
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-iodine-promoted metal-catalyst-free reactions of [60]fullerene with β-keto esters for the selective formation of [60]fullerene derivatives†
Han-Lin Yanga,
Li-Jun Xubc,
Wen-Zhong Lia,
Tao Sund,
Bao-Rong Duana,
Si Chen *a and
Xiang Gaob
*a and
Xiang Gaob
aCollege of Chemistry and Chemical Engineering, Yantai University, 30 Qingquan Road, Yantai, Shandong 264005, China. E-mail: sichen@ytu.edu.cn; chemchensi@163.com
bState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, Jilin 130022, China
cUniversity of Science and Technology of China, Hefei, Anhui 230026, China
dSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore
First published on 26th June 2020
Abstract
In this study, methanofullerenes and 2′,3′-dihydrofuran C60 derivatives were selectively synthesized in high yields via the reactions of C60 with β-keto esters under mild conditions by controlling the addition sequence and molar ratio of iodine and base. The structures of the products were determined by spectroscopic characterization. Moreover, a possible reaction mechanism for the selective formation of fullerene derivatives was proposed.
Introduction
Fullerenes have attracted significant attention from chemists due to their unique geometric and electronic structures.1–3 To date, the functionalization of fullerenes is of great importance for fullerene chemistry since fullerenes can be synthesized and isolated in macroscopic quantities.4–7 Various fullerene derivatives are promising for the development of diverse fullerene-based materials possessing unique properties for application in materials and biological sciences.8–11 Accordingly, methanofullerenes and 2′,3′-dihydrofuran C60 derivatives (dihydrofuran-fused C60 derivatives), as shown in Fig. 1, are classical fullerene derivatives containing a fused three- or five-membered ring, which have been synthesized in moderate yields via the Bingel–Hirsch reaction12,13 and transition-metal catalyst-mediated14,15 or high-speed vibration milling (HSVM).16 However, these methods frequently suffer from drawbacks such as the requirements of high temperatures, long reaction time, the use of transition-metal catalysts, and a heterogeneous reaction. Thus, the development of more simple and efficient methods for the synthesis of methanofullerenes and 2′,3′-dihydrofuran C60 derivatives is highly desirable. | ||
| Fig. 1 Structures of methanofullerenes and 2′,3′-dihydrofuran C60 derivatives. | ||
Recently, we have selectively synthesized methanofullerenes and 2′,3′-dihydrofuran C60 derivatives as a single product in each reaction, respectively, in an iodine-base system by controlling the addition sequence and molar ratio of iodine and base, as shown in Scheme 1.
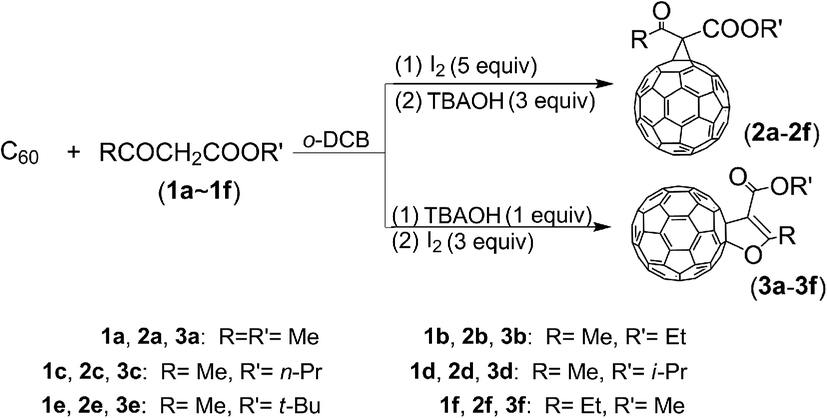 | ||
| Scheme 1 Synthesis of the compounds 2a–2f and 3a–3f. | ||
Results and discussion
Table 1 presents the screening of the reaction conditions. Ethyl acetoacetate was used for reaction screening. The experiment was designed to control the selective formation of a single product in a relatively high yield. It was divided into two types of reactions according to the addition sequence of base and iodine as follows: (1) reaction involving the addition of base first and iodine later (entries from 1 to 11) and (2) reaction involving the addition of iodine first and base later (entries from 12 to 15). Both the compounds 2b and 3b were obtained when 3 equiv. of OH− with respect to C60 was first added to the reaction mixture followed by the addition of 5 equiv. of iodine with respect to C60 (entries from 1 to 7). Interestingly, only 3b was obtained when the reaction was conducted at 0 °C by first adding 1 equiv. of OH− followed by the addition of 3 equiv. of iodine (entry 8). Upon further increasing the amount of base to 5 equiv. and that of iodine to 8 equiv., a crude product was obtained, which was quite complex and difficult to separate, as indicated by the HPLC measurement performed using a Buckyprep column (entry 11). However, a reduction in the amount of iodine would result in bis-2′,3′-dihydrofuran C60 derivatives.17 In contrast, only 2b was obtained when the reaction was carried out at room temperature by first adding 5 equiv. of iodine followed by the addition of 3 or 4 equiv. of OH− (entries 12 and 13). To achieve 2b in a higher yield, it was better to use 3 equiv. of OH− (52% isolated yield, 65% yield based on the consumed C60) (entry 12). Similarly, upon further increasing the amount of base to 5 equiv. and that of iodine to 5 or 8 equiv., a crude product was obtained, which was quite complex and difficult to purify, as shown by the HPLC measurement conducted using the Buckyprep column (entries 14 and 15).| Entry | OH− (equiv.) | I2 (equiv.) | Temp (°C) | Timeb (min) | Timec (min) | Product (% yield)d | |
|---|---|---|---|---|---|---|---|
| 2b | 3b | ||||||
| a Reaction conditions: (1) for the entries from 1 to 11, C60 (36 mg, 50 μmol) and ethyl acetoacetate (500 μL, 79 equiv.) were put in o-DCB (o-dichlorobenzene) (15 mL). The mixture was stirred for 15 min under argon or nitrogen at a preset temperature. Then, OH− (TBAOH, tetra-n-butylammonium hydroxide, 1.0 M in CH3OH) was added to the solution, and the reaction was allowed to proceed for a definite timeb. The reaction was then quenched with I2 for a definite timec. (2) For the entries from 12 to 15, C60 (36 mg, 50 μmol), ethyl acetoacetate (500 μL, 79 equiv.), and I2 were put into o-DCB (15 mL). The mixture was stirred for 15 min under argon or nitrogen at a preset temperature. Then, OH− (TBAOH, 1.0 M in CH3OH) was added to the solution, and the reaction was allowed to proceed for a definite timec.b Reaction time after base addition and before the addition of iodine.c Reaction time after iodine addition.d Yield in parentheses based on the consumed C60.e Addition sequence is different from the entries 1 to 11, that is, iodine is added first followed by the addition of the base. | |||||||
| 1 | 3 | 5 | 0 | 30 | 30 | 6 (16) | 27 (71) |
| 2 | 3 | 5 | 0 | 60 | 60 | 9 (22) | 28 (69) |
| 3 | 3 | 5 | rt | 30 | 30 | 32 (36) | 18 (20) |
| 4 | 3 | 5 | rt | 30 | 60 | 29 (36) | 18 (22) |
| 5 | 3 | 5 | rt | 30 | 5 | 33 (39) | 23 (27) |
| 6 | 3 | 5 | 50 | 30 | 30 | 28 (32) | 16 (18) |
| 7 | 3 | 5 | 50 | 30 | 60 | 26 (34) | 19 (25) |
| 8 | 1 | 3 | 0 | 30 | 30 | Trace | 28 (70) |
| 9 | 1 | 3 | rt | 30 | 30 | 7 (19) | 27 (73) |
| 10 | 1.2 | 3 | rt | 30 | 30 | 9 (23) | 27 (69) |
| 11 | 5 | 8 | rt | 30 | 30 | — | — |
| 12e | 3 | 5 | rt | — | 60 | 52 (65) | — |
| 13e | 4 | 5 | rt | — | 60 | 46 (48) | — |
| 14e | 5 | 5 | rt | — | 60 | — | — |
| 15e | 5 | 8 | rt | — | 60 | — | — |
The reactions were further examined using other β-keto ester substrates (1a, 1c–1f) listed in Table 2. It was found that the methods were effective for the selective preparation of methanofullerenes (2a–2f) and 2′,3′-dihydrofuran C60 derivatives (3a–3f) in relatively high yields. Methanofullerenes (2a–2f) could be obtained with the isolated yield of 38–66% (56–74% based on the consumed C60) by adding iodine first and base later. However, the formation of 2′,3′-dihydrofuran C60 derivatives (3a–3f) with the isolated yield of 26–32% (60–73% based on the consumed C60) could be controlled by adding base first and iodine later. Generally, 0 °C was good for the synthesis of 2′,3′-dihydrofuran C60 derivatives, and room temperature was good for the synthesis of methanofullerenes, except for 2e and 2f. Consequently, we found that 50 °C was better for the preparation of 2e and 2f.
| Entry | RCOCH2COOR′ | OH− (equiv.) | I2 (equiv.) | Temp (°C) | Product (% yield)a | |
|---|---|---|---|---|---|---|
| 2a–2f | 3a–3f | |||||
| a Yield in parentheses based on the consumed C60.b Addition sequence is different from that for the other entries (the addition sequence of the entries 1, 3, 5, 7, 9, and 11 involves the addition of base first and iodine later), that is, the addition of iodine first and base later. | ||||||
| 1 | 1a: R = R′ = CH3 | 1 | 3 | 0 | — | 3a: 29 (73) |
| 2b | 1a: R = R′ = CH3 | 3 | 5 | rt | 2a: 66 (74) | — |
| 3 | 1b: R = CH3, R′ = CH2CH3 | 1 | 3 | 0 | — | 3b: 28 (70) |
| 4b | 1b: R = CH3, R′ = CH2CH3 | 3 | 5 | rt | 2b: 52 (65) | — |
| 5 | 1c: R = CH3, R′ = (CH2)2CH3 | 1 | 3 | 0 | — | 3c: 27 (66) |
| 6b | 1c: R = CH3, R′ = (CH2)2CH3 | 3 | 5 | rt | 2c: 50 (63) | — |
| 7 | 1d: R = CH3, R′ = CH(CH3)2 | 1 | 3 | 0 | — | 3d: 26 (60) |
| 8b | 1d: R = CH3, R′ = CH(CH3)2 | 3 | 5 | rt | 2d: 51 (62) | — |
| 9 | 1e: R = CH3, R′ = C(CH3)3 | 1 | 3 | 0 | — | 3e: 32 (66) |
| 10b | 1e: R = CH3, R′ = C(CH3)3 | 3 | 5 | 50 | 2e: 38 (57) | — |
| 11 | 1f: R = CH2CH3, R′ = CH3 | 1 | 3 | 0 | — | 3f: 27 (61) |
| 12b | 1f: R = CH2CH3, R′ = CH3 | 3 | 5 | 50 | 2f: 45 (56) | — |
The structures of the compounds 2a–2f and 3a–3f were characterized by MALDI-TOF MS and 1H NMR, 13C NMR, and UV-vis spectroscopies. The MALDI-TOF MS spectrum of each compound exhibits a molecular ion peak that well matches with the theoretical value (see figures in ESI†). The resonances of the alkyl protons are shown in the 1H NMR spectra. The UV-vis spectra of all the compounds exhibit peaks at around 428 nm, which is attributed to the diagnostic absorption of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cycloadduct of C60 at the 6,6-junction.16 For example, the MALDI-TOF MS spectrum of the compound 2f (Fig. S42†) shows a molecular ion peak at m/z 848.0437. The 13C NMR spectrum of 2f (Fig. S44†) exhibits 22 peaks for the sp2 carbon atoms of C60, in agreement with the Cs symmetry of the molecule, two peaks at 195.93 ppm and 164.22 ppm due to the two carbonyl carbons in β-keto ester, respectively, and one peak at 72.31 ppm due to the two sp3 carbons of the C60 cage. The MALDI-TOF MS spectrum of the compound 3f (Fig. S46†) similarly shows a molecular ion peak at m/z 848.0425. However, the 13C NMR spectrum of 3f (Fig. S48†) displays 30 peaks for the sp2 carbon atoms of C60, consistent with the Cs symmetry of the molecule, one peak at 173.88 pm due to the 5′-carbon atom of the heterocycle, one signal at 164.52 ppm due to the carbonyl carbon atom, one peak at 104.26 ppm due to the 4′-carbon atom of the heterocycle, and two peaks at 102.87 ppm and 72.31 ppm assigned to the two sp3 carbons of the C60 cage.
:1 cycloadduct of C60 at the 6,6-junction.16 For example, the MALDI-TOF MS spectrum of the compound 2f (Fig. S42†) shows a molecular ion peak at m/z 848.0437. The 13C NMR spectrum of 2f (Fig. S44†) exhibits 22 peaks for the sp2 carbon atoms of C60, in agreement with the Cs symmetry of the molecule, two peaks at 195.93 ppm and 164.22 ppm due to the two carbonyl carbons in β-keto ester, respectively, and one peak at 72.31 ppm due to the two sp3 carbons of the C60 cage. The MALDI-TOF MS spectrum of the compound 3f (Fig. S46†) similarly shows a molecular ion peak at m/z 848.0425. However, the 13C NMR spectrum of 3f (Fig. S48†) displays 30 peaks for the sp2 carbon atoms of C60, consistent with the Cs symmetry of the molecule, one peak at 173.88 pm due to the 5′-carbon atom of the heterocycle, one signal at 164.52 ppm due to the carbonyl carbon atom, one peak at 104.26 ppm due to the 4′-carbon atom of the heterocycle, and two peaks at 102.87 ppm and 72.31 ppm assigned to the two sp3 carbons of the C60 cage.
The proposed reaction mechanism for the formation of the compounds 2a–2f and 3a–3f is shown in Scheme 2. The reaction is initiated by the abstraction of an α-proton from β-keto esters (1a–1f) to generate the enolate anion A, which can either react with I2 to afford the intermediate B when I2 is present in the mixture at the beginning of the reaction, or attack C60 to form the intermediate E (RC60−) when I2 is absent. The subsequent abstraction of an α′-proton from the β-keto ester moiety in B would take place to generate the intermediate C when extra OH− is present. Then, the intermediate C would attack C60 and afford the intermediate D, followed by intramolecular nucleophilic substitution to provide methanofullerenes (2a–2f). In contrast, the intermediate E is completely oxidized by I2 or partially oxidized by another molecule of neutral C60 to obtain the radical F in the presence of less OH−. Intramolecular cyclization of the radical F with the release of a hydrogen radical affords the 2′,3′-dihydrofuran C60 derivatives (3a–3f).
 | ||
| Scheme 2 Proposed mechanism for the formation of the compounds 2a–2f and 3a–3f. | ||
Conclusions
The chemistry presented herein provides rapid access to methanofullerenes and 2′,3′-dihydrofuran C60 derivatives in high yields. The addition sequence of iodine and base is crucial for the control over the formation of these two types of products. Methanofullerenes can be selectively afforded by adding iodine first and base later. However, the 2′,3′-dihydrofuran C60 derivatives were obtained by adding base first and iodine later. These simple and benign methods are highly selective, have short reaction times, and do not require transition metal catalysts.Experimental
General methods
All reactions were carried out under an argon or nitrogen atmosphere. All reagents were commercially obtained and used without further purification.Preparation and spectral characterization of 2a
Typically, 36 mg of C60 (0.05 mmol), 89 equiv. of 1a (4.47 mmol, 500 μL), and 5 equiv. of I2 (0.25 mmol, 63 mg) were added to o-DCB (15 mL) followed by stirring under an argon or nitrogen atmosphere for 15 min at room temperature. Then, 3.0 equiv. of TBAOH (1.0 M in methanol, 150 μL, 0.15 mmol) was added to the abovementioned solution, and the reaction was allowed to proceed for 60 min. The solvent of the reaction mixture was removed using a rotary evaporator under vacuum, and the residue was separated using a silica gel column with CS2/toluene (4:1 v/v) as the eluent to obtain methanofullerene 2a (27.5 mg, 74% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (4.0 mg, 11%). MALDI-TOF MS m/z calcd for C65H6O3+ [M]+ 834.0311, found 834.0305. 1H NMR (600 MHz, DMSO) δ 3.84 (s, 3H), 2.56 (s, 3H). 13C NMR (151 MHz, DMSO) δ 190.60 (1C, COCH3), 163.23 (1C, CO2CH2CH3), 145.24 (2C), 144.99 (6C), 144.92 (5C), 144.83 (2C), 144.71 (2C), 144.55 (2C), 144.52 (1C), 144.47 (4C), 144.37 (4C), 143.62 (2C), 143.60 (2C), 142.92 (1C), 142.87 (1C), 142.80 (8C), 142.02 (4C), 141.62 (2C), 141.59 (2C), 140.83 (2C), 140.79 (2C), 139.10 (2C), 137.98 (2C), 72.25 (2C, sp3-C of C60), 59.06 (1C, CCOCH3), 53.21 (1C, OCH3), 27.86 (1C, COCH3). UV-vis (in toluene) λmax: 328, 428 nm.
Preparation and spectral characterization of 3a
Typically, 36 mg of C60 (0.05 mmol) and 89 equiv. of 1a (4.47 mmol, 500 μL) were added to o-DCB (15 mL) followed by stirring under an argon or nitrogen atmosphere for 15 min at 0 °C. Then, 1.0 equiv. of TBAOH (1.0 M in methanol, 50 μL, 0.05 mmol) was added to the abovementioned solution, and the reaction was allowed to proceed for 30 min. The reaction mixture was then quenched with 3 equiv. of I2 (0.15 mmol, 38 mg) for 30 min. The solvent of the reaction mixture was removed using a rotary evaporator under vacuum, and the residue was separated using a silica gel column with CS2/toluene (4:1 v/v) as the eluent to afford 2′,3′-dihydrofuran fullerene 3a (12.1 mg, 73% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (21.6 mg, 60%). MALDI-TOF MS m/z calcd for C65H6O3+ [M]+ 834.0311, found 834.0285. 1H NMR (600 MHz, DMSO) δ 3.58 (s, 3H), 2.62 (s, 3H). 13C NMR (151 MHz, DMSO) δ 168.71 (1C, OCCH3), 163.39 (1C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 148.39 (2C), 147.82 (1C), 147.16 (2C), 147.11 (1C), 146.24 (2C), 146.03 (2C), 145.97 (2C), 145.83 (2C), 145.76 (2C), 145.45 (2C), 145.20 (2C), 144.98 (2C), 144.83 (2C), 144.59 (2C), 144.35 (2C), 144.24 (2C), 144.03 (2C), 142.63 (2C), 142.60 (2C), 142.51 (2C), 142.38 (2C), 142.28 (2C), 142.15 (2C), 142.10 (2C), 141.42 (2C), 141.29 (2C), 139.71 (2C), 139.32 (2C), 137.25 (2C), 135.06 (2C), 105.03 (1C, CCO), 50.84 (1C, OCH3), 15.40 (1C, CH3), (sp3-C of C60 not found). UV-vis (in toluene) λmax: 319, 428 nm.
O), 148.39 (2C), 147.82 (1C), 147.16 (2C), 147.11 (1C), 146.24 (2C), 146.03 (2C), 145.97 (2C), 145.83 (2C), 145.76 (2C), 145.45 (2C), 145.20 (2C), 144.98 (2C), 144.83 (2C), 144.59 (2C), 144.35 (2C), 144.24 (2C), 144.03 (2C), 142.63 (2C), 142.60 (2C), 142.51 (2C), 142.38 (2C), 142.28 (2C), 142.15 (2C), 142.10 (2C), 141.42 (2C), 141.29 (2C), 139.71 (2C), 139.32 (2C), 137.25 (2C), 135.06 (2C), 105.03 (1C, CCO), 50.84 (1C, OCH3), 15.40 (1C, CH3), (sp3-C of C60 not found). UV-vis (in toluene) λmax: 319, 428 nm.
Preparation and spectral characterization of 2b
The procedure was similar to that used for the preparation of 2a, except that 1b (79 equiv., 3.95 mmol, 500 μL) was used instead of 1a. The reaction afforded 2b (22.0 mg, 65% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (7.2 mg, 20%). MALDI-TOF MS m/z calcd for C66H8O3+ [M]+ 848.0468, found 848.0438. 1H NMR (600 MHz, DMSO) δ 4.31 (q, J = 7.2 Hz, 2H), 2.61 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 190.62 (1C, COCH3), 162.71 (1C, CO2CH2CH3), 145.35 (2C), 145.03 (2C), 145.00 (3C), 144.92 (5C), 144.86 (2C), 144.78 (2C), 144.54 (4C), 144.50 (1C), 144.47 (3C), 144.35 (4C), 143.63 (2C), 143.61 (2C), 142.93 (1C), 142.88 (1C), 142.81 (6C), 142.78 (2C), 142.03 (4C), 141.60 (4C), 140.83 (2C), 140.78 (2C), 139.13 (2C), 137.95 (2C), 72.26 (2C, sp3-C of C60), 63.06 (1C, OCH2CH3), 59.23 (1C, CCOCH3), 27.83 (1C, COCH3), 14.46 (1C, CH2CH3). UV-vis (in toluene) λmax: 326, 429 nm.Preparation and spectral characterization of 3b
The procedure was similar to that used for the preparation of 3a, except that 1b (79 equiv., 3.95 mmol, 500 μL) was used instead of 1a. The reaction afforded 3b (11.8 mg, 70% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (21.6 mg, 60%). MALDI-TOF MS m/z calcd for C66H8O3+ [M]+ 848.0468, found 848.0495. 1H NMR (600 MHz, DMSO) δ 4.05 (q, J = 7.2 Hz, 2H), 2.61 (s, 3H), 1.07 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 168.57 (1C, OCCH3), 162.99 (1C, CO), 148.43 (2C), 147.82 (1C), 147.32 (2C), 147.11 (1C), 146.25 (2C), 146.02 (2C), 145.97 (2C), 145.82 (2C), 145.76 (2C), 145.47 (2C), 145.20 (2C), 144.98 (2C), 144.82 (2C), 144.62 (2C), 144.36 (2C), 144.32 (2C), 144.05 (2C), 142.64 (2C), 142.60 (2C), 142.52 (2C), 142.40 (2C), 142.29 (2C), 142.15 (2C), 142.11 (2C), 141.39 (2C), 141.30 (2C), 139.72 (2C), 139.15 (2C), 137.24 (2C), 135.06 (2C), 104.80 (1C, CCO), 102.50 (1C, sp3-C of C60, C60–O), 72.07 (1C, sp3-C of C60, C60–C), 60.28 (1C, OCH2CH3), 15.36 (1C, CH3), 14.52 (1C, CH2CH3). UV-vis (in toluene) λmax: 320, 429 nm.
Preparation and spectral characterization of 2c
The procedure was similar to that used for the preparation of 2a, except that 1c (70 equiv., 3.50 mmol, 500 μL) was used instead of 1a. The reaction afforded 2c (21.6 mg, 63% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (7.6 mg, 21%). MALDI-TOF MS m/z calcd for C67H10O3+ [M]+ 862.0624, found 862.0601. 1H NMR (500 MHz, CDCl3) δ 4.48 (t, J = 7.0 Hz, 2H), 2.87 (s, 3H), 1.92 (m, 2H), 1.13 (t, J = 7.5 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 191.83 (1C, COCH3), 163.10 (1C, CO2CH2CH2CH3), 145.20 (2C), 144.92 (3C), 144.91 (2C), 144.85 (5C), 144.76 (2C), 144.54 (2C), 144.48 (3C), 144.41 (3C), 144.38 (4C), 144.28 (2C), 144.27 (2C), 143.54 (1C), 143.51 (2C), 142.84 (1C), 142.80 (1C), 142.72 (6C), 142.67 (2C), 141.93 (4C), 141.53 (2C), 141.50 (2C), 140.75 (2C), 140.69 (1C), 139.12 (2C), 137.73 (2C), 72.11 (2C, sp3-C of C60), 68.59 (1C, CO2CH2CH2CH3), 59.07 (1C, CCOCH3), 28.03 (1C, COCH3), 22.24 (1C, CH2CH2CH3), 10.54 (1C, CH2CH2CH3). UV-vis (in toluene) λmax: 329, 429 nm.Preparation and spectral characterization of 3c
The procedure was similar to that used for the preparation of 3a, except that 1c (70 equiv., 3.50 mmol, 500 μL) was used instead of 1a. The reaction afforded 3c (11.6 mg, 66% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (21.2 mg, 59%). MALDI-TOF MS m/z calcd for C67H10O3+ [M]+ 862.0624, found 862.0609. 1H NMR (600 MHz, CDCl3) δ 4.25 (t, J = 7.2 Hz, 2H), 2.89 (s, 3H), 1.73 (m, 2H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 169.01 (1C, OCCH3), 164.00 (1C, CO), 148.44 (2C), 147.93 (1C), 147.23 (3C), 146.35 (2C), 146.13 (2C), 146.07 (2C), 145.93 (2C), 145.87 (2C), 145.56 (2C), 145.29 (2C), 145.09 (2C), 144.93 (2C), 144.67 (2C), 144.44 (2C), 144.33 (2C), 144.14 (2C), 142.72 (2C), 142.69 (2C), 142.61 (2C), 142.47 (2C), 142.36 (2C), 142.24 (2C), 142.20 (2C), 141.48 (2C), 141.37 (2C), 139.81 (2C), 139.31 (2C), 137.38 (2C), 135.20 (2C), 105.01 (1C, CCO), 66.12 (1C, CO2CH2CH2CH3), 22.42 (1C, CH2CH2CH3), 15.52 (1C, CH3), 10.93 (1C, CH2CH2CH3), (sp3-C of C60 not found). UV-vis (in toluene) λmax: 320, 429 nm.
Preparation and spectral characterization of 2d
The procedure was similar to that used for the preparation of 2a, except that 1d (69 equiv., 3.44 mmol, 500 μL) was used instead of 1a. The reaction afforded 2d (22.0 mg, 62% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (6.5 mg, 18%). MALDI-TOF MS m/z calcd for C67H10O3+ [M]+ 862.0624, found 862.0645. 1H NMR (600 MHz, CDCl3) δ 5.42 (m, 1H), 2.85 (s, 3H), 1.52 (d, J = 6.6 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 192.60 (1C, COCH3, overlapped with the peak of CS2), 163.01 (1C, CO2CH(CH3)2), 145.59 (2C), 145.33 (2C), 145.29 (2C), 145.28 (2C), 145.22 (4C), 145.16 (2C), 144.96 (2C), 144.84 (2C), 144.79 (2C), 144.75 (4C), 144.64 (4C), 143.91 (2C), 143.89 (2C), 143.22 (1C), 143.18 (1C), 143.09 (6C), 143.04 (2C), 142.30 (4C), 141.91 (2C), 141.86 (2C), 141.11 (2C), 141.05 (2C), 139.44 (2C), 138.14 (2C), 72.52 (2C, sp3-C of C60), 71.79 (1C, OCH(CH3)2), 59.54 (1C, CCOCH3), 28.43 (1C, COCH3), 21.93 (2C, CH(CH3)2). UV-vis (in toluene) λmax: 330, 429 nm.Preparation and spectral characterization of 3d
The procedure was similar to that used for the preparation of 3a, except that 1d (69 equiv., 3.44 mmol, 500 μL) was used instead of 1a. The reaction afforded 3d (11.2 mg, 60% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (20.5 mg, 57%). MALDI-TOF MS m/z calcd for C67H10O3+ [M]+ 862.0624, found 862.0660. 1H NMR (600 MHz, CDCl3) δ 5.24 (m, 1H), 2.88 (s, 3H), 1.33 (d, J = 6.0 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 169.04 (1C, OCCH3), 163.81 (1C, CO), 148.68 (2C), 148.13 (1C), 147.55 (2C), 147.41 (1C), 146.55 (2C), 146.32 (2C), 146.26 (2C), 146.12 (2C), 146.06 (2C), 145.77 (2C), 145.49 (2C), 145.29 (2C), 145.12 (2C), 144.88 (2C), 144.65 (2C), 144.58 (2C), 144.34 (2C), 142.92 (2C), 142.88 (2C), 142.80 (2C), 142.67 (2C), 142.56 (2C), 142.44 (2C), 142.40 (2C), 141.68 (2C), 141.57 (2C), 140.00 (2C), 139.42 (2C), 137.58 (2C), 135.38 (2C), 105.24 (1C, CCO), 102.80 (1C, sp3-C of C60, C60–O), 72.34 (1C, sp3-C of C60, C60–C), 68.28 (1C, OCH(CH3)2), 22.31 (1C, OCCH3), 15.67 (2C, CH(CH3)2). UV-vis (in toluene) λmax: 320, 429 nm.
Preparation and spectral characterization of 2e
The procedure was similar to that used for the preparation of 2a, except that 1e (62 equiv., 3.10 mmol, 500 μL) was used instead of 1a, and a temperature of 50 °C was used. The reaction afforded 2e (16.6 mg, 57% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (11.9 mg, 33%). MALDI-TOF MS m/z calcd for C68H12O3+ [M]+ 876.0781, found 876.0743. 1H NMR (600 MHz, CDCl3) δ 2.85 (s, 3H), 1.73 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 191.82 (1C, COCH3), 162.17 (1C, CO2C(CH3)3), 145.55 (2C), 145.18 (2C), 145.08 (4C), 145.01 (6C), 144.97 (3C), 144.62 (4C), 144.56 (4C), 144.41 (4C), 143.71 (3C), 143.03 (1C), 142.99 (1C), 142.89 (6C), 142.86 (3C), 142.12 (3C), 141.74 (2C), 141.69 (2C), 140.90 (2C), 140.85 (2C), 139.17 (2C), 137.90 (2C), 84.87 (1C, CO2C(CH3)3), 72.59 (2C, sp3-C of C60), 60.32 (1C, CCOCH3), 28.14 (1C, COCH3), 27.92 (3C, C(CH3)3). UV-vis (in toluene) λmax: 330, 429 nm.Preparation and spectral characterization of 3e
The procedure was similar to that used for the preparation of 3a, except that 1e (62 equiv., 3.10 mmol, 500 μL) was used instead of 1a. The reaction afforded 3e (14.0 mg, 66% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (18.7 mg, 52%). MALDI-TOF MS m/z calcd for C68H12O3+ [M]+ 876.0781, found 876.0753. 1H NMR (600 MHz, CDCl3) δ 2.81 (s, 3H), 1.52 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 168.33 (1C, OCCH3), 163.25 (1C, CO), 148.53 (2C), 147.92 (1C), 147.48 (2C), 147.20 (1C), 146.35 (2C), 146.10 (2C), 146.06 (2C), 145.90 (2C), 145.85 (2C), 145.60 (2C), 145.28 (2C), 145.07 (2C), 144.90 (2C), 144.71 (2C), 144.52 (2C), 144.44 (2C), 144.16 (2C), 142.73 (2C), 142.68 (2C), 142.60 (2C), 142.50 (2C), 142.35 (2C), 142.21 (4C), 141.45 (2C), 141.38 (2C), 139.79 (2C), 139.17 (2C), 137.32 (2C), 135.19 (2C), 105.67 (1C, CCO), 81.18 (1C, CO2C(CH3)3), 28.35 (3C, C(CH3)3), 15.55 (1C, CH3), (sp3-C of C60 not found). UV-vis (in toluene) λmax: 320, 430 nm.
Preparation and spectral characterization of 2f
The procedure was similar to that used for the preparation of 2a, except that 1f (78 equiv., 3.90 mmol, 500 μL) was used instead of 1a, and a temperature of 50 °C was used. The reaction afforded 2f (19.1 mg, 56% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (7.2 mg, 20%). MALDI-TOF MS m/z calcd for C66H8O3+ [M]+ 848.0468, found 848.0437. 1H NMR (600 MHz, CDCl3) δ 4.11 (s, 3H), 3.26 (q, J = 7.2 Hz, 2H), 1.39 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 195.93 (1C, COCH2CH3), 164.22 (1C, CO2CH3), 145.62 (2C), 145.31 (4C), 145.29 (4C), 145.24 (4C), 145.11 (2C), 144.99 (2C), 144.88 (2C), 144.76 (4C), 144.65 (4C), 143.92 (2C), 143.90 (2C), 143.22 (1C), 143.17 (1C), 143.09 (6C), 143.04 (2C), 142.29 (4C), 141.95 (2C), 141.91 (2C), 141.11 (2C), 141.07 (2C), 139.34 (2C), 138.16 (2C), 72.31 (2C, sp3-C of C60), 58.96 (1C, CCOCH2CH3), 53.79 (1C, OCH3), 34.77 (1C, CH2CH3), 8.54 (1C, CH2CH3). UV-vis (in toluene) λmax: 330, 429 nm.Preparation and spectral characterization of 3f
The procedure was similar to that used for the preparation of 3a, except that 1f (78 equiv., 3.90 mmol, 500 μL) was used instead of 1a. The reaction afforded 3f (11.4 mg, 61% based on the consumed C60) as an amorphous brown solid along with the recovered C60 (20.2 mg, 56%). MALDI-TOF MS m/z calcd for C66H8O3+ [M]+ 848.0468, found 848.0425. 1H NMR (600 MHz, CDCl3) δ 3.87 (s, 3H), 3.34 (q, J = 7.8 Hz, 2H), 1.64 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 173.88 (1C, OCCH2CH3), 164.52 (1C, CO), 148.68 (2C), 148.15 (1C), 147.42 (1C), 147.31 (2C), 146.56 (2C), 146.34 (2C), 146.26 (2C), 146.14 (2C), 146.07 (2C), 145.77 (2C), 145.50 (2C), 145.29 (2C), 145.14 (2C), 144.90 (2C), 144.64 (2C), 144.50 (2C), 144.34 (2C), 142.92 (2C), 142.89 (2C), 142.81 (2C), 142.66 (2C), 142.57 (2C), 142.45 (2C), 142.40 (2C), 141.71 (2C), 141.58 (2C), 140.01 (2C), 139.64 (2C), 137.59 (2C), 135.39 (2C), 104.26 (1C, CCO), 102.87 (1C, sp3-C of C60, C60–O), 72.31 (1C, sp3-C of C60, C60–C), 51.26 (1C, OCH3), 30.17 (1C, CH2CH3), 11.83 (1C, CH2CH3). UV-vis (in toluene) λmax: 320, 429 nm.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21602192) and Shandong Provincial Government-Sponsored Study Abroad Program “Provincial School Joint Training Plan” (2019).Notes and references
- H. W. Kroto, J. R. Heath, S. O. Obrien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- H. W. Kroto, Pure Appl. Chem., 1990, 62, 407 CAS.
- H. W. Kroto, A. W. Allaf and S. P. Balm, Chem. Rev., 1991, 91, 1213 CrossRef CAS.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354 CrossRef.
- R. Taylor and D. R. Walton, Nature, 1993, 363, 685 CrossRef CAS.
- Y. Matsuo and E. Nakamura, Chem. Rev., 2008, 108, 3016 CrossRef CAS PubMed.
- H.-S. Lin and Y. Matsuo, Chem. Commun., 2018, 54, 11244 RSC.
- E. Nakamura and H. Isobe, Acc. Chem. Res., 2003, 36, 807 CrossRef CAS PubMed.
- M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262 CrossRef CAS PubMed.
- C. Tian, M. Chen, H. Tian, Z. Nan, Y. Liang and Z. Wei, J. Mater. Chem. C, 2019, 7, 12688 RSC.
- D. Iohara, F. Hirayama, M. Anraku and K. Uekama, ACS Appl. Nano Mater., 2019, 2, 716 CrossRef CAS.
- C. Bingel, Chem. Ber., 1993, 126, 1957 CrossRef CAS.
- X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997,(11), 1595 RSC.
- C. Li, D. Zhang, X. Zhang, S. Wu and X. Gao, Org. Biomol. Chem., 2004, 2, 3464 RSC.
- G.-W. Wang and F.-B. Li, Org. Biomol. Chem., 2005, 3, 794 RSC.
- T.-H. Zhang, G.-W. Wang, P. Lu, Y.-J. Li, R.-F. Peng, Y.-C. Liu, Y. Murata and K. Komatsu, Org. Biomol. Chem., 2004, 2, 1698 RSC.
- S. Chen, Z.-J. Li and X. Gao, J. Org. Chem., 2016, 81, 121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra of all the synthesised compounds. See DOI: 10.1039/d0ra04996d |
| This journal is © The Royal Society of Chemistry 2020 |