
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo and in vitro studies of antisense oligonucleotides – a review
Anna Kilanowska * and
Sylwia Studzińska
* and
Sylwia Studzińska
Chair of Environmental Chemistry and Bioanalytics, Faculty of Chemistry, Nicolaus Copernicus University in Toruń, 7 Gagarin Str., PL-87-100 Toruń, Poland. E-mail: k.anna@doktorant.umk.pl; Fax: +48 56 6114837; Tel: +48 56 6114308
First published on 17th September 2020
Abstract
The potential of antisense oligonucleotides in gene silencing was discovered over 40 years ago, which resulted in the growing interest in their chemistry, mechanism of action, and metabolic pathways. This review summarizes the selected mechanisms of antisense drug action, as well as therapeutics which are to date approved by the Food and Drug Administration and European Medicines Agency. Moreover, bioanalytical methods used for ASO pharmacokinetics and metabolism studies are briefly summarized. Special attention is paid to the primary pharmacokinetic properties of the different chemistry classes of antisense oligonucleotides. Moreover, in vivo and in vitro metabolic pathways of these compounds are widely described with the emphasis on the different animal models as well as in vitro models, including tissues homogenates, enzyme solutions, and human liver microsomes.
1. Introduction
The increasing interest in antisense therapies was initiated by Zamecnik and Stephenson, who discovered that introduction of the exogenous fragment of nucleic acid, complementary to the mRNA of the Rous Sarcoma virus, may be an efficient inhibitor of the translation process.1,2 Such DNA or RNA fragments are called “antisense oligonucleotides” (ASOs) since they bind via Watson–Crick base pairing to the sense strand of target RNA.3,4 ASOs are synthetic, single-stranded compounds, typically built of several dozen nucleotides.5The ASO structure should be chemically modified since phosphodiester oligonucleotides are rapidly digested by intracellular enzymes such as endo- and exonucleases.3–5 Moreover, native oligonucleotides have a very small affinity to proteins present in the blood (e.g. albumin), which resulted in their fast elimination from the bloodstream.6 To increase their stability, enhance tissue distribution, and binding affinity to the target sequences, modifications are introduced into sugar moieties, bases or phosphodiester linkages (Fig. 1).3,7 Oligonucleotides with modified phosphodiester linkages belong to the first generation of ASOs. Such modification involves the replacement of one of the non-bridging oxygen atoms by other atom or chemical group such as e.g. methyl one. Phosphorothioate oligonucleotides (PS) with the oxygen substituted by sulfur atom are the most commonly investigated ASOs.8–11 The second generation of these compounds includes modification within sugar moieties. In this case, the hydroxyl group at 2′ position of ribose is replaced with a fluorine atom or methyl and methoxyethyl groups (ME and MOE), which significantly reduce polarity.3,12–14 Third generation ASOs usually contains different modification either in phosphate groups, sugar moieties as well as in nucleobases. N3′ → N5′ phosphoramidates, peptide nucleic acids (PNA), morpholino phosphoroamidates (PMOs) as well as locked nucleic acid (LNA) are examples of this generation of ASOs.13,15
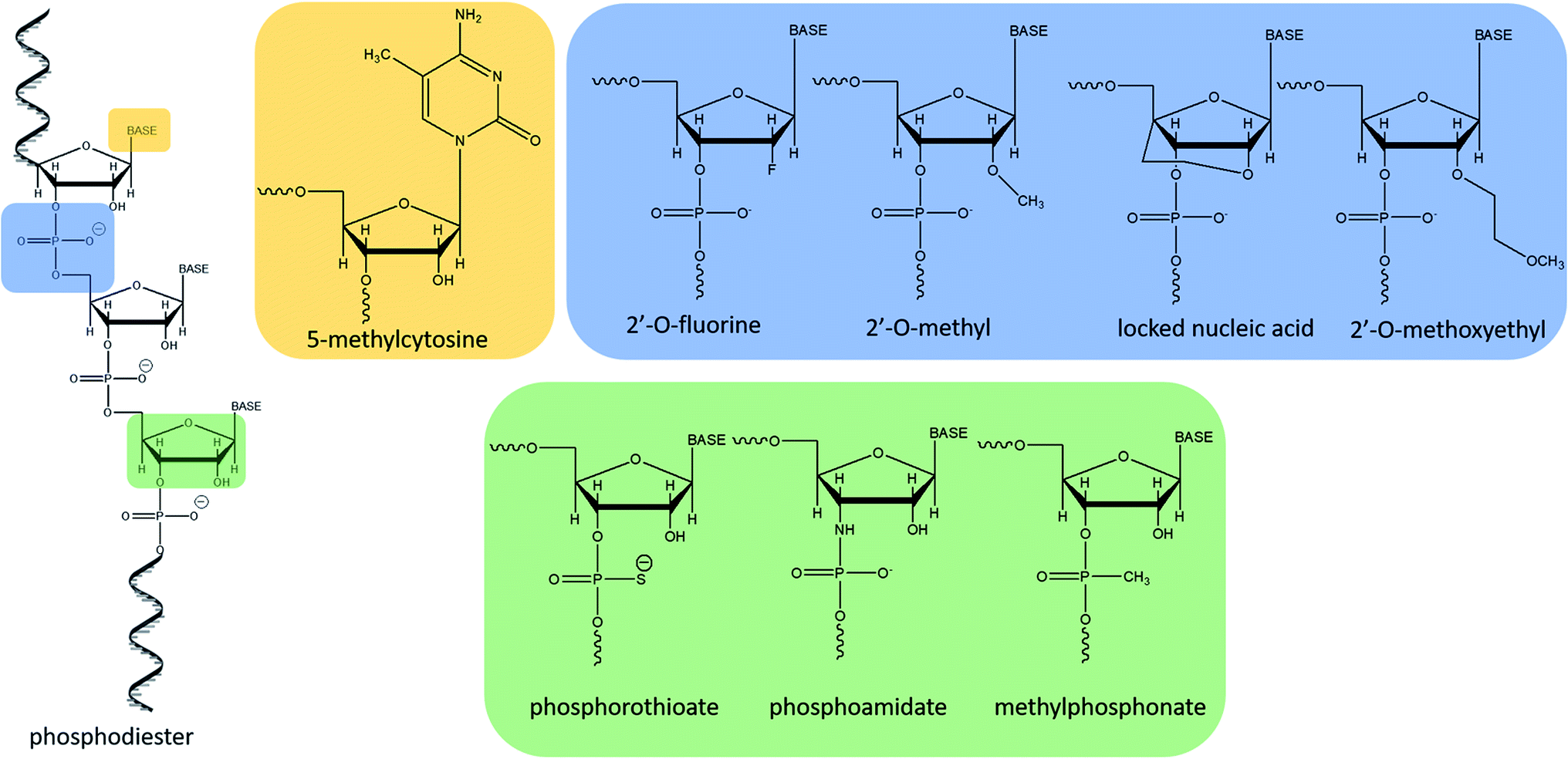 | ||
| Fig. 1 Most common structural modifications of oligonucleotides. | ||
The study of ASOs biotransformation is especially important since some of their metabolism products may be toxic.6,16–18 For this reason, the evaluation of nonclinical toxicology with the use of animal models is essential to understand the undesirable effects of potential antisense drugs. The main disadvantage of such an approach is limited knowledge of whether the potentially harmful effects in animals will be analogous in the case of patients. However, in vitro metabolism studies with the use of human tissues or enzymes may be carried out in order to improve the knowledge. Non-clinical metabolism studies are especially important for further clinical trials.17,19,20
The resent review will concern mainly the in vitro and in vivo investigations of the ASOs with the use of the different analytical methods. Special attention will be also paid to the ASOs different mechanisms of action and antisense drugs, which to date was approved by Food and Drugs Administration (FDA), as well as European Medicines Agency (EMA).
2. Mechanisms of action of oligonucleotide therapeutics
The antisense therapy concept is based on the probability that all RNA or DNA sequences longer than 13 and 17 nucleotides occur only once in the human genome.15,21 ASOs may be designed to bind not only to the RNA but also DNA, proteins, or other molecules.22 Based on the mechanism of action, the oligonucleotide therapeutics may be divided into ASOs, siRNA, miRNA as well as aptamers. However, the most popular mechanism of their action includes gene silencing with the use of ASOs. Inhibition of translation may be achieved in various ways, including RNA degradation by RNase H. Moreover, another approaches include splicing inhibition and translational arrest.6,15,23–25 Gene silencing may also be induced by the activity of small RNA fragments including siRNA and miRNA. These RNA fragments bind with RNA-induced silencing complex (RISC), naturally occurring in cells that possess enzymatic activity due to the presence of Ago2 protein. An active RISC complex with an incorporated single strand of siRNA or miRNA can recognize target mRNA, which is then destroyed by Ago2.6,15,22,26,27A most important ASOs mechanism of action is based on RNase H enzyme activity. It is present mainly in the nucleus, however, it can exist also in cytoplasm and mitochondria. This endonuclease can destroy the RNA strand in mRNA/ASO duplex by hydrolytic mechanism.5,15 Therefore, released ASO is then able to bind with another copy of mRNA. Thus, the number of targeted RNA is reduced, which consequently leads to a decrease in the level of the target protein.6 It should be noted that modification type significantly influences the mechanism of ASOs activity and only some modifications are able to activate enzymatic cleavage mediated by RNase H. One of the ASOs which promotes RNase H cleavage of target sequences are PS ASO.28 Such modification not only increases ASOs' resistance against nucleases, allowing them to reach target RNA sequence but also increases their stability in tissue and plasma.29 Moreover, they are able to destabilize heteroduplex with RNA.4 However, PS ASOs have some limitations, regarding specificity, cellular uptake toxicology, and binding affinity to the target sequences.21,30,31
Another approach of the downregulation of mRNA expression by ASOs, is the translational arrest of the targeted protein. ASOs are designed to bind with the translation initiation codon of mRNA and inhibit protein translation.15,28,32 It should be noted that in this case, chemical modification of ASOs also influences the mechanism of action. Most ASOs which are capable to create a steric block are RNase H independent. This group of modification includes changes in the furanose ring structure, such as 2′-O-methyl and 2′-O-methoxyethyl ASOs, and LNAs, PNAs, and morpholino.5,30 They increase ASO stability and cellular uptake as well as the binding affinity to the target sequences.30 Fig. 2 presents selected mechanisms of action of oligonucleotide-based therapeutics.
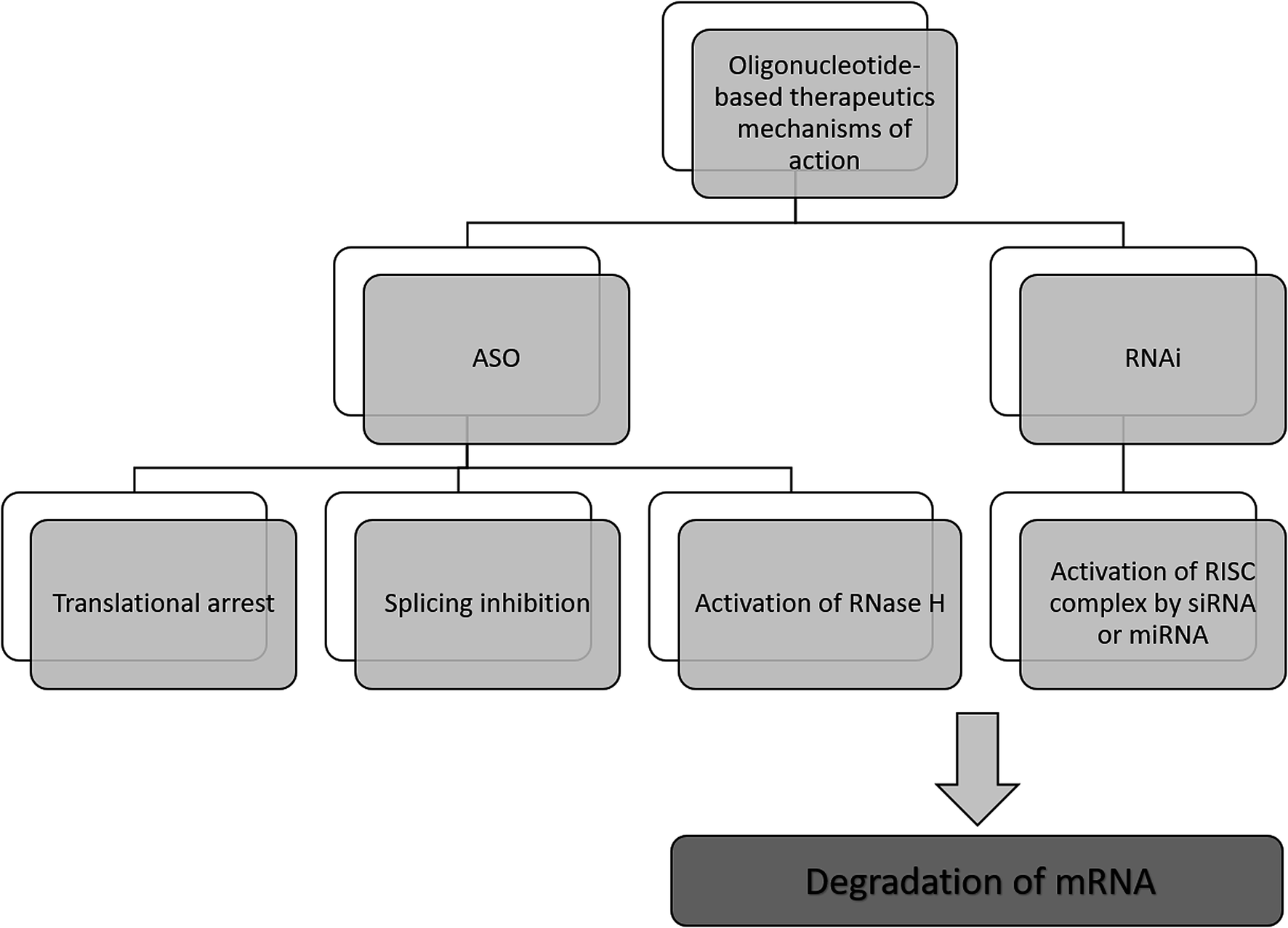 | ||
| Fig. 2 Selected mechanisms of the gene silencing based on the activity of oligonucleotide-based drugs. | ||
3. FDA/EMA approved antisense therapies
Although over 40 years have passed since Zamecnik and Stephenson discovery, antisense therapy is applied in clinical practice for 22 years.33 Such a phenomenon results from certain limitations related to the characteristic structure of ASOs, whereby they break all the rules regarding small-molecule potential drugs proposed by Lipinski.23,34 Due to the high molecular mass of ASOs as well as solubility in lipids, new rules, that antisense drugs should fulfill to be active in vivo were defined.23 These rules include appropriate pharmacological activity, resistance for nucleases, appropriately long circulation, and accessibility to a target site of activity.23,35,36There is a significant increase in the number of antisense drugs that received marketing authorization over the last 5 years, which may suggest a significant development in antisense therapy. Nowadays, there are ten antisense therapeutics available on market.29 Table 1 presents FDA and/or EMA approved antisense therapies. It should be noted that according to the website http://www.clinicaltrials.gov, 187 antisense therapeutics are now under different stages of clinical trials. However, only 17 of them entered the third phase. This group includes potential therapeutics for the treatment of e.g. leukemia, lung cancer, prostate cancer, Crohn's disease, plasma cell myeloma, Leber congenital amaurosis, and Huntington's disease.
| Drug name | Sequence length | Modification | Mechanism of action | Administration | Target | Use for treatment | Approved by | Year of approval by EMA/FDA |
|---|---|---|---|---|---|---|---|---|
| Fomivirsen (Vitravene™) | 21 | Phosphorothioate | RNase H-mediated degradation of mRNA | Intravitreal injection | IE-2 mRNA | CMV retinitis | FDA/EMA | 1998/1999 |
| Mipomersen (Kynamro™) | 20 | Phosphorothoiate/2′O-methoxyethyl | RNase H-mediated degradation of mRNA | Subcutaneous injection | ApoB-100 mRNA | Homozygous familial hypercholesterolemia | FDA | 2013 |
| Eteplirsen (Exondys 51™) | 30 | Phosphorodiamidate morpholino oligomer | Exon skipping | Intravenous injection | Exon 51 | Duchenne muscular dystrophy | FDA | 2016 |
| Defibrotide (Defitelio™) | Polydisperse mixture (9–80 mer, average 50-mer) | Unmodified | Incompletely known, probably based on charge–charge interactions | Intravenous infusion | Probably plasminogen activator | Veno-occlusive disease | FDA/EMA | 2016/2013 |
| Nusinersen (Spinraza™) | 18 | Phosphorothioate/2′O-methoxyethylated | Exon skipping | Intrathecal injection | SMN2 | Spinal muscular atrophy | FDA/EMA | 2016/2017 |
| Inotersen (Tegsedi™) | 20 | Phosphorothioate/2′O-methoxyethylated | RNase H-mediated degradation of RNA | Subcutaneous injection | TTR mRNA | Polyneuropathy in patients with hereditary transthyretin amyloidosis | FDA/EMA | 2018/2018 |
| Patisiran (Onpattro™) | 21 nucleotides per strand | A part of riboses are 2′O-methylated | RISC-mediated degradation of mRNA | Intravenous infusion of nanolipid particle solution | TTR mRNA | Polyneuropathy in patients with hereditary transthyretin amyloidosis | FDA/EMA | 2018/2018 |
| Volanesorsen (Waylivra™) | 20 | Phosphorothioate/2′O-methoxyethyl | RNase H mediated mRNA degradation | Subcutaneous injection | Apo C-III mRNA | Familial chylomicronaemia syndrome | EMA | 2019 |
| Givosiran (Givlaari™) | 21/23 nucleotides | Phosphorothioate/2′O-fluorine/2′O-methoxyethylated ASO, conjugated with N-acetylgalactosamine | RISC-mediated degradation of mRNA | Subcutaneous injection | ALAS1 mRNA | Acute hepatic porphyria | FDA/EMA | 2019/2020 |
| Golodirsen (Vyondys 53™) | 25 | Phosphorodiamidate morpholino oligomer | Exon skipping | Intravenous injection | Exon 53 | Duchenne muscular dystrophy | FDA | 2019 |
3.1 Fomivirsen (Vitravene™)
The first approved antisense drug was Fomivirsen, commercially named Vitravene. This ASO was developed for the treatment of retinitis caused by cytomegalovirus (CMV) in patients with AIDS.35 Fomivirsen is 21-mer phosphorothioate ASO with CpG motif near its 5′ end, with the sequence 5′-GCG TTT GCT CTT CTT CTT GCG-3′, resulting in mRNA degradation by RNase H-mediated mechanism.37,383.2 Mipomersen (Kynamro™)
Mipomersen (commercialized as Kynamro) was approved by the FDA in 2013 for the treatment of homozygous familial hypercholesterolemia. Kynamro is an ASO designed to degrade targeted mRNA, by activation of RNase H enzyme.35,36,39 Mipomersen is a second-generation ASO, composed of 20 nucleotides with the following sequence 5′-GCCUCAGTCTGCTTCGCACC-3′. Inter nucleotide linkages in its structure are modified with the use of phosphorothioate groups and a part of nucleotides is 2′-O-methoxyethylated.33,343.3 Eteplirsen (Exondys 51™)
Eteplirsen, commercially available as Exondys 51, was the first drug approved in 2016 for the treatment of Duchenne muscular dystrophy (DMD).40 Mechanism of this morpholino phosphorodiamidate oligomer with the sequence 5′-CTCCAACATCAAGGAAGATGGCATTTCTAG-3′, is based on splicing modulation.35,40,41 Similarly, as in the case of Mipomersen, EMA refused its marketing authorization due to limitations such as a small group of investigated patients.3.4 Defibrotide (Defitelio™)
Another antisense drug Defibrotide (commercialized as Defitelio) is used in the treatment of veno-occlusive disease of the liver, occurring mainly in patients after bone marrow transplant.35,42,43 Contrary to the other antisense drugs, Defibrotide has a natural origin, obtained from controlled polymerization of porcine intestinal mucosal DNA. It is composed of a double and single-stranded phosphodiester mixture with an average length of 50 nucleotides. Interestingly, although Defitelio is unmodified ASO, it is not digested by nucleases, probably due to its ability to form higher-order structures.35,42,433.5 Nusinersen (Spinraza™)
Spinraza is another antisense drug, whose mechanism of action is based on splicing modulation. It was approved in 2016 and 2017 by FDA and EMA respectively, for the treatment of Spinal Muscular Atrophy (SMA) mainly in infants.35,36,44,45 Intrathecal injection of Spinraza, which is 18-mer phosphorothioate 2′-O-methoxyethylated ASOs with all methylated cytidines at 5′ position, results in modulation of SMN2 gene splicing and consequently leads to its conversion into SMN1 gene and increasing of SMN protein production.463.6 Inotersen (Tegsedi™)
Tegsedi is an antisense drug, used for the treatment of nerve damage caused by hereditary transthyretin amyloidosis (hATTR), which involves the accumulation of proteins (amyloids) in tissues. Inotersen is a phosphorothioate 2′O-methoxyethylated gapmer with all methylated pyrimidines and following sequence 5′-UCUUGGTTACATGAAAUCCC-3′.36,47 It is designed to bind to mRNA, which consequently leads to its degradation by RNase H. Concentration of circulating proteins is then reduced.473.7 Patisiran (Onpattro™)
Onpattro was approved by the FDA and EMA in August 2018 as the first RNAi based therapy.36,48–50 Similarly to Inotersen, it is used for the treatment of polyneuropathy in patients with hATTR. The main difference between these two drugs results from the structure of the active substance and consequently, mechanism of action. Patisiran is siRNA, consisting of complementary strands with 21 nucleotides per strand, encapsulated in lipid nanoparticle to protect against nucleases, and for delivery to hepatocytes.49 The mechanism of its action is based on the RNAi approach. Its main limitation is the high cost of the treatment.3.8 Volanesorsen (Waylivra™)
Waylivra is an antisense drug, used for the treatment of familial chylomicronaemia syndrome (FSC), which is a genetic disorder, leading to the high concentration of triglycerides in the blood, related with overexpression of apolipoprotein (Apo) C-III.51,52 Volanesorsen is second-generation phosphorothioate, 2′O-methoxyethylated antisense drug with the following sequence 5′-AGCTTCTTGTCCAGCTTTAT-3′. It is designed for mRNA degradation via is RNase H activation.53,543.9 Givosiran (Givlaari™)
Givosiran is another drug used in antisense therapy based on the RNAi mechanism for the treatment of acute hepatic porphyria (AHP).55,56 Contrary to Onpattro™, its stability against nucleases results from modification of its structure using terminal phosphorothioate backbone linkages and the introduction of 2′O-fluorine and 2′O-methyl groups in the pentose structure.55 Givosiran was approved in November 2019 by FDA, while by EMA in March 2020.3.10 Golodirsen (Vyondys 53™)
Golodirsen is another antisense drug targeted for the treatment of DMD.57,58 Similarly, as in the case of Exondys51™, it was developed by Sarepta Therapeutics and its mechanism of action is based on the splicing modulation. Golodirsen is a 25-nucleotide phosphorodiamidate morpholino oligomer with the sequence of 5′-GTTGCCTCCGGTTCTGAAGGTGTTC-3′ and triethylene glycol chain incorporated at 5′end.58,594. In vitro and in vivo studies of antisense oligonucleotides
Similarly, as in the case of a traditional small-molecule drug, it is important to determine the safety and efficacy of the potential antisense therapeutic during the initial phase of its development. It may be achieved by drug metabolism and pharmacokinetics in vivo investigation often called ADMET (Administration, Disposition, Metabolism, Excretion, Toxicity) studies.60 Such an approach allows understanding if a drug has an ability to reach a target and provides knowledge of the metabolism pathway and potential toxicity of its products.18,60 It is suggested that in vitro studies before in vivo investigations allow for simple and fast determination of ASOs efficacy. Moreover, it gives the possibility of testing several different ASOs to select the best one for in vivo studies. Additionally, metabolite profiling with the use of human in vitro models such as tissue homogenates, subcellular fractions, or enzymes, may be useful in the development of the analytical methods of its separation and identification. Moreover, they allow for testing the influence of the drug on the hepatocytes, which is crucial in terms of potential toxicity in patients' prediction. Additionally, such an approach requires lower dosages of ASOs, which reduces costs.33,614.1 Analytical methods for ASOs pharmacokinetic, in vivo and in vitro studies
In order to choose an appropriate bioanalytical method for pharmacokinetics and metabolism study of ASOs in the different biological matrix (including plasma, tissues, and urine), various factors should be taken into consideration.62 Firstly, for the study of ASOs concentration changes in plasma, an appropriate method sensitivity is required. This is especially important in terms of local administration or elimination phase after 24 hours of drug administration since at that time ASOs concentration usually does not exceed nanomolar levels.63 Considering metabolism studies, a method providing differentiation between parent compounds and their metabolites is required.The most commonly used bioanalytical methods for ASOs analysis include different chromatographic and electromigration techniques, such as high-performance liquid chromatography (HPLC) coupled with ultraviolet detection (UV) and/or mass spectrometry (MS), capillary gel electrophoresis with UV detection. Moreover, ligand-binding assays (hybridization-based enzyme-linked immunosorbent assay, HELISA) are also used. In general, HELISA is an appropriate approach for the quantification of parent ASO in different biological samples (including plasma, urine, and tissue homogenates). This technique is important in pharmacokinetics studies, especially for the post-distribution (elimination) phase plasma concentrations (>24 h after administration).60,62,63 This method is characterized by relatively high sensitivity (LOQ < 2 ng mL−1), thereby it enables to monitor the very low concentration of ASOs in the elimination phase, as well as provides information about ultimate tissue exposure of the administered drug.62 Moreover, it provides minimal matrix effect and lack of necessity of sample clean-up.64 HELISA may be also used for the determination of full-length ASOs in urine over 24 h after administration. The main drawback of HELISA is the lack of ability to differentiate between truncated metabolites and parent compound. Moreover, there is the risk that shorter metabolites may also interact with an analytical probe via Watson–Crick base pairing.60 However, an appropriate selection of hybridization procedures may significantly decrease the cross-reactivity of shortened metabolites.65–67 Such results were presented for pharmacokinetics studies of PS and PS-MOE ASOs in rat, human, and monkey plasma.65–67 Application of ultrasensitive noncompetitive hybridization–ligation heterogeneous enzyme-linked immunosorbent assay, allowed for the determination of the different PS and PS-MOE ASOs plasma half-lives with the minimal cross-reactivity for 3′end truncated metabolites. These results demonstrated high selectivity regarding parent compound detection and allowed to obtain high sensitivity with the limit of quantification for PS ASOs equal 50 pM, while for PS-MOE ASOs – 0.78 ng mL−1.65–67 However, these methods had high selectivity only for 3′end metabolites, neither 5′end metabolites, which might still interact with the analytical probe.67 ELISA approach was also used for the determination of peptide-conjugated PMOs plasma half-lives, as well as their determination in mouse plasma and tissue lysates (kidney, liver, muscle, brain) at LLOQ value of 5 pM.68
Chromatographic methods coupled with different detectors, used for ASOs bioanalysis have been previously widely reviewed.3,4,29,69,70 HPLC coupled with triple quadrupole MS (MS/MS) or quadrupole time-of-flight (Q-TOF-MS) is commonly used for the separation, identification, and quantification of the full-length ASOs and their metabolites. This technique is more suitable for monitoring of plasma distribution phase (<24 hours) and determination of ASOs in tissues and urine since their concentrations in such samples are significantly greater compared to plasma concentration in the elimination phase. Although different modes of LC including hydrophilic interaction liquid chromatography (HILIC) and ion-exchange chromatography (IEC), were applied for ASOs analysis, MS coupled with ion pair chromatography (IPC-MS) is the most commonly used technique for ASOs bioanalysis, since it provides an appropriate compromise between method sensitivity and separation capacity.3,4,63,71–73 IEC with UV or fluorescent detector and HILIC coupled with MS are other techniques used for ASOs analysis. However, the main drawback of IEC is limited compatibility with MS detection, while in the case of HILIC - insufficient separation efficiency.3,4,74,75 The main drawbacks of IPC-MS compared to HELISA approach, are the higher LOQ (between 4–75 ng mL−1 depending on the sample type) and the necessity of sample clean-up before analysis.60,62,70,73,76,77 Lower sensitivity is derived from some technical issues during ASOs bioanalysis, including matrix effect, limited effectiveness of ASOs ionization, and signal suppression caused by the cation adducts.70,78,79 However, careful selection of sample preparation method as well as analysis conditions including ion pair reagent, chromatographic column, and source parameters allows for effective separation of parent ASOs from truncated metabolites with relatively low limits of quantification. For example, Deng et al.73 separated parent PS ASOs from its 3′N-1, 5′N-2 and, 5′N-3 metabolites in rat plasma with the LLOQ of 4 ng mL−1 for each compound with the use of IPC-MS/MS with electrospray ionization. However, this method did not allow for separation and distinguishing between 5′N-1 and 3N-1 metabolites, which possess the same number of charges. A similar issue has been encountered by Zhang et al.,80 who determined PS ASOs in rat plasma at relatively low LLOQ value (5 ng mL−1), but without complete separation. Such a problem has been largely solved by Ewles et al.81 by the application of accurate sample clean up, careful optimization of MS parameters, and selection of chromatographic parameters. The separation of 20 mer PS and 3′N-1–3′N-3 as well as 5′N-1–5′N-3, and their quantification with LLOQ values in the range of 2–1000 ng mL−1 were obtained.81
4.2 Pharmacokinetics of ASOs
In vivo pharmacokinetics study allows establishing if ASO is stable enough to reach the target cells and to determine its therapeutic effect.61,62,82,83 Such a study involved different routes of ASOs administration, including parenteral injections (intravenous infusion, subcutaneous, intradermal, intrathecal injections) and local applications.15,83 First-generation ASOs are commonly administered intravenously, due to their limited resistance against nucleases, whereby they may be rapidly degraded after subcutaneous injection.61 Oral administration is rarely used due to low adsorption of administered dose into the systemic circulation, which not exceeded 1%. However, the application of permeation enhancers such as sodium caprate allowed to achieve optimal first and second-generation ASOs' plasma bioavailability.84 Table 2 presents some pharmacokinetic properties of different ASOs.| Modification | Sequence (5′ → 3′) | Species | Dose (mg kg−1) | Route of administration | Cmax | Tmax (min) | Distribution t1/2 (min) | Clearance | Elimination t1/2 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a NA – not analyzed; ND – not detected; NM – not monitored. | ||||||||||
| PS ASO | TCC GTC ATC GCT CCT CAG GG | Mice/monkey | 1–50/1–10 | Intravenous | 15.1–647/3.4–65 μm mL−1 | 2/120 | 2.15–13.7/19.5–64.4 | 5.50–7.32/0.9–3.1 (mL min−1 kg−1) | 23.7–37.7 min/NA | 81 |
| PS or phosphodiester/MOE modified ASOs | GCG TTT GCT CTT CTT CTT GCG TTT TTT | Monkey | 10 | Intravenous | 76.7 | 30–120 | 14 | 54 | 83 min | 82 |
| 51.7 | 85 | 66 min | ||||||||
| 10.5 mg mL−1 | 524 (mL h−1 kg−1) | 23 min | ||||||||
| PS ASO | TCC CGC CTG TGA CAT GCA TT CGP | Mice | 4–100 | Intravenous | 39–542 μg mL−1 | — | 2.3–4.4 | 14.3–9.3 mL min−1 kg−1 | 29–64 min | 83 |
| PEG conjugated aptamer | CGG AAU CAG UGA AUG CUU AUA CAU CCG | Monkey | 1 | Subcutaneous | 3.4–7.1 μg mL−1 | 8–12 | — | 5.4–11.4 (mL h−1 kg−1) | 624–750 min | 84 |
| Intravenous | 20.8–27.1 μg mL−1 | — | — | 4.9–7.2 (mL h−1 kg−1) | 402–642 min | |||||
| Unmodified and modified siRNA (2′O-ME/2′-O-fluorine) | Sense: 5′-CGU ACG CGG AAU ACU UCG AUU-3’; antisense: 5′-UCG AAG UAU UCC GCG UAC GUU-3′ | Rat | 14.6 | Intravenous | — | — | ND | 22.4 | — | 85 |
| 15.4 | ND | 22.8 | ||||||||
| 11.3 | 3.4 | 6.0 (mL min−1) | ||||||||
| PS/2′O-MOE | GCT GAT TAG AGA GAG GTC CC | Human | 0.1–6 | Intravenous | 0.8–39.6 mg mL−1 | — | 22–109 | 23.6–122 (mL h−1 kg−1) | 11.9–27 days | 86 |
| PS/2′O-MOE | GCC TCA GTC TGC TTC GCA ACC | Mice | 5 | Subcutaneous | 3.8 μg mL−1 | 30 | 0.33 h | 674 mL h−1 kg−1 | NM | 87 |
| Rat | 5 | Intravenous bolus | 73.9 μg mL−1 | 2 | 0.39 h | 181 mL h−1 kg−1 | 4.7 days | |||
| Monkey | 4 | Intravenous infusion | 39.8 μg mL−1 | 60 | 0.68 h | mL h−1 kg−1 | 16 days | |||
| Human | 200 | Intravenous infusion | 21.5 μg mL−1 | 119 | 1.26 h | 40.9 mL h−1 kg−1 | 32 days | |||
Several factors have an impact on the in vivo pharmacokinetic properties of ASOs, including their resistance against nucleases, affinity to bind with proteins, plasma clearance, tissue distribution, and cellular uptake.85 Pharmacokinetic properties of these compounds are sequence-independent, however, they depend on ASOs backbone modification, which is related to their protein binding capacity.61,86 Generally, oligonucleotides with an unmodified backbone have low affinity to binding with the protein present in the blood (such as albumin and α-2 macroglobulin) and for this reason, they are rapidly eliminated from the blood by glomerular filtration (plasma half-lives below 5 minutes), degraded, and excreted.6 Enhancing their pharmacokinetic properties is obtained by the introduction of the backbone modification, conjugation with some lipophilic groups (including e.g. PEG, cholesterol, or trivalent N-acetyl galactosamine) or their encapsulation with the use of lipid nanoparticle technology.23,83,87 The conjugation of the different groups via a covalent bond prolongs the circulation of ASOs by the increasing of molecular weight above the threshold of renal clearance as well as prevention of nonspecific interactions between ASOs and plasma components.23 Another strategy using for enhancing of pharmacokinetic properties of ASOs is the use of nanoparticle formulations. Such an approach provides an appropriate resistance against nucleases as well as promotes cellular uptake.23,88
The introduction of phosphorothioate modification to the backbone of the oligonucleotide results in the increasing of their affinity to the plasma proteins and consequently increases their half-life and promotes uptake into systemic tissues.23,61,83,85 However, it should be noted that other chemical modifications, such as 2′O-methyl or 2′O-methoxyethyl ASOs influence only their stability against nucleases. It is supposed that increasing protein binding affinity is a unique feature of phosphorothioates. The highest concentration of ASOs after parenteral administration may be found in the liver, kidney, bone marrow, and lymph nodes.76,86,89 Such tendencies were observed for the different ASOs modifications including PS, PS-2′O-ME, PS-2′O-MOE, and PS-LNA. Study concerning tissue disposition of two different 2′O-methoxyethyl modified phosphorothioate ASOs in monkeys after intravenous infusion of 10 mg kg−1 dose has demonstrated the highest ASOs concentrations in the kidney cortex, kidney medulla, as well as in liver (Fig. 3).90 The investigation conducted by Yu et al.91 has shown similar tendencies for PS ASO. The greatest concentration of ASOs after intravenous infusion of 10 mg kg−1 dose and a bolus injection of 20 mg kg−1 dose in mice and monkeys was observed in the kidney, liver, spleen, and lymph nodes. Moreover, they observed that the tissue disposition of the tested ASO was not altered by the length of administration, however, it was dose-dependent.91 Approximately the same tissue distribution was noted for PS ASOs with LNA modification after intravenous injection of 5–25 mg kg−1 doses to mice. The highest ASOs concentration was found in the kidney cortex, liver, bone marrow, spleen, ovary, uterus, and adrenal cortex.92
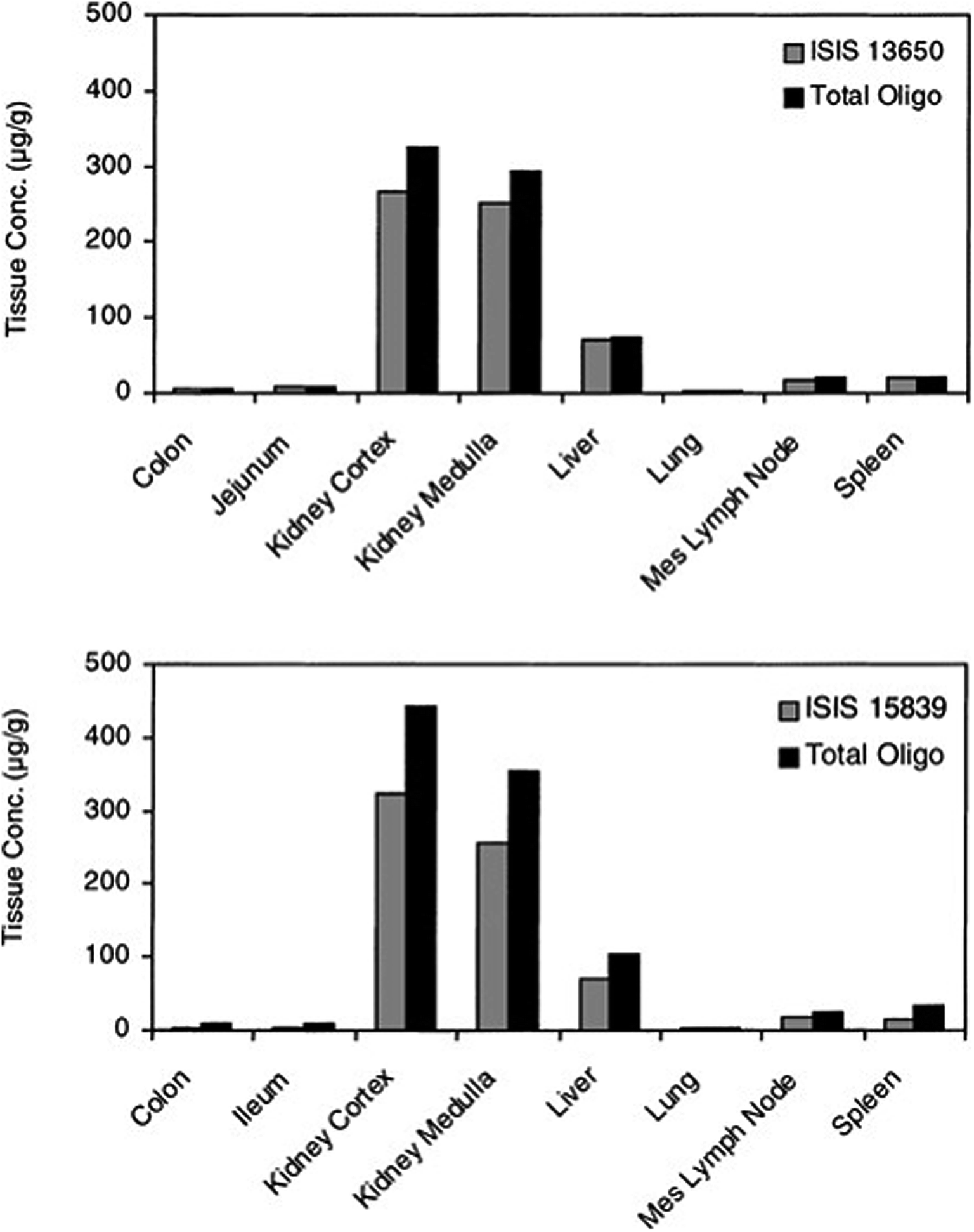 | ||
| Fig. 3 Concentrations of ASOs in tissues at 24 h after single 2 h intravenous infusion of 10 mg kg−1 to monkeys (n = 2). Reprinted from ref. 90 with permission from Elsevier (license number 4817110408883). | ||
It should be noted that systemic administered ASOs do not cross the brain–blood barrier, which is related to their size and charge. For this reason, for the treatment of central nervous system diseases, intrathecal injection to cerebrospinal fluid (CSF) is required.83,88 It has been shown that Spinraza (ISIS 396443) used for the treatment of SMA, demonstrated significant maintained splicing correction after intrathecal administration in rodents, compared to intraperitoneal bolus injection. Conducted experiments have shown that after administration directly to CSF, duration of ASOs action equaled up to 6 months, while after intraperitoneal bolus injection up to 8 weeks after administration.93 Pharmacokinetics of ASOs in the central nervous system is characterized by a steep distribution phase with the prolonged tissue half-life (over 100 days after administration in the spinal cord and brain of monkeys) and slow elimination phase from central nervous system tissues to the systemic circulation.93 Similar pharmacokinetic properties were observed in humans, for which CSF half-life approximated 163 days.94 Fig. 4 presents simulated pharmacokinetic profiles of Nusinersen in humans for the different compartments.
 | ||
| Fig. 4 Simulated median PK profiles of Nusinersen in the cerebrospinal fluid, central nervous system tissue, plasma, and systemic tissue following a single 12 mg fixed-dose. Reprinted from ref. 94 with permission of John Wiley and Sons (license number: 4822440774979). | ||
In general, first and second-generation ASOs bind to the plasma proteins with the affinity in the range 77–99% (ref. 17) and then are quickly transferred to target tissues and cells via endocytosis.83 However, it should be noted that despite protein binding affinity is comparable between species, the highest was observed for humans, while the lowest for mice or guinea pig.15,95,96 Bosgra et al.97 performed plasma incubation experiments with Drisapersen and concluded that this ASO binds to albumin and γ-globulin with the affinity more than 99% for different species (mouse, rat, monkey, human). The distribution half-lives (t1/2) usually does not exceed 1 hour for phosphorothioates, while for second-generation ASOs – 4–6 hours.15,61–63,95,98 Elimination half-lives for phosphorothioate ASOs range between 40 to 60 hours, while for second-generation ASOs extends over a dozen days.17,62,83,99,100 For example, studies with ISIS 104838, phosphorothioate ASOs with 2′O-methoxyethyl modification at 3′ and 5′ end (complementary to tumor necrosis factor mRNA) show that terminal plasma elimination approximated 25 days,101 while in the case of Mipomersen and Inotersen with the same modification it equaled 31 days and 27 days respectively.102,103 Considering fully modified PS 2′O-ME ASO Drisapersen used for the treatment of Duchenne Muscular Dystrophy this parameter ranged from 19 to 56 days.104,105 The prolongation of the clearance and elimination phase of second-generation ASOs results from their greater resistance against nucleases and for this reason, they are metabolized more slowly compared to phosphorothioates. As a consequence, the frequency of a drug administration may be reduced.
First-generation ASOs are cleaved by nucleases into fragments of lower molecular masses, thus losing their ability to bind to plasma proteins, resulting in their filtration and renal excretion.83 Geary et al.96 conducted investigations concerning plasma protein binding of 20-mer phosphorothioate ASO targeted to ICAM-1 and its shorter metabolites. They observed comparable binding to plasma proteins (>90%) between the parent compound and its N-1–N-8 metabolites. For the metabolites with the shorter sequence (<N-10), a significant reduction of plasma protein binding affinity was noticed.96 Similar conclusions were drawn for second-generation ASOs (PS-2′O-MOE).67
Interestingly, third-generation ASOs, such as unconjugated PNA or phosphorodiamidate morpholinos, have significantly lower affinity to plasma proteins (below 25%), whereby they are rapidly filtrated and excreted, resulting in their low target bioavailability.17,62,86,106 However as has been mentioned above, their conjugation significantly improves its bioavailability. PMO ASOs show an increased resistance against nucleases.107 Pharmacokinetic profiles of these compounds are dose-dependent and similar for the different routes of administration, including intravenous, transdermal or subcutaneous routes.40,106,108,109 These compounds are characterized by rapid tissue distribution phase (between 1 to 4 hours) and plasma half-lives usually up to 20 hours. PMOs are enzymatically stable and may be detected in tissues even after six half-lives.106 For example, plasma half-lives for positively charged PMO used for the treatment of Marburg virus approximated 3 hours for humans and nonhuman primates.108,110 A study conducted by Amantana et al.109 has shown rapid distribution to tissues of PMOs administered intravenously, with the initial distribution half-life ranging from 0.40 to 1.56 hours, while the elimination half-live did not exceed 9 hours. The highest ASOs concentration was detected in the kidney and liver with lower concentrations observed in the lungs, spleen, heart, and brain.109
It should be noted that pharmacokinetic plasma parameters for the different ASOs are similar between different species, including mouse, rat, dog, monkey, and are independent of a route of administration as well as ASOs sequence. Moreover, these parameters may be simply transferred to human applications based on body weight.61,83
4.3 In vitro metabolism
In vitro metabolism investigation of ASOs was performed with the use of the different models, including human and mouse liver microsomes, human, rat, and mouse liver homogenates as well as purified enzymes (3′exonuclease solution, DNase I and Exonuclease I).111–117 Different ASOs modifications were tested during these studies, such as unmodified deoxyribonucleotides, first-generation PS ASOs, partially 2′O-ME, and 2′O-MOE modified phosphorothioates, as well as fully modified one with 2′-O-ME groups in sugar moieties. Metabolic pathways obtained in vitro for ASOs are generally in accordance with those obtained during in vivo studies and are based on the hydrolysis of phosphate and phosphorothioate backbones via exonucleases with some endonucleolytic activity.86,111,112,114 However, some differences may be observed between animal models and in vitro incubation, which was confirmed during investigations conducted by Kim et al.111 They observed 3′ exonucleases degradation products after incubation of PS fully 2′-O-ME modified ASOs with mouse liver homogenates, while in the case of in vivo-generated metabolites, also 5′ exonucleases contributed to ASOs biotransformation. Rodents and human liver homogenates were also used for the incubation of phosphodiesters, first-generation ASOs, and partially 2′O-MOE modified phosphorothioates.113,114Phosphorothioates are metabolized to a lower extend in vitro, compared to in vivo conditions. The full-length ASOs nuclease stability equals several hours in the liver. For this reason, careful optimization of incubation time is required.113 Their main metabolic pathway is a result of 3′ exonucleases degradation. The number and amount of produced metabolites depend on the in vitro model. The greater number of PS metabolites may be found in liver homogenates, compared to liver microsomes and exonucleases solutions.112,115,117 The optimization of incubation parameters with liver microsomes seems to be an important factor, which was confirmed for PS ASOs.112 Studzińska et al.112 proved that HLM concentration, incubation time, ASOs concentration, and concentration of NADP/NADPH regeneration system components had a significant impact on the number of formed metabolites. Such results were also confirmed in our recent study for the different ASOs modifications, including 2′O-ME, 2′O-MOE, and LNA ASOs.
Partially 2′O-MOE modified PS ASO are metabolized in the same way as in the case of in vivo studies (initial endonuclease cleavage followed by exonucleases). Moreover, similar metabolic pathways may be identified after ASOs incubation with human, rat, and mouse liver homogenates.114,115 Interestingly, recent studies showed that these ASOs were not metabolized after 7 days incubation with the human liver microsomes, which points out that such compounds are not substrates for CYP450 mediated oxidative metabolism.115 Moreover, it indicated that human liver microsomes show a lower degree of metabolism compared to liver homogenates. Similar conclusions may be drawn in the case of pure enzyme solutions. As an example, Kim et al.111,115 did not observe any metabolites after 48 and 24 hours of second-generation ASOs incubation (fully and partially modified PS ASOs with 2′O-ME and 2′O-MOE groups) with exo- and endonucleases solutions (RNase A, DNase I, Exonuclease I and Exo-T), which was related with their greater enzymatic stability. Interestingly, in our recent investigation concerning metabolism studies of different ASOs generations with human liver microsomes, we observed exonucleolytic degradation products even for the ASOs modification, characterized by the greatest nuclease resistance (ME, MOE and LNA modifications). These results indicated that the optimization of incubation parameters is one of the crucial factors during metabolism studies with HLM.
Although the modification of ASOs mainly influences their biotransformation pathways, different factors may also have an impact on the metabolism rate, including nucleotides position and sequence length. It was reported, that pyrimidines (U, C, T) are metabolized faster than purines (G, A).96,109 Moreover, longer ASOs are metabolized more slowly than shorter ones. Studzińska et al.112 conducted in vitro metabolism study with human liver microsomes for 18 mer and 20 mer PS ASOs and identified 3′ exonuclease degradation products for shorter sequences after 8 hours of incubation, while for a longer one – after 12 hours.112
4.4 In vivo metabolism of ASOs
In vivo metabolism studies of ASOs were performed with the use of different models, including mice, guinea pigs, rats, dogs, pigs, monkeys, and humans.62,91,95,102,111,117 Several various ASOs differing in length, sequence and chemical modifications, such as PS, PS-MOE, PS-ME, PS-LNA, PMOs, GaInac-conjugates, were analyzed in different biological samples, including plasma, tissues (liver, kidney cortex, and medulla, lungs, lymph nodes, spleen), urine, and cerebrospinal fluid.65,89,101,102,116,118–122 Tissues and plasma clearance of ASOs resulted from nucleases activity since these compounds are not susceptible to cytochrome P450-mediated oxidative metabolism.60,72,90,116,122,123 Depending on the nuclease type, the ASOs sequence may be truncated in two different ways. Firstly, consecutive sequential deletion of nucleotides present at the 3′ and 5′ end of the ASOs sequence is caused by exonucleases. Moreover, endonuclease-mediated hydrolysis of ASOs at various sequence position is also possible.62,70,86,98,101,119,122,124As stated above, unmodified oligonucleotides are rapidly degraded mainly by 3′ exonucleases, which are present in the blood and tissues.107,122 Similarly, as in the case of unmodified oligonucleotides, 3′exonucleolytic degradation is the main metabolic pathway of phosphorothioates, however, products of 5′exonuclease-mediated degradation may be also present, especially considering PS ASOs metabolism in tissues.73,86,98,113,116,119 Noll et al.119 used MALDI-TOF for the ASOs metabolism study in different tissues and plasma. They identified 3′exonucleases-mediated N-1–N-6 metabolites and metabolism products formed by 5′ exonucleases deletion of one and two nucleotides (Fig. 5). Moreover, a small amount of 3′ exonucleolytic degradation products was detected in plasma samples (mainly 3′N-1 metabolites). Geary et al.125 observed that the metabolism pattern for PS ASOs in plasma samples was dose- and time-independent. Obtained results showed that after ASOs administration, their metabolism in plasma was very rapid (with 65% of the full compound after 10 minutes). However, it becomes slower over time (about 35–65% of parent ASOs detected after 300 minutes of intravenous administration). The authors indicated that such results may be related to stereoisomeric selectivity of exonucleases as well as inhibition of exonucleases metabolism by the generation of monomer nucleosides or nucleotides.125
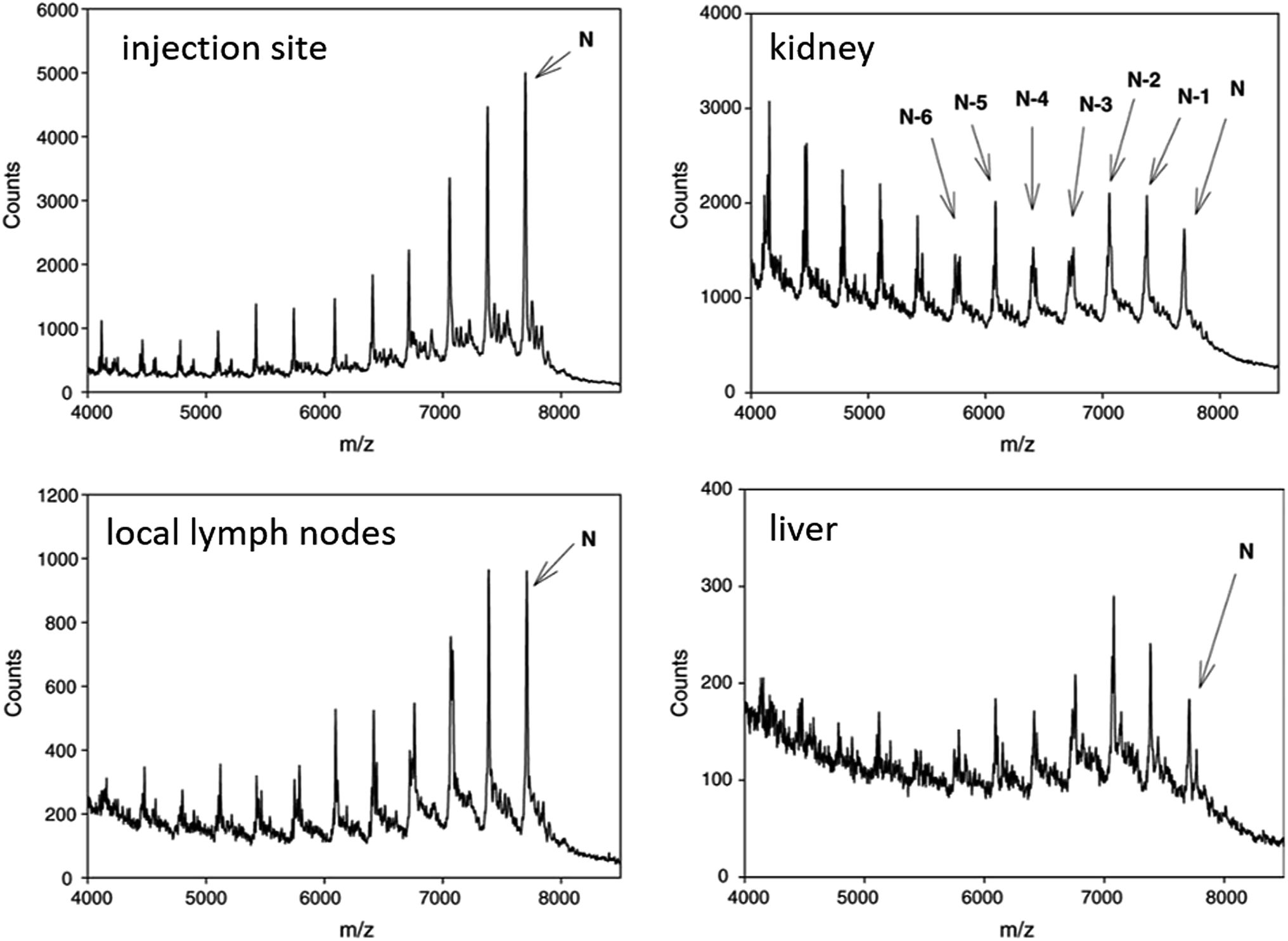 | ||
| Fig. 5 MALDI-TOF spectra of CPG 7909 and metabolites in rat tissues after SC administration of 9.0 mg kg−1 parent compound. Reprinted from ref. 119 with permission from Elsevier (license number 4817111143580). | ||
Metabolism routes of the first and second-generation ASOs seem to be similar across species, which was confirmed in several studies, in which similar metabolic pathways for mice, monkeys, rats, and humans were observed.65,91,119 Geary et al.65 observed several metabolites of 20 mer PS ASO, partially modified with MOE groups, ranging from 8 to 12 nucleotides in length in the different biological samples including plasma, urine, and tissues. Interestingly, similar metabolites were detected in human, rat, and mouse urine after six hours of administration with the use of CGE method.65 More sensitive methods (LC/radiometric and LC-MS) gave the same results for plasma, for which metabolites concentration was significantly lower, as well as for tissues.65 Yu et al.89 identified 5′endonucleolytic degradation products of 2′O-MOE modified PS ASO in human and monkey urine with the use of LC/MS method. Moreover, the same metabolites were detected in tissues with the use of CGE method, which indicated that after metabolites production in tissues, they were extracted in urine.89
The introduction of terminal ribose modifications (including ME, MOE, and LNA) to the PS ASOs structure significantly prolongs their plasma and tissues half-lives (usually to several days) through increasing resistance against nucleases.107,124,126–128 For this reason, PS-2′-O modified ASOs are initially cleaved by endonucleases at the central sequence region, resulting in two shorter sequences with the unprotected terminus, which are substrates for 3′ and 5′-exonucleases-mediated metabolism (Fig. 6).62,89,124 Such a metabolism pathway was confirmed in several different studies.39,90,124 Yu et al.,89 detected N-6–N-10 metabolites of 20-mer parent phosphorothioate 2′-O-MOE ASOs in urine by LC-MS, resulting from PS backbone hydrolysis by endonuclease at various positions of deoxyribonucleotide gap. Moreover, some shorter metabolites (N-10–N-13) were further produced by exonucleolytic degradation. It should be noted that the metabolism of such compounds in plasma is minimal, compared to PS ASOs.90
 | ||
| Fig. 6 Schematic presentation of metabolism pathways for chimeric antisense oligonucleotides. Reprinted from ref. 124. This figure was published by ASPET under the CC BY-NC Attribution 4.0 International license. | ||
There are some reports concerning in vivo metabolism of fully 2′ribose modified PS ASOs.111,129 The authors indicated that these modifications had an impact on the reduction of metabolic rate and significantly increased plasma and tissue half-lives. Degradation of PS-2′O-MOE and PS-2′O-ME ASOs is a result of 3′ and 5′ exonuclease activity, however, such compounds are metabolized more slowly compared to partially modified ASOs. Spinraza terminal elimination half-life is estimated to be 63 to 87 days in plasma,129 while for partially modified Volanesorsen ranged from 11.7 to 31.2 days respectively.124
To date, no reports are presenting successful detection of PMOs metabolites in the different samples, including plasma, tissues, urine, or cerebrospinal fluid, which proved their high resistance against nucleases degradation.106,107 The metabolism of ASOs, conjugated with the different lipophilic groups, is the same as in the case of PS or PS-ribose modified ASOs, however, in this case, metabolism of the linkers was mainly considered.118
5. Conclusions and remarks for the future
A significant advancement in antisense drugs development may be observed for the last five years, resulting in eight new therapeutics recently approved by FDA and/or EMA. Moreover, a notable number of these compounds are in clinical trials due to their great potential for the treatment of a wide range of various diseases. Although chemical modifications of ASOs significantly improved ASOs bioavailability and ability to reach the target sequences in cells, their chemistry should be still developed through novel modifications or formulations, which may potentially result in the increase of ASOs therapeutic effects. In addition, the same emphasis should be paid on further improvement in the bioanalytical methods used for ASO pharmacokinetics and metabolism studies, since ligand-binding assays and LC-MS methods are characterized by several limitations such as lack of ability to the differentiation between parent ASO and metabolites as well as limited sensitivity.The pharmacokinetics of ASOs depends on chemical modification and is similar for different species. Improvements in the pharmacokinetic properties of ASOs caused by the introduction of ribose modification result in the increase of tissue distribution which is mainly important in terms of reduction of frequency of drug administration. Metabolism of ASOs is based on exonucleases degradation of subsequent nucleotides, with the activity of endonucleases in the case of some modifications. In vivo and in vitro biotransformation studies at an early stage of drug development are especially important for the prediction of the potential toxicity of ASOs, which is crucial in the further clinical trials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Science Center (Cracow, Poland) under Sonata Bis project (2016/22/E/ST4/00478).References
- M. L. Stephenson and P. C. Zamecnik, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 285–288 CrossRef CAS.
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280–284 CrossRef CAS.
- A. Kaczmarkiewicz, Ł. Nuckowski, S. Studzińska and B. Buszewski, Crit. Rev. Anal. Chem., 2019, 49, 256–270 CrossRef CAS.
- S. Studzińska, Talanta, 2018, 176, 329–343 CrossRef.
- N. Dias and C. A. Stein, Cancer Res., 2002, 1, 347–355 CAS.
- H. S. Younis, M. Templin, L. O. Whiteley, D. Kornbrust, T. W. Kim and S. P. Henry, Overview of the Nonclinical Development Strategies and Class-Effects of Oligonucleotide-Based Therapeutics, Elsevier Inc., 2nd edn, 2017 Search PubMed.
- Ł. Nuckowski, A. Kaczmarkiewicz and S. Studzińska, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1090, 90–100 CrossRef.
- S. Studzińska, R. Rola and B. Buszewski, J. Pharm. Biomed. Anal., 2017, 138, 146–152 CrossRef.
- P. A. Lobue, M. Jora, B. Addepalli and P. A. Limbach, J. Chromatogr. A, 2019, 1595, 39–48 CrossRef CAS.
- K. J. Fountain, M. Gilar and J. C. Gebler, Rapid Commun. Mass Spectrom., 2003, 17, 646–653 CrossRef CAS.
- W. Zhang, N. Leighl, D. Zawisza, M. J. Moore and E. X. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 829, 45–49 CrossRef CAS.
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef CAS.
- E. Urban and C. R. Noe, Farmaco, 2003, 58, 243–258 CrossRef CAS.
- A. Zimmermann, R. Greco, I. Walker, J. Horak, A. Cavazzini and M. Lämmerhofer, J. Chromatogr. A, 2014, 1354, 43–55 CrossRef CAS.
- M. Mansoor and A. J. Melendez, Gene Regul. Syst. Biol., 2008, 2008, 275–295 Search PubMed.
- C. F. Bennett and E. E. Swayze, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259–293 CrossRef CAS.
- E. K. Mustonen, T. Palomäki and M. Pasanen, Regul. Toxicol. Pharmacol., 2017, 90, 328–341 CrossRef CAS.
- S. A. Wrighton, B. J. Ring and M. VandenBranden, Toxicol. Pathol., 1995, 23, 199–208 CrossRef CAS.
- M. Szultka-Mlynska and B. Buszewski, Anal. Bioanal. Chem., 2016, 408, 8273–8287 CrossRef CAS.
- M. Szultka, R. Krzeminski, M. Jackowski and B. Buszewski, Chromatographia, 2014, 77, 1027–1035 CrossRef CAS.
- Y. C. Zhang, M. M. Taylor, W. K. Samson and M. I. Phillips, Methods Mol. Med., 2005, 106, 11–35 CAS.
- J. W. Gaynor and R. Cosstick, Methods Mol. Biol., 2011, 764, 17–30 CrossRef CAS.
- D. B. Rozema, The Chemistry of Oligonucleotide Delivery, Elsevier Inc., 1st edn, 2017, vol. 50 Search PubMed.
- A. Sehgal, A. Vaishnaw and K. Fitzgerald, J. Hepatol., 2013, 59, 1354–1359 CrossRef CAS.
- S. T. Crooke, Adv. Pharmacol., 1997, 40, 1–49 CAS.
- N. M. Dean and C. Frank Bennett, Oncogene, 2003, 22, 9087–9096 CrossRef CAS.
- G. Kher, S. Trehan and A. Misra, Antisense Oligonucleotides and RNA Interference, Elsevier Inc., 1st edn, 2011 Search PubMed.
- S. T. Crooke, Biochim. Biophys. Acta, Gene Struct. Expression, 1999, 1489, 31–43 CrossRef CAS.
- A. Goyon, P. Yehl and K. Zhang, J. Pharm. Biomed. Anal., 2020, 113105 CrossRef CAS.
- M. A. Havens and M. L. Hastings, Nucleic Acids Res., 2016, 44, 6549–6563 CrossRef.
- T. P. Prakash, Chem. Biodiversity, 2011, 8, 1616–1641 CrossRef CAS.
- T. A. Vickers and S. T. Crooke, PLoS One, 2014, 9(10), e108625 CrossRef.
- R. Lakhia, A. Mishra and V. Patel, Manipulation of renal gene expression using oligonucleotides, Elsevier Inc., 1st edn, 2019, vol. 154 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- C. A. Stein and D. Castanotto, Mol. Ther., 2017, 25, 1069–1075 CrossRef CAS.
- J. Rüger, S. Ioannou, D. Castanotto and C. A. Stein, Trends Pharmacol. Sci., 2020, 41, 27–41 CrossRef.
- G. B. Mulamba, A. Hu, R. F. Azad, K. P. Anderson and D. M. Coen, Antimicrob. Agents Chemother., 1998, 42, 971–973 CrossRef CAS.
- K. P. Anderson, M. C. Fox, V. Brown-Driver, M. J. Martin and R. F. Azad, Antimicrob. Agents Chemother., 1996, 40, 2004–2011 CrossRef CAS.
- E. Wong and T. Goldberg, Pressocolata Tecnol., 2014, 39, 119–122 Search PubMed.
- K. R. Q. Lim, R. Maruyama and T. Yokota, Drug Des., Dev. Ther., 2017, 11, 533–545 CrossRef CAS.
- K. Anthony, L. Feng, V. Arechavala-Gomeza, M. Guglieri, V. Straub, K. Bushby, S. Cirak, J. Morgan and F. Muntoni, Hum. Gene Ther: Methods., 2012, 23, 336–345 CrossRef CAS.
- C. Stein, D. Castanotto, A. Krishnan and L. Nikolaenko, Mol. Ther.--Nucleic Acids, 2016, 5, e346 CrossRef CAS.
- L. Morgan, Am. Health Drug Benef., 2017, 10, 51–53 Search PubMed.
- M. Pacione, C. E. Siskind, J. W. Day and H. K. Tabor, J. Neuromuscul. Dis., 2019, 6, 119–131 Search PubMed.
- S. Simoens and I. Huys, Gene Ther., 2017, 24, 539–541 CrossRef CAS.
- C. Zanetta, M. Nizzardo, C. Simone, E. Monguzzi, N. Bresolin, G. P. Comi and S. Corti, Clin. Ther., 2014, 36, 128–140 CrossRef.
- L. Gales, Pharmaceuticals, 2019, 12, 10–15 CrossRef.
- A. V. Kristen, S. Ajroud-Driss, I. Conceição, P. Gorevic, T. Kyriakides and L. Obici, Neurodegener Dis Manag., 2019, 9, 5–23 CrossRef.
- X. Zhang, V. Goel and G. J. Robbie, J. Clin. Pharmacol., 2020, 60(5), 573–585 CAS.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 CrossRef CAS.
- A. Digenio, R. L. Dunbar, V. J. Alexander, M. Hompesch, L. Morrow, R. G. Lee, M. J. Graham, S. G. Hughes, R. Yu, W. Singleton, B. F. Baker, S. Bhanot and R. M. Crooke, Diabetes Care, 2016, 39, 1408–1415 CrossRef CAS.
- M. J. Graham, R. G. Lee, T. A. Bell, W. Fu, A. E. Mullick, V. J. Alexander, W. Singleton, N. Viney, R. Geary, J. Su, B. F. Baker, J. Burkey, S. T. Crooke and R. M. Crooke, Circ. Res., 2013, 112, 1479–1490 CrossRef CAS.
- N. A. Rocha, C. East, J. Zhang and P. A. McCullough, Curr. Atheroscler. Rep., 2017, 19(62), 1–9 CAS.
- FDA, FDA Briefing Document – Endocrinologic and Metabolic Drugs Advisory Committee Meeting, 2018 Search PubMed.
- L. J. Scott, Drugs, 2020, 80, 335–339 CrossRef.
- P. Kumar, R. Degaonkar, D. C. Guenther, M. Abramov, G. Schepers, M. Capobianco, Y. Jiang, J. Harp, C. Kaittanis, M. M. Janas, A. Castoreno, I. Zlatev, M. K. Schlegel, P. Herdewijn, M. Egli and M. Manoharan, Nucleic Acids Res., 2020, 1–13 Search PubMed.
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2020, 13, 1–16 CrossRef.
- Y. A. Heo, Drugs, 2020, 80, 329–333 CrossRef.
- L. Echevarría, P. Aupy and A. Goyenvalle, Hum. Mol. Genet., 2018, 27, R163–R172 CrossRef.
- S. Andersson, M. Antonsson, M. Elebring, R. Jansson-Löfmark and L. Weidolf, Drug Discovery Today, 2018, 23, 1733–1745 CrossRef CAS.
- R. Z. Yu, R. S. Geary and A. A. Levin, Pharmacokinet. Pharmacodyn. Biotech Drugs Princ. Case Stud. Drug Dev., 2006, 93–120 Search PubMed.
- R. Z. Yu, J. S. Grundy and R. S. Geary, Expert Opin. Drug Metab. Toxicol., 2013, 9, 169–182 CrossRef CAS.
- D. A. Norris, N. Post, R. Z. Yu, S. Greenlee and Y. Wang, Bioanalysis, 2019, 11, 1909–1912 CrossRef CAS.
- H. Legakis and S. Carriero, Handbook of analysis of oligonucleotides and related products, 2012, vol. 32 Search PubMed.
- R. S. Geary, R. Z. Yu, T. Watanabe, S. P. Henry, G. E. Hardee, A. Chappell, J. Matson, H. Sasmor, L. Cummins and A. A. Levin, Drug Metab. Dispos., 2003, 31, 1419–1428 CrossRef CAS.
- X. Wei, G. Dai, G. Marcucci, Z. Liu, D. Hoyt, W. Blum and K. K. Chan, Pharm. Res., 2006, 23, 1251–1264 CrossRef CAS.
- R. Z. Yu, B. Baker, A. Chappell, R. S. Geary, E. Cheung and A. A. Levin, Anal. Biochem., 2002, 304, 19–25 CrossRef CAS.
- U. Burki, J. Keane, A. Blain, L. O'Donovan, M. J. Gait, S. H. Laval and V. Straub, Nucleic Acid Ther., 2015, 25, 275–284 CrossRef CAS.
- A. C. McGinnis, E. C. Grubb and M. G. Bartlett, Rapid Commun. Mass Spectrom., 2013, 27, 2655–2664 CrossRef CAS.
- Z. J. Lin, W. Li and G. Dai, J. Pharm. Biomed. Anal., 2007, 44, 330–341 CrossRef CAS.
- R. Wheller, S. Summerfield and M. Barfield, Int. J. Mass Spectrom., 2013, 345–347, 45–53 CrossRef CAS.
- G. Dai, X. Wei, Z. Liu, S. Liu, G. Marcucci and K. K. Chan, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 825, 201–213 CrossRef CAS.
- P. Deng, X. Chen, G. Zhang and D. Zhong, J. Pharm. Biomed. Anal., 2010, 52, 571–579 CrossRef CAS.
- A. C. McGinnis, B. Chen and M. G. Bartlett, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 883–884, 76–94 CrossRef CAS.
- S. Noga, S. Bocian and B. Buszewski, J. Chromatogr. A, 2013, 1278, 89–97 CrossRef CAS.
- J. L. Johnson, W. Guo, J. Zang, S. Khan, S. Bardin, A. Ahmad, J. X. Duggan and I. Ahmad, Biomed. Chromatogr., 2005, 19, 272–278 CrossRef CAS.
- V. K. Sharma, J. Glick and P. Vouros, J. Chromatogr. A, 2012, 1245, 65–74 CrossRef CAS.
- B. Basiri and M. G. Bartlett, Bioanalysis, 2014, 6, 1525–1542 CrossRef CAS.
- W. D. Van Dongen and W. M. A. Niessen, Bioanalysis, 2011, 3, 541–564 CrossRef CAS.
- G. Zhang, J. Lin, K. Srinivasan, O. Kavetskaia and J. N. Duncan, Anal. Chem., 2007, 79, 3416–3424 CrossRef CAS.
- M. Ewles, L. Goodwin, A. Schneider and T. Rothhammer-Hampl, Bioanalysis, 2014, 6, 447–464 CrossRef CAS.
- S. T. Crooke, Med. Res. Rev., 1996, 16, 319–344 CrossRef CAS.
- C. F. Bennett, B. F. Baker, N. Pham, E. Swayze and R. S. Geary, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 81–105 CrossRef CAS.
- L. G. Tillman, R. S. Geary and G. E. Hardee, J. Pharm. Sci., 2008, 97, 225–236 CrossRef CAS.
- S. T. Crooke, Handbook of Experimental Pharmacology, 1998, vol. 258 Search PubMed.
- R. S. Geary, D. Norris, R. Yu and C. F. Bennett, Adv. Drug Delivery Rev., 2015, 87, 46–51 CrossRef CAS.
- W. Brad Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef.
- K. M. Bishop, Neuropharmacology, 2017, 120, 56–62 CrossRef CAS.
- R. Z. Yu, T. W. Kim, A. Hong, T. A. Watanabe, H. J. Gaus and R. S. Geary, Drug Metab. Dispos., 2007, 35, 460–468 CrossRef CAS.
- R. Z. Yu, R. S. Geary, D. K. Monteith, J. Matson, L. Truong, J. Fitchett and A. A. Levin, J. Pharm. Sci., 2004, 93, 48–59 CrossRef CAS.
- R. Z. Yu, H. Zhang, R. S. Geary, M. Graham, L. Masarjian, K. Lemonidis, R. Crooke, N. M. Dean and A. A. Levin, J. Pharmacol. Exp. Ther., 2001, 296, 388–395 CAS.
- E. M. Straarup, N. Fisker, M. Hedtjärn, M. W. Lindholm, C. Rosenbohm, V. Aarup, H. F. Hansen, H. Ørum, J. B. R. Hansen and T. Koch, Nucleic Acids Res., 2010, 38, 7100–7111 CrossRef CAS.
- F. Rigo, S. J. Chun, D. A. Norris, G. Hung, S. Lee, J. Matson, R. A. Fey, H. Gaus, Y. Hua, J. S. Grundy, A. R. Krainer, S. P. Henry and C. F. Bennett, J. Pharmacol. Exp. Ther., 2014, 350, 46–55 CrossRef.
- K. T. Luu, D. A. Norris, R. Gunawan, S. Henry, R. Geary and Y. Wang, J. Clin. Pharmacol., 2017, 57, 1031–1041 CrossRef CAS.
- C. A. Stein, L. Benimetskaya and S. Mani, Semin. Oncol., 2005, 32, 563–572 CrossRef CAS.
- T. A. Watanabe, R. S. Geary and A. A. Levin, Oligonucleotides, 2006, 16, 169–180 CrossRef CAS.
- S. Bosgra, J. Sipkens, S. De Kimpe, C. Den Besten, N. Datson and J. Van Deutekom, Nucleic Acid Ther., 2019, 29, 305–322 CrossRef CAS.
- R. S. Geary, R. Z. Yu, J. M. Leeds, T. A. Watanabe, S. P. Henry and A. A. Levin, in Antisense Drugs Technology: Principle, Strategies, and Applications, 2001 Search PubMed.
- A. Y. Edwards, A. Elgart, C. Farrell, O. Barnett-Griness, L. Rabinovich-Guilatt and O. Spiegelstein, Br. J. Clin. Pharmacol., 2017, 83, 1932–1943 CrossRef CAS.
- H. Xu, X. Tong, G. Mugundu, M. L. Scott, C. Cook, C. Arfvidsson, E. Pease, D. Zhou, P. Lyne and N. Al-Huniti, J. Pharmacokinet. Pharmacodyn., 2019, 46, 65–74 CrossRef CAS.
- K. L. Sewell, R. S. Geary, B. F. Baker, J. M. Glover, T. G. K. Mant, R. Z. Yu, J. A. Tami and F. A. Dorr, J. Pharmacol. Exp. Ther., 2002, 303, 1334–1343 CrossRef CAS.
- R. S. Geary, B. F. Baker and S. T. Crooke, Clin. Pharmacokinet., 2015, 54, 133–146 CrossRef CAS.
- R. Z. Yu, S. Hall, R. S. Geary, B. P. Monia, S. P. Henry, Y. Wang, J. W. Collins and E. J. Ackermann, Nucleic Acid Ther., 2020, 30, 153–163 CrossRef CAS.
- N. M. Goemans, M. Tulinius, J. T. Van Den Akker, B. E. Burm, P. F. Ekhart, N. Heuvelmans, T. Holling, A. A. Janson, G. J. Platenburg, J. A. Sipkens, J. M. A. Sitsen, A. Aartsma-Rus, G. J. B. Van Ommen, G. Buyse, N. Darin, J. J. Verschuuren, G. V. Campion, S. J. D. Kimpe and J. C. Van Deutekom, N. Engl. J. Med., 2011, 364, 1513–1522 CrossRef.
- N. M. Goemans, M. Tulinius, M. Van Den Hauwe, A. K. Kroksmark, G. Buyse, R. J. Wilson, J. C. Van Deutekom, S. J. De Kimpe, A. Lourbakos and G. Campion, PLoS One, 2016, 11, 1–20 Search PubMed.
- A. Amantana and P. L. Iversen, Curr. Opin. Pharmacol., 2005, 5, 550–555 CrossRef CAS.
- M. Dirin and J. Winkler, Expert Opin. Biol. Ther., 2013, 13, 875–888 CrossRef CAS.
- A. E. Heald, P. L. Iversen, J. B. Saoud, P. Sazani, J. S. Charleston, T. Axtelle, M. Wong, W. B. Smith, A. Vutikullird and E. Kaye, Antimicrob. Agents Chemother., 2014, 58, 6639–6647 CrossRef.
- A. Amantana, H. M. Moulton, M. L. Cate, M. T. Reddy, T. Whitehead, J. N. Hassinger, D. S. Youngblood and P. L. Iversen, Bioconjugate Chem., 2007, 18, 1325–1331 CrossRef CAS.
- J. H. Beigel, J. Voell, P. Muñoz, P. Kumar, K. M. Brooks, J. Zhang, P. Iversen, A. Heald, M. Wong and R. T. Davey, Br. J. Clin. Pharmacol., 2018, 84, 25–34 CrossRef CAS.
- J. Kim, B. Basiri, C. Hassan, C. Punt, E. van der Hage, C. den Besten and M. G. Bartlett, Mol. Ther.--Nucleic Acids, 2019, 17, 714–725 CrossRef CAS.
- S. Studzińska, R. Rola and B. Buszewski, Anal. Bioanal. Chem., 2016, 408, 1585–1595 CrossRef.
- R. M. Crooke, M. J. Graham, M. J. Martin, K. M. Lemonidis, T. A. D. Wyrzykiewiecz and L. L. Cummins, J. Pharmacol. Exp. Ther., 2000, 292, 140–149 CAS.
- M. S. Baek, R. Z. Yu, H. Gaus, J. S. Grundy and R. S. Geary, Oligonucleotides, 2010, 20, 309–316 CrossRef CAS.
- J. Kim, N. M. El Zahar and M. G. Bartlett, Biomed. Chromatogr., 2020, e4839 CAS.
- M. Gilar, A. Belenky, D. L. Smisek, A. Bourque and A. S. Cohen, Nucleic Acids Res., 1997, 25, 3615–3620 CrossRef CAS.
- X. Wei, G. Dai, Z. Liu, H. Cheng, Z. Xie, R. Klisovic, G. Marcucci and K. K. Chan, Drug Metab. Dispos., 2008, 36, 2227–2233 CrossRef CAS.
- C. S. Shemesh, R. Z. Yu, H. J. Gaus, S. Greenlee, N. Post, K. Schmidt, M. T. Migawa, P. P. Seth, T. A. Zanardi, T. P. Prakash, E. E. Swayze, S. P. Henry and Y. Wang, Mol. Ther.--Nucleic Acids, 2016, 5, e319 CrossRef CAS.
- B. O. Noll, M. J. McCluskie, T. Sniatala, A. Lohner, S. Yuill, A. M. Krieg, C. Schetter, H. L. Davis and E. Uhlmann, Biochem. Pharmacol., 2005, 69, 981–991 CrossRef CAS.
- E. Winquist, J. Knox, J. P. Ayoub, L. Wood, N. Wainman, G. K. Reid, L. Pearce, A. Shah and E. Eisenhauer, Invest. New Drugs, 2006, 24, 159–167 CrossRef CAS.
- J. A. Phillips, S. J. Craig, D. Bayley, R. A. Christian, R. Geary and P. L. Nicklin, Biochem. Pharmacol., 1997, 54, 657–668 CrossRef CAS.
- R. S. Geary, Expert Opin. Drug Metab. Toxicol., 2009, 5, 381–391 CrossRef CAS.
- R. Z. Yu, R. S. Geary, J. D. Flaim, G. C. Riley, D. L. Tribble, A. A. VanVliet and M. K. Wedel, Clin. Pharmacokinet., 2009, 48, 39–50 CrossRef CAS.
- N. Post, R. Yu, S. Greenlee, H. Gaus, E. Hurh, J. Matson and Y. Wang, Drug Metab. Dispos., 2019, 47, 1164–1173 CrossRef CAS.
- R. S. Geary, J. M. Leeds, J. O. N. Fitchett, T. Burckin, L. Truong, C. Spainhour, M. Creek, A. A. Levin, I. P. R. S. G., C. S. Chrysalis and S. R. I. I. M. C., Am. Soc. Pharmacol. Exp. Ther., 1997, 25, 1272–1281 CAS.
- N. Gupta, N. Fisker, M. C. Asselin, M. Lindholm, C. Rosenbohm, H. Ørum, J. Elmén, N. G. Seidah and E. M. Straarup, PLoS One, 2010, 5, 1–9 Search PubMed.
- K. Fluiter, A. L. M. A. ten Asbroek, M. B. de Wissel, M. E. Jakobs, M. Wissenbach, H. Olsson, O. Olsen, H. Oerum and F. Baas, Nucleic Acids Res., 2003, 31, 953–962 CrossRef CAS.
- S. T. Crooke, B. F. Baker, J. L. Witztum, T. J. Kwoh, N. C. Pham, N. Salgado, B. W. McEvoy, W. Cheng, S. G. Hughes, S. Bhanot and R. S. Geary, Nucleic Acid Ther., 2017, 27, 121–129 CrossRef CAS.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf.
| This journal is © The Royal Society of Chemistry 2020 |