. Liu, A. Mace, Z. Bacsik, J. Sun, A. Laaksonen and N. Hedin, Chem. Commun., 2010, 46, 4502–4504 RSC.
- O. Cheung, Z. Bacsik, Q. Liu, A. Mace and N. Hedin, Appl. Energy, 2013, 112, 1326–1336 CrossRef CAS.
- A. Goj, D. S. Sholl, E. D. Akten and D. Kohen, J. Phys. Chem. B, 2002, 106, 8367–8375 CrossRef CAS.
- S. Izzaouihda, H. Abou El Makarim, D. M. Benoit and N. Komiha, J. Phys. Chem. C, 2017, 121, 20259–20265 CrossRef CAS.
- Q. Liu, T. Pham, M. D. Porosoff and R. F. Lobo, ChemSusChem, 2012, 5, 2237–2242 CrossRef CAS PubMed.
- Zeolite Structures, http://www.iza-structure.org/databases/, accessed June 13, 2020.
- C.-Y. Sung, S. Al Hashimi, A. McCormick, M. Cococcioni and M. Tsapatsis, Microporous Mesoporous Mater., 2013, 172, 7–12 CrossRef CAS.
- C.-Y. Sung, S. Al Hashimi, A. McCormick, M. Tsapatsis and M. Cococcioni, J. Phys. Chem. C, 2012, 116, 3561–3575 CrossRef CAS.
- W. Shen, Microporous Mesoporous Mater., 2017, 247, 136–144 CrossRef CAS.
- W. Zhang, Y. Chu, Y. Wei, X. Yi, S. Xu, J. Huang, M. Zhang, A. Zheng, F. Deng and Z. Liu, Microporous Mesoporous Mater., 2016, 231, 216–229 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- Y. Chu, B. Han, A. Zheng and F. Deng, J. Phys. Chem. C, 2012, 116, 12687–12695 CrossRef CAS.
- T. T. Trinh, X. Rozanska, F. Delbecq, A. Tuel and P. Sautet, Phys. Chem. Chem. Phys., 2016, 18, 14419–14425 RSC.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- P. Kumar, C.-Y. Sung, O. Muraza, M. Cococcioni, S. Al Hashimi, A. McCormick and M. Tsapatsis, Microporous Mesoporous Mater., 2011, 146, 127–133 CrossRef CAS.
- D.-Z. Li, C.-C. Dong and S.-G. Zhang, J. Mol. Model., 2013, 19, 3219–3224 CrossRef CAS PubMed.
- Z. Bazhanova, Y. I. Tarasov, D. Kovtun, A. Boltalin, B. Novosadov and I. Kochikov, J. Struct. Chem., 2008, 49, 810–817 CrossRef CAS.
- K. Ahmad, B. A. Khan, T. Akhtar, J. Khan and S. K. Roy, New J. Chem., 2019, 43, 19200–19207 RSC.
- K. N. Son, G. E. Cmarik, J. C. Knox, J. A. Weibel and S. V. Garimella, J. Chem. Eng. Data, 2018, 63, 1663–1674 CrossRef CAS.
- J. Dunne, M. Rao, S. Sircar, R. Gorte and A. Myers, Langmuir, 1996, 12, 5896–5904 CrossRef CAS.
- Y. Xie, J. Zhang, J. Qiu, X. Tong, J. Fu, G. Yang, H. Yan and Y. Tang, Adsorption, 1997, 3, 27–32 CrossRef CAS.
- J. W. Yoon, T.-U. Yoon, E.-J. Kim, A.-R. Kim, T.-S. Jung, S.-S. Han and Y.-S. Bae, J. Hazard. Mater., 2018, 341, 321–327 CrossRef PubMed.
- F. B. van Duijneveldt, J. G. C. M. van Duijneveldt-van de Rijdt and J. H. van Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS.
- R. S. Pillai, M. Jorge and J. R. B. Gomes, Microporous Mesoporous Mater., 2014, 190, 38–45 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Naka, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09; Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- B. Bonelli, B. Civalleri, B. Fubini, P. Ugliengo, C. O. Areán and E. Garrone, J. Phys. Chem. B, 2000, 104, 10978–10988 CrossRef CAS.
- R. S. Pillai, M. Jorge and J. R. Gomes, Z. für Kristallogr. - Cryst. Mater., 2019, 234, 483–493 CAS.
- G. Maurin, P. L. Llewellyn and R. G. Bell, J. Phys. Chem. B, 2005, 109, 16084–16091 CrossRef CAS PubMed.
- M. Fischer and R. G. Bell, J. Phys. Chem. C, 2013, 117, 24446–24454 CrossRef CAS.
- T. D. Pham, M. R. Hudson, C. M. Brown and R. F. Lobo, ChemSusChem, 2017, 10, 946–957 CrossRef CAS PubMed.
- W. Wong-Ng, J. A. Kaduk, Q. Huang, L. Espinal, L. Li and J. Burress, Microporous Mesoporous Mater., 2013, 172, 95–104 CrossRef CAS.
- L. Grajciar, J. Čejka, A. Zukal, C. Otero Arean, G. Turnes Palomino and P. Nachtigall, ChemSusChem, 2012, 5, 2011–2022 CrossRef CAS PubMed.
- J. Yang, Q. Zhao, H. Xu, L. Li, J. Dong and J. Li, J. Chem. Eng. Data, 2012, 57, 3701–3709 CrossRef CAS.
- S.-T. Yang, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2010, 135, 90–94 CrossRef CAS.
- F. N. Ridha and P. A. Webley, J. Colloid Interface Sci., 2009, 337, 332–337 CrossRef CAS PubMed.
- S. Park, Y. Yun, S. Kim, S. Yang, W. Ahn, G. Seo and W. Kim, J. Porous Mater., 2010, 17, 589–595 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Han Sun,
Lingchuang Meng,
Bing Wang,
Chenxi Zhan and
Bin Zhao
*,
Han Sun,
Lingchuang Meng,
Bing Wang,
Chenxi Zhan and
Bin Zhao