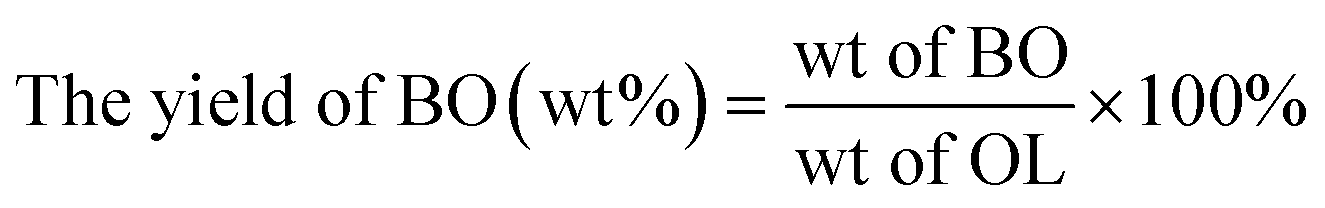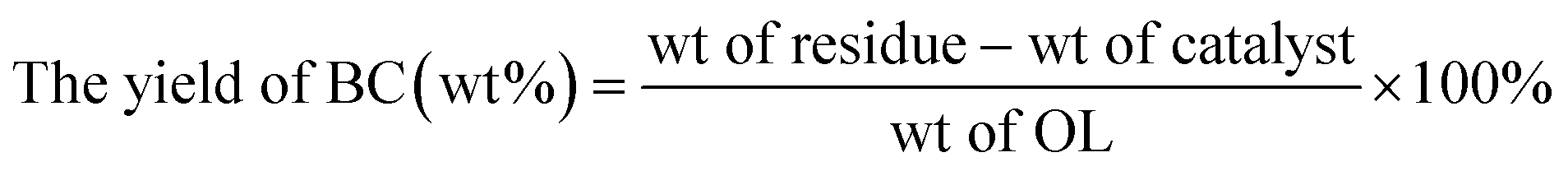DOI:
10.1039/D0RA05069E
(Paper)
RSC Adv., 2020,
10, 31479-31494
Efficient and controllable ultrasound-assisted depolymerization of organosolv lignin catalyzed to liquid fuels by MCM-41 supported phosphotungstic acid†
Received
7th April 2020
, Accepted 14th July 2020
First published on 26th August 2020
Abstract
In this study, effects of catalyst types, reaction temperatures, reaction times, reaction solvents and ultrasound frequencies were carefully investigated to improve the yields and characteristics of various depolymerization products of organosolv lignin. Generally, both catalyst types and ultrasound frequencies played important roles in promoting lignin depolymerization and reducing char yield. In particular, the yield and distribution of phenolic monomer (PM) products were greatly influenced by pore structure and acidity of the catalyst. The optimal reaction condition was got in isopropanol at 310 °C for 6 h with 30% ultrasound frequency and 50% phosphotungstic acid (PTA)/MCM-41 catalyst. The highest yields of PM, bio-oil, liquid fuels and lignin conversion were reached as 8.63 wt%, 86.89 wt%, 95.52 wt% and 98.54 wt%, respectively. The results showed that ultrasound acoustic cavitation could enhance the depolymerization of lignin, thus greatly enhancing production of liquid fuels. Simultaneously, the hydrogen composition and high heating value of various lignin depolymerization products improved, and the oxygen content decreased, indicating that hydrogenation and/or hydrodeoxygenation happened during the depolymerization process. Finally, we also found that the 50% PTA/MCM-41 catalyst had high stability; it could be reused for up to five cycles without loss of catalytic activity.
1. Introduction
With the world's population increasing over the past 50 years, the increasing energy demand of such a population has resulted in depletion of fossil resources, and thus accelerating the huge demand for sustainable resources.1,2 Thus, looking for new avenues towards sustainable and commercial biofuels and bio-products to mitigate the stress of existing fossil resources becomes now a critical theme.3,4 Lignocellulose contains cellulose, hemicellulose and lignin as the main fractions, and it is the most renewable, abundant and cleanest energy of the Earth-based biomass.5 But most of the biomass conversion is mainly cellulose and hemicellulose, and the conversion of lignin is not yet mature.6,7 In this context, lignin is the second most abundant biomass component in nature and is attracting increasing attention of the research community on account of its global abundance and availability.8 Lignin is the only renewable source of aromatics. It is a natural amorphous three-dimensional polymer consisting of methoxylated phenylpropane structures, non-linearly and randomly crosslinked by C–O–C and C–C bonds.9 However, tremendous challenges (such as liquid bio-oil fuels and high-value aromatic chemicals) for lignin valorization via series of biorefinery processes have remained owing to its intrinsic structural complexity and heterogeneity.10,11 Accordingly, there is an urgent need to find effective methods to promote the high value utilization of lignin.
With such an intense research focus in recent years, much effort has been invested toward conversion of lignin into low-molecular-weight high-value aromatic chemicals and liquid bio-oil fuels, which can potentially compete with traditional products derived from petrochemistry. These efforts have led to a number of effective lignin depolymerization approaches, including pyrolysis,12,13 hydrocracking,14,15 depolymerization,16,17 oxidation,1,18 hydrolysis19,20 etc. Among these methods, it has been demonstrated that lignin depolymerization can effectively convert high molecular weight lignin to low molecular weight liquid fuels.16,17 S. Van den Bosch et al. presented a biorefinery process that selectively converts wood sawdust into a carbohydrate pulp and phenolic monomer-rich lignin oil in the presence of a Ru/C catalyst in methanol under H2 atmosphere.21 Chen et al. reported that when using Ru/TiO2 catalyst for lignin depolymerization, the higher temperatures led to higher levels of hydrogenation (71.9 wt%), but more solid coke formed on the catalyst surface for hydrogenation.22 In another report, Song et al. studied birch lignin degradation over Ni catalyst supported on activated carbon in methanol at 200 °C, for 6 h in the presence of 1 bar Ar.23 Under these reaction conditions aryl methoxide undergoes β-elimination over Ni catalyst forming aryl hydrates as primary products. And in the next step, C–O bond cleavage occurred in the aryl hydrates resulting in formation of propylguaiacol and propylsyringol being the final monomeric product totally with 89 wt% selectivity at 54 wt% conversion. Besides, Song et al. also developed an efficient lignin hydrogenolysis process with controllable product distribution using Pd/C catalyst in combination with metal chloride.23 Results showed that lignin produced a high amount of phenols in the presence of CrCl3. Despite some recent progress, many of these reports focus on the noble metal-based catalysis of lignin into small-molecule fragments, and little attention has been paid to the cost of the large-scale use of catalysts used in the reaction. At the same time, the yield of low-molecular-weight liquid bio-oil fuels is only about 50–80 wt%, and some lignin depolymerization products can form more bio-char products, both of which reduce the efficiency of the lignin depolymerization reaction. Hence, in order to solve the cost and efficiency problems of lignin depolymerization, we seek to optimize this process. By comparison, as low-cost noble metal-based catalyst replacements, a number of non-noble metals catalysts, such as W, Fe, Ni–Mo, heteropolyacids and Ni–W2C/AC, have been reported for the lignin depolymerization reaction.24–26 In particular, using heteropolyoxometalates as lignin depolymerization reaction catalysts is considered a good alternative solution. Phosphotungstic acid (PTA) has been used widely in catalysis and homogeneous catalysis due to its good solubility in polar solvents, high proton mobility, strong acidity, and thermal and oxidation stability besides its capability to activate oxidants.27,28 However, owing to the good solubility of PTA in the reaction system, the separation and recovery of PTA will be more difficult. So, the heterogeneity of this catalyst with a solid support will be very important for facile catalyst/product separation as well as for improving the apportionment of the active sites for high catalytic activity.29,30 MCM-41 zeolite is applied as a support for the Keggin phosphomolybdic and vanadium-containing phosphomolybdic acids, which enhances the activity of the prepared catalysts owing to its ability to control both electron- and charge-transfer processes.31,32 In addition, the application of MCM-41 zeolite also stabilizes the charge-transfer state of transient species, and its acidity also plays an important role in the stability of the heteropolyoxometalate structure.33,34 Jha et al. reported that the PTA/MCM-41 catalyst could promoted the formation of phenolphthalein and found that PTA evenly distributed on MCM-41 was a highly active catalyst with high acidity.27 Thereupon, the exploration of the heterogeneity of heteropolyoxometalate catalysts with solid support technologies will advance the impact of lignin susceptibly towards the catalytic depolymerization reaction. Furthermore, among a number of sustainable approaches, ultrasound-assisted technology has proved to be a green process with higher reaction efficiency and lower production cost.35,36 When ultrasound with sufficient intensity is transmitted through a liquid medium, micron-sized bubbles are generated that oscillate violently in response to acoustic pressure fluctuations, i.e. acoustic cavitation, thereby promoting chemical reactions. Many studies have shown potential benefits of ultrasound technology in various processes, such as hydrolysis of biomass,37,38 cleavage of chemical bonds such as those find in lignocellulosic biomass and thermal degradation.39,40 Other studies have also shown that due to its cavitation effects, the use of ultrasound can reduce biomass hydrolysis time by 80%,41 which is useful for the production of biofuels. More interestingly, ultrasound has been shown to be able to cleave chemical bonds in biomasses, thereby facilitating the extraction of compounds of interest, such as cellulose, hemicellulose or lignin.36,37
In the current work, we aim to effectively convert organosolv lignin (OL) to liquid fuels (LF) with a higher heating value and a higher yield. We first prepared MCM-41-supported PTA solid acid catalysts with different contents of PTA. Then, effects of the catalyst types, reaction temperatures, reaction times, reaction solvents and ultrasound frequencies were carefully investigated to improve the yields and characteristics of various depolymerization products. Finally, we also found that the 50% PTA/MCM-41 catalyst had high stability, and it could be reused for up to five cycles without loss of catalytic activity. Based on the results of the study, this method is more promising than other previous methods of depolymerization, the ultrasound acoustic cavitation enhancing the depolymerization of lignin and greatly enhancing production of liquid fuels. And the correlations amongst the catalyst types and phenolic monomer distributions will also furnish a helpful theoretical basis for lignin valorization in future biorefinery processes.
2. Materials and methods
2.1 Materials
In this study, all chemicals were used directly without any treatment. The OL was prepared from bagasse using our previously reported method.42 Methanol, ethanol, isopropanol, cetyltrimethylammonium bromide (CTAB), tetrahydrofuran (THF), and hydrochloric acid (HCl) were purchased from Aladdin Reagent Company (Shanghai, China). PTA, tetraethyl orthosilicate (TEOS), HZSM-5, SBA-15, MCM-22, NaOH, dimethyl sulfoxide (DMSO) and dichloromethane (DCM) were purchased from Sinopharm Chemical Reagent Co. Ltd.
2.2 Preparation of catalysts
The MCM-41 catalyst was produced by hydrothermal synthesis as described in previous reports.27,28 In a typical synthesis, firstly, 2.4 g NaOH, 146 g deionized water and 5.96 g CTAB were mixed together for 0.5 h at 25 °C. Then, after a homogeneous mixture was formed, a 14 g portion of TEOS was added drop by drop under stirring. The resulting reaction mixture was stirred for 5 h and was transferred into a Teflon-lined autoclave which was subsequently heated to 100 °C for 48 h. The resulting solid product was recovered by filtration, washed several times with deionized water and dried at 70 °C overnight. Finally, the template was removed by calcination of the as-synthesized sample at 550 °C in air for 6 h.
The different contents of PTA (10 wt%, 30 wt% and 50 wt%) impregnated on the MCM-41 support were prepared by a wet impregnation method. Firstly, the calculated amount of PTA was dissolved in 40 mL of methanol, and MCM-41 was slowly added to the mixed solution. Secondly, the mixture was kept for 12 h at 25 °C under stirring conditions and the mixture was evaporated on a rotary evaporator. Finally, the solid product was calcined at 350 °C for 5 h. According to the initial weight concentration of PTA in the MCM-41 support, the finally obtained different PTA/MCM-41 catalysts were named as 10% PTA/MCM-41, 30% PTA/MCM-41 and 50% PTA/MCM-41, respectively.
2.3 Characterization of catalysts
X-ray diffraction (XRD) was performed using a Shimadzu XRD-6100 diffractometer with Cu-Kα radiation (λ = 1.5406 nm) of 40 kV and 30 mA.43 Diffuse reflectance UV-visible spectra of the different catalysts were recorded using a PerkinElmer Lambda 18 spectrophotometer equipped with a Praying-Mantis diffuse reflectance attachment.44 Infrared spectroscopic analysis was performed using a Fourier transform infrared (FT-IR) spectrometer over the range of 400–4000 cm−1 at a resolution of 4 cm−1 with 64 scans.44 Pyridine-FT-IR analysis was conducted using a Shimadzu FT-IR-8700 spectrometer.45 The total acidity of the different catalysts was measured by temperature-programmed desorption of ammonia (NH3-TPD, Micromeritics, USA) with an on-line thermal conductivity detector.44 The microstructure and elemental distribution of the different catalysts were characterized by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) (JEOL JSM 7800F).5
2.4 Ultrasound-assisted depolymerization of organosolv lignin
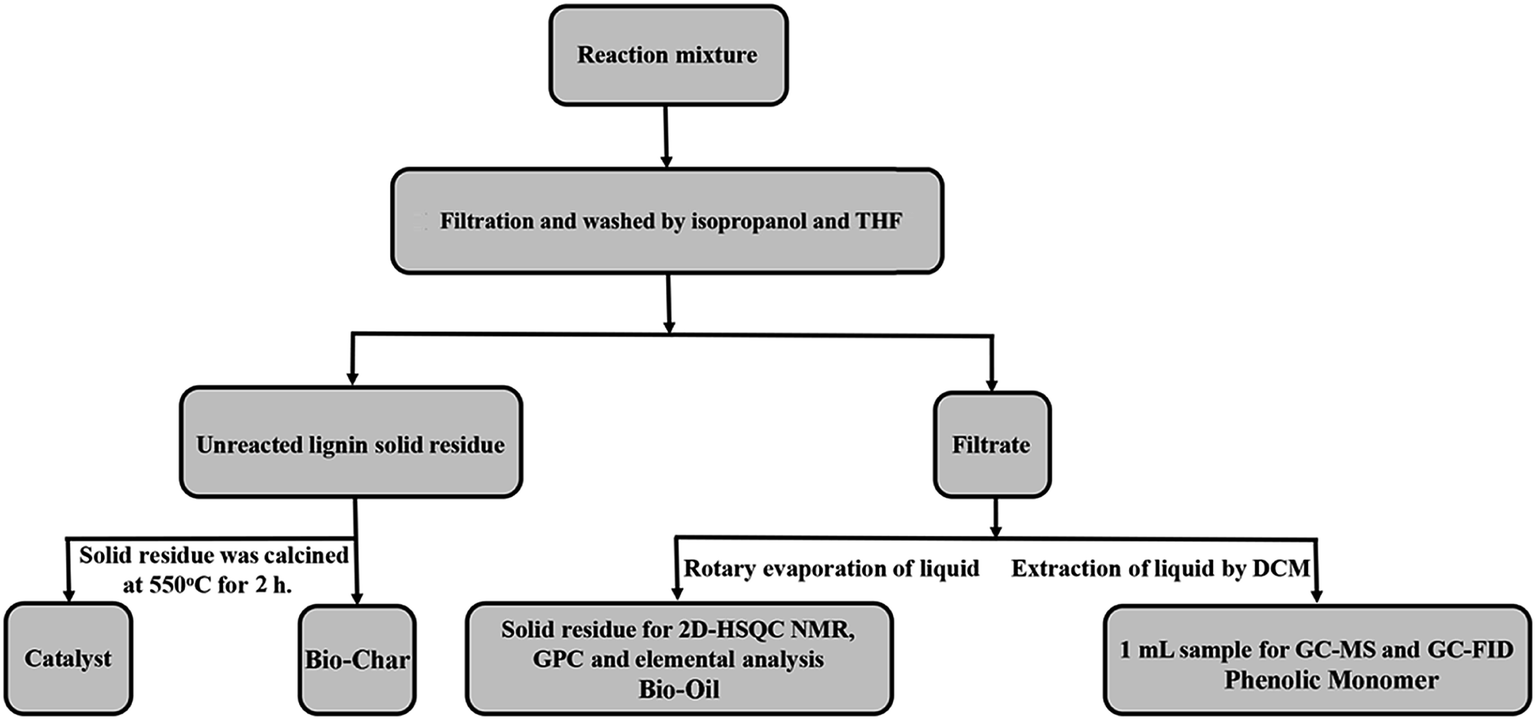
The ultrasonic-assisted depolymerization of OL experiment was conducted in a 100 mL stainless steel autoclave reactor equipped with a pressure gauge, a pressure relief valve, an ultrasound wave generator and a stirrer. 0.5 g OL, 0.25 g catalyst and 60 mL isopropanol/water solvent (v/v = 30/30 mL) were loaded into the reactor. The ultrasonic-assisted depolymerization was conducted under different reaction conditions. After the reaction, the reactor was rapidly quenched to room temperature in a water bath. A scheme of the depolymerization product separation is provided in Fig. 1. The detailed steps are shown in the ESI.†
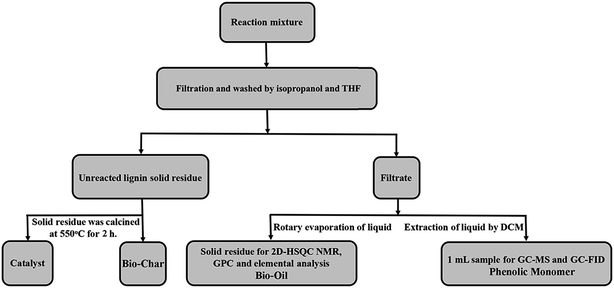 |
| | Fig. 1 Procedure for mixture separation after lignin ultrasound-assisted depolymerization reaction. | |
2.5 Characterization of organosolv lignin and various depolymerization products
The phenolic monomer (PM) depolymerization products were identified by gas chromatography-mass spectrometry (GC-MS) (Agilent 7890/5978, USA) using a 30 m × 0.25 mm × 0.25 μm capillary column (HP-5MS).1,46 The quantitative analysis was based on a 1 mL gas chromatography-flame ionization detector (GC-FID).1,46 The yields of PM, bio-oil (BO), bio-char (BC), LF and lignin conversion (LC) were calculated using the following equations:| |
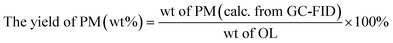 | (1) |
| |
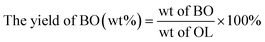 | (2) |
| |
 | (3) |
| | |
The yield of LF (wt%) = (1) + (2)
| (4) |
| | |
The yield of LC (wt%) = (3) + (4)
| (5) |
The 2D heteronuclear single quantum coherence nuclear magnetic resonance (2D-HSQC NMR) spectra were obtained with a Bruker Biospin (Billerica, MA) AVANCE 500 MHz spectrometer fitted with a cryogenically cooled 5 mm DCH gradient probe.8 THF was used as the mobile phase and the average molecular weight was recorded by gel permeation chromatography (GPC).47 Elemental analysis for the content of carbon, hydrogen, nitrogen, sulfur and oxygen was conducted using a Flash 2000 series CHNSO analyzer (Thermo Scientific, MA, USA).25
3. Results and discussion
3.1 Analysis of catalysts
The physicochemical properties of different types of catalysts are listed in Table 1. Compared with HZSM-5, MCM-22 and SBA-15, since the pore size of MCM-41 is larger, PTA can be better considered as present inside the pores and on the surfaces of the MCM-41 support.27,44,45 Furthermore, the BET surface areas, pore volumes and pore sizes of all the PTA/MCM-41 catalysts are reduced. This indicates that some pores of the MCM-41 support are blocked by PTA. In particular, as the content of PTA increases, the specific surface areas of the different PTA/MCM-41 catalysts become progressively smaller. As is shown in the Table 1, the order of catalyst specific surface area from large to small is MCM-41 > SBA-15 > MCM-22 > HZSM-5 > 10% PTA/MCM-41 > 30% PTA/MCM-41 > 50% PTA/MCM-41 > PTA.
Table 1 The physicochemical properties and acidities of different catalysts
| Entry |
Catalyst |
SBETa (m2 g−1) |
Pore volumeb (cm3 g−1) |
Pore sizec (nm) |
Total acidityd (mmol g−1) |
Brønsted aciditye (μmol g−1) |
Lewis aciditye (μmol g−1) |
B/Le |
| Specific surface area calculated by the BET method. Total pore volume determined by N2 adsorption at a relative pressure of 0.99. Mean pore diameter obtained from the desorption isotherm by the BJH method. The amount of acid sites was calculated by quantifying the desorbed NH3 from NH3-TPD. The amount of acid sites was determined by quantifying the desorbed pyridine from pyridine FT-IR spectra. The structural changes of the catalysts after being regenerated 1, 3, and 5 times, respectively. |
| 1 |
PTA |
13 |
— |
— |
0.36 |
64.94 |
1.02 |
63.55 |
| 2 |
MCM-41 |
663 |
0.86 |
7.85 |
0.08 |
0.44 |
4.88 |
0.09 |
| 3 |
HZSM-5 |
332 |
0.137 |
2.18 |
0.04 |
0.38 |
3.97 |
0.09 |
| 4 |
MCM-22 |
400 |
0.16 |
2.84 |
0.05 |
0.23 |
3.32 |
0.07 |
| 5 |
SBA-15 |
630 |
0.85 |
5.63 |
0.05 |
0.27 |
3.41 |
0.08 |
| 6 |
10% PTA/MCM-41 |
495 |
0.28 |
5.92 |
0.16 |
41.58 |
6.92 |
6.01 |
| 7 |
30% PTA/MCM-41 |
348 |
0.23 |
4.57 |
0.29 |
53.17 |
5.67 |
9.38 |
| 8 |
50% PTA/MCM-41 |
218 |
0.19 |
3.39 |
0.42 |
61.94 |
4.31 |
14.38 |
| 9 |
50% PTA/MCM-41 (1)f |
201 |
0.17 |
3.28 |
0.39 |
59.85 |
4.48 |
13.36 |
| 10 |
50% PTA/MCM-41 (3)f |
186 |
0.12 |
3.01 |
0.36 |
54.38 |
4.71 |
11.55 |
| 11 |
50% PTA/MCM-41 (5)f |
165 |
0.09 |
2.61 |
0.32 |
46.24 |
4.73 |
9.77 |
Table 1 also tabulates the detailed concentration of Brønsted and Lewis acidic sites of different types of catalysts. It can be seen that all the PTA/MCM-41 catalysts have higher Brønsted acidity than MCM-41, HZSM-5, MCM-22 and SBA-15, and their Lewis acidity is greater than that of PTA. Simultaneously, we also find that PTA mainly contains Brønsted acidity, while MCM-41, HZSM-5, MCM-22 and SBA-15 mostly have Lewis acidity. Because of the presence of different content of PTA on the MCM-41 support, the different PTA/MCM-41 catalysts have acidities of both Brønsted acid sites and Lewis acid sites. Especially, according to the detailed pyridine FT-IR spectra of the different PTA/MCM-41 catalysts in Fig. S1(a),† the peak at 1548 cm−1 is attributed to Brønsted acidic sites, and the Lewis acidic sites have two peaks at 1595 and 1445 cm−1.27,44,45 And the peak at about 1486 cm−1 is ascribed to both Brønsted and Lewis acidic sites.27,44,45 Besides, in order to calculate the total strength of acid sites in all catalysts, NH3-TPD analysis results are also reported in Table 1. MCM-41, HZSM-5, MCM-22 and SBA-15 show a low acidity. And PTA has strong acidic sites. In Fig. S1(b),† the different PTA/MCM-41 catalysts demonstrate two broad desorption peaks at a temperature ranging between 130 °C and approximately 650 °C (including low-temperature and high-temperature desorption peaks). Obviously, the different PTA/MCM-41 catalysts have the acid sites of both PTA and MCM-41. But with further increase of PTA content to 50 wt%, the 50% PTA/MCM-41 catalyst shows the highest total amount of acidity. In general, the size of acidic sites is an important indicator determining the activity of the catalyst. Indeed, catalysts with higher acidity also have much better catalytic performance.27,44,45
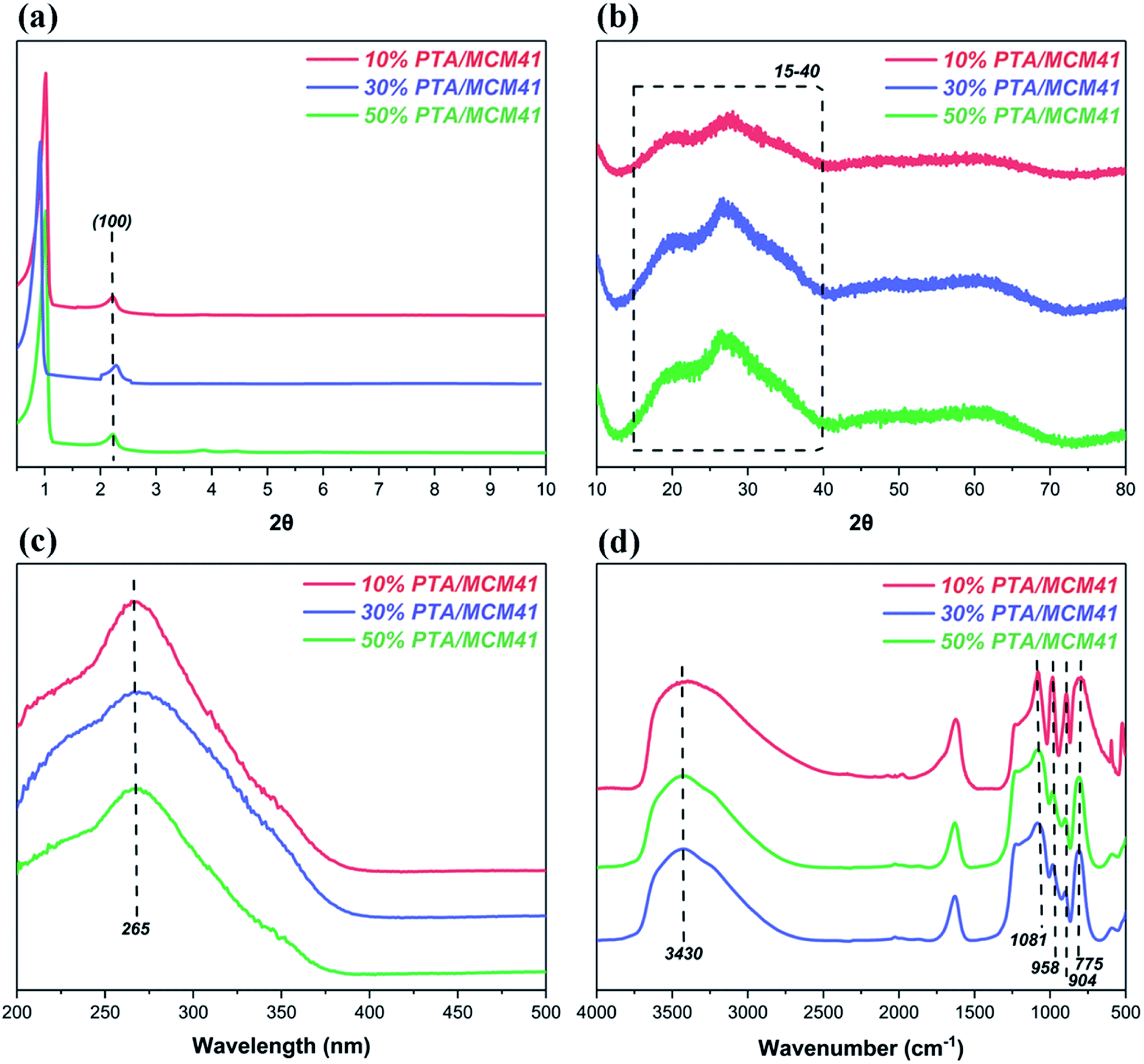
Finally, the XRD patterns, diffuse reflectance UV-visible spectra and FT-IR spectra of the different PTA/MCM-41 catalysts are given in Fig. 2. As shown in Fig. 2a, all PTA/MCM-41 catalysts show low-angle XRD patterns similar to that of MCM-41.48,49. But the (100) diffraction intensity of PTA/MCM-41 catalysts declines with increasing PTA. This is because an increase in PTA loading develops disorder into the hexagonal structure of the MCM-41 support.49 The wide-angle XRD patterns of the different PTA/MCM-41 catalysts are presented in Fig. 2b. It is clear that all PTA/MCM-41 samples also show wide-angle XRD patterns similar to that of MCM-41.49,50 No separate crystal phase characteristic of PTA exists in any of the PTA/MCM-41 catalysts due to the larger surface areas of the MCM-41 support. This also confirms that PTA can better absorb into the MCM-41 support and the different PTA/MCM-41 catalysts are successfully synthesized.27,50 Fig. 2c illustrates the UV-visible diffuse reflectance spectra of all PTA/MCM-41 catalysts. The different PTA/MCM-41 catalysts show an absorption peak at 265 nm, proving the existence of PTA on the MCM-41 support.45 And Fig. 2d depicts the FT-IR spectra of all PTA/MCM-41 catalysts. Comparing PTA/MCM-41 catalysts with PTA reveals that the different PTA/MCM-41 catalysts have vibration bands at 1081, 958, 904 and 775 cm−1 similar to PTA, indicating that PTA has been incorporated into the MCM-41 framework.45,50 However, the addition of PTA to the MCM-41 support shifts and reduces the intensity of these characteristic vibration bands, and some of these PTA peaks overlap with those of MCM-41. Thus, we can conclude that the different PTA/MCM-41 catalysts are successfully synthesized.
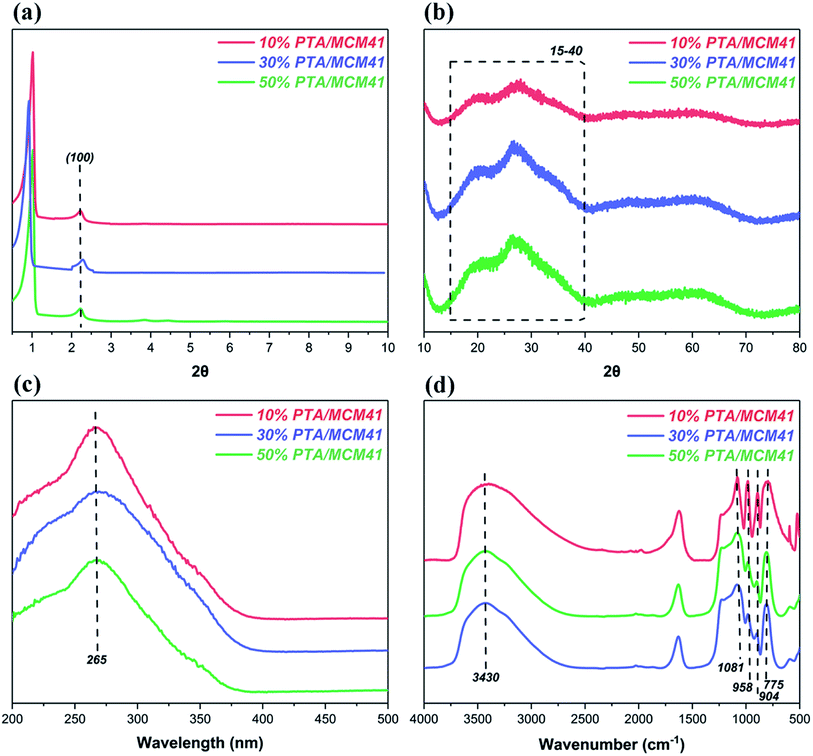 |
| | Fig. 2 The characterization of different PTA/MCM-41 catalysts: (a) low-angle XRD patterns, (b) wide-angle XRD patterns, (c) UV-visible spectra and (d) FT-IR spectra. | |
3.2 Analysis of organosolv lignin
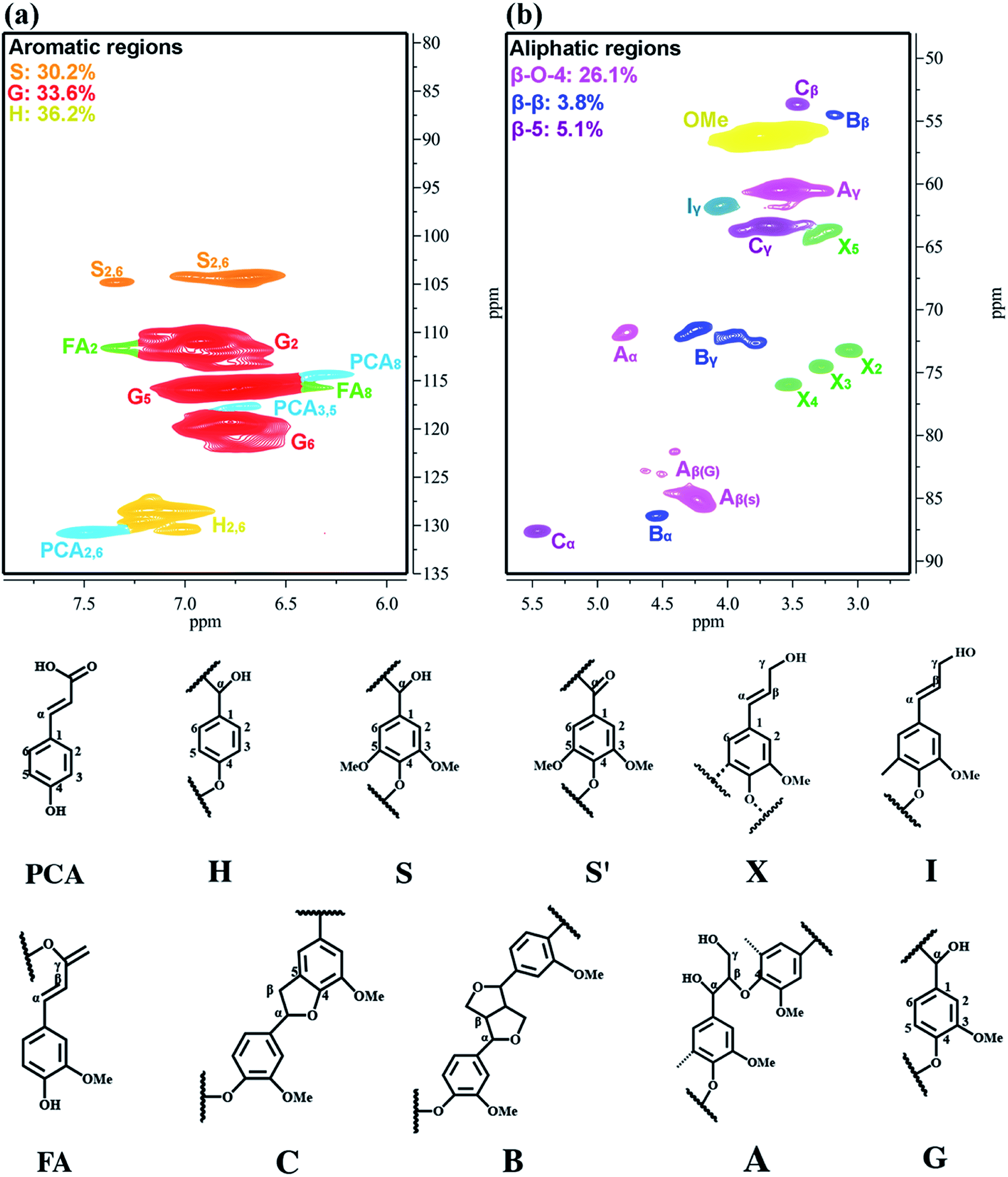
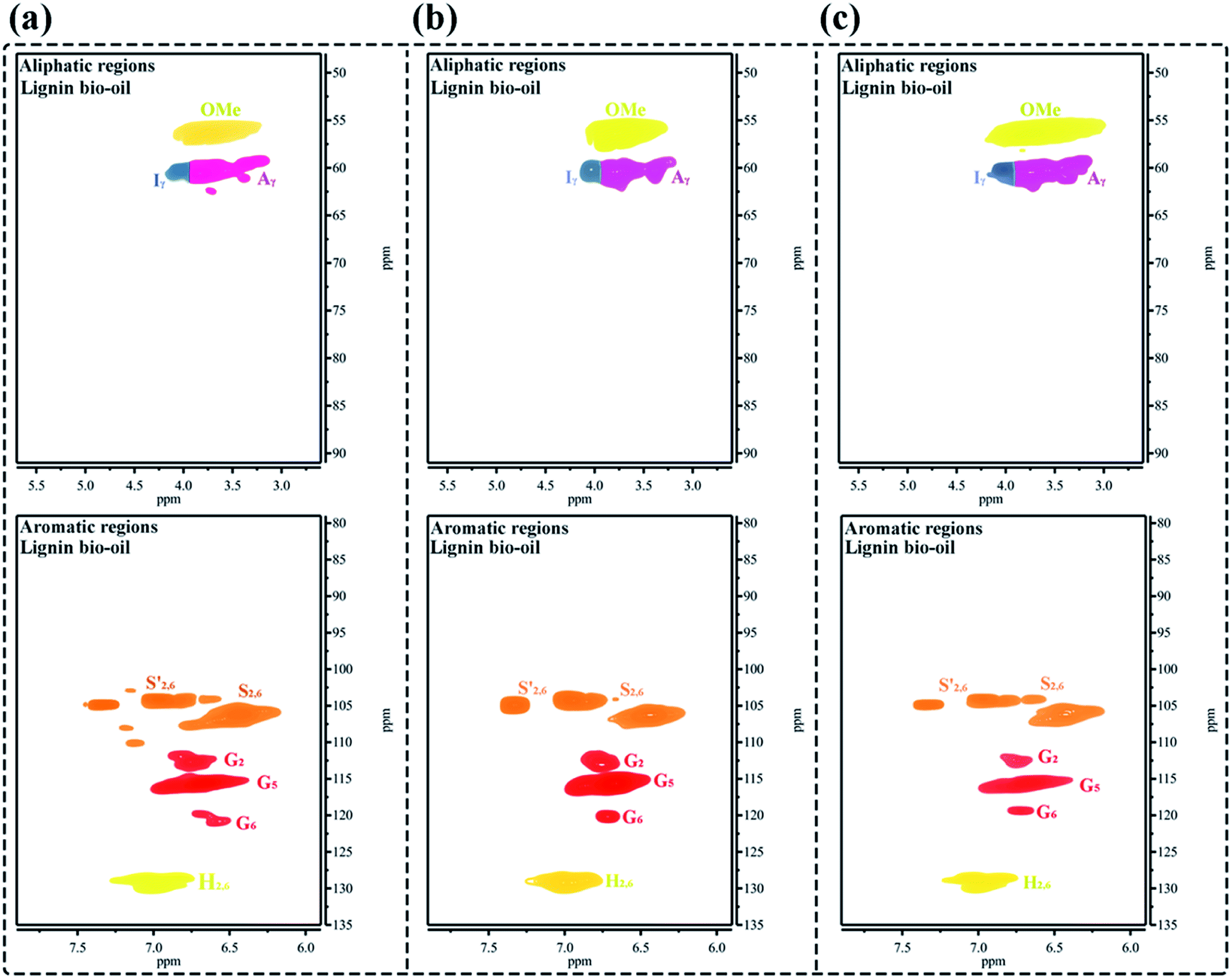
The 2D-HSQC NMR analysis of OL in this study is given in Fig. 3 and the spectra are annotated with peak assignments based on previous publications (Table S1†).51,52 The amount of H, G and S units significantly observed in the aromatic region are 36.2%, 33.6%, and 30.2%, respectively, suggesting that OL is a typical S–G–H type grass lignin.42 Moreover, the methoxy groups (OMe), β-O-4 aryl ether linkages (A), β–β linkages (B) and β–5 linkages (C) also show prominent correlations in the aliphatic regions at δC/δH = 56.4/3.71 (OMe), 85.8/4.12 (Aβ(S)), 83.4/4.38 (Aβ(G)), 53.5.7/3.07 (Bβ) and 53.1/3.46 (Cβ), respectively. According to the computing method of the literature, the different internal linkages and aromatic units also can be expressed by a comparative mode.5 The content of β-O-4 aryl ether linkages in OL is 26.1/100 Ar, while the contents of β–β linkages and β–5 linkages are 3.8/100 Ar and 5.1/100 Ar, respectively. Wang et al. reported that lignin had more β-O-4 aryl ether bonds, which could mean relatively easier preparation of PM and low-molecular-weight lignin bio-oil.43 Additionally, the carbon, hydrogen, oxygen and nitrogen contents of OL are presented in Table S2,† which indicates that the OL contains a higher amount of oxygen (44.95 wt%) and lower hydrogen content (5.36 wt%). It has a relatively low heating value HHV (19.08 MJ kg−1) with a higher O/C ratio (0.82) and lower H/C ratio (0.08), due to the high oxygen levels. This result indicates that the value-added utilization of low heating value OL is urgently needed to produce liquid fuels or fine chemicals.53,54 Table S3† also shows the molecular weight (Mw) of OL determined by GPC analysis. In general, the higher the contents of β-O-4 aryl ether bonds, the larger the Mw of lignin.54,55 As expected, the OL has a relatively high Mw of 4750 g mol−1, and the polydispersity index (PDI) of OL is 3.08.
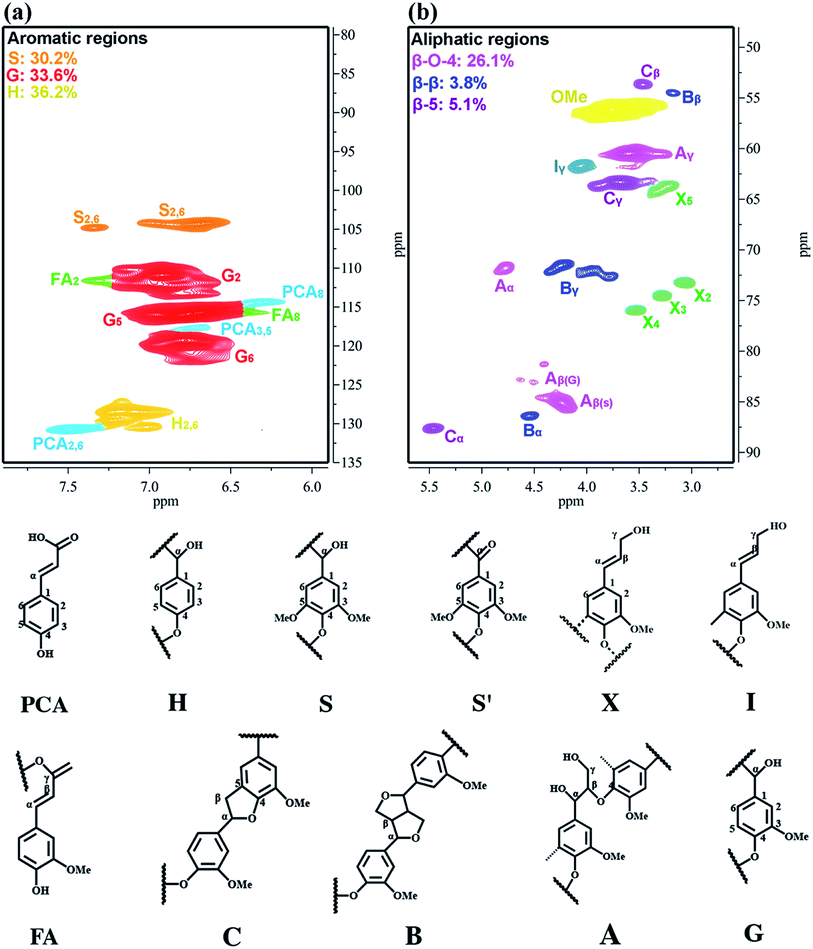 |
| | Fig. 3 The 2D-HSQC NMR spectra of organosolv lignin: (a) aromatic regions and (b) aliphatic regions. | |
3.3 Catalytic depolymerization of organosolv lignin in isopropanol
After the effective preparation of the different PTA/MCM-41 catalysts, they were applied to catalytic depolymerization of OL to produce PM and low-molecular-weight BO depolymerization products. Firstly, as shown in Table 2, the catalytic depolymerization of OL is performed at 310 °C for 6 h in isopropanol in order to study the effect of different catalysts on the various depolymerization product yields and distribution regularities. It is found that the decomposition of OL generates three main products: volatile products (namely the PM), nonvolatile products (namely the BO) and BC. GC-MS and GC-FID are used to analyze the volatile products. The blank experiment carried out without catalysts shows relatively low PM and BO yields of only 0.41 wt% and 40.27 wt%, respectively. Most of the lignin is converted to BC, yielding 45.08 wt%, which is considered insufficient to produce low-molecular-weight LF efficiently. In contrast, under the same reaction conditions, results shown in Table 2 indicate that all catalysts provide better results comparing with the blank runs (without catalyst), which indicate the positive effect of catalysts on the lignin depolymerization, such as higher yields of PM and BO depolymerization products. A superior OL conversion exceeding 80% is observed for molecular sieve catalysts (Table 2, entries 3–6). In the presence of MCM-41, HZSM-5, MCM-22 and SBA-15 catalysts, PM is formed with yields of 4.57 wt%, 4.31 wt%, 3.94 wt% and 4.18 wt%, respectively, although the liquefaction degree changes a little. This indicates that the larger mesoporous surface area of molecular sieve catalysts is beneficial to the conversion of lignin. It indicates that the larger mesoporous surface area of molecular sieve is not only conducive to the dispersion of the catalyst, but also beneficial to the conversion of lignin (Tables 1 and 2). However, the different content of PTA supported on MCM-41 zeolite supports has been shown to be very active in the catalytic lignin depolymerization process (Table 2, entries 7–9). In particular, with increasing PTA content, lignin depolymerization becomes much easier. LC and LF yields both exceeding 85 wt% are observed for 10% PTA/MCM-41, 30% PTA/MCM-41 and 50% PTA/MCM-41 catalysts. This can be interpreted in that the larger surface area of the MCM-41 support is not only conducive to the dispersion of the catalyst, but also beneficial to the liquefaction conversion of lignin (Tables 1 and 2).44,45,55 The highest LF and LC yields are obtained for the 50% PTA/MCM-41 catalyst (90.99 wt% and 96.25 wt%), followed closely by the 30% PTA/MCM-41 catalyst (88.09 wt% and 94.15 wt%) > 10% PTA/MCM-41 catalyst (86.61 wt% and 93.59 wt%). These results indicate that the trends of various depolymerization product yields (PM and BO) are positively correlated with the total acidity of all catalysts, which may be due to the higher total acidity of the catalyst promoting the continuity reaction of the lignin macromolecular fragments,27,55 eventually forming more small-molecule fragments, PM and BO depolymerization products. Zhou et al. reported the effect of different types of solid acid catalysts on the yields of aromatic monomers from lignin.55 Their results indicated that most of the aromatic monomers were obtained inside the higher acidity sites of catalysts. On the other hand, compared with different PTA/MCM-41 catalysts, the molecular sieve support has almost negligible activity, which is attributed to its extremely low concentration of acidic sites since the number of acidic sites of the catalyst is a more powerful factor than its surface area (Table 1).27,45 Simultaneously, PTA shows the lowest activity compared to the supported PTA catalysts despite its strong Brønsted acidity since its particularly small surface area can lead to the limited availability of acidic sites on the surface of the catalyst (Table 1).27,45 Thus, when PTA is dispersed on the MCM-41 support, the number of active sites on the surface of the catalyst considerably increases, thereby leading to the higher activity of the different PTA/MCM-41 catalysts.
Table 2 The effect of different catalysts on lignin depolymerization product yields and distributionsa
| Entry |
Catalyst |
YPMb (wt%) |
YBO (wt%) |
YLF (wt%) |
YBC (wt%) |
YLC (wt%) |
| H-types |
G-types |
S-types |
Total |
| Reaction conditions: 0.5 g OL, 0.25 g catalysts, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h. Determined by GC-MS and GC-FID, where n-tetradecane was used as internal standard. The catalytic performance of the catalysts after being regenerated 1, 3, and 5 times, respectively. Reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C, 6 h and 30% ultrasonic frequency. |
| 1 |
None |
0.12 |
0.15 |
0.14 |
0.41 |
40.27 |
40.68 |
45.08 |
85.76 |
| 2 |
PTA |
1.41 |
2.44 |
1.31 |
5.16 |
80.56 |
85.72 |
5.12 |
90.84 |
| 3 |
MCM-41 |
1.62 |
1.69 |
1.26 |
4.57 |
77.16 |
81.73 |
8.03 |
89.76 |
| 4 |
HZSM-5 |
1.53 |
1.43 |
1.35 |
4.31 |
76.24 |
80.55 |
9.24 |
89.79 |
| 5 |
MCM-22 |
1.17 |
1.26 |
1.51 |
3.94 |
70.85 |
74.79 |
11.48 |
86.27 |
| 6 |
SBA-15 |
1.49 |
1.38 |
1.31 |
4.18 |
73.13 |
77.31 |
10.47 |
87.78 |
| 7 |
10% PTA/MCM-41 |
1.93 |
2.43 |
1.32 |
5.68 |
80.93 |
86.61 |
6.98 |
93.59 |
| 8 |
30% PTA/MCM-41 |
2.08 |
2.94 |
1.39 |
6.41 |
81.68 |
88.09 |
6.06 |
94.15 |
| 9 |
50% PTA/MCM-41 |
2.53 |
3.07 |
1.52 |
7.12 |
83.87 |
90.99 |
5.26 |
96.25 |
| 10 |
50% PTA/MCM-41 (1)c |
2.48 |
2.99 |
1.48 |
6.95 |
82.01 |
88.96 |
5.53 |
94.49 |
| 11 |
50% PTA/MCM-41 (3)c |
2.43 |
2.95 |
1.45 |
6.83 |
81.24 |
88.07 |
5.62 |
93.69 |
| 12 |
50% PTA/MCM-41 (5)c |
2.41 |
2.84 |
1.42 |
6.67 |
80.52 |
87.19 |
5.98 |
93.17 |
| 13 |
50% PTA/MCM-41d |
2.93 |
3.76 |
1.94 |
8.63 |
86.89 |
95.52 |
3.02 |
98.54 |
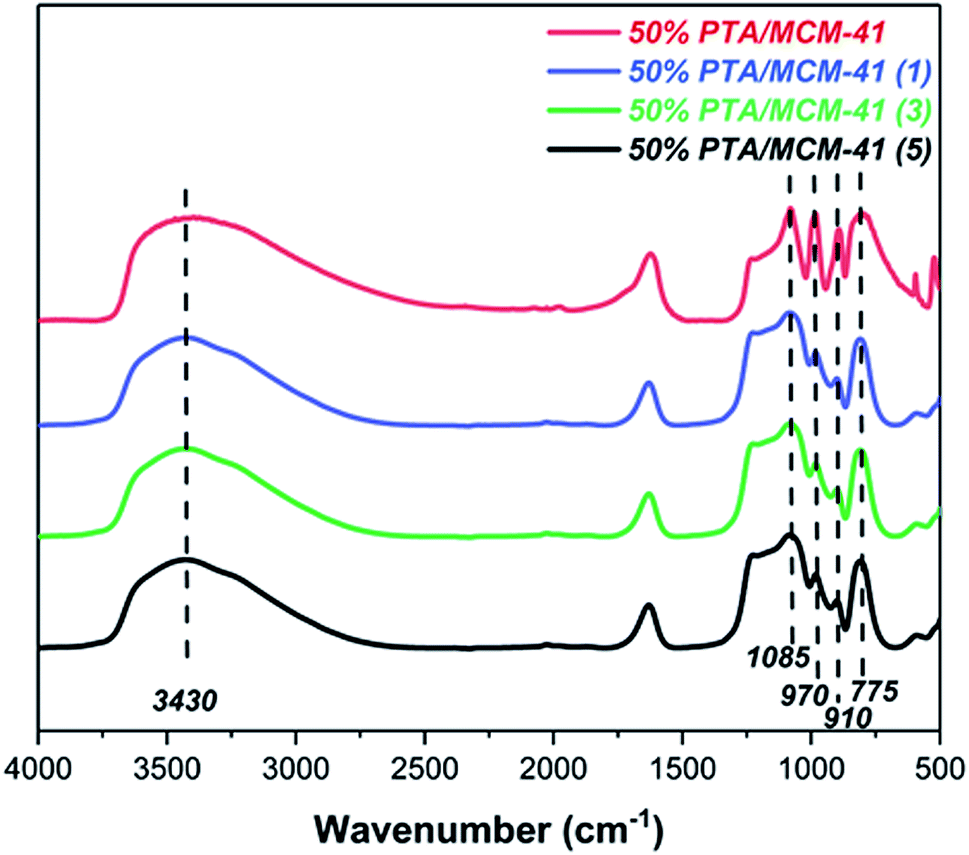
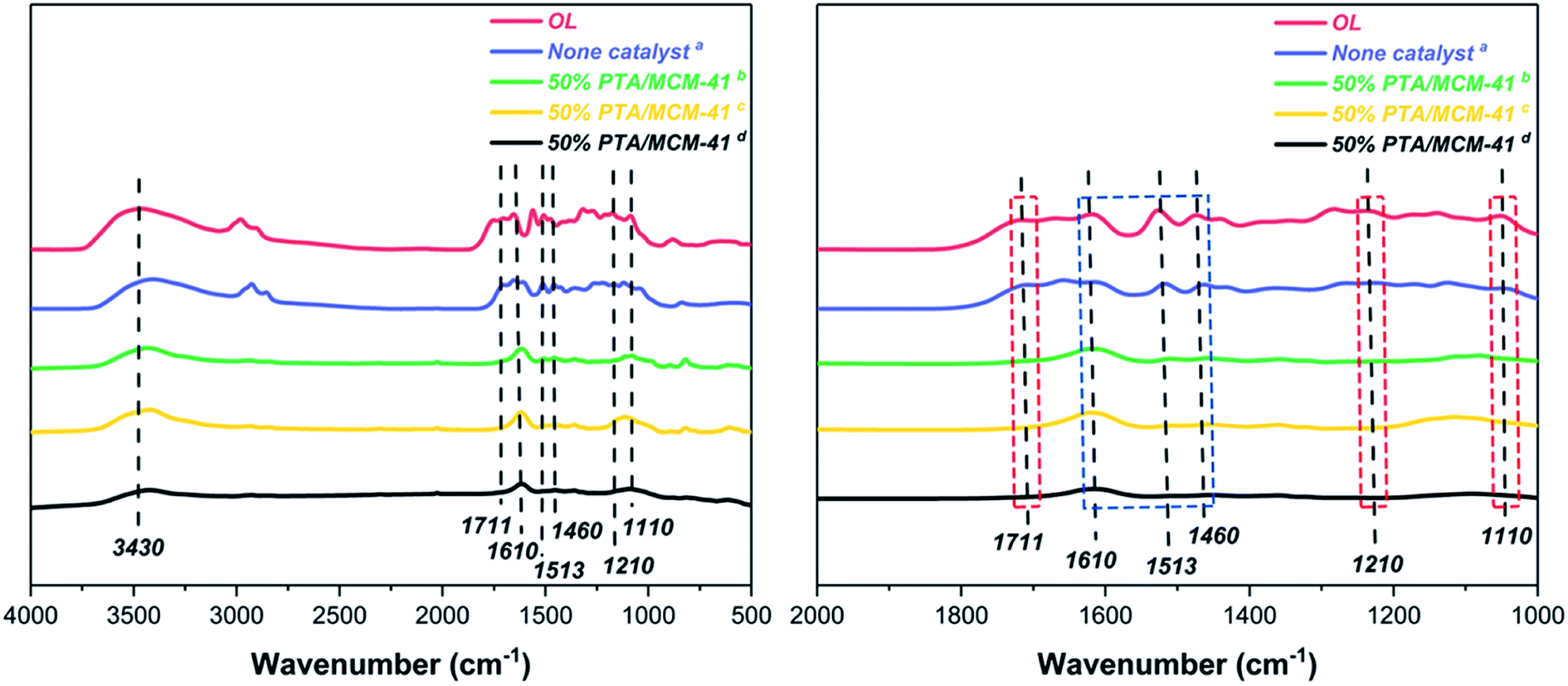
One of the greatest advantages of heterogeneous catalysts is their ability to be recycled. The reusability of the 50% PTA/MCM-41 catalyst is studied for five cycles under identical experimental conditions. The catalyst is filtered, washed, and calcined at 550 °C for 2 h after completing each reaction. As can be seen in Table 2, the regenerated catalyst displayed no significant loss of catalytic activity for the catalytic depolymerization reaction of OL, and the LF and LC yields obtained for the five consecutive runs are 88.96 wt% (1, LF), 94.49 wt% (1, LC), 88.07% (3, LF), 93.69 wt% (3, LC), 87.19 wt% (5, LF), and 93.17 wt% (5, LC). In order to clarify the cause of the slight decrease in catalytic performance, the composition and texture of the catalyst after every recovery are investigated. As shown in Fig. 4, the regenerated catalyst is characterized by the same FT-IR spectrum. All different regenerated catalysts are found to be similar to that of the fresh catalyst (50% PTA/MCM-41) with no appreciable shift in band positions, which indicates the retention of the PTA ions still in the regenerated catalysts even after three cycles.44,56 Moreover, in Fig. S3,† the SEM images and EDS images of the regenerated catalysts also show no significant changes in the morphologies and element distributions after five cycles. Meanwhile, the pore structure of the regenerated catalysts is also found not to show a significant collapse. Therefore, the reason for the no significant decrease of the 50% PTA/MCM-41 catalytic performance is presumed to be due to the preservation of the active components and the pore structure.57
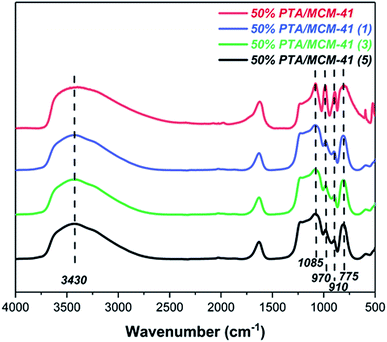 |
| | Fig. 4 The FT-IR spectra of original catalyst and different regenerated catalysts. | |
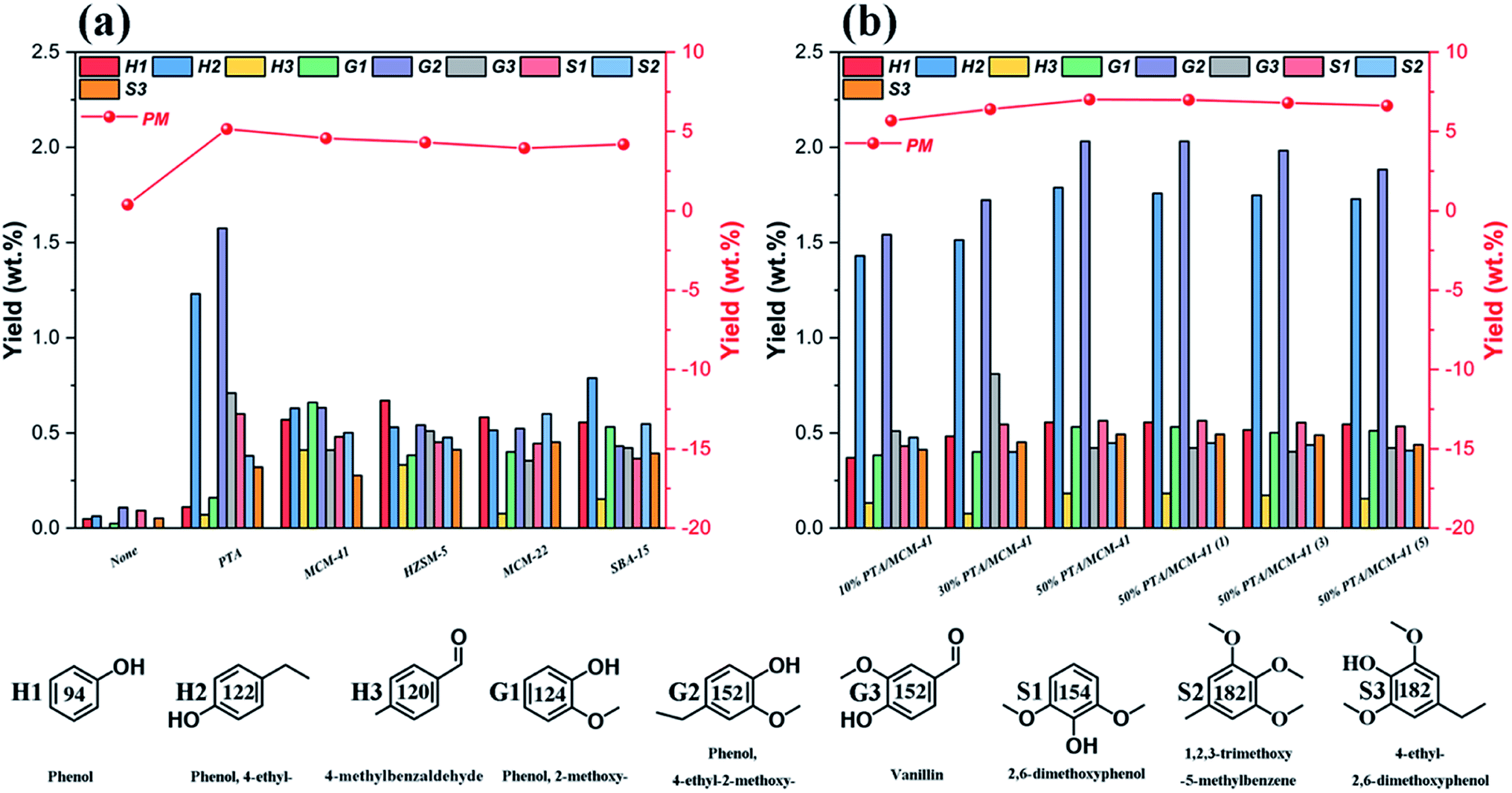
Fig. 5 summarizes the detailed component distributions of the PM depolymerization products obtained from catalytic depolymerization of OL in isopropanol at 310 °C for 6 h in the presence of various catalysts. We find that the PM depolymerization products are mainly aromatic compounds including phenol-type compounds (H-type phenols), guaiacol-type compounds (G-type phenols), syringol-type compounds (S-type phenols) and other aromatic compound derivatives with some methyl or ethyl substituents. This indicates that lignin alkylation reaction occurs during the lignin catalytic depolymerization process.58 Low yields of PM depolymerization products are obtained in the absence of catalyst. Only phenol (0.04 wt%), 4-ethylphenols (0.06 wt%), 2-methoxyphenols (0.02 wt%), 4-ethyl-2-methoxyphenols (0.11 wt%) and 2,6-dimethoxyphenol (0.09 wt%) are detected as the major products. Then, it seems that the acidity of various catalysts can change the reaction pathway in the hydrodeoxygenation process, thus affecting the PM depolymerization product yields and distributions. In particular, the 50% PTA/MCM-41 catalyst has been shown to be very active in increasing the yields and distributions toward PM depolymerization products in the lignin catalytic depolymerization reaction. We find that under the 50% PTA/MCM-41 catalyst, the PM depolymerization product yield produced by the hydrodeoxygenation process is the highest at up to 7.12 wt%. And 4-ethylphenols (1.78 wt%) and 4-ethyl-2-methoxyphenols (2.03 wt%) are detected as the main products, with phenol, 2-methoxyphenol, vanillin, 2,6-dimethoxyphenol, 4-ethyl-2,6-dimethoxyphenol, etc., as the minor products. This can be explained in that the total acidity of the 50% PTA/MCM-41 catalyst is conducive to the dehydration of lignin fragment intermediates into more alkane aromatic monomer products, such as 4-ethylphenols, 4-ethyl-2-methoxyphenols, etc.58 Simultaneously, the existence of more active sites of the 50% PTA/MCM-41 catalyst also contributes to efficient in situ hydrogen preparation by the isopropanol solvent.55 Therefore, the efficient hydrodeoxygenation of the catalytic depolymerization process can boost lignin conversion and lead to better yields and distributions of PM depolymerization products in the presence of the 50% PTA/MCM-41 catalyst. This is also the same as the results of elemental analysis and GPC analysis, where BO depolymerization products with smaller O/C ratios and smaller Mw are obtained. Besides, based on our study, we find that the content of S units in the OL is almost same as G units (Fig. 5). Theoretically, the lignin catalytic depolymerization reaction can produce similar G-type phenol and S-type phenol yields. However, in all cases, the yield of the G-type phenols is higher than that of the S-type phenols (Table 2). It is possible that the S-type intermediates can undergo demethoxylation, dealkylation and demethylation reactions, and yield phenolic compounds with simpler structures such as guaiacol, phenol, cresols and catechols, as has been demonstrated previously.5,42
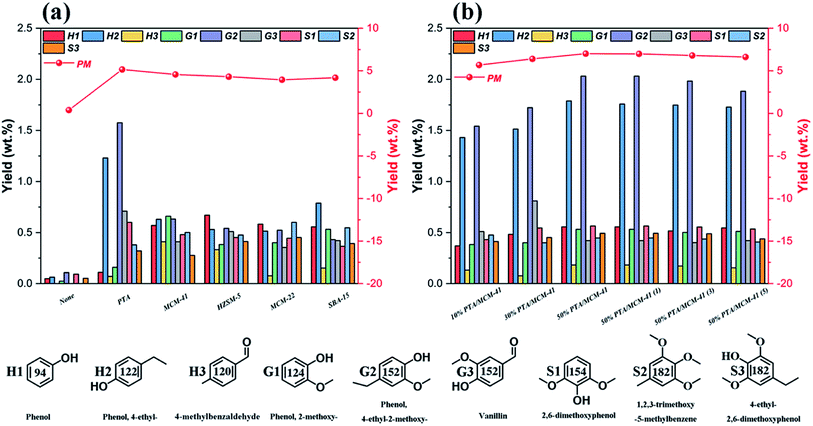 |
| | Fig. 5 The GC-MS results and products distributions of phenolic monomers from different catalysts: (a) different types of catalysts and (b) different PTA contents of catalysts. (Reaction condition: 0.5 g OL, 0.25 g catalysts, 30 mL isopropanol, 30 mL deionized-water, 310 °C and 6 h.) | |
The PM depolymerization product detailed yields and distributions obtained using the different regenerated 50% PTA/MCM-41 catalysts during the catalytic depolymerization process are shown in Fig. 5b. According to the data, the distributions of PM depolymerization products are not significantly changed in all five successive recycles, demonstrating that the 50% PTA/MCM-41 catalyst has good stability and reusability. However, as the number of recycles increases, the yields of PM depolymerization products decrease slightly. This may be due to slight reduction of the hydrogenolysis activity for each regenerated catalyst.56
3.4 Effect of reaction temperatures, reaction times, reaction solvents and ultrasonic frequencies on catalytic depolymerization of organosolv lignin
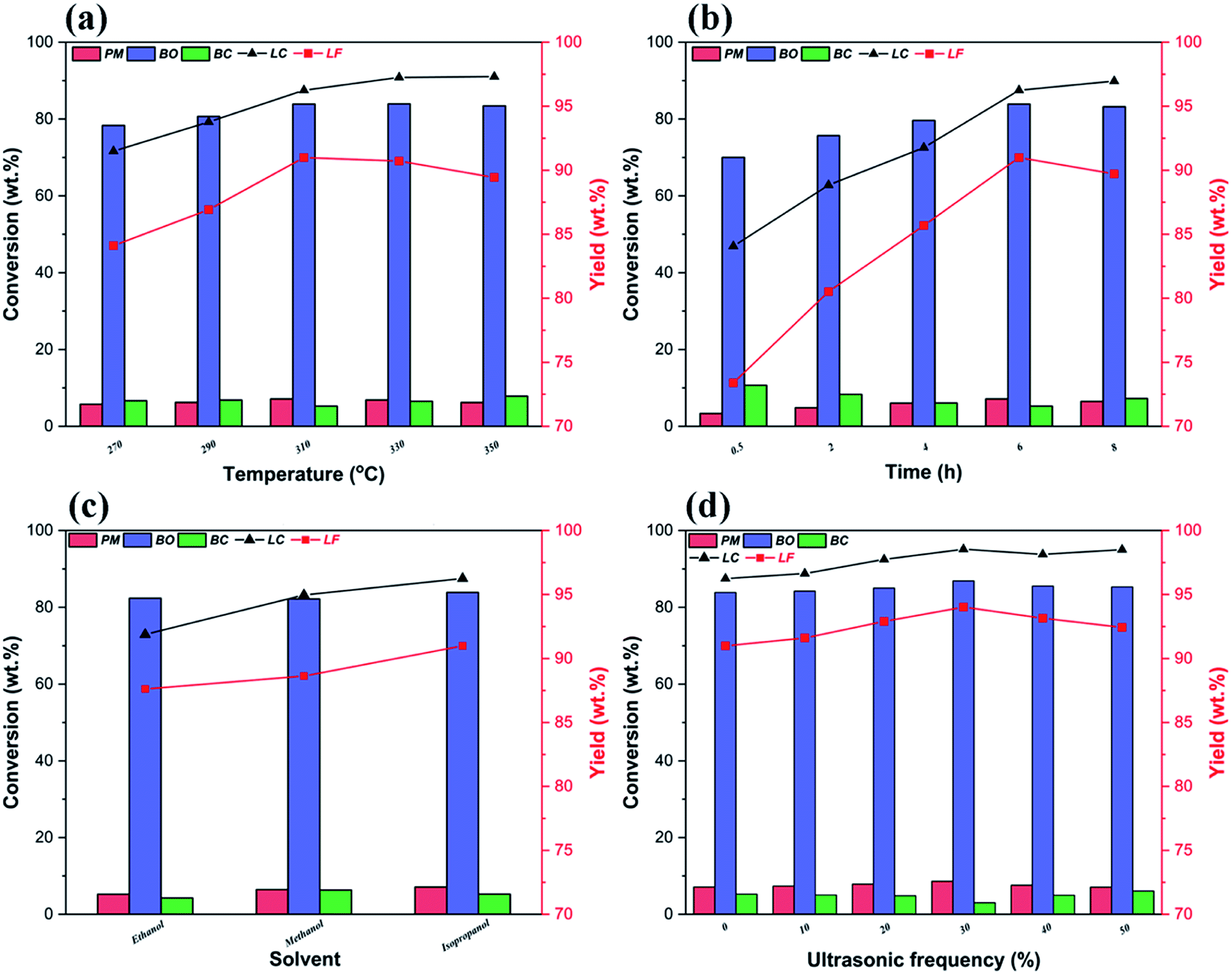
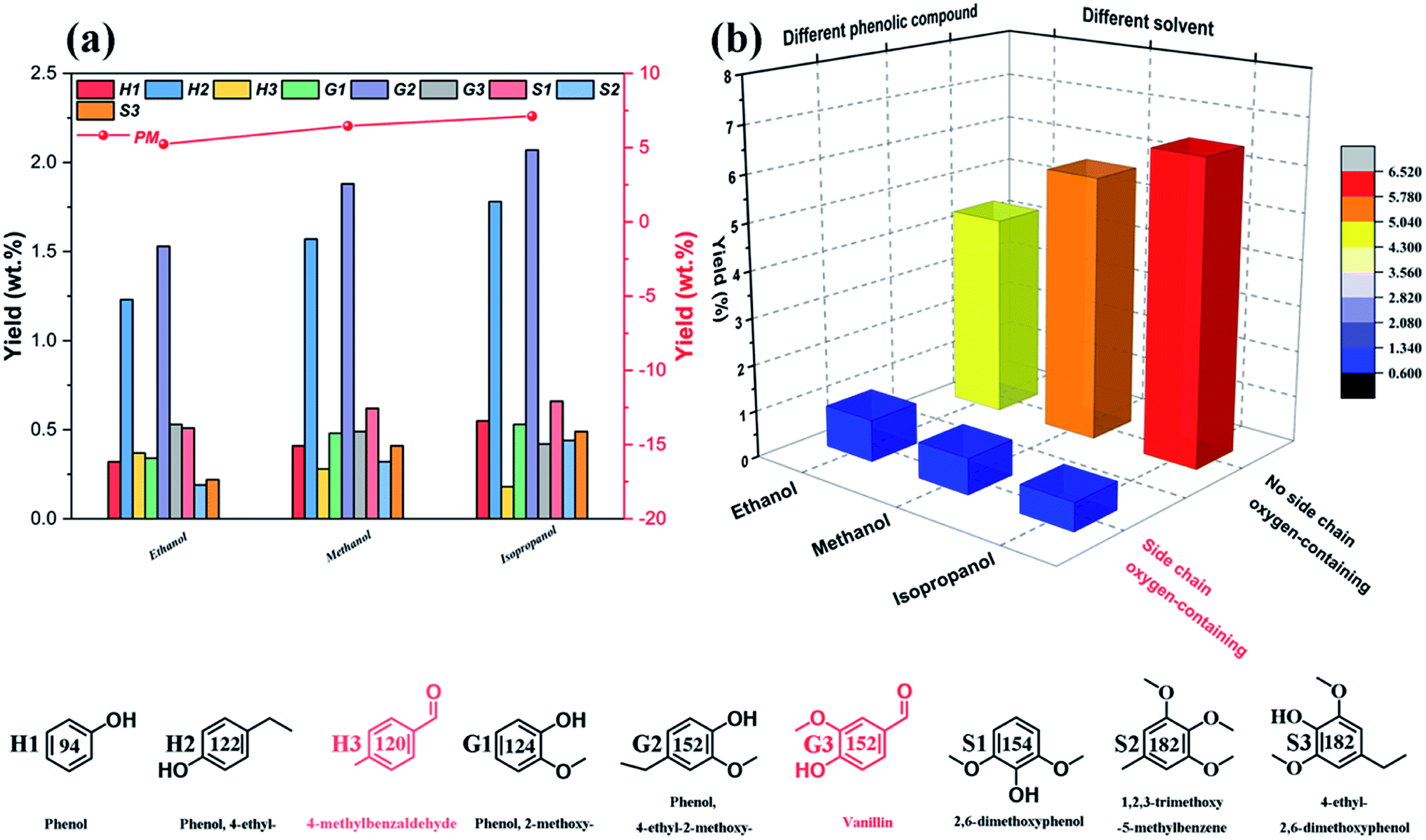
Fig. 6a illustrates the influence of reaction temperatures on the yields and distributions of various depolymerization products of OL. A significant change is seen in the yields and distributions of the various depolymerization products in the reaction temperature range of 270–350 °C. In fact, as the reaction temperature rises from 270 °C to 310 °C, the yields of PM and BO increase from 5.76 wt% and 78.35 wt% to 7.12 wt% and 83.87 wt%, respectively, while the yield of BC drops from 7.38 wt% to 5.26 wt%. But when the reaction temperature further increases to 350 °C, the PM and BO depolymerization product yields decline to 6.01 wt% and 83.43 wt% respectively; meanwhile, the production rate of BC improves, which indicates that the recombination of low-molecular-weight small lignin fragments can occur at higher reaction temperature during the catalytic depolymerization process (leading to formation of more BC depolymerization product).46,59 Thus, the catalytic depolymerization reaction of OL is not favored at high reaction temperature. A similar trend can be seen in the yields and distributions of the various depolymerization products of catalytic depolymerization of OL when the reaction time varies (Fig. 6b). For example, the yields of PM and BO depolymerization products improve from 3.36 wt% and 70.03 wt% to 7.12 wt% and 83.87 wt% respectively by increasing the reaction time from 0.5 to 6 h, while the BC depolymerization product yield drops from 10.68 wt% to 5.26 wt%. As long as the reaction time increases, the yield of BC rises (7.24 wt%), which is probably owing to the condensation of low-molecular-weight small lignin fragments on the previous reaction time basis (leading to formation of more BC depolymerization product).60 Besides, the reaction solvent system is also one of the important factors affecting the yields and distributions of various depolymerization products. So in this study, three kinds of alcohols (ethanol, methanol and isopropanol) are chosen as both ultrasound receptor and hydrogen-donor solvents to study their effect during the catalytic depolymerization of OL. Fig. 6c exhibits the lignin depolymerization results catalyzed by the 50% PTA/MCM-41 catalyst in different reaction solvent systems. We find that the yields of various PM, BO, BC and LC depolymerization products obtained from ethanol solvent are only 5.24 wt%, 82.38 wt%, 4.26 wt% and 91.88 wt%, respectively. Most of the OL is transformed into oligomers and residues due to the poor polarity in ethanol solvent. By comparison, methanol shows a higher yield and better activity than ethanol. Although the yield of BO depolymerization product is not changed significantly at 82.17 wt%, the yields of PM and LC depolymerization products in methanol are increased to 6.46 wt% and 94.97 wt%, respectively. Compared with ethanol, the relatively higher yields of PM and LC depolymerization products in methanol are due to the higher polarity of the methanol, as a better solvent polarity can increase the catalytic depolymerization efficiency.55 The best performance for the lignin catalytic depolymerization process is exhibited in isopropanol. Especially, highest conversion (96.25 wt%), monomer yield (7.12 wt%) and BO yield (83.87 wt%) are achieved, which is ascribed to the better hydrogen transfer performance of isopropanol.55 Kong et al. found that strong hydrogen donor solvents had coordination function in promoting hydrodeoxygenation of aromatic monomers and reducing char formation of small-molecule lignin fragments in the catalytic conversion of lignin to liquid bio-oil.53 Additionally, as shown in Fig. 7, different alcohol solvents also have a greater effect on the composition of PM products. More concretely, 0.91 wt% and 0.77 wt% yields of side chain oxygen-containing phenolic compounds are obtained in ethanol and methanol solvents, respectively, which indicates that the side chain oxygen-containing phenolic compounds cannot undergo a deep hydrodeoxygenation process due to the lack of hydrogen supply. Instead, the lowest yield of side chain oxygen-containing phenolic compounds is observed in isopropanol system as 0.59 wt%, which further indicates that good hydrogen supply ability of isopropanol is beneficial to the hydrodeoxygenation of lignin-derived phenol monomers. Thence, it is essential to optimize reaction temperatures, reaction times and reaction solvents for the efficient catalytic conversion of lignin into low-molecular-weight LF (including PM and BO depolymerization products). Based on the above results, the following best reaction conditions are obtained: reaction temperature of 310 °C, reaction time of 6 h and reaction solvent of isopropanol.
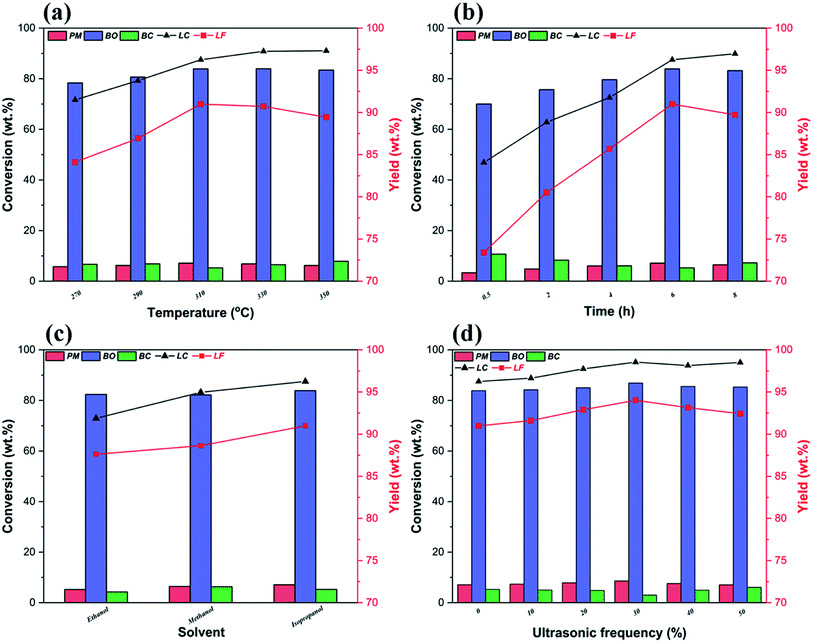 |
| | Fig. 6 Effect of different reaction conditions on the yields and distributions of various depolymerization products: (a) reaction temperatures (reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 6 h and 0% ultrasonic frequency); (b) reaction times (reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 0% ultrasonic frequency); (c) reaction solvents (reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL deionized water, 310 °C, 6 h and 0% ultrasonic frequency); and (d) ultrasonic frequencies (reaction conditions: 0.5 g OL, 0.25 g catalyst, 310 °C, 6 h, 30 mL isopropanol and 30 mL deionized water). | |
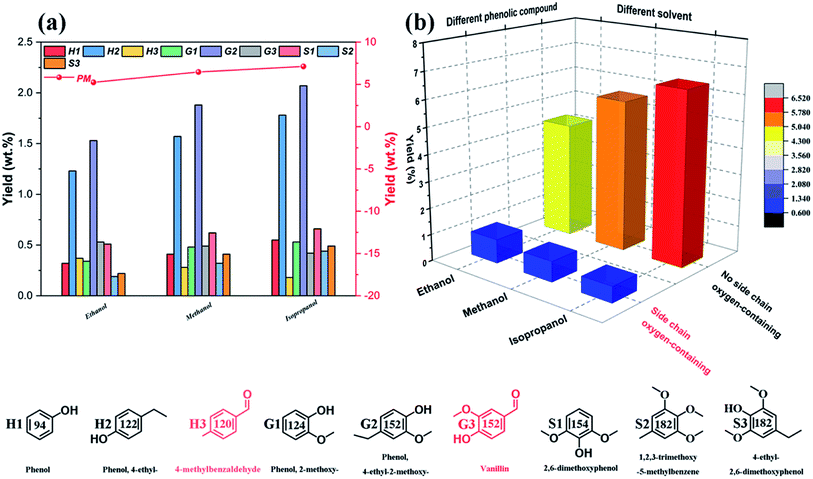 |
| | Fig. 7 Effect of different solvent systems on the phenolic monomer products: (a) phenolic monomer distributions; (b) phenolic monomer types. | |
Apart from different reaction temperature, reaction time and reaction solvent, ultrasound frequency also plays important roles during the ultrasound-assisted depolymerization process. Liu et al. showed that certain ultrasound receptors should be introduced into the ultrasound-assisted depolymerization system, as lignin is one kind of poor ultrasound receptor during the degradation. Therefore, under the best reaction conditions, we further discuss the effect of different ultrasound frequencies on the lignin catalytic depolymerization process. In Fig. 6d, when ultrasound at a frequency of 30% is applied to the ultrasound-assisted depolymerization reaction of OL over 50% PTA/MCM-41 catalyst, the highest yields of various PM, BO and BC depolymerization products reach 8.63 wt%, 86.89 wt% and 3.02 wt%, and the yields of LF and LC are equal to 95.02 wt% and 98.54 wt% respectively. The improvement in the activity and selectivity of the catalytic depolymerization reaction in the presence of ultrasound can be attributed to the collapse of ultrasonic cavitation bubbles generating extreme local conditions such as determined temperatures up to approximately 5000 K and estimated pressures around 50–1000 atm.36,37 Actually, the hot spots created by ultrasonic cavitation are able to dramatically accelerate the chemical reactivity of the medium, thereby further accelerating the lignin depolymerization reaction.36,37 So we need to vary the ultrasound frequency to find the optimum ultrasonic acoustic cavitation conditions. Compared with the control depolymerization not using ultrasound, the presence of ultrasound leads to a gradual increase in PM and BO depolymerization products and a reduction in the yield of BC depolymerization product. As the ultrasound frequency increases from 0% to 30%, the yields of PM and BO depolymerization products rise steadily. When the ultrasound frequency reaches 30%, the highest yields of PM and BO depolymerization products are equal to 8.23 wt% and 86.05 wt%, respectively, and the yield of BC depolymerization product reaches the lowest value of 4.24 wt%. The increase in the yields of PM and BO depolymerization products may be due to a relatively higher degree of chemical bond scission under intense ultrasonic acoustic cavitation at high ultrasound frequencies.37 However, further increasing the ultrasound frequency to 50% decreases the yields of PM and BO depolymerization products to 7.11 wt% and 85.32 wt% respectively, but obviously increases the yield of BC depolymerization product to 6.08 wt%. This result indicates that excessive ultrasonic acoustic cavitation can cause the low-molecular-weight oligomers obtained from lignin ultrasound-assisted depolymerization to repolymerize.37 Our experimental results confirm that the ultrasonic acoustic cavitation promotes not only the lignin catalytic depolymerization, but also the repolymerization of the depolymerization products. Only a suitable ultrasonic acoustic cavitation can promote the ultrasound-assisted OL depolymerization into a greater yield of low-molecular-weight LF. Furthermore, based on the current study, OL has a similar content of S and G units (Fig. 3); hence, theoretically, lignin depolymerization may lead to similar yields of G-type and S-type phenols. However, in the ultrasound-assisted depolymerization process, the yields of H-type phenols and G-type phenols are higher than that of S-type phenols (Table 2). This indicates that the S-type intermediates can more easily undergo demethoxylation, dealkylation, and demethylation reactions and yield phenolic compounds with simpler structures, such as guaiacol, phenol, cresols, and catechols, as also reported elsewhere.5,42 Thus, it also leads to a sharp decrease in the oxygen content of the various ultrasound-assisted depolymerization products (Table 4).
3.5 Analysis of bio-oil depolymerization products
To further investigate the fundamental chemistry of the lignin bio-oil obtained after the depolymerization reaction, the Mw, Mn and PDI of various BO products are analyzed using the GPC technique as shown in Table 3. Firstly, we find that the BO product obtained without catalyst has the largest Mw. This can be caused by lignin not being completely depolymerized into aromatic monomers and low-molecular-weight lignin fragments.53,55 Comparatively speaking, the 50% PTA/MCM-41 catalyst-obtained BO product after ultrasound-assisted depolymerization (30% ultrasound frequency) has the lowest Mw. This is probably because of the enhancement of depolymerization due to the total acidity increase of the catalyst and ultrasound frequency increase of reaction, and further effectively converted into PM and low-molecular-weight BO.37,53 Secondly, the Mw of BO product produced in the presence of PTA is distinctly higher than that of products obtained using the 50% PTA/MCM-41 catalyst, this observation further illustrating that the very low surface area makes finite availability of acidic sites of the catalyst preventing the complete conversion of OL into PM and low-molecular-weight BO.53,55 Finally, in addition to the acidities and active sites of different catalysts, the pore sizes of catalysts can also affect the LB depolymerization product performances. It is found that BO obtained using MCM-41 exhibited a much lower molecular weight than those obtained using either HZSM-5, MCM-22 or SBA-15. Chen et al. also reported that a large pore size of a catalyst could allow more unsaturated intermediates to enter in, reducing the contact with each other according to space constraints and promoting further decomposition of lignin into PM and low-molecular-weight BO.60 Besides, in this study, taking 50% PTA/MCM-41 as an example, compared with the 50% PTA/MCM-41 (5) regenerated catalyst, in addition to the difference in acidity, the larger pore size of the 50% PTA/MCM-41 catalyst can make it more conductive, thereby inhibiting repolymerization reaction and improving the effective conversion of OL into PM and low-molecular-weight BO. Thus, the Mw and PDI of BO depolymerization products (50% PTA/MCM-41 catalyst) are significantly reduced, which is also the same as the low yield of BC depolymerization product and the enhancement of PM and BO depolymerization product yields (Table 2). The Mw of BO depolymerization product is also closely associated with reaction temperatures, reaction times, reaction solvents and ultrasound frequencies. The Mw of various BO depolymerization products from OL under different reaction conditions are presented in Table S4.† Due to the different lignin depolymerization efficiency and lignin condensation efficiency, the various BO depolymerization products obtained under different reaction conditions have different Mw and PDI. In particular, for different reaction solvents, the lowest Mw of BO depolymerization product is observed in isopropanol, which is not dependent on the reaction temperatures, reaction times and ultrasound frequencies. It is found that BO depolymerization product obtained in isopropanol exhibits much lower Mw and PDI than those obtained from either ethanol or methanol, which is in accordance with the better hydrogen transfer performances and polarities of different reaction solvents.55 And this phenomenon also corresponds to the results of the effect of different reaction solvents on the yields and distributions of various depolymerization products (Fig. 6 and 7).
Table 3 Average molecular weight of various bio-oil depolymerization products obtained from different reaction conditions
| Entry |
Catalyst |
Mwa |
Mna |
PDIa |
| Mw: weight-average molecular weight; Mn: number-average molecular weight; PDI: polydispersity index, PDI = Mw/Mn. Reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h. The catalytic performance of the catalysts after being regenerated 1, 3, and 5 times, respectively. Reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C, 6 h and 30% ultrasonic frequency. |
| 1 |
Noneb |
2330 |
930 |
2.51 |
| 2 |
PTAb |
1240 |
710 |
1.76 |
| 3 |
MCM-41b |
1380 |
740 |
1.87 |
| 4 |
HZSM-5b |
1640 |
820 |
2.01 |
| 5 |
MCM-22b |
1570 |
790 |
1.98 |
| 6 |
SBA-15b |
1460 |
760 |
1.91 |
| 7 |
10% PTA/MCM-41b |
1150 |
660 |
1.74 |
| 8 |
30% PTA/MCM-41b |
1020 |
600 |
1.71 |
| 9 |
50% PTA/MCM-41b |
810 |
510 |
1.58 |
| 10 |
50% PTA/MCM-41 (1)b,c |
830 |
520 |
1.61 |
| 11 |
50% PTA/MCM-41 (3)b,c |
890 |
540 |
1.65 |
| 12 |
50% PTA/MCM-41 (5)b,c |
940 |
560 |
1.69 |
| 13 |
50% PTA/MCM-41d |
650 |
520 |
1.24 |
In the above results, it is shown that the yields of LC and LF are highly dependent on catalyst types, reaction temperatures, reaction times, reaction solvents and ultrasonic frequencies. And the conclusion is that the highest yields of LC and LF from OL are obtained over 50% PTA/MCM-41 catalyst and at 30% ultrasonic frequency in isopropanol at 310 °C for 6 h, because of the better hydrogen transfer performance and polarity of isopropanol, better ultrasonic frequency, better reaction temperature, better reaction time and higher acidity and active sites of the 50% PTA/MCM-41 catalyst. Therefore, we use elemental analysis to determine the elemental distribution of different BO depolymerization products obtained under different reaction conditions. As shown in Table 4, the oxygen contents of different BO depolymerization products obtained using PTA (33.58 wt%), MCM-41 (38.34 wt%), HZSM-5 (40.53 wt%), MCM-22 (39.46 wt%), SBA-15 (38.98 wt%), 10% PTA/MCM-41 (29.27 wt%), 30% PTA/MCM-41 (26.56 wt%) and 50% PTA/MCM-41 (21.58 wt% and 15.31 wt%) are much lower than that of blank reaction BO depolymerization product (42.24 wt%) and OL (44.95 wt%). Simultaneously, the H content and C content are also enhanced with the use of the different catalysts. This indicates that the introduction of different catalysts can increase the ability of hydrodeoxygenation in the lignin depolymerization reaction. This phenomenon is also consistent with the results of GC-MS analysis, where H-type phenols and G-type phenols are the main PM depolymerization products, and S-type phenols with high oxygen content are rarely detected (Table 2 and Fig. 5). In particular, the O/C atomic ratio of BO depolymerization product is reduced evidently and the HHV of BO depolymerization product is enhanced evidently in the presence of 50% PTA/MCM-41 catalyst, with 30% ultrasonic frequency and isopropanol solvent. This can be attributed to the better hydrogenation ability and polarity of isopropanol solvent, the stronger total acidity concentration of the 50% PTA/MCM-41 catalyst and the better ultrasonic frequency of reaction.53,55 Furthermore, the HHV of BO depolymerization product obtained using the regenerated 50% PTA/MCM-41 (5) catalyst is 31.55 MJ kg−1. Compared with the BO depolymerization product obtained with the 50% PTA/MCM-41 catalyst (34.41 MJ kg−1), the HHV of this BO depolymerization product is slightly reduced. This indicates that the regenerated catalyst after five cycles is still suitable for the high value conversion process of lignin. As can be seen in Table S5,† a significant promotion in hydrogen content and HHV of various BO depolymerization products from different depolymerization reactions is also observed under the 50% PTA/MCM-41 catalyst. The same as the yield and distribution results of various BO depolymerization products, a common rule can be summarized from the elemental distribution results. The best reaction conditions are observed at the maximum of hydrogen content and HHV (reaction temperature: 310 °C; reaction time: 6 h; reaction solvent: isopropanol; and ultrasonic frequency: 30%). Understanding these optimal reaction conditions will allow us to design more efficient ultrasound-assisted depolymerization methods for greater production of liquid fuels and fine chemicals from lignin.
Table 4 Effect of different reaction conditions on elemental compositions and heating value of various bio-oil depolymerization products
| Entry |
Catalyst |
Elemental composition (wt%) |
HHVa |
| C |
H |
O |
N |
O/C |
H/C |
| HHV (MJ kg−1) = (34C + 124.3H + 6.3N + 19.3S − 9.8O)/100, where C, H, N, S, and O are the weight percentages of carbon, hydrogen, nitrogen, sulfur, and oxygen.53,55 Reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h. The catalytic performance of the catalysts after being regenerated 1, 3, and 5 times, respectively. Reaction conditions: 0.5 g OL, 0.25 g catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C, 6 h and 30% ultrasonic frequency. |
| 1 |
Noneb |
49.94 |
5.73 |
42.24 |
0.11 |
0.89 |
0.13 |
19.78 |
| 2 |
PTAb |
60.24 |
6.05 |
33.58 |
0.13 |
0.56 |
0.11 |
24.72 |
| 3 |
MCM-41b |
55.97 |
5.58 |
38.34 |
0.11 |
0.69 |
0.09 |
22.21 |
| 4 |
HZSM-5b |
53.36 |
6.02 |
40.53 |
0.09 |
0.76 |
0.11 |
21.65 |
| 5 |
MCM-22b |
54.02 |
6.41 |
39.46 |
0.11 |
0.73 |
0.12 |
22.47 |
| 6 |
SBA-15b |
54.87 |
6.03 |
38.98 |
0.12 |
0.71 |
0.11 |
22.34 |
| 7 |
10% PTA/MCM-41b |
61.65 |
8.23 |
29.97 |
0.15 |
0.49 |
0.13 |
28.26 |
| 8 |
30% PTA/MCM-41b |
63.87 |
9.41 |
26.56 |
0.16 |
0.42 |
0.15 |
30.81 |
| 9 |
50% PTA/MCM-41b |
67.39 |
10.94 |
21.58 |
0.09 |
0.32 |
0.16 |
34.41 |
| 10 |
50% PTA/MCM-41 (1)b,c |
67.29 |
9.92 |
22.67 |
0.12 |
0.34 |
0.15 |
32.95 |
| 11 |
50% PTA/MCM-41 (3)b,c |
68.32 |
8.64 |
22.93 |
0.11 |
0.34 |
0.13 |
31.72 |
| 12 |
50% PTA/MCM-41 (5)b,c |
67.56 |
8.75 |
23.54 |
0.15 |
0.35 |
0.13 |
31.55 |
| 13 |
50% PTA/MCM-41d |
73.79 |
10.84 |
15.31 |
0.06 |
0.26 |
0.15 |
37.07 |
Fig. 8 shows the FT-IR spectra of various BO depolymerization products. It can be found that there is little change of the structure of OL and BO depolymerization product obtained without catalyst. In contrast, with the 50% PTA/MCM-41 catalyst and 30% ultrasound frequency reaction conditions, the structures of various BO depolymerization products are changed remarkably after different depolymerization reaction processes. Among them, the band at 3430 cm−1 is assigned to the formation of phenolic compounds.26,47 It also can be observed that the band at 1711 cm−1 of carbonyl group gradually decreases which is considered to be caused by the reduction of degree of unsaturation, due to the occurrence of hydrogenation.26,47 In addition, the bands observed at 1210 and 1110 cm−1 are assigned to aromatic-O structures, which are present in all spectra with different absorption intensities, indicating that the –OCH3 bonds of various BO depolymerization products are effectively cleaved.26,47 This phenomenon is consistent with GC-MS results (demethoxy reaction), where a lower yield of S-type phenols is detected (Table 2 and Fig. 5).
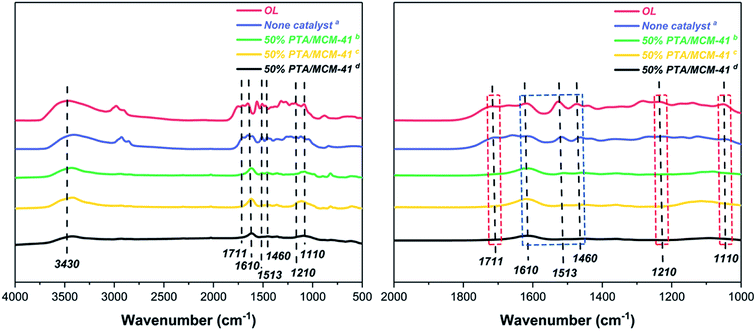 |
| | Fig. 8 The FT-IR spectra of OL and various BO depolymerization products under different reaction conditions: (a) reaction conditions: 0.5 g OL, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h; (b) reaction conditions: 0.5 g OL, 0.25 g 50% PTA/MCM-41 catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h; (c) reaction conditions: 0.5 g OL, 0.25 g 50% PTA/MCM-41 catalyst, 30 mL isopropanol, 30 mL deionized water, 30% ultrasonic frequency, 310 °C and 6 h; and (d) reaction conditions: 0.5 g OL, 0.25 g regenerated 50% PTA/MCM-41 (5) catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h. | |
Finally, to better demonstrate the structural characteristics of the small-molecule BO products after ultrasound-assisted depolymerization of OL, as shown in Fig. 9a–c, the 2D-HSQC NMR structural evolution spectra of BO obtained under different reaction conditions are compared. After the catalytic depolymerization of OL, the structure of BO depolymerization product significantly changes. The G, S, and H structural units can be clearly observed in the fraction, but the characteristic signals of aryl ether bonds, such as β-O-4 and α-O-4, disappear in the aliphatic regions, denoting the effective breakage of β-O-4 aryl ether linkages in the lignin.50,52 Additionally, a strong 56.18/3.76 ppm signal is attributed to OMe, indicating the retention of the lignin “nuclear” structure. Hence, 2D-HSQC NMR spectra confirm that the 50% PTA/MCM-41 catalyst under ultrasound-assisted catalytic depolymerization reaction conditions can cleave β-O-4 aryl ether bonds of OL to efficiently produce small-molecule liquid fuels.
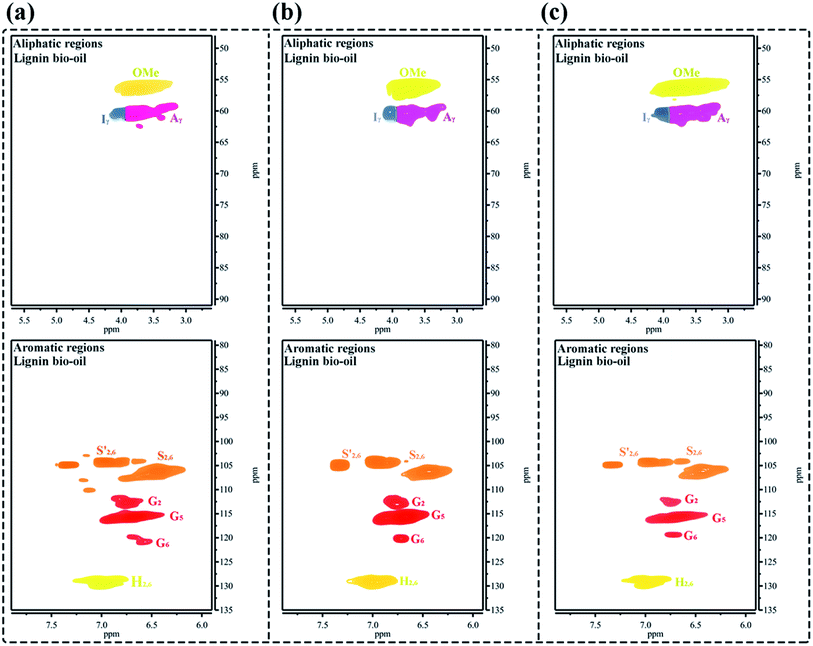 |
| | Fig. 9 The 2D-HSQC NMR spectra of various BO depolymerization products obtained under different reaction conditions: (a) reaction conditions: 0.5 g OL, 0.25 g 50% PTA/MCM-41 catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h; (b) reaction conditions: 0.5 g OL, 0.25 g 50% PTA/MCM-41 catalyst, 30 mL isopropanol, 30 mL deionized water, 30% ultrasonic frequency, 310 °C and 6 h; and (c) reaction conditions: 0.5 g OL, 0.25 g regenerated 50% PTA/MCM-41 (5) catalyst, 30 mL isopropanol, 30 mL deionized water, 310 °C and 6 h. | |
4. Conclusion
This contribution has discussed the efficient ultrasound-assisted depolymerization of OL performed over 50% PTA/MCM-41 catalyst. Both catalyst type and ultrasound frequency played important roles in promoting the lignin depolymerization and reducing char yield. Generally, the yields and distributions of PM products were greatly influenced by pore structure and acidity of the catalyst. In addition, the depolymerization efficiency of OL was also closely dependent on reaction temperatures, reaction times and reaction solvents. Thus, in this study, the optimal reaction conditions were found to be in isopropanol at 310 °C for 6 h with 30% ultrasound frequency using 50% PTA/MCM-41 catalyst. The highest yields of PM, BO, LF and LC were reached as 8.63 wt%, 86.89 wt%, 95.52 wt% and 98.54 wt%, respectively. The results showed that ultrasound acoustic cavitation could enhance the depolymerization of lignin, thus greatly enhancing production of LF. In particular, the hydrogen composition and HHV of various lignin depolymerization products improved, and the oxygen content decreased, indicating that hydrogenation and/or hydrodeoxygenation happened during the depolymerization process. Finally, we also illustrated that the 50% PTA/MCM-41 catalyst had high stability, and it could be reused for up to five cycles without loss of catalytic activity.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the National Key R&D Program of China (no. 2017YFB0307900), National Natural Science Foundation of China (no. 21908014 and 31470604), Liaoning Providence Science and Technology Project (no. 20180550759), Opening Project of Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control (KF201803-5) and State Key Laboratory of Pulp and Paper Engineering (no. 201803).
References
- C. Liu, S. Wu, H. Zhang and R. Xiao, Fuel Process. Technol., 2019, 191, 181–201 CrossRef CAS
 .
. - L. Cherpozat, E. Loranger and C. Daneault, Biomass Bioenergy, 2019, 124, 54–63 CrossRef CAS .
- C. Liu, J. Hu, H. Zhang and R. Xiao, Fuel, 2016, 182, 864–870 CrossRef CAS .
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS .
- B. Du, C. Liu, X. Wang, Y. Han, Y. Guo, H. Li and J. Zhou, Renewable Energy, 2020, 147, 1331–1339 CrossRef CAS .
- C. Liu, Y. Deng, S. Wu, H. Mou, J. Liang and M. Lei, J. Anal. Appl. Pyrolysis, 2016, 118, 123–129 CrossRef CAS .
- J. Yang, L. Zhao, S. Liu, Y. Wang and L. Dai, Bioresour. Technol., 2016, 212, 302–310 CrossRef CAS PubMed .
- B. Du, Y. Sun, B. Liu, Y. Yang, S. Gao, Z. Zhang, X. Wang and J. Zhou, Polym. Test., 2020, 81, 106207 CrossRef CAS .
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed .
- J. Sun, H. Li, L.-P. Xiao, X. Guo, Y. Fang, R.-C. Sun and G. Song, ACS Sustainable Chem. Eng., 2019, 7, 4666–4674 CrossRef CAS .
- M. H. El-barbary, P. H. Steele and L. Ingram, Appl. Biochem. Biotechnol., 2009, 154, 3–13 CrossRef PubMed .
- C. Sáiz-Jiménez and J. De Leeuw, Org. Geochem., 1986, 10, 869–876 CrossRef .
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS .
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS .
- Z. Tang, Y. Zhang and Q. Guo, Ind. Eng. Chem. Res., 2010, 49, 2040–2046 CrossRef CAS .
- H. Ma, H. Li, W. Zhao, L. Li, S. Liu, J. Long and X. Li, Green Chem., 2019, 21(3), 658–668 RSC .
- G. Zhu, X. Qiu, Y. Zhao, Y. Qian, Y. Pang and X. Ouyang, Bioresour. Technol., 2016, 218, 718–722 CrossRef CAS PubMed .
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed. Engl., 2015, 54, 258–262 CrossRef CAS PubMed .
- N. Mahmood, Z. Yuan, J. Schmidt and C. C. Xu, Bioresour. Technol., 2013, 139, 13–20 CrossRef CAS PubMed .
- K. Öhgren, R. Bura, J. Saddler and G. Zacchi, Bioresour. Technol., 2007, 98(13), 2503–2510 CrossRef PubMed .
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. Huijgen, W. Dehaen and C. Courtin, Energy Environ. Sci., 2015, 8, 1748–1763 RSC .
- W. Chen, D. J. McClelland, A. Azarpira, J. Ralph, Z. Luo and G. W. Huber, Green Chem., 2016, 18, 271–281 RSC .
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC .
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 RSC .
- B. Du, B. Liu, Y. Yang, X. Wang and J. Zhou, Catalysts, 2019, 9(5), 399 CrossRef .
- H. Guo, B. Zhang, Z. Qi, C. Li, J. Ji, T. Dai, A. Wang and T. Zhang, ChemSusChem, 2017, 10, 523–532 CrossRef PubMed .
- A. Jha, A. C. Garade, S. P. Mirajkar and C. V. Rode, Ind. Eng. Chem. Res., 2012, 51, 3916–3922 CrossRef CAS .
- A. Liu, Z. Zhang, Z. Fang, B. Liu and K. Huang, J. Ind. Eng. Chem., 2014, 20, 1977–1984 CrossRef CAS .
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC .
- G. Marcì, E. García-López, V. Vaiano, G. Sarno, D. Sannino and L. Palmisano, Catal. Today, 2017, 281, 60–70 CrossRef .
- Y. Zhou, G. Chen, Z. Long and J. Wang, RSC Adv., 2014, 4, 42092–42113 RSC .
- D. Carriazo, C. Domingo, C. Martín and V. Rives, J. Solid State Chem., 2008, 181, 2046–2057 CrossRef CAS .
- G. Karthikeyan and A. Pandurangan, J. Mol. Catal. A: Chem., 2009, 311, 36–45 CrossRef CAS .
- B. R. Jermy and A. Pandurangan, Appl. Catal., A, 2005, 295, 185–192 CrossRef CAS .
- W. Wang, X. Wu, T. Chantapakul, D. Wang, S. Zhang, X. Ma, T. Ding, X. Ye and D. Liu, Food Res. Int., 2017, 102, 101–110 CrossRef CAS PubMed .
- W.-Q. Guo, R.-L. Yin, X.-J. Zhou, J.-S. Du, H.-O. Cao, S.-S. Yang and N.-Q. Ren, Ultrason. Sonochem., 2015, 22, 182–187 CrossRef CAS PubMed .
- B. Liu, B. Du, Y. Sun, M. Zhu, Y. Yang, X. Wang and J. Zhou, Fuel Process. Technol., 2020, 203, 106387 CrossRef CAS .
- R. Mettin, C. Cairós and A. Troia, Ultrason. Sonochem., 2015, 25, 24–30 CrossRef CAS PubMed .
- Z. M. Bundhoo and R. Mohee, Ultrason. Sonochem., 2018, 40, 298–313 CrossRef CAS PubMed .
- H. Han, S. Wang, M. Rakita, Y. Wang, Q. Han and Q. Xu, Food Nutr. Sci., 2018, 09, 1034–1045 CAS .
- J. Luo, Z. Fang and R. L. Smith Jr, Prog. Energy Combust. Sci., 2014, 41, 56–93 CrossRef .
- B. Du, B. Liu, X. Wang and J. Zhou, ChemistryOpen, 2019, 8, 643–649 CrossRef CAS PubMed .
- X. Wang, B. Du, L. Pu, Y. Guo, H. Li and J. Zhou, J. Anal. Appl. Pyrolysis, 2018, 129, 13–20 CrossRef CAS .
- T. Ma, J. Ding, R. Shao, W. Xu and Z. Yun, Chem. Eng. J., 2017, 316, 797–806 CrossRef CAS .
- S. S. Sakate, S. B. Kamble, R. C. Chikate and C. V. Rode, New J. Chem., 2017, 41, 4943–4949 RSC .
- B. Du, C. Chen, Y. Sun, B. Liu, Y. Yang, S. Gao, Z. Zhang, X. Wang and J. Zhou, Catal. Lett., 2019, 1–14 Search PubMed .
- S. Gao, J. Zhao, X. Wang, Y. Guo, Y. Han and J. Zhou, Polymers, 2018, 10(9), 967 CrossRef PubMed .
- I. Kozhevnikov, A. Sinnema, R. Jansen, K. Pamin and H. Van Bekkum, Catal. Lett., 1994, 30, 241–252 CrossRef CAS .
- G. Kamalakar, K. Komura and Y. Sugi, Appl. Catal., A, 2006, 310, 155–163 CrossRef CAS .
- B. Du, C. Chen, Y. Sun, M. Yu, M. Yang, X. Wang and J. Zhou, Fuel Process. Technol., 2020, 206, 106479 CrossRef CAS .
- J.-L. Wen, B.-L. Xue, F. Xu, R.-C. Sun and A. Pinkert, Ind. Crops Prod., 2013, 42, 332–343 CrossRef CAS .
- B. Du, C. Chen, Y. Sun, M. Yang, M. Yu, B. Liu, X. Wang and J. Zhou, Int. J. Biol. Macromol., 2020, 156, 669–680 CrossRef CAS PubMed .
- L. Kong, C. Liu, J. Gao, Y. Wang and L. Dai, Bioresour. Technol., 2019, 276, 310–317 CrossRef CAS PubMed .
- X. Liu, Z. Jiang, S. Feng, H. Zhang, J. Li and C. Hu, Fuel, 2019, 244, 247–257 CrossRef CAS .
- M. Zhou, B. K. Sharma, J. Li, J. Zhao, J. Xu and J. Jiang, Fuel, 2019, 239, 239–244 CrossRef CAS .
- A. M. Hussein, A. H. Mady, S. Mahmoud, J. J. Shim and F. Z. Yehia, J. Photochem. Photobiol., A, 2019, 377, 173–181 CrossRef CAS .
- S. Xu, D. Pan, Y. Wu, N. Xu, H. Yang, L. Gao, W. Li and G. Xiao, Ind. Eng. Chem. Res., 2019, 58, 9276–9285 CrossRef CAS .
- X. Huang, X. Ouyang, B. M. S. Hendriks, O. M. M. Gonzalez, J. Zhu, T. I. Koranyi, M. D. Boot and E. J. M. Hensen, Faraday Discuss., 2017, 202, 141–156 RSC .
- X. Huang, T. I. Korányi and E. J. Hensen, Nanoporous catalysts for biomass conversion, 2017, pp. 231–251 Search PubMed .
- P. Chen, Q. Zhang, R. Shu, Y. Xu, L. Ma and T. Wang, Bioresour. Technol., 2017, 226, 125–131 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05069e |
| ‡ Present Address: Liaoning Key Laboratory of Pulp and Papermaking Engineering, Dalian Polytechnic University, Dalian. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a