DOI:
10.1039/D0RA05168C
(Paper)
RSC Adv., 2020,
10, 32490-32496
Synthesis, characterization and computational studies of Zn complex based on the 8-hydroxyquinoline group containing benzimidazole†
Received
12th June 2020
, Accepted 17th August 2020
First published on 2nd September 2020
Abstract
A novel fluorescent zinc complex with 8-hydroxyquinoline containing benzimidazole ligands has been designed and synthesized. Its emission, IR spectroscopy, thermo-gravimetric analysis as well as electrochemical properties have been studied. The solid-state structures were determined via single crystal X-ray diffraction and powder X-ray diffraction. It was found that the ligands around the Zn atoms are distorted by the constrained coordination environment. Computational studies have also been performed to provide insights into the electronic transitions, excited state origins and electrochemical properties of the complex. Based on the observed luminescence phenomena and the quantum chemical calculated results, it was also investigated that the energy transfer mechanism for the luminescence of the complex, which indicated that the ligand structural distortion could cause the blue shift in the emission profile of the complex.
Introduction
Since the reports by Tang et al.,1 organic light-emitting diodes (OLEDs) have received considerable attention due to their application in various displays.2 In the field of luminescence, metal complexes have attracted intensive research interest because of displaying a double role of electron transport and light emission, higher environmental stability, and a much greater diversity of tunable electronic properties by virtue of the coordinated metal center and the ligands. In general, both main group metals and transition metals can be used as the metal centers of the complexes in numerous OLED materials.3 8-Hydroxyquinoline was originally used as a gravimetric reagent to analyse the concentration of metal ions owing to its excellent coordinating abilities.4 Because of the intense luminescence and good stabilities of the resultant complexes such as aluminium tris(8-hydroxyquinolinate) and zinc bis(8-hydroxyquinolinate), they are incorporated in OLED devices as emissive layers.5 For the full-colour display, red, green, and blue emitting materials are essential to apply in OLED devices. Unfortunately, due to the high energy of blue light, those materials developed so far are inferior to green and red in terms of electroluminescence (EL) efficiency and device lifetime. Therefore, the development of materials that emit blue light with a pure colour and high efficiency is still required. Although numerous molecules have been designed and synthesized as the blue emitting materials,6 the metal complexes were rarely reported because of the large conjugation in the molecule and the low energy in the triplet excited state.
In this study, we report the design, synthesis, characterization and crystal structure of a new zinc complex with 8-hydroxyquinoline containing benzimidazole ligands, [Zn2L12]. Compared with the similar complex Zn(BIBQ)2 reported by us,7 the photophysical and electrochemical properties of [Zn2L12] and its ligand have also been studied and supported by computational studies, which have revealed that the structural distortion results in the blue shift of emission.
Experimental
Materials and general methods
All chemicals used were of AR grade. Solvents used for synthesis were distilled over appropriate drying reagents. The FTIR spectra were obtained on a Nicolet AV-360 spectrometer (KBr pellet). NMR spectra were recorded on a Bruker DRX-400 NMR spectrometer at room temperature in CDCl3 using TMS as the internal reference. Lower resolution mass spectra were obtained on an Applied Biosystems Mariner time-of-flight mass spectrometer using an electrospray ionization technique (ESI). The elemental analysis is achieved on a PerkinElmer 2400 microanalyser. Thermogravimetric analysis (TGA) was conducted under nitrogen atmosphere at a heating rate 20 K min−1 with a NETZSCH TG 209 F3 Tarsus. The fluorescence spectra were conducted on a Hitachi F-4600 luminescence spectrometer with a xenon lamp as the light source. Cyclic voltammetry were performed using a CH Instrument, Inc. model CHI 620A electrochemical analyzer. Electrochemical measurements were performed with 0.1 M nBu4NPF6 (TBAH) as a supporting electrolyte at room temperature. The reference electrode used was a Ag/AgNO3 (0.1 M in acetonitrile) electrode and the working electrode used was a glassy carbon electrode (CH Instruments, Inc.) with a platinum wire as the counter electrode. The redox potentials are based on values measured from differential pulsed voltammetry and are reported relative to a ferrocene/ferrocenium (Fc/Fc+) redox couple used as an internal reference.
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) patterns were recorded on an X'pert Philips diffractometer with CuKα radiation (λ = 0.15406 nm). All data were collected at room temperature over the angular 2θ range 5–50° with a step of 0.01° and a counting time of 1.5 s per step.
Single-crystal X-ray diffraction
The diffraction data from a selected single crystal were collected at room temperature on a MAR diffractometer with a 300 mm image plate detector using graphite monochromatized MoKα radiation (λ = 0.71073 Å). The structure was solved by direct methods employing the SHELXS-97 program8 on PC. Zn and many non-H atoms were located according to the direct methods. The positions of the other non-hydrogen atoms were found after successful refinement by full-matrix least-squares using program SHELXL-97 (ref. 9) on PC.
Computational details
All calculations were carried out using Gaussian 09 software package.10 The molecular structure optimization and molecular orbital calculations were conducted using density functional theory (DFT) calculations with a B3LYP functional and 6-31G* basis set. The electronic properties such as HOMO and LUMO energies were determined by time-dependent DFT (TD-DFT) approach.
Syntheses
Synthesis of 2-((2-(8-hydroxyquinolin-2-yl)-1H-benzo[d]imidazol-1-yl)methyl)quinolin-8-ol, L1. Thionyl chloride (1.5 mL, 20 mmol) was added to silica gel (4.5 g) dissolved in 15 mL of anhydrous DCM. The mixture was stirred for 1 h at room temperature under an inert atmosphere of nitrogen. Then, o-phenylenediamine (1.5 g, 14.5 mmol) and 8-hydroxyquinoline-2-carbaldehyde (2.1 g, 7.5 mmol) were added to this suspension. The reaction mixture was stirred overnight and then it was poured into water and made neutral with saturated aqueous sodium hydrogencarbonate solution. The precipitate filtered and washed with EA. The solvent was extracted with EA (3 × 100 mL). The combined organic washed with brine and were dried over anhydrous Na2SO4, filtered and the solvent removed in vacuo. The obtained off-white product was subjected to column chromatography with EA/PE as the eluent to give off-white crystals (yield 60%, 1.65 g). Mp: 161–162 °C; 1H NMR (400 MHz, CDCl3): δ 8.56 (2H, d), 8.28 (2H, d), 8.01 (2H, d), 7.61 (1H, d), 7.50–7.27 (7H, m), 7.21 (1H, d, 7.6), 7.10 (1H, d), 6.40 (2H, s); ESI-MS (m/z): 419 (M + H)+. Elemental analyses (C, H, N) anal. calc. for C26H18N4O2: C, 74.63; H, 4.34; N, 13.39, found: C, 74.55; H, 4.32; N, 13.27.
Synthesis of [Zn2L12], complex 1. A solution of zinc acetate (0.08 g, 0.43 mmol) in methanol (20 mL) was added dropwise to a solution of L1 (0.04 g, 0.095 mmol) in methanol (20 mL) and was kept stirring and refluxed under an inert atmosphere of nitrogen for 0.5 h. This mixture was refluxed for 8 h under constant stirring. On cooling the reaction mixture to 4 °C, a yellow-colored product was precipitated out. It was filtered and washed with excess methanol and water. The product was isolated as yellow solid and further purified by recrystallization to afford crystals. (Yield 52%, 0.023 g). The molecular structures of it was determined via single crystal X-ray diffraction.
Results and discussion
Synthesis and characterization
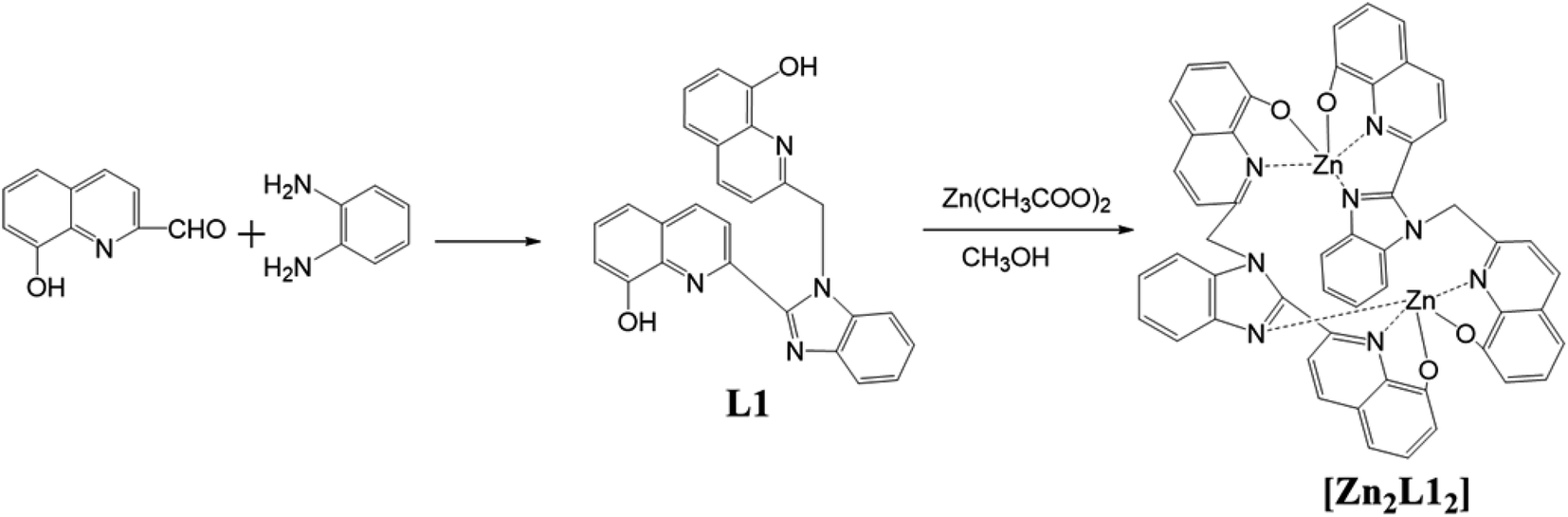
Synthetic route to the ligand, L1, and the complex, [Zn2L12], is shown in Scheme 1. In all these cases, off-white or yellow-colored products were obtained and purified either via column chromatography or by the recrystallization method. The products were characterized via elemental analyses, IR, 1H NMR, mass and X-ray crystallography. Furthermore, these results agree with the proposed structures for the bimetallic complex, which is a coordination dimer featuring each L1 ligand involved in the two Zn(II) atoms both in solution and in the solid phase.
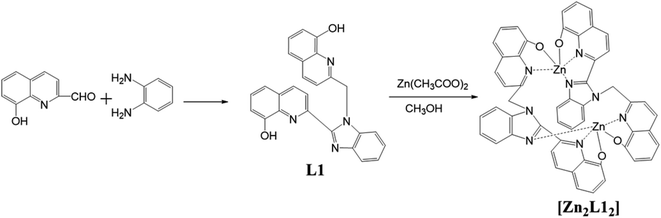 |
| | Scheme 1 Synthesis of the L1 and complex [Zn2L12]. | |
Single crystal X-ray crystallography
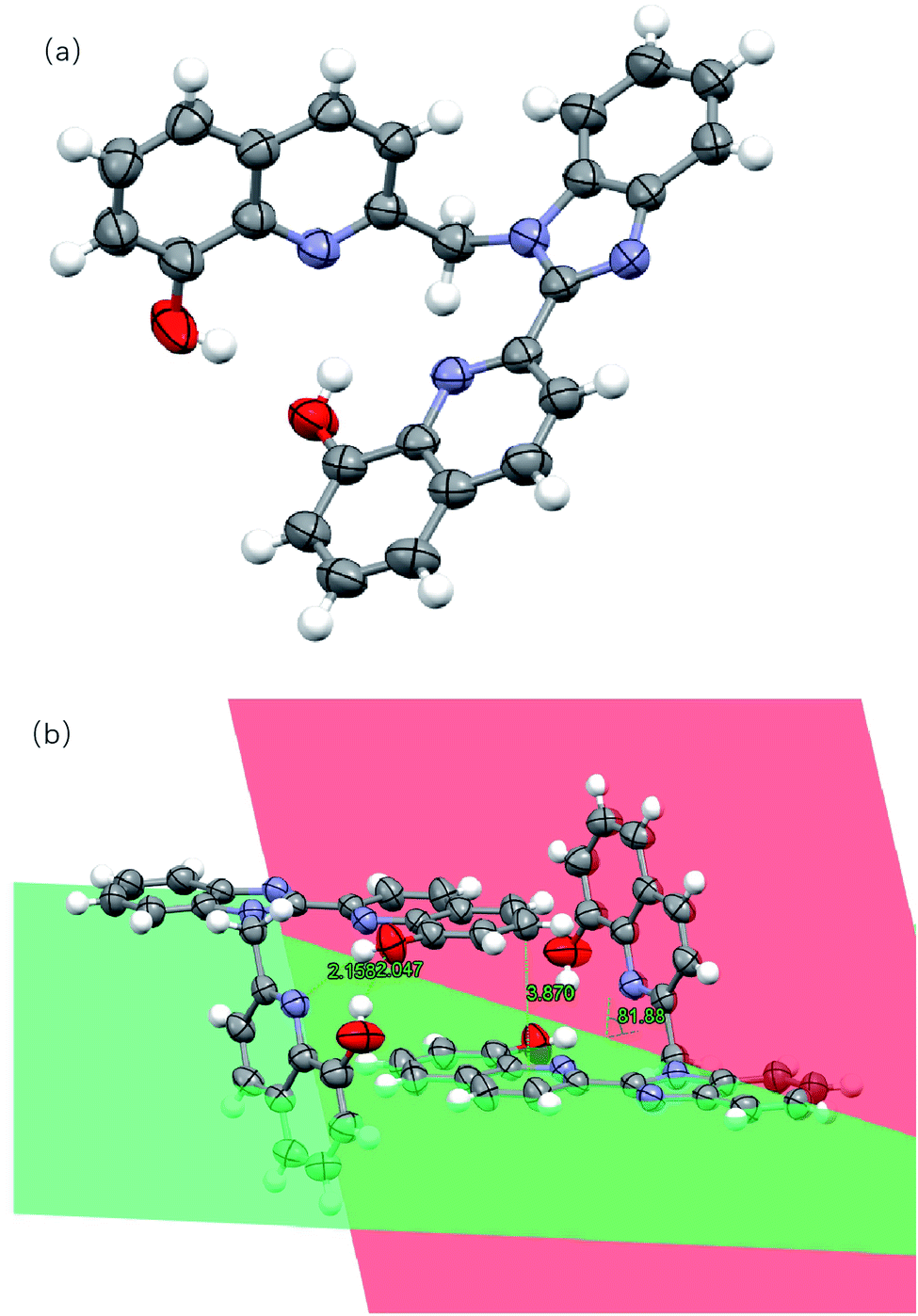
The molecular structures of two compounds were determined via single crystal X-ray diffraction. There are some disorders of solvent molecules in both crystals. The single crystal analysis of L1 shows that it belongs to the monoclinic crystal system with the space group P21/n. The crystallographic and structure refinement data are shown in Table S1,† and the selected bond distances and angles are shown in Table S2.† As shown in Fig. 1, for ligand L1, it was noticed that the benzimidazole ring and one 8-hydroxyquinoline ring are co-planar (represented in green color). The other 8-hydroxyquinoline ring attached to the methylene group (represented in red color) is almost perpendicular to the benzimidazole plane with a dihedral angle of 81.88°. Significant intramolecular hydrogen bonding between O–H⋯O (2.047 Å) and O–H⋯N (2.158 Å) were present. The two monomeric units of L1 are associated to each other through π–π stacking with the distance of 3.870 Å.
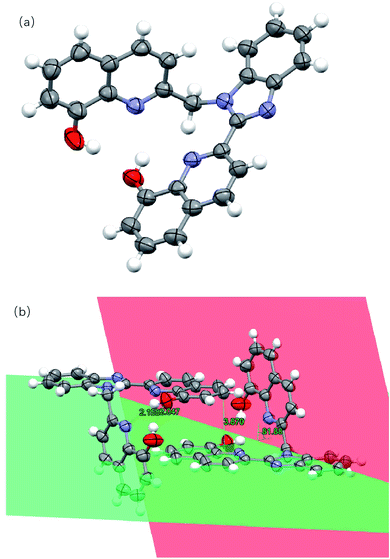 |
| | Fig. 1 (a) Crystal structure of L1. (b) Hydrogen bonds, π–π stacking and dihedral angles between the planes in L1. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are shown at the 30% probability level. (The plane containing the benzimidazole ring and the 8-hydroxyquinoline ring is shown in green, and the plane containing the other 8-hydroxyquinoline ring is shown in red.) | |
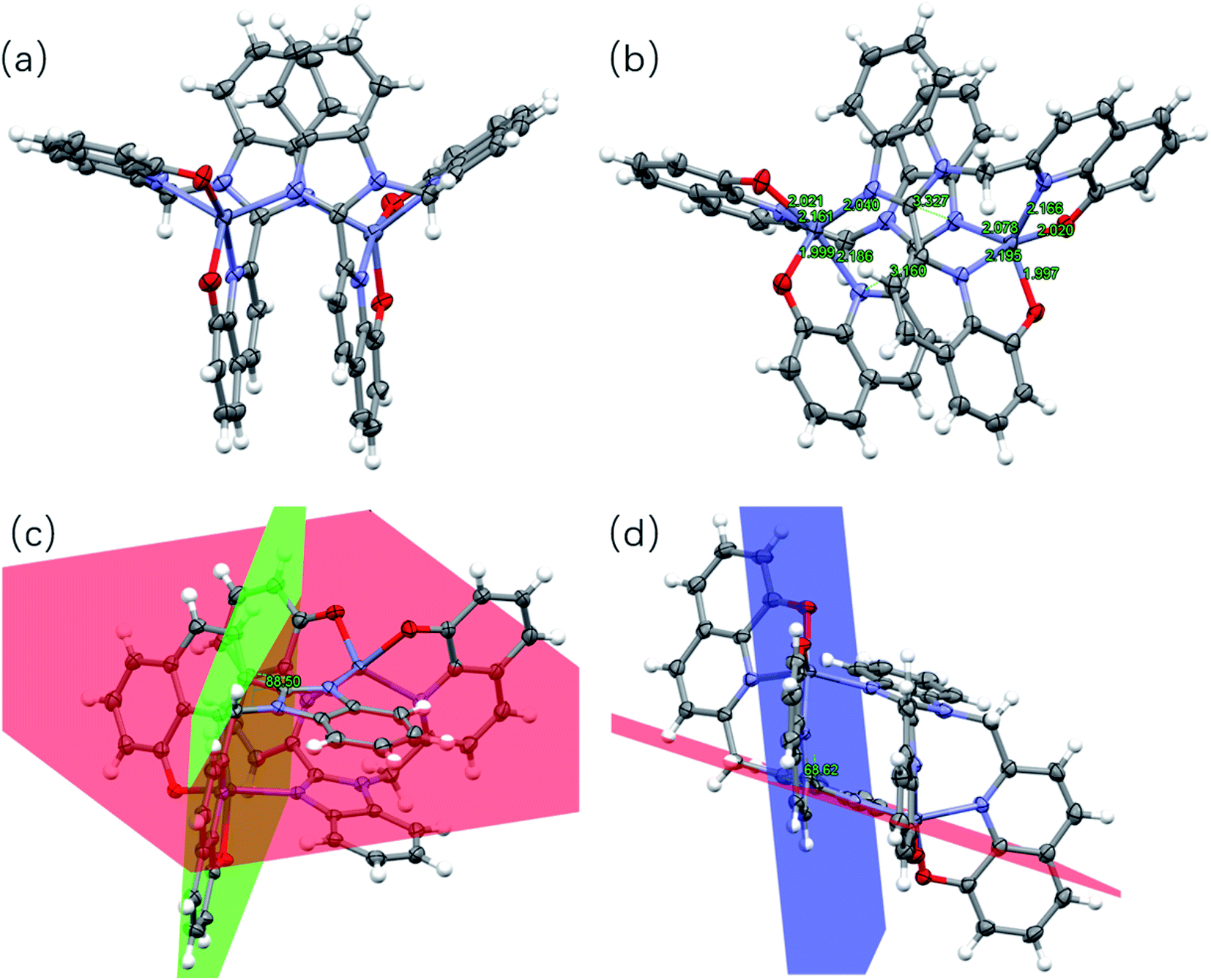
The bimetallic complex [Zn2L12] crystallizes in the triclinic system in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The crystal and structure refinement data are collected in Table S3,† while selected bond distances and angles are tabulated in Table S4.† The X-ray crystal structure of the complex also is shown in Fig. 2. The zinc complex exists as a dimer. Moreover, the bimetallic complex molecule corresponds to a butterfly-shaped with a near C2 rotation axis. Both of the L1 ligands are involved in the Zn atoms. Each Zn center is coordinated by two N atoms and two O atoms from two 8-hydroxyquinoline moieties of the same L1 ligand and one N atom from the benzimidazole of the other ligand as a fifth coordination. The Zn–N and Zn–O bond lengths range from 2.040(3) to 2.195(3) Å and from 1.997(3) to 2.021(3) Å, respectively. These values are within the range of bond distances for similar systems.11 Compared with the L1 crystal, there is a significant dihedral angle of 68.62° (Fig. 2d) between the benzimidazole plane (represented in red color) and the 8-hydroxyquinoline ring (represented in blue color) in the complex. Such a structural distortion is presumably induced by the constrained coordination environment around the Zn atom and led to the benzimidazole and the 8-hydroxyquinoline (no longer co-planar). Moreover, it is notable that the complex involves strong intramolecular π–π stacking interactions. As shown in Fig. 2b, the aromatic rings of pairs of benzimidazoles as well as hydroxyquinolines possess significant offset face-to-face π–π stacking interactions, where the centroid-to-centroid distances are in 3.327 Å and 3.160 Å, respectively.
space group. The crystal and structure refinement data are collected in Table S3,† while selected bond distances and angles are tabulated in Table S4.† The X-ray crystal structure of the complex also is shown in Fig. 2. The zinc complex exists as a dimer. Moreover, the bimetallic complex molecule corresponds to a butterfly-shaped with a near C2 rotation axis. Both of the L1 ligands are involved in the Zn atoms. Each Zn center is coordinated by two N atoms and two O atoms from two 8-hydroxyquinoline moieties of the same L1 ligand and one N atom from the benzimidazole of the other ligand as a fifth coordination. The Zn–N and Zn–O bond lengths range from 2.040(3) to 2.195(3) Å and from 1.997(3) to 2.021(3) Å, respectively. These values are within the range of bond distances for similar systems.11 Compared with the L1 crystal, there is a significant dihedral angle of 68.62° (Fig. 2d) between the benzimidazole plane (represented in red color) and the 8-hydroxyquinoline ring (represented in blue color) in the complex. Such a structural distortion is presumably induced by the constrained coordination environment around the Zn atom and led to the benzimidazole and the 8-hydroxyquinoline (no longer co-planar). Moreover, it is notable that the complex involves strong intramolecular π–π stacking interactions. As shown in Fig. 2b, the aromatic rings of pairs of benzimidazoles as well as hydroxyquinolines possess significant offset face-to-face π–π stacking interactions, where the centroid-to-centroid distances are in 3.327 Å and 3.160 Å, respectively.
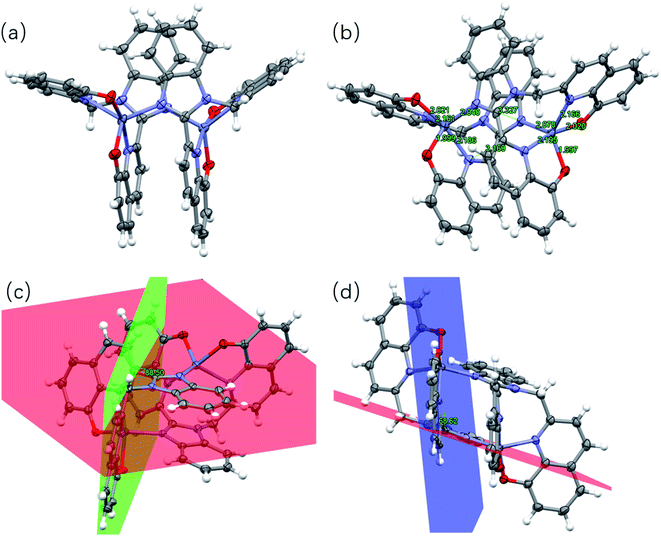 |
| | Fig. 2 (a) Crystal structure of [Zn2L12]. (b) Intramolecular π–π interaction and Zn–N and Zn–O bond in the complex. (c) and (d) Dihedral angles between the planes in the complex. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are shown at the 30% probability level. (The plane containing the benzimidazole ring is shown in red. The plane containing one 8-hydroxyquinoline ring attached to the methylene group is shown in red. The plane containing the other 8-hydroxyquinoline ring is shown in blue.) | |
Powder X-ray diffraction and IR
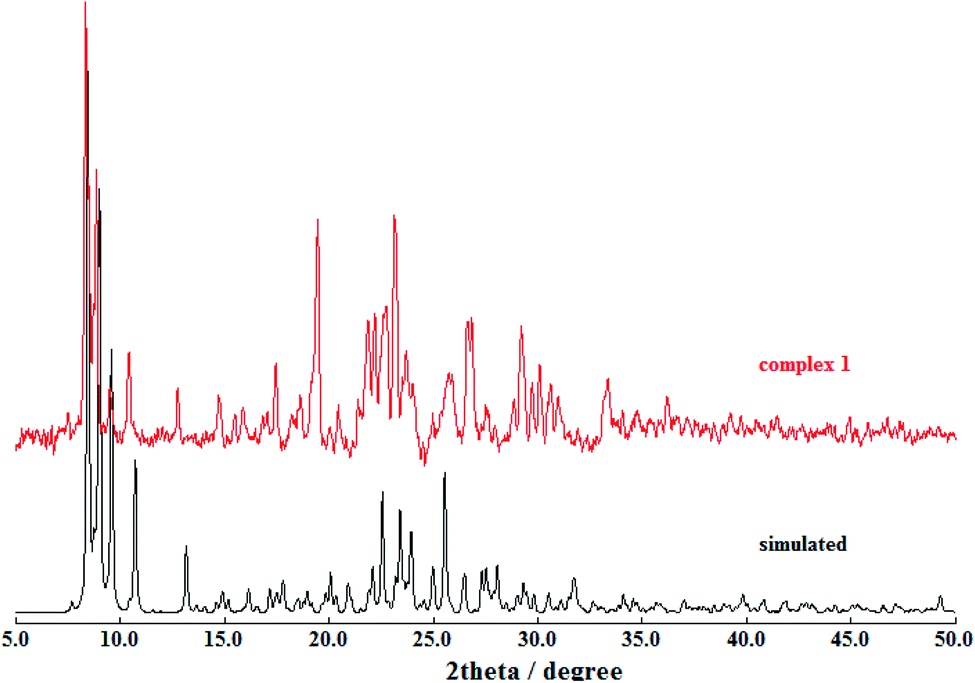
Powder X-ray diffraction (PXRD) patterns were used to verify the phase purity of the bulk sample in the solid state (Fig. 3). The PXRD pattern of the as-synthesized sample of the complex [Zn2L12] and the simulated one are subtly different, which is presumably induced by the solvent molecules in the crystal and similar to the other reports.12
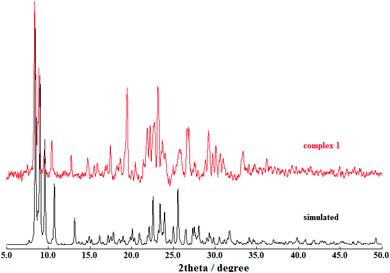 |
| | Fig. 3 Powder XRD patterns of the complex [Zn2L12] (red) compared with the simulated (black). | |
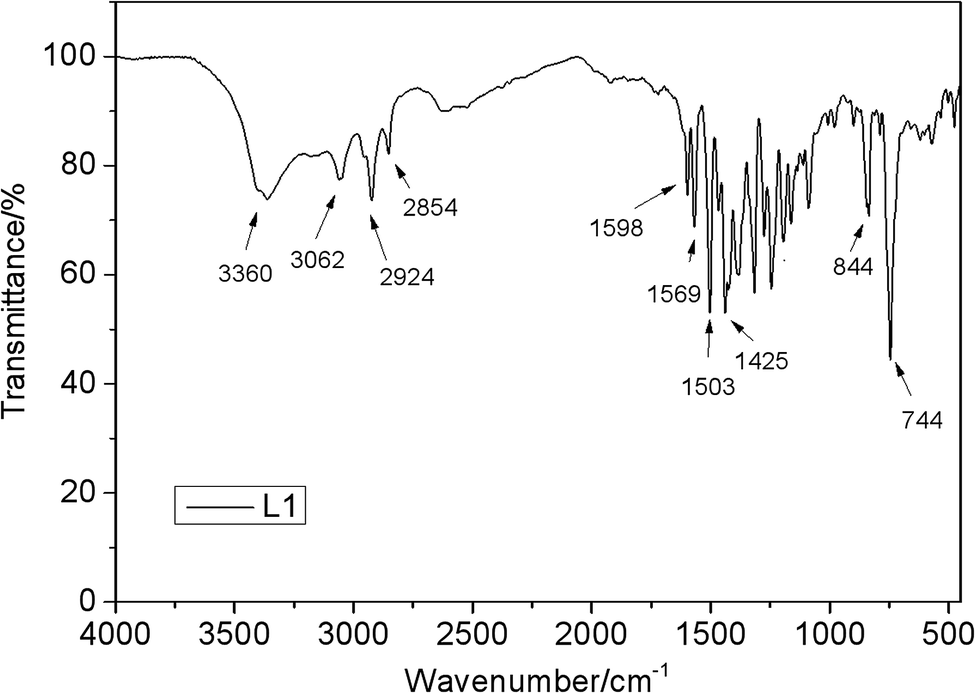
The IR spectra of L1 (Fig. 4) shows one characteristic absorption peak at 844 cm−1, thus verifying the presence of N–H bending vibration. In addition, the absorption peak of O–H is observed at 3360 cm−1. A sharp peak at 744 cm−1 can be assigned to the out-of-plane bending mode of the transvinylene groups, suggesting that the generated double bond is mainly in the aromatic ring configuration.
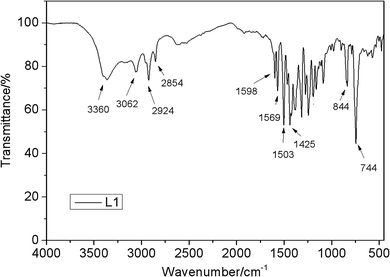 |
| | Fig. 4 FTIR spectra of L1. | |
Electrochemical studies
Electrochemical properties of the complex [Zn2L12] and its ligand L1 were studied by cyclic voltammetry in a dimethyl sulfoxide solution. The electrochemical data for the compounds are given in Table 1, Fig. S2 and S3.† In order to understand the energy levels of molecules, the HOMO (highest occupied molecular orbital) and the LUMO (lowest unoccupied molecular orbital) energy levels of the ligand and the complex were estimated via cyclic voltammetry. The ferrocene/ferricenium redox couple ion (Fc/Fc+) was used as an internal standard.13 The cyclic voltammogram of L1 exhibits one irreversible anodic peak at Epa = 1.07 V and one irreversible cathodic peak at Epc = −2.12 V, which are equal to EHOMO = −5.47 eV and ELUMO = −2.28 eV, respectively. The HOMO and LUMO energy levels were calculated using the equations, EHOMO = −[(EOX − E1/2, ferrocene) + 4.8] eV and ELUMO = −[(Ere − E1/2, ferrocene) + 4.8] eV. It was found from the experiment that one irreversible oxidation event at Epa = 0.75 V and one irreversible reduction event at Epc = −2.06 V for [Zn2L12]. The HOMO and LUMO energy levels of the Zn complex are determined as −5.15 and −2.46 eV, respectively. It is indicated that the energy band gaps of L1 and [Zn2L12] are 3.19 eV and 2.69 eV, respectively. These energy levels are similar to that of the other Zn(II)-hydroxyquinoline systems, BIBQ and Zn(BIBQ)2, which are reported by our group earlier.7
Table 1 Electrochemical data and quantum chemical calculated data for L1 and [Zn2L12]
| |
Epa ox |
EHOMOa |
EHOMOb |
Epc re |
ELUMOc |
ELUMOb |
| The energy levels were calculated via EHOMO = −[(EOX − E1/2, ferrocene) + 4.8] eV. The energy levels were calculated using density functional theory calculations. The energy levels were calculated via ELUMO = −[(Ere − E1/2, ferrocene) + 4.8] eV. |
| L1 |
1.07 V |
−5.47 eV |
−5.66 eV |
−2.12 V |
−2.28 eV |
−1.86 eV |
| [Zn2L12] |
0.75 V |
−5.15 eV |
−4.65 eV |
−2.06 V |
−2.46 eV |
−1.93 eV |
Emission spectra
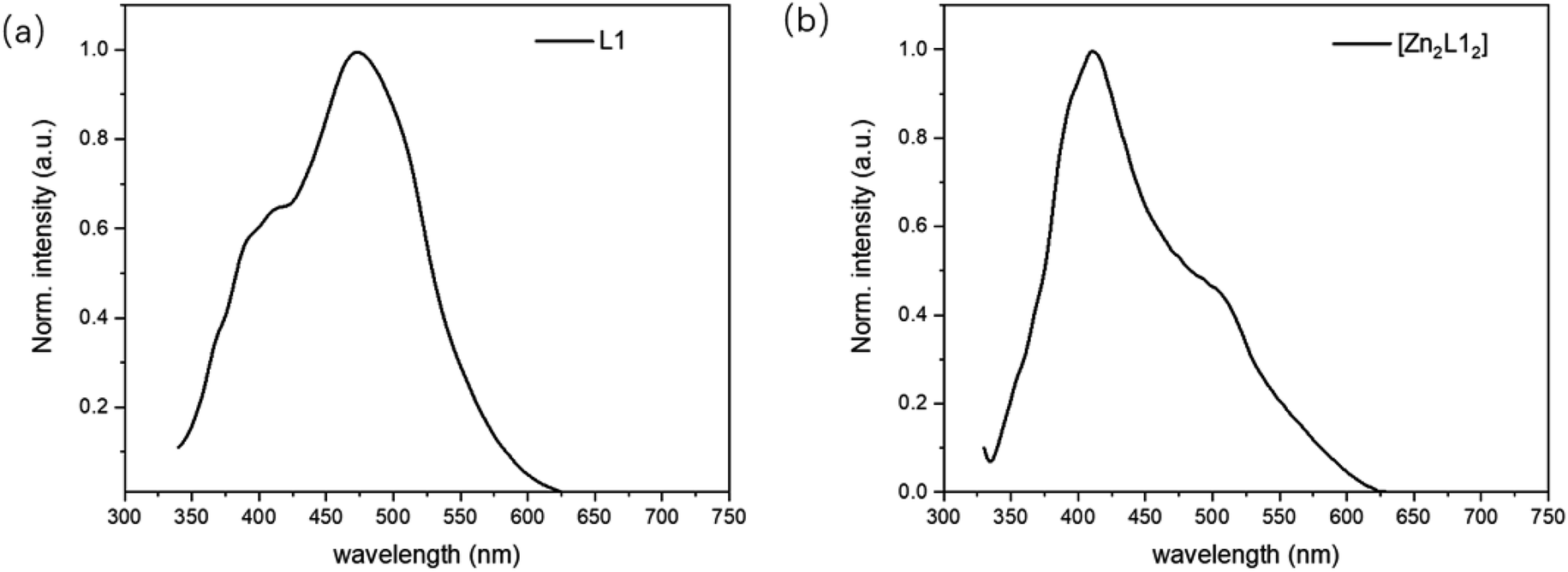
The emission properties of L1 and [Zn2L12] were studied at room temperature in the CH2Cl2 solution (10−5 M) and the fluorescence emission spectra are shown in Fig. 5. The emission spectra of L1 exhibits one emission peak and one shoulder at 472 nm and 406 nm, respectively. Compared with the L1, the corresponding complex [Zn2L12], the energy gap of which is smaller, exhibits the maximum emission peak at 413 nm with a shoulder at 507 nm. This fluorescence property is different from the similar complex Zn(BIBQ)2, which shows a red shift of the emission peak (493 nm) when compared with the ligand BIBQ (404 nm).7 According to the result of computational studies, the origin of the different luminescence behavior of [Zn2L12] has been ascribed to the molecular structural distortion.
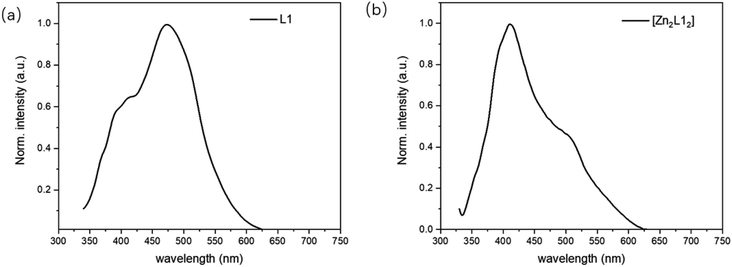 |
| | Fig. 5 PL spectra of (a) L1 and (b) [Zn2L12]. | |
Computational studies
Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations have been performed to provide further insight into the properties and the interrelationship of molecular structures, molecular orbitals, electrochemical and emission of L1, [Zn2L12] and Zn(BIBQ)2.
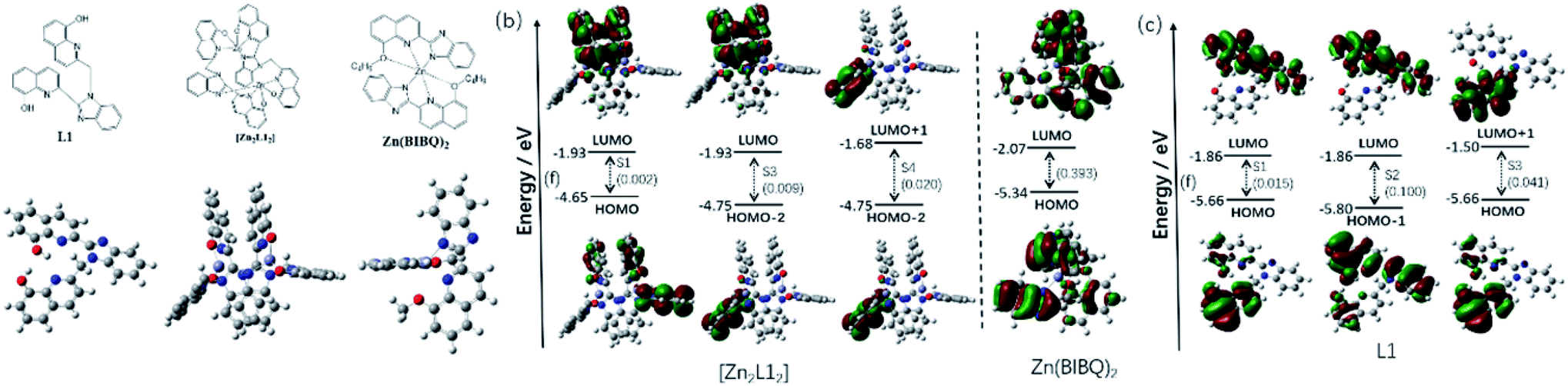
The frontier molecular orbitals of those compounds are shown in Fig. 6b–d. The contours for the HOMO and LUMO obtained from the calculations of the ligand L1 (Fig. 6c) show the HOMO is primarily localized on the 8-hydroxyquinoline, which is attached to the methylene and is non-conjugate with other parts, whereas the LUMO is predominantly on the other conjugation group consisted of the benzimidazole and the other 8-hydroxyquinoline. Therefore, the electronic CT transition in the L1 between the HOMO and LUMO (S1) is restricted with a small oscillator strength (f = 0.015). Being different from the HOMO, the HOMO−1 is mainly localized on the benzimidazole and the coplanar 8-hydroxyquinoline, which is nearly same as LUMO. Therefore, the electronic transition is stronger between the HOMO−1 and LUMO (S2, f = 0.100) than the S1. It was also found that the LUMO+1 is originated from the π* orbital of the 8-hydroxyquinoline, which is non-conjugate with the benzimidazole and coincides with HOMO, resulting in the easier electronic CT transition between the HOMO and LUMO (S3, f = 0.041) than the S1. According to the oscillator strengths of the S1, S2 and S3, the emission peaks at 472 nm and 406 nm of L1 is considered to be comprised of the LUMO to HOMO−1 (f = 0.100) and the LUMO+1 to HOMO (f = 0.041), respectively. The energy values of HOMO and LUMO of L1 also are computed via DFT, which are −5.66 and −1.86 eV respectively.
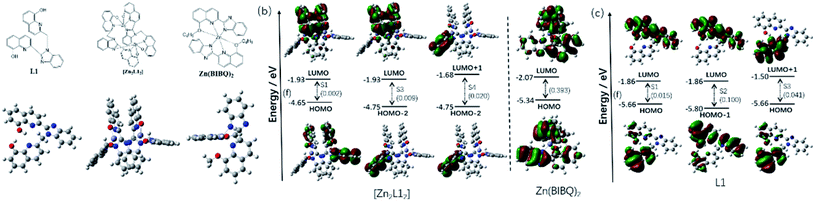 |
| | Fig. 6 (a) Optimized ground-state geometry of L1, [Zn2L12] and Zn(BIBQ)2; (b) excited states of [Zn2L12] and Zn(BIBQ)2 with oscillator strengths and contributing orbitals (B3LYP/6-31G*); (c) excited states of L1 with oscillator strengths and contributing orbitals (B3LYP/6-31G*). | |
As shown in Fig. 6a, there is a significant dihedral angle (67.8°) between the benzimidazole ring and the attached 8-hydroxyquinoline ring in the optimized structure of the complex [Zn2L12], which is also in good agreement with those found in the X-ray crystal structure. The computed energy values of HOMO and LUMO of [Zn2L12] are −4.65 and −1.93 eV respectively. The filled π orbitals and the unfilled orbitals of the complex [Zn2L12] are mainly dominated by orbitals, originating from the L1 ligands (Fig. 6b). The contribution of Zn atoms of all these orbitals is negligible. Due to the significant dihedral angle distorted by the coordination environment in [Zn2L12], the electron cloud in the orbital is difficult to delocalize from the 8-hydroxyquinoline ring to the directly linked benzimidazole ring, which is different from the L1. The electron density of the HOMO is localized mainly on one 8-hydroxyquinoline ring attached to the methylene and inversely the LUMO is localized on another two 8-hydroxyquinoline rings, which are stacked face-to-face to each other. The HOMO−1 and HOMO−2 are localized on the different 8-hydroxyquinoline rings respectively which are in both sides of the complex. Similarly, the LUMO+1 and LUMO+2 are also localized on those 8-hydroxyquinoline rings respectively. It was found that the electronic transition is weak between the HOMO and LUMO (S1) due to the structural distortion and non-conjugation, resulting in a small oscillator strength (f = 0.002) consistent with the weak emission in the long wavelength region. Similarly, the electronic CT transition between the HOMO−2 and LUMO is also restricted (f = 0.009) for the same reason. Due to the fact that the LUMO+1 and HOMO−2 are localized on the same 8-hydroxyquinoline ring, the oscillator strength of the electronic transition between LUMO+1 and HOMO−2 in [Zn2L12] is higher (f = 0.020) than the S1. According to these oscillator strengths of [Zn2L12] and the energy bandgap between LUMO+1 and HOMO−2 (3.07 eV) by the computational study, the short wavelength (413 nm) fluorescence emission peak comprised of the LUMO+1 to HOMO−2 transition for [Zn2L12]. As a comparison, the optimized structure and the frontier molecular orbitals of Zn(BIBQ)2 are shown in Fig. 6. It was found that the benzimidazole ring and the 8-hydroxyquinoline ring are co-planar in the ligand BIBQ, which is different from the complex [Zn2L12]. In addition, the HOMO mainly consists of a π orbital of the benzimidazole moieties and the LUMO is mainly contributed by the π* orbital of the 8-hydroxyquinoline moieties. Therefore, the charge transfer (CT) at λem = 493 nm of Zn(BIBQ)2 (ref. 7) corresponds to the excited state relaxation from LUMO to HOMO with a significant oscillator strength (f = 0.393). Those computed results are in reasonable agreement with the luminescence experiments of the difference of emission peaks wavelength in [Zn2L12], Zn(BIBQ)2 and L1 and support that the structural distortion of the ligand in [Zn2L12] results in a blue shift of the emission.
Conclusions
A novel bimetallic Zn complex [Zn2L12] has been prepared with five-dentate 8-hydroxyquinoline-derived ligands. The molecular structures of [Zn2L12] and ligand L1 obtained by crystallization at room temperature were determined by X-ray diffraction. The single crystal structure of [Zn2L12] shows that the ligands around the Zn atoms are distorted by the constrained coordination environment. The luminescence and the electrochemical properties of L1 and [Zn2L12] have been studied. Compared with the L1, the maximum emission peak of the complex [Zn2L12] shows a blue shift, which is ascribed to the structural distortion. DFT and TDDFT calculation results are also consistent with the experimental observations on X-ray crystallography, emission and electrochemical studies, further confirming their corresponding assignments. This study provides further understanding on the photoluminescence properties of the zinc(II) 8-hydroxyquinoline system and suggests that the complex [Zn2L12] could be exploited for further application as a functional material. Moreover, these results indicated that the ligand structural distortion in the complex is a promising candidate for designing deep blue luminescent complex materials. However, the relationship between the extent of the structure distortion and the luminescence properties of the complex should be known and further studies about those are underway.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Innovation Team Project in Universities of Guangdong Province (Natural Science) (No. 2017GKCXTD006).
References
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
-
(a) S. Welter, K. Brunner, J. W. Hofstraat and L. D. Cola, Nature, 2003, 421, 54 CrossRef CAS;
(b) L. Hung and C. Chen, Mater. Sci. Eng. R., 2002, 39, 143–222 CrossRef;
(c) D. Li, W. Y. Lai, Y. Z. Zhang and W. Huang, Adv. Mater., 2018, 30, 1704738 CrossRef.
-
(a) X. Ma, G. K. Lim, K. D. M. Harris, D. C. Apperley, P. N. Horton, M. B. Hursthouse and S. L. James, Cryst. Growth Des., 2012, 12, 5869 CrossRef CAS;
(b) D. Zhou, W.-P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C.-M. Che, Angew. Chem., Int. Ed., 2020, 59, 6375 CrossRef CAS;
(c) X. Ouyang, G. Wang, H. Zeng, W. Zhang and J. Li, J. Organomet. Chem., 2009, 694, 3511 CrossRef CAS.
- R. A. Chalmers and M. A. Basit, Analyst, 1967, 92, 680 RSC.
-
(a) V. A. Montes, R. Pohl, J. Shinar and P. Anzenbacher, Jr., Chem.–Eur. J., 2006, 12, 523 Search PubMed;
(b) V. A. Montes, C. Pérez-Bolívar, L. A. Estrada, J. Shinar and P. Anzenbacher, Jr., J. Am. Chem. Soc., 2007, 129, 12598 CrossRef CAS;
(c) T. A. Hopkins, K. Meerholz, S. Shaheen, M. L. Anderson, A. Schmidt, B. Kippelen, A. B. Padias, H. K. Hall, Jr., N. Peyghambarian and N. R. Armstrong, Chem. Mater., 1996, 8, 344 CrossRef CAS.
-
(a) H. Lee, H. Jung, S. Kang, J. H. Heo, S. H. Im and J. Park, J. Org. Chem., 2018, 83, 2640 CrossRef CAS;
(b) J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329 CrossRef CAS;
(c) H. Shin, H. Jung, B. Kim, J. Lee, J. Moon, J. Kim and J. Park, J. Mater. Chem. C, 2016, 4, 3833 RSC;
(d) Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678 CrossRef CAS.
- S.-J. Li, Y. Li and J.-A. Zhang, Inorg. Chem. Commun., 2012, 20, 334 CrossRef CAS.
- G. M. Sheldrick and SHELXS97, SHELX97, Programs for Crystal Structure Analysis (Release 97-2), University of Göetingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick and SHELXL97, SHELX97, Programs for Crystal Structure Analysis (Release 97-2), University of Göetingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
-
(a) Y. Mikata, K. Kawata, S. Iwatsuki and H. Konno, Inorg. Chem., 2012, 51, 1859 CrossRef CAS;
(b) F. Gross and H. Vahrenkamp, Inorg. Chem., 2005, 44, 3321 CrossRef CAS;
(c) H. Yan-Ping, Z. Shi-Zheng and H. Sheng, Tetrahedron, 2010, 66, 8635 CrossRef.
-
(a) K. M. Zhao, S. Q. Liu, G. Y. Ye, Q. M. Gan, Z. Zhou and Z. He, J. Mater. Chem. A, 2018, 6, 2166 RSC;
(b) M. T. Zhao, Q. P. Lu, Q. L. Ma and H. Zhang, Small Methods, 2017, 1, 1600030 CrossRef.
-
(a) S. Trasatti, Pure Appl. Chem., 1986, 58, 955 CAS;
(b) G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461 Search PubMed;
(c) Y. Liu, M. S. Liu and A. K.-Y. Jen, Acta Polym., 1999, 50, 105 CrossRef CAS;
(d) B. Dandrade, S. Datta, S. Forrest, P. Djurovich, E. Polikarpov and M. Thompson, Org. Electron., 2005, 6, 11 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further details are given in Fig. S1 and Tables S1–S4. CCDC 2007031 and 2007032. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05168c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Ningning Yuan,
Pengbo Xie,
Jinlan Qin and
Zhengfang Wang
*,
Ningning Yuan,
Pengbo Xie,
Jinlan Qin and
Zhengfang Wang