
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryldiazoquinoline based multifunctional small molecules for modulating Aβ42 aggregation and cholinesterase activity related to Alzheimer's disease†
Monika Ranaa,
Abhishek Pareekb,
Shivani Bhardwaja,
Geeta Aryac,
Surendra Nimesh c,
Hemant Aryac,
Tarun K. Bhattc,
Srinivasarao Yaragorla*b and
Anuj K. Sharma*a
c,
Hemant Aryac,
Tarun K. Bhattc,
Srinivasarao Yaragorla*b and
Anuj K. Sharma*a
aDepartment of Chemistry, Central University of Rajasthan, NH-8, Bandarsindri, Ajmer, Rajasthan 305817, India. E-mail: anuj.sharma@curaj.ac.in; aks.iitk@gmail.com
bSchool of Chemistry, University of Hyderabad, P.O. Central University, Hyderabad, 5000046, Telangana State, India. E-mail: Srinivas.yaragorla@uohyd.ac.in
cDepartment of Biotechnology, Central University of Rajasthan, NH-8, Bandarsindri, Ajmer, Rajasthan 305817, India
First published on 4th August 2020
Abstract
Research continues to find a breakthrough for the treatment of Alzheimer's Disease (AD) due to its complicated pathology. Presented herein is a novel series of arydiazoquinoline molecules investigated for their multifunctional properties against the factors contributing to Alzheimer's disease (AD). The inhibitory properties of fourteen closely related aryldiazoquinoline derivatives have been evaluated for their inhibitory effect on Aβ42 peptide aggregation. Most of these molecules inhibited Aβ42 fibrillation by 50–80%. Selected molecules were also investigated for their binding behaviour to preformed Aβ40 aggregates indicating a nanomolar affinity. In addition, these compounds were further investigated as cholinesterase inhibitors. Interestingly, some of the compounds turned out to be moderate in vitro inhibitors for AChE activity with IC50 values in low micro molar range. The highest anti-AChE activity was shown by compound labelled as 2a with an IC50 value of 6.2 μM followed by 2b with IC50 value of 7.0 μM. In order to understand the inhibitory effect, binding of selected molecules to AChE enzyme was studied using molecular docking. In addition, cell toxicity studies using Neuro2a cells were performed to assess their effect on neuronal cell viability which suggests that these molecules possess a non-toxic molecular framework. Overall, the study identifies a family of molecules that show good in vitro anti-Aβ-aggregation properties and moderately inhibit cholinesterase activity.
1. Introduction
Alzheimer's disease (AD), the most common cause of dementia, affects around 50 million people worldwide. Its prevalence is expected to double every 20 years, rising to more than 130 million by 2050. Impact in countries varies with the proportion of elderly individuals in the population.1 In the absence of an effective treatment for this, AD remains one of the most feared consequences of aging and requires better diagnostic tools, management and effective therapies.2–5The aetiology of AD is very complicated. Several pathological factors such as loss of acetylcholine (ACh) and butyrylcholine (BuCh) neurotransmitters, amyloid beta (Aβ) aggregation into toxic oligomers and plaques, metal ion dyshomeostasis and oxidative stress etc. are suggested for AD pathogenesis.5–12 The classical amyloid cascade hypothesis suggests that the most important contributor to AD is amyloid plaques which are deposits of Aβ peptide aggregates.13–15 However, recent studies suggest that in addition to the plaques, smaller soluble aggregates of Aβ peptides are much more neurotoxic.16–19 Molecules that reduces the aggregation of Aβ peptides and formation of such neurotoxic species are in demand.7 Several inhibitors of Aβ aggregation are reported in literature; some of these inhibitors lead to soluble neurotoxic oligomers and some to non-toxic aggregates.2,18,20–22
On the other hand, inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) to protect acetylcholine in AD patients has been the most attractive therapeutic strategy. The cholinesterase (ChE) enzymes AChE and BuChE are responsible for the hydrolysis of acetylcholine.23,24 Tacrine and donepezil based scaffolds are so far the most potent type of AChE and BuChE inhibitors.25,26 However, alternative molecular scaffolds (e.g., coumarins, quinolines, xanthones etc.) are also known to inhibit AChE and BuChE, albeit with lower potency.27–29
AD is truly a multifaceted disease and various groups have been working on developing suitable multitargeting molecules for its treatment.20–22,30–35 For example, quinolines are natural alkaloids and their derivatives possess a number of biological and medical applications including antioxidation, anti-osteoporosis, anti-influenza, antimalarial and anticancer activities.36–38 Hydroxychloroquine is getting popularity again for its possible role in COVID-19 treatment. Quinoline moiety has been widely explored for its application in treatment for AD. The molecular framework of quinoline leads to the discovery of a small lipophilic molecule clioquinolo (5-chloro-7-iodo-8-hydroxyquinoline, CQ).20,39,40 Recent studies reported the utility of quinoline derivatives as AChE and monoamineoxidase inhibitors.41–43 Fu et al. explored a series of quinoline derivatives with dithiocarbamate moiety and reported their strong inhibition to AChE as well as Aβ aggregation. Sang and co-workers have determined that quinoline derivatives effectively inhibit cholinesterse and monoamineoxidase.41,44 Considering their wide range of biological and medical importance, our group also recently only reported an attractive synthetic strategy of a series of 3-iodo-6-(aryldiazenyl)quinolines.45
In the current work, we report the potential application of selected aryldiazoquinoline derivatives towards the factors contributing to AD. The inhibitory effect of these compounds was investigated for Aβ aggregation using Aβ42 as a model peptide. In addition, these molecules were also investigated for inhibition of cholinergic enzymes AChE and BuChE. The molecular docking studies were carried out to get detailed insights for understanding underlying mechanisms for the observed in vitro inhibitory effect of these compounds on AChE. The cell viability studies show that these molecules are non-toxic in nature and can be used in vivo for further applications. The free radical scavenging assay suggested that these compounds do not enhance free radical species and can be safely used in biological settings. Overall, these aryldiazoquinoline derivatives possess a nontoxic molecular framework, effective cholinesterase inhibition together with anti-aggregation properties towards Aβ42 peptide suggesting these as promising multifunctional agents for AD.
2. Results and discussion
2.1. Molecular design
The design strategy for these compounds consists of a hybrid of quinoline molecule with the diazoaryl-group and to vary the substituents to equip compounds with better intercalating ability to inhibit the Aβ aggregation and interact with cholinesterase enzymes. The design is novel and partly inspired by the properties of Congo Red (CR), an azo-dye known to stain ex vivo amyloid plaques.31,46 Half CR type molecules, resveratrol-stilbene framework have been adopted since these are known for their amyloid binding affinities along with antioxidant properties.46–48 On the other hand, in recent studies, quinolines have demonstrated potential anti-AD activity, in terms of inhibitors to peptide aggregation along with AChE and monoamineoxidase inhibitors and molecular docking studies indicated that it can serve as PAS binding moiety through π–π stacking interaction.36,41,42 Unlike tacrine and donepezil, quinoline moiety is still less explored as cholinesterase and aggregation inhibitor. The design of the molecules used in the current studies, is an extension of our efforts in this direction.31,45 A total of >50 molecules were synthesized (Fig S3 and S4†), using all possible substitutions and combinations, but we have chosen only selective 14 compounds for the current studies to understand their structure–activity relationship from prepared compounds which represent all types of substitution patterns and functional groups (Fig. 1).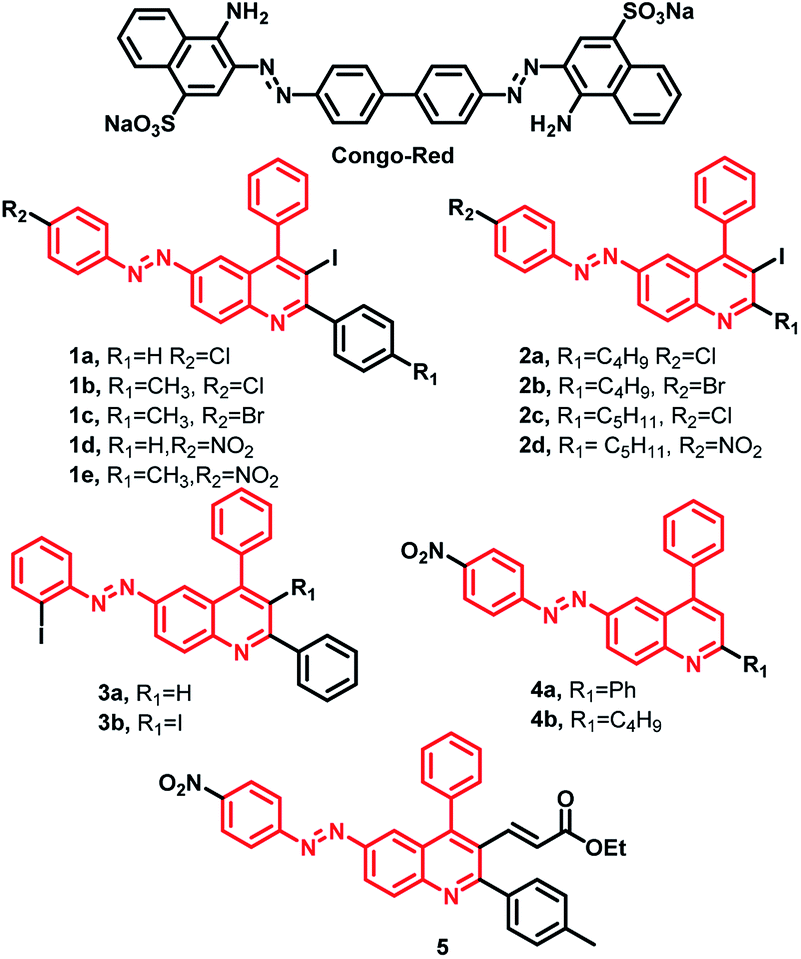 | ||
| Fig. 1 Schematic representation of the inhibitors under study. | ||
Most of these compounds are deep red in color and to understand their conjugation and aromatic properties, the electronic spectra for all the compounds were recorded at room temperature in methanol from 200 to 800 nm range (ESI Fig. S2†). All the compounds show mainly two intense absorption bands with high molar absorptivity values (∼104 M−1 cm−1). One absorption band is observed in the range of 330 to 360 nm which may be attributed to n → π* transition. In most of the compounds, another absorption band is also present in the range of 240 to 300 nm which can be assigned to π → π* transition. When these molecules were excited at their respective λmax values, no fluorescence was observed. The non-fluorescent nature may be due to the presence of lone pairs of azo-group quenching the fluorescence. In addition, rotation about the double bond may be another factor contributing to non-fluorescent nature of these molecules.
To consider a compound as an anti-AD drug, good permeability through blood–brain barrier (BBB) and its ability to reach the central nervous system (CNS) is an essential criterion.49 The verification of Lipinski's rule of five which includes parameters such as calculated octanol–water partition coefficient (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), number of hydrogen bond donor–acceptors, polar surface area provides a good estimate of the BBB permeability of any compound.50 The logBB value is a common measure of the degree of BBB penetration and can be calculated using eqn (1).51
P), number of hydrogen bond donor–acceptors, polar surface area provides a good estimate of the BBB permeability of any compound.50 The logBB value is a common measure of the degree of BBB penetration and can be calculated using eqn (1).51
|
logBB = −0.0148(TPSA) + 0.152(clogP) + 0.139
| (1) |
Drug likeness properties were calculated using program molinspiration property engine v2014.11 (available on http://www.molinspiration.com/cgi-bin/properties). Overall the calculated logBB parameter for all the compounds has the values within the acceptable range (−1.0 to 0.3),52 albeit some individual parameters are found to be on a higher or lower side for some of these compounds. A table of such parameters has been now included in ESI (Table S2†).
2.2. Aβ fibrillation inhibition
The studies on the effect of the compounds in the inhibition of peptide aggregation were carried out upon incubating Aβ42 in the presence and absence of the compounds. The affinity of the compounds toward Aβ fibrillation inhibition was investigated by fluorescence spectroscopy. The model peptide used for fibrillation was Aβ42 which is believed to play most important role in the AD pathogenesis. Thioflavin T (ThT) is used as the most common probe for fibril formation.53,54 When ThT binds to beta sheet-rich structures, more than 20-fold enhancement in its fluorescence intensity is observed which is used for detecting the presence of fibrils.55 In our experimental settings, Aβ fibrillation (in absence of any aryldiazoquinoline additives) starts showing increase in ThT fluorescence within 2–4 hours of incubation. The inhibitory effect of aryldiazoquinoline molecules was observed to be very prominent from beginning itself. The increase in ThT fluorescence occurred after 6–10 hours of incubation in presence of aryldiazoquinoline molecules and remained the same after. Results of ThT fluorescence inhibition assay are presented in Fig. 2 and S33.† Analysis of the result summarized in Table 1 show that all the compounds induced a decrease in ThT fluorescence indicating the inhibition of Aβ fibrillation in a range of 50 to 77%. Compounds 1d, 1e, 2d and 5 are the most effective inhibitors with ∼75% inhibition. In general, the inhibitory effect of these molecules can be attributed to their capability to intercalate and act as β-sheet disruptors. Presence of certain functional groups like substitution of an unsaturated ester functionality at 3-position of quinoline ring in molecule 5 are expected to increase the bioavailability and binding interactions of these molecules with Aβ peptide and likely the reason for observed inhibitory effect. | ||
| Fig. 2 Normalised ThT fluorescence for inhibition of Aβ42 fibrillation measured upon incubation at 37 °C for 24 h. Samples are as indicated on top of bar graphs. Conditions: (PBS; [Aβ] = 25 μM; [compounds] = 25 μM). | ||
| S. no. | Compound | % inhibition for Aβ aggregation | IC50 ± SD (μM)b for AChE | % inhibition for BuChE (100 μM) |
|---|---|---|---|---|
| a AChE from electric eel, Aβ42 peptide from Anaspec, BuChE from equine serum.b Results are expressed as mean of three experiments.c nd indicates not determined. | ||||
| 1 | 1a | 49.6 ± 5.4 | 15.9 ± 1.1 | 48.7 ± 6.3 |
| 2 | 1b | 61.7 ± 3.9 | 11.9 ± 1.1 | 18.8 ± 2.4 |
| 3 | 1c | 59.0 ± 3.0 | 12.0 ± 1.1 | 14.9 ± 12.3 |
| 4 | 1d | 75.2 ± 3.0 | 9.3 ± 1.1 | 54.8 ± 5.8 |
| 5 | 1e | 74.4 ± 2.0 | 8.4 ± 1.1 | No inhibition |
| 6 | 2a | 60.9 ± 1.9 | 6.2 ± 1.2 | 52.3 ± 8.6 |
| 7 | 2b | 62.8 ± 3.4 | 7.0 ± 1.2 | 32.2 ± 2.1 |
| 8 | 2c | 64.5 ± 3.1 | 14.3 ± 1.2 | 36.3 ± 0.5 |
| 9 | 2d | 74.7 ± 2.3 | 16.8 ± 1.2 | 40.5 ± 6.3 |
| 10 | 3a | 66.0 ± 1.8 | 13.1 ± 1.2 | 32.5 ± 6.3 |
| 11 | 3b | 70.1 ± 2.7 | 16.0 ± 1.2 | No inhibition |
| 12 | 4a | 66.0 ± 2.2 | 16.8 ± 1.1 | 66.9 ± 7.3 |
| 13 | 4b | 69.9 ± 2.3 | 17.0 ± 1.1 | 44.5 ± 6.1 |
| 14 | 5 | 77.2 ± 2.5 | 18.1 ± 1.1 | 39.7 ± 7.8 |
| 15 | RTG | ndc | 11.0 ± 1.3 | ndc |
| 16 | DZ | ndc | 0.006 ± 1.1 | ndc |
In order to gain further insights for inhibition of Aβ42 aggregation, transmission electron microscopy (TEM) technique was employed for selected molecules. One inhibitor among each series was selected as representative compound which showed best IC50 values (1e, 2a, 3a, 4a and 5). TEM images show the presence of dense fibrillar aggregates of Aβ (Fig. 3, panel 1). A change in morphology of these aggregates was observed in the presence of inhibitors (Fig. 3, panel 2–6). Fewer Aβ fibrils were detected as compared to Aβ alone when compounds 1e and 5 (Fig. 3, panel 2 & 6) were incubated with the sample, whereas 2a, 3a and 4a (Fig. 3, panel 3–5) demonstrated larger amorphous aggregates. These results were consistent with the ThT assay showing a good inhibition of self-induced Aβ aggregation.
 | ||
| Fig. 3 TEM images of inhibition of Aβ42 aggregation for representative compounds from each series (PBS; [Aβ42] = 25 μM; [compounds] = 25 μM, 37 °C for 24 h, scale bar = 200 nm). | ||
2.3. Binding affinity with preformed Aβ40 fibrils
In order to measure the binding affinity of the molecular framework of the compounds studied here, ThT fluorescence competition assays were performed. Since, these molecules consist of azo-stilbene type fragments and likely to intercalate in β-sheets and may replace ThT. The binding affinity of the selected three compounds (1e, 3a and 5) toward Aβ fibrils was investigated. For binding studies, Aβ40 was used since it forms well defined amyloid fibrils.21,56 For the experiment a solution of compound under study was added sequentially in nanomolar amounts to a solution of fixed concentration of Aβ40 fibrils (5 μM) in presence of ThT (2 μM).57 A significant decrease in ThT fluorescence intensity was observed upon the addition which indicates the replacement of pre-bound ThT to Aβ fibrils through competitive binding. One site competitive-binding model was used for data fitting and yields a Ki value of 204 ± 19 nM, 177 ± 15 nm and 168 ± 17 nm respectively for 1e, 3a and 5 (Fig. 4). Overall the result suggests that this molecular framework exhibits a strong affinity for the Aβ fibrils. The strong binding of these molecules also indirectly support the inhibitory effect on Aβ fibril formation as observed above. | ||
| Fig. 4 ThT fluorescence competition assays of Aβ fibrils with 1e, 3a and 5 respectively ([Aβ] = 5 μM; [ThT] = 2 μM; λex/λem = 435/485 nm). | ||
2.4. AChE and BuChE inhibitory activity
The inhibitory effect of the designed compounds were analyzed in vitro for AChE using modified Ellman's method.58 The experimental results were presented as IC50 in Table 1. In comparison to the known AChE inhibitors donepezil (DZ) and rivastigmine (RTG), the IC50 values (Table 1) show a moderate AChE inhibitory activity of compounds studied here. Out of the total fourteen compounds, good anti-AChE activity was exhibited by compounds 2a, 2b, 1d and 1e with IC50 values in the low micromolar range (6.2, 7.0, 9.3 and 8.4 μM, respectively). It is worth noting that 2a which has highest AChE activity, contains a chloro group at the para position of to diazoaryl group in combination with an aliphatic group (C4H9) at 2-position of quinoline ring with iodo substitution at 3-position. Interestingly, the presence of these functional groups in other molecules but in different combinations does not exhibit similar activity. Even replacement of chloro group by a bromo group in the aryldiazo moiety decreased the AChE inhibitory activity. The compounds 2c and 2d, where C4H9 group is replaced by C5H11 group exhibited remarkable decline in inhibitory activity. The compounds with best AChE activity belong to series 1 and 2 containing iodo group at 3-position of quinoline ring. In general, molecules without a donepezil molecular framework show IC50 values in very high micro molar range. Therefore, it is interesting to see that the chosen set of compounds showed good AChE inhibitory activity, which is comparable to reference inhibitor RTG in the same experimental settings (Table 1).These compounds were further tested for their inhibitory activity against BuChE following the similar protocol. Inspired by good AChE results; first, the activity for these compounds were checked at low concentration range (2–100 μM). The compounds were found to be very less active in these settings for BuChE inhibition. Studies at their higher concentrations (up to 300 μM) showed moderate inhibition, but a regular pattern of inhibition could not be observed. It is likely due to the inherent absorption by the compounds themselves in the monitoring absorption wavelength range (∼405 nm). For BuChE inhibition, compound 4a exhibited comparatively well with more than 65% inhibition at 100 μM concentration. As a regular pattern of inhibition was not observed, IC50 values were not calculated from data. The volume of the catalytic site in BuChE is much larger than that of AChE and also, BuChE does not have a functional peripheral site, these structural differences might contribute towards the different activity of a molecule against AChE and BuChE.59–62 Overall, these results suggest that selected compounds from the series are good inhibitors of AChE activity but not for BuChE activity.
2.5. Molecular docking
Molecular docking is a powerful bioinformatics approach to predict the binding mode of a ligand molecule with the active site of a target protein. Molecular docking provides information such as binding pattern, binding orientation, hydrophobic interaction, hydrogen bond, van der Waals force, electrostatic interaction between the ligand and active site of the target protein.63,64 In this study, the acetylcholinesterase enzyme of Tetronarce californica (TcAChE) and human (hAChE) was used to dock with the designed molecules to understand the binding mechanism between them.65 The active site of TcAChE is almost identical to the hAChE. The only minor difference lies in the mutation of tyr337 in human to phe330 in the TcAChE.66,67 The crystal structure of TcAChE was superimposed with the hAChE and it is observed that these are structurally similar with 0.6 Å RMSD between them (Fig. S34†). Structurally, AChE possesses a catalytic active site (CAS) and a peripheral anionic site (PAS), the commonly understood two binding sites. Molecular docking studies were performed with Maestro v11.9 of Schrödinger software. The X-ray crystal structure of torpedo AChE (TcAChE) in complex with tacrine (PDB ID: 1ACJ) was downloaded from PDB and docked with the designed compounds (1a, 1b, 1c, 2a, 2b, 2c and 3a). Three compounds (2a, 2b and 3a) were able to interact with target protein TcAChE. Molecular docking results revealed that these compounds were able to interact via hydrophobic interaction (within the range of 5 Å) with the CAS and PAS residues of the TcAChE.68 In general, these compounds were enclosed by the active site residues – Asp72, Trp84, Tyr121, Tyr334, Trp279, His440 and Tyr442, which are similar to the native crystal structure interactions. Compound 3a was having a docking score of −10.20, glide energy of −37.93 kcal mol−1 and binding free energy of −30.36 kcal mol−1 and was making π–π interaction with Trp84, Trp279, Phe330 and Tyr334 (Fig. S35†). Similarly, compound 2b was having a docking score of −9.35, glide energy of −46.23 kcal mol−1 and binding free energy of −41.83 kcal mol−1 and was making three π–π interaction with Trp84, Trp279 and Phe330 (Table S3† and Fig. S35†). Compound 2a was docked in the active site the TcAChE with a docking score of −9.34, glide energy of −46.65 kcal mol−1 and binding free energy of −42.29 kcal mol−1 (Fig. 5 and S35†). The quinoline ring, chlorophenyl and phenyl group of 2a compound was interacted via π–π interactions with Trp84 and Phe330 residue of CAS and Trp279 (of PAS), respectively (Fig. 5 and S35†). Rivastigmine, an FDA approved drug is used as control for the in silico study. It was observed that rivastigmine also interacts with active site residues of TcAChE via hydrophobic interaction (Trp84 and Phe330) and one halogen bond interaction (Asp72) with a docking score of −5.59, glide energy of −44.27 kcal mol−1 and binding free energy of −40.51 kcal mol−1 (Table S3†). The careful analysis of molecular docking results of the designed compounds with TcAChE suggests that compound 2a has significant interactions with TcAChE with better binding free energy and glide energy as compared to the other designed compounds and the FDA approved drug rivastigmine which is in good agreement with experimental results (IC50 of 2a 6.2 ± 1.2 μM). | ||
| Fig. 5 (A) Surface representation of the binding mode of compound 2a within the active site of the torpedo AChE and (B) 3D binding mode (via π–π stacking) of compound 2a within the active site of residues of the torpedo AChE. | ||
Further, 2a and rivastigmine (as control) was also docked into the active site of human acetylcholinesterase (hAChE, PDB ID: 6F25) using Maestro v11.9 since AChE of human and torpedo are structurally similar. On a comparative note, a docking score of −8.63, −4.64, glide energy of −71.75 kcal mol−1, −47.71 kcal mol−1 and binding free energy of −70.74 kcal mol−1, -52.22 kcal mol−1, were observed for compound 2a and rivastigmine, respectively. Compound 2a forms one hydrogen bond with Gly82 and π–π stacking with Trp286 and Tyr341 (Fig. 6), whereas rivastigmine made two hydrogen bonds with Phe295 and Arg296 (Fig. S35†). These studies revealed that 2a could be a better AChE inhibitor with hAChE than rivastigmine as it is capable to interact simultaneously with residues of both PAS and CAS of target enzyme.
 | ||
| Fig. 6 (A) Surface representation of the binding mode of compound 2a within the active site of the human AChE and (B) 3D binding mode (via π–π stacking and halogen bond) of compound 2a within the active site of residues of the human AChE. | ||
2.6. Inhibition of AChE-induced Aβ42 aggregation
As previously mentioned, cholinesterase enzyme holds a pivotal role in AD research and these enzymes work in controlling the levels of neurotransmitters in brain. AChE is also known to play a key role in accelerating Aβ aggregation by interaction through PAS of enzyme to form stable AChE–Aβ complex along with presence of BuChE also.69,70 Therefore, inhibitors with dual interactions with both PAS and CAS simultaneously appears to be a better therapeutic strategy as these can combat cognitive dysfunctions as well as prevent complications related to Aβ aggregation.10,29,71–73 To further understand the role of these compounds as dual inhibitors as indicated by the chosen scaffold and also supported by docking results, AChE induced Aβ42 aggregation inhibitory activity was examined using the ThT fluorometric assay. For this study, again same compound from each sub-series was selected (1e, 2a, 3a, 4a and 5) and results of the assay is presented in Fig. 7. Results indicate that these compounds are capable of inhibiting the Aβ42 aggregation in presence of AChE also (inhibition in the range from 40 to 70%). Molecule 5 was the most efficient one in inhibiting the Aβ fibrillation as well as AChE-induced Aβ aggregation. As discussed above also, the ester functionality is expected to increase the bioavailability and binding interactions of molecule 5 with Aβ (and may be AChE) and likely reason for observed inhibitory effect. These results validate the fact that the compound which binds both CAS and PAS of AChE simultaneously could inhibit AChE induced Aβ aggregation. | ||
| Fig. 7 Normalised ThT fluorescence for inhibition of AChE–Aβ fibrillation measured upon incubation at 37 °C for 24 h. Samples are as indicated on top of bar graphs. Conditions: (PBS; [Aβ] = 25 μM; [Compounds] = 25 μM, AChE = 1 unit). | ||
Overall, the study identifies a family of fourteen aryldiazoquinoline molecules that modestly inhibit cholinesterase activity as well as show in vitro anti-aggregation properties. These extended polyaromatic compounds are expected to intercalate in a hydrophobic binding pocket to disrupt aggregation pathway. These probes may be useful for studying the consequences of cholinesterase inhibition for AD progression. Molecular docking results and inhibition of AChE-induced Aβ aggregation demonstrates their dual-site binding with both CAS and PAS which can simultaneously improve cognition and slow the rate of Aβ-elicited neurodegeneration and could be used in future to design potential therapeutics for AD considering its multifactorial nature.
2.7. Cytotoxicity studies
Cell viability can be greatly affected by the subtle changes in the structures of molecules; so studying all the fourteen molecules for their effect on cell survival was attempted. Cell viability studies were performed using Alamar blue assay using Neuro2A cells. Toxicity effects were investigated in a range of 2–20 μM concentration. Interestingly; almost all the molecules exhibit very low cell toxicity. Only two compounds, 3a and 4a showed slight toxicity at higher concentration (20 μM) (Fig. 8 and S36†), all other compounds exhibited a cell survival of >90%. Overall, the cell studies suggested that the series of compounds studied here are almost nontoxic in nature towards Neuro2a cells. | ||
| Fig. 8 Cell viability (% control) upon incubation of Neuro2A cells for representative compounds from each series at 20, 10, 5 and 2 μM concentration. | ||
Since the molecules studied here possess a redox-active azo-function, it may enhance the levels of neurotoxic radical species. Proper management of reactive oxygen/nitrogen species and disposal of damaged cellular components is essential for maintaining regular neuronal function.74,75 To evaluate the antioxidant activities of compounds, DPPH and ABTS assay were performed. Trolox, a vitamin E analogue, is used as a standard in assay protocol. Analysis of results from the DPPH and ABTS assay suggested that these molecules did not lead to any enhancement of free radicals. Overall, the studies suggest that molecular framework may be expected to be used safely in biological settings.
3. Conclusion
A series of fourteen aryldiazoquinoline compounds have been evaluated for their biological potential particularly relevant to Alzheimer's pathology. Interestingly, the molecular framework used here show good inhibition of Aβ aggregation as observed by ThT fluorescence assay and TEM analysis. It was interesting that these molecules are effective inhibitors to fibrillation process, a major key player in AD development. Most of molecules possess a central aryldiazoquinoline moiety, likely responsible for intercalation and inhibition to aggregation. It was also very interesting to observe that most of the molecules exhibited an inhibitory effect on the esterase activity. These compounds inhibited AChE with a moderate activity in low micro molar range. Compound 2a was most potent with IC50 value of 6.2 μM which is remarkable for a non-donepezil or tacrine molecular framework. Variation of substituents suggested that minor change in structure of the molecules could enhance or decrease the AChE activity or Aβ inhibition. It was also satisfying that compounds are quite non-toxic in nature against neuronal cell line and suitable for further investigation in AD research.4. Experimental section
4.1. Materials and methods
All reagents were purchased from commercial sources and used as received unless stated otherwise. Solvents were purified by distillation prior to use. All aqueous solutions and buffers were prepared using commercially available distilled water. The UV-visible spectra were recorded on a Carry-60 UV-Visible Spectrophotometer and data are reported as λmax (nm) absorbance. For the preparation of fibrils and inhibition studies, a laboratory thermomixer from Genetix Biotech Asia was used. All the AChE, BuChE, DPPH and ABTS studies were performed on Elisa reader from ThermoFisher Scientific (MultiSkan Go). Fluorescence measurements were done on Cary Eclipse Fluorescence Spectrophotometer from Agilent. TEM Images were captured using a FEI G2 Spirit Twin, MNIT, Jaipur. Ca(OTf)2 and Bu4NPF6 catalyst were obtained from Sigma-Aldrich and used without further purification. Reactions were performed in flame-dried or oven-dried glassware with magnetic stirring. Reactions were monitored using thin-layer chromatography (TLC) with aluminium sheets silica gel 60 F254 from Merck. TLC plates were visualized with UV light (254 nm), iodine treatment or using ninhydrin stain. Column chromatography was carried out using silica gel 60–120 mesh as stationary phase. NMR spectra were recorded at 500 MHz and 400 MHz (H) and at 125 MHz and 100 MHz (C), respectively on Avance Bruker spectrometer. Chemical shifts (δ) are reported in ppm, using the residual solvent peak in CDCl3 (H: δ = 7.26 and C: δ = 77.0 ppm) as internal standard, and coupling constants (J) are given in Hz.4.2. General procedure for the synthesis
:95, v/v) to obtain the desired product 1d in 67% yield.4.3. Cytotoxicity studies
Mouse neuroblastoma Neuro2a (N2A) cell lines were purchased from the American Type Culture Collection (ATCC). Cells were grown in Dulbecco's Modified Eagle's medium (DMEM)/10% FBS, which is the regular growth media for N2A cells. N2A cells were plated to each well of a 96 well plate with DMEM/10% FBS. The media was changed to serum-free medium. After 1 h, the compounds were added into each well (final volume: 100 μL) with different concentrations (2, 5, 10 and 20 μM), followed by incubation for 24 h at 37 °C. Due to the poor solubility of compounds in water or media, their stock solutions were prepared in DMSO and diluted in media. The final DMSO concentration in this way was less than 1%. Our control experiments suggest that there is no toxicity caused by 2% DMSO. At last, each well was treated with 10 μL of Alamar blue reagent and the cells were incubated for 1 h. The fluorescence (excitation: 560 nm, emission: 590 nm) was measured using an Elisa Reader.4.4. AChE and BuChE assay
Acetylcholinesterase (AChE, Type V-S, lyophilized powder, from electric eel, 1000 unit), acetylthiocholine iodide (ATCI), and 5,5-dithiobis-(2-nitrobenzoic acid) (DTNB) were purchased from Sigma-Aldrich. Potassium dihydrogen phosphate and dipotassium hydrogen phosphate were obtained from Fisher Scientific. To measure, AChE activity, modified Ellman's method was performed.58 The assay was performed using a 96 well plate reader (Thermo Scientific, MultiSkan Go) at 25 °C. The assay was carried out in 0.1 M KH2PO4/K2HPO4 buffer, pH 7.4. The assay solution consists of total 200 μL of solution. The solution contains 20 μL of AChE enzyme of 5 units per mL solution (final conc. 0.5 units per mL), 1.5 μM of dithiobisnitrobenzoate (DTNB) in solution, 0.3 μM of substrate (acetylthiocholine) in solution, 20 μL of samples of various concentrations and 0.1 M potassium phosphate buffer (pH 7.4, 25 °C). A control was also measured containing all other reagents except enzyme and substrate. The activity of AChE was determined spectrophotometrically by the formation of 4-nitrophenol at 405 nm.Enzyme and samples were incubated first for 15 minutes, followed by addition of DTNB and ATCI and the final volume was reached by the addition of buffer and incubated for 20 minutes before the absorbance was recorded. The data was analyzed and IC50 value was calculated using Graph Pad Prism software. Multiple assays were performed and results reported are at least from the data collected in triplicate. BuChE activity was also determined following the same protocol where BuChE enzyme was used in place of AChE.
4.5. Molecular docking studies
The chemical structure of the designed molecules was drawn using Marvin Sketch v19.20 (https://chemaxon.com/products/marvin) computational tool and the 3D orientations were saved in structure-data file (.sdf) file format. “LigPrep” module of Maestro v11.9 of Schrödinger software v2019 was used to prepare the ligand library.76 The ligands were prepared at pH 7.0 ± 0.2 using Optimized Potentials for Liquid Simulations (OPLS) force field.Next, the crystal structures of the target protein acetylcholinesterase of Tetronarce californica (TcAChE, PDB ID: 1ACJ)77 and human (hAChE, PDB ID: 6F25)8 were downloaded from the Protein Data Bank (PDB). The downloaded proteins were prepared individually by the multilevel process by adding hydrogen atoms, assigning bond orders, adding missing amino acids and removing water molecule using the “Protein preparation wizard” in the Prime module of Maestro v11.9.78 The protein structures were optimized and minimized with OPLS force filed at neutral pH. Molecular docking of the designed compounds with TcAChE and hAChE were carried out using Glide extra precision (XP) approach implemented in Maestro v11.9.79 For this purpose, the native ligands (tacrine in TcAChE and C-35 in hAChE), which were present in the active site of the target proteins, were used to generate a receptor grid box around the active site using “Receptor Grid Generation” wizard of Maestro v11.9. Next, the designed compounds were allowed to dock in the defined active site of the target protein with the Glide XP default parameter. Later, the docked file was subjected to calculate binding free energy (ΔG) using Prime MM-GBSA.78 The top-ranked protein-ligand complexes were analyzed carefully by studying docking score, glide energy, MM-GBSA score, hydrogen and hydrophobic interactions.
4.6. Amyloid β peptide experiments
Aβ monomeric films were prepared by dissolving commercial Aβ42 peptide (Anaspec, USA) in HFIP and incubating for 1 h at room temperature. The solution was then aliquoted out and allowed it to evaporate at room temperature. The aliquots were vacuum centrifuged and the resulting monomeric films stored at −80 °C. Aβ fibrils were generated by dissolving monomeric Aβ films in DMSO, diluting into the PBS buffer (pH 7.4), and incubating for 24 h at 37 °C with continuous agitation of 400 rpm (final DMSO concentration was <2%). For inhibition studies, compounds (DMSO stock solutions) 25 μM (final concentration) were added to Aβ solutions (25 μM) along with reference (Aβ alone) and incubated for 24 h at 37 °C with constant agitation of 400 rpm. The percent inhibition of the Aβ aggregation due to the presence of the inhibitor was calculated by the following formula: 100 − (IFi/IFo × 100), where IFi and IFo are the fluorescence intensities obtained for Aβ in the presence and in the absence of the inhibitor, respectively, minus the fluorescence intensities due to the respective blanks.4.7. Inhibition of AChE-induced Aβ aggregation
Inhibition of AChE-induced Aβ aggregation was measured employing a ThT assay. Monomeric Aβ42 film was dissolved in DMSO and diluted into the PBS buffer (pH 7) to a final concentration of 25 μM. For co-incubation experiments, 25 μM solution of Aβ42, and AChE (20 μL, 1 U mL−1, final concentration) in presence and absence of inhibitors (4 μL, 25 μM, final concentration) were incubated for 24 h at 37 °C with continuous agitation of 400 rpm (final DMSO concentration was <2%). Each inhibitor was examined in triplicate. Fluorescence intensities of respective blanks were subtracted in inhibition plot.4.8. Fluorescence measurements
All fluorescence measurements were performed using Agilent Cary Eclipse Fluorescence Spectrophotometer. For ThT fluorescence studies, samples were diluted to 2.5 μM Aβ in PBS containing 10 μM ThT and the fluorescence measured at 485 nm (λex = 435 nm).4.9. DPPH assay
The DPPH assay is a well-known, simple and sensitive technique for determination of radical-scavenging ability. Antioxidant activity of compounds were evaluated using DPPH (1,1-diphenyl-2-picrylhydrazyl) assay. Different concentrations of test compounds in DMSO were prepared and the compound solution was added to the methanolic DPPH solution (0.1 mM). The mixture was kept in the dark for 30 min. Then, the absorbance at 517 nm was measured using a microplate UV/Visible spectrophotometer (ThermoFisher, MultiScan Go).4.10. ABTS assay
ABTS (2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt), potassium persulfate (di-potassium peroxodisulfate), Trolox (6-hydroxy-2,5,7,8-tetramethychroman-2-carboxylic acid), and ascorbic acid were obtained from Sigma-Aldrich (St. Louis, MO, USA). Aqueous solutions were prepared using deionized water obtained from Central Drug House Pvt Ltd. New Delhi, India. Trolox was used as the antioxidant standard. A 4 mM solution of Trolox was prepared in deionized water. ABTS was dissolved in deionized water to a 7 mM concentration. The solution of ABTS radical cation (ABTS˙+) was produced by mixing 7 mM ABTS stock solution with 2.45 mM potassium persulfate aqueous solution in equal quantities and allowing them to react for 12–16 h at room temperature in the dark. For the working solution ABTS˙+ solution was diluted to get final absorbance around 1 at λmax 734 nm. Fresh working ABTS˙+ solution was prepared for each assay. The radical scavenging capacity of the compounds was analyzed by mixing 20 μL of compounds (different concentrations, ranging from 2 to 500 μm) with 80 μL of working solution of ABTS˙+. The decrease in absorbance was measured spectrophotometrically at 734 nm after 1 h of mixing the solutions using a microplate UV/VIS spectrophotometer (ThermoFisher, MultiScan Go).4.11. Transmission electron microscopy (TEM)
Glow-discharged grids (Formar/Carbon 300-mesh, Electron Microscopy Sciences) were treated with preformed Aβ aggregates in absence and presence of compounds for 30 min at room temperature. Excess solution was removed using filter paper and grids were rinsed with distilled H2O. Grids were stained with ammonium molybdate (1% w/v, H2O) for 1 min, blotted with filter paper, and dried overnight at room temperature. Images were captured using a FEI G2 Spirit Twin microscope (200 kV). TEM analysis was performed at the Material Research Centre at MNIT, Jaipur.4.12. Binding with Aβ fibrils
Aβ40 (Anaspec, USA) was used for Aβ-fibril-binding studies. ThT competition assay was followed. For ThT competition assays, a 5 μM Aβ fibril solution with 2 μM ThT was titrated with small amounts of compound and the ThT fluorescence measured (λex/λem = 435/485 nm). For calculation of Ki values, a Kd value of 1.17 μM was used for the binding of ThT to Aβ fibrils.4.13. UV-Vis spectroscopy
UV-Vis spectra for all compounds were recorded on Cary-60 UV-Vis spectrophotometer. The solution for the compounds were prepared from the stock solution of DMSO (2 mM) and diluted in distilled methanol.Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
A. K. S. acknowledges the Department of Science and Technology (DST, Govt. of India) and Science and Engineering Research Board (SERB) for financial support (grant reference number EMR/2016/001452). MR acknowledges CSIR SRF for fellowship (File No. 09/1131(0030)-19-EMR-I).Notes and references
- Y. Guan, Z. Du, N. Gao, Y. Cao, X. Wang, P. Scott, H. Song, J. Ren and X. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- N. Bandara, A. K. Sharma, S. Krieger, J. W. Schultz, B. H. Han, B. E. Rogers and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 12550–12558 CrossRef CAS PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- L. Blaikie, G. Kay and P. Kong Thoo Lin, MedChemComm, 2019, 10, 2052–2072 RSC.
- J. L. Cummings, T. Morstorf and K. Zhong, Alzheimer's Res. Ther., 2014, 6, 37 CrossRef PubMed.
- A. J. Doig, M. P. del Castillo-Frias, O. Berthoumieu, B. Tarus, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen, N. M. Hooper, P. Faller and P. Derreumaux, ACS Chem. Neurosci., 2017, 8, 1435–1437 CrossRef CAS PubMed.
- I. Zueva, J. Dias, S. Lushchekina, V. Semenov, M. Mukhamedyarov, T. Pashirova, V. Babaev, F. Nachon, N. Petrova and L. Nurullin, Neuropharmacology, 2019, 155, 131–141 CrossRef CAS PubMed.
- K. Chand, R. Rajeshwari, E. Candeias, S. M. Cardoso, S. Chaves and M. A. Santos, Metallomics, 2018, 10, 1460–1475 RSC.
- P. Anand and B. Singh, Arch. Pharmacal Res., 2013, 36, 375–399 CrossRef CAS PubMed.
- S. Ayton, P. Lei and A. I. Bush, Free Radicals Biol. Med., 2013, 62, 76–89 CrossRef CAS PubMed.
- N. Giulia, S. Simona, D. Maria and R. Simona, Curr. Top. Med. Chem., 2017, 17, 3062–3079 CrossRef CAS PubMed.
- J. T. Jarrett, E. P. Berger and P. T. Lansbury, Ann. N. Y. Acad. Sci., 1993, 695, 144–148 CrossRef CAS PubMed.
- T. Hartmann, S. C. Bieger, B. Brühl, P. J. Tienari, N. Ida, D. Allsop, G. W. Roberts, C. L. Masters, C. G. Dotti, K. Unsicker and K. Beyreuther, Nat. Med., 1997, 3, 1016 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- S. Sadigh-Eteghad, B. Sabermarouf, A. Majdi, M. Talebi, M. Farhoudi and J. Mahmoudi, Med. Princ. Pract., 2015, 24, 1–10 CrossRef PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, Metallomics, 2013, 5, 1529–1536 RSC.
- M. Rana and A. K. Sharma, Metallomics, 2019, 11, 64–84 RSC.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308–2332 CrossRef.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- A. K. Sharma, J. Kim, J. T. Prior, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Inorg. Chem., 2014, 53, 11367–11376 CrossRef CAS PubMed.
- R. Bartus, R. Dean, B. Beer and A. Lippa, Science, 1982, 217, 408–414 CrossRef CAS PubMed.
- A. Kochi, T. J. Eckroat, K. D. Green, A. S. Mayhoub, M. H. Lim and S. Garneau-Tsodikova, Chem. Sci., 2013, 4, 4137–4145 RSC.
- B. Sameem, M. Saeedi, M. Mahdavi and A. Shafiee, Eur. J. Med. Chem., 2017, 128, 332–345 CrossRef CAS PubMed.
- P. B. Watkins, H. J. Zimmerman, M. J. Knapp, S. I. Gracon and K. W. Lewis, JAMA, J. Am. Med. Assoc., 1994, 271, 992–998 CrossRef CAS PubMed.
- Y. Yun, J. Yang, Y. Miao, X. Wang and J. Sun, Bioorg. Med. Chem. Lett., 2020, 30, 126900 CrossRef CAS PubMed.
- Y. Xu, M.-M. Jian, C. Han, K. Yang, L.-g. Bai, F. Cao and Z.-Y. Ma, Bioorg. Med. Chem. Lett., 2020, 30, 126985 CrossRef CAS PubMed.
- A. Nunes, S. M. Marques, C. Quintanova, D. F. Silva, S. M. Cardoso, S. Chaves and M. A. Santos, Dalton Trans., 2013, 42, 6058–6073 RSC.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed.
- M. Rana, H.-J. Cho, T. K. Roy, L. M. Mirica and A. K. Sharma, Inorg. Chim. Acta, 2018, 471, 419–429 CrossRef CAS PubMed.
- M. L. Bolognesi, A. Cavalli, L. Valgimigli, M. Bartolini, M. Rosini, V. Andrisano, M. Recanatini and C. Melchiorre, J. Med. Chem., 2007, 50, 6446–6449 CrossRef CAS PubMed.
- X. Zhang, X. He, Q. Chen, J. Lu, S. Rapposelli and R. Pi, Bioorg. Med. Chem., 2018, 26, 543–550 CrossRef CAS PubMed.
- P. Ambure, J. Bhat, T. Puzyn and K. Roy, J. Biomol. Struct. Dyn., 2019, 37, 1282–1306 CrossRef CAS PubMed.
- D. Kris Simone Tranches and V. Claudio, Curr. Neuropharmacol., 2014, 12, 239–255 CrossRef PubMed.
- M. R. Jones, C. Dyrager, M. Hoarau, K. J. Korshavn, M. H. Lim, A. Ramamoorthy and T. Storr, J. Inorg. Biochem., 2016, 158, 131–138 CrossRef CAS PubMed.
- A. A. Joshi and C. Viswanathan, Bioorg. Med. Chem. Lett., 2006, 16, 2613–2617 CrossRef CAS.
- D. Audisio, S. Messaoudi, S. Cojean, J.-F. Peyrat, J.-D. Brion, C. Bories, F. Huteau, P. M. Loiseau and M. Alami, Eur. J. Med. Chem., 2012, 52, 44–50 CrossRef CAS PubMed.
- L. Cahoon, Nat. Med., 2009, 15, 356–359 CrossRef CAS PubMed.
- A. M. Mancino, S. S. Hindo, A. Kochi and M. H. Lim, Inorg. Chem., 2009, 48, 9596–9598 CrossRef CAS PubMed.
- Z. Sang, W. Pan, K. Wang, Q. Ma, L. Yu and W. Liu, Bioorg. Med. Chem., 2017, 25, 3006–3017 CrossRef CAS PubMed.
- R. Farina, L. Pisani, M. Catto, O. Nicolotti, D. Gadaleta, N. Denora, R. Soto-Otero, E. Mendez-Alvarez, C. S. Passos and G. Muncipinto, J. Med. Chem., 2015, 58, 5561–5578 CrossRef CAS PubMed.
- J. Zhu, L.-N. Wang, R. Cai, S.-Q. Geng, Y.-F. Dong and Y.-M. Liu, Bioorg. Med. Chem. Lett., 2019, 29, 1325–1329 CrossRef CAS PubMed.
- J. Fu, F. Bao, M. Gu, J. Liu, Z. Zhang, J. Ding, S.-S. Xie and J. Ding, J. Enzyme Inhib. Med. Chem., 2020, 35, 118–128 CrossRef CAS PubMed.
- S. Yaragorla and A. Pareek, Eur. J. Org. Chem., 2018, 1863–1871 CrossRef CAS.
- I. Maezawa, H.-S. Hong, R. Liu, C.-Y. Wu, R. H. Cheng, M.-P. Kung, H. F. Kung, K. S. Lam, S. Oddo, F. M. LaFerla and L.-W. Jin, J. Neurochem., 2008, 104, 457–468 CAS.
- M. M. Picken, Arch. Pathol. Lab. Med., 2010, 134, 545–551 Search PubMed.
- C. Lu, Y. Guo, J. Yan, Z. Luo, H.-B. Luo, M. Yan, L. Huang and X. Li, J. Med. Chem., 2013, 56, 5843–5859 CrossRef CAS.
- D. E. Clark and S. D. Pickett, Drug Discovery Today, 2000, 5, 49–58 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- M. A. Telpoukhovskaia, B. O. Patrick, C. Rodríguez-Rodríguez and C. Orvig, Mol. BioSyst., 2013, 9, 792–805 RSC.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. H. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663–16665 CrossRef CAS.
- N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS PubMed.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- C. Rodriguez-Rodriguez, N. S. de Groot, A. Rimola, A. Alvarez-Larena, V. Lloveras, J. Vidal-Gancedo, S. Ventura, J. Vendrell, M. Sodupe and P. Gonzalez-Duarte, J. Am. Chem. Soc., 2009, 131, 1436–1451 CrossRef CAS PubMed.
- A. Lockhart, L. Ye, D. B. Judd, A. T. Merritt, P. N. Lowe, J. L. Morgenstern, G. Z. Hong, A. D. Gee and J. Brown, J. Biol. Chem., 2005, 280, 7677–7684 CrossRef CAS PubMed.
- A. K. Sharma, J. W. Schultz, J. T. Prior, N. P. Rath and L. M. Mirica, Inorg. Chem., 2017, 56, 13801–13814 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- A. Asghar, M. Yousuf, G. Fareed, R. Nazir, A. Hassan, A. Maalik, T. Noor, N. Iqbal and L. Rasheed, RSC Adv., 2020, 10, 19346–19352 RSC.
- S.-Y. Li, N. Jiang, S.-S. Xie, K. D. Wang, X.-B. Wang and L.-Y. Kong, Org. Biomol. Chem., 2014, 12, 801–814 RSC.
- X. Zuwei, Z. Ning, S. Yu and L. Kunlan, Science, 2001, 292, 1139–1141 CrossRef CAS PubMed.
- X. Li, H. Wang, Z. Lu, X. Zheng, W. Ni, J. Zhu, Y. Fu, F. Lian, N. Zhang, J. Li, H. Zhang and F. Mao, J. Med. Chem., 2016, 59, 8326–8344 CrossRef CAS PubMed.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2011, 7, 146–157 CrossRef CAS PubMed.
- H. Arya, S. B. Syed, S. S. Singh, D. R. Ampasala and M. S. Coumar, Interdiscip. Sci.: Comput. Life Sci., 2018, 10, 792–804 CrossRef CAS PubMed.
- F. Mao, H. Wang, W. Ni, X. Zheng, M. Wang, K. Bao, D. Ling, X. Li, Y. Xu and H. Zhang, ACS Chem. Neurosci., 2018, 9, 328–345 CrossRef CAS.
- H. Dvir, I. Silman, M. Harel, T. L. Rosenberry and J. L. Sussman, Chem.-Biol. Interact., 2010, 187, 10–22 CrossRef CAS PubMed.
- J. Wiesner, Z. Kříž, K. Kuča, D. Jun and J. Koča, J. Enzyme Inhib. Med. Chem., 2007, 22, 417–424 CrossRef CAS PubMed.
- M. A. Telpoukhovskaia, B. O. Patrick, C. Rodríguez-Rodríguez and C. Orvig, Mol. BioSyst., 2013, 9, 792–805 RSC.
- N. C. Inestrosa, A. Alvarez, C. A. Perez, R. D. Moreno, M. Vicente, C. Linker, O. I. Casanueva, C. Soto and J. Garrido, Neuron, 1996, 16, 881–891 CrossRef CAS PubMed.
- A. Alvarez, R. Alarcón, C. Opazo, E. O. Campos, F. J. Muñoz, F. H. Calderón, F. Dajas, M. K. Gentry, B. P. Doctor and F. G. De Mello, J. Neurosci., 1998, 18, 3213–3223 CrossRef CAS PubMed.
- Z. Najafi, M. Mahdavi, M. Saeedi, E. Karimpour-Razkenari, R. Asatouri, F. Vafadarnejad, F. H. Moghadam, M. Khanavi, M. Sharifzadeh and T. Akbarzadeh, Eur. J. Med. Chem., 2017, 125, 1200–1212 CrossRef CAS PubMed.
- M. Harel, L. K. Sonoda, I. Silman, J. L. Sussman and T. L. Rosenberry, J. Am. Chem. Soc., 2008, 130, 7856–7861 CrossRef CAS PubMed.
- A. Castro and A. Martinez, Curr. Pharm. Des., 2006, 12, 4377–4387 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- P. C. Trippier, K. Jansen Labby, D. D. Hawker, J. J. Mataka and R. B. Silverman, J. Med. Chem., 2013, 56, 3121–3147 CrossRef CAS PubMed.
- Schrödinger Release 2019-4, LigPrep, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- M. Harel, I. Schalk, L. Ehret-Sabatier, F. Bouet, M. Goeldner, C. Hirth, P. Axelsen, I. Silman and J. Sussman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9031–9035 CrossRef CAS PubMed.
- Schrödinger Release 2019-4: Prime, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- Schrödinger Release 2019-4: Glide, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05172a |
| This journal is © The Royal Society of Chemistry 2020 |