
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterzeolite conversion of a micronsized FAU to a nanosized CHA zeolite free of organic structure directing agent with a high CO2 capacity†
Kristoffer H. Møllerab,
Maxime Debostb,
Louwanda Lakiss b,
Søren Kegnæs*a and
Svetlana Mintova*b
b,
Søren Kegnæs*a and
Svetlana Mintova*b
aTechnical University of Denmark, Department of Chemistry, Kemitorvet 207, 2800 Kongens Lyngby, Denmark. E-mail: skk@kemi.dtu.dk
bENSICAEN, Laboratoire Catalyse & Spectrochimie, 6 Boulevard Maréchal Juin, 14050 Caen Cedex 4, France. E-mail: svetlana.mintova@ensicaen.fr
First published on 25th November 2020
Abstract
The interzeolite transformation of a micronsized FAU zeolite to a nanosized CHA zeolite via alkali treatment is presented. The impact of the selection of the FAU zeolite starting material on the properties of the produced CHA zeolite was analyzed by XRD, ICP, SEM, TEM, N2 and CO2 adsorption, and in situ FT-IR. The analysis showed that the choice of starting FAU zeolite had a large impact on the chemical composition, size, morphology, and porosity of the produced CHA zeolite. The as prepared CHA samples show high capacity toward CO2 (4.26 mmol g−1) and it was demonstrated that the chemisorbed vs. physisorbed CO2 was controlled by varying the amount of alkali cations in the CHA zeolite.
Introduction
Zeolites are unique ordered microporous aluminosilicates which have found many uses in the chemical industry.1 Zeolites have pores, and cages of molecular dimensions and can be used for molecular sieving. Molecular sieving is a very useful property in both catalysis2–6 and molecules separation in gas and liquid phases.7–9 With more than 240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 different zeolite framework structures, a key parameter is finding the zeolite structure that fits the right needs for the desired application. One zeolite structure that has received much attention lately is the small pore chabazite (CHA).11–16 The chabazite zeolite consists of double six ring (d6r) rings and cha composite building units (cages). The cha composite building units are connected through 8 ring openings making the channels of about 3.8 Å that run in three dimensions.17 One of the applications where the chabazite zeolite has shown to be useful is in the automotive industry where it acts as a catalyst support for the selective catalytic reduction (SCR) reaction in diesel vehicles due to its high stability and activity.14,18–20 Other studies have shown that chabazite can be useful in gas adsorption and separation, where it can be used for natural gas or flue gas purification and for carbon capture due to the size selectivity for gasses.7,21,22
10 different zeolite framework structures, a key parameter is finding the zeolite structure that fits the right needs for the desired application. One zeolite structure that has received much attention lately is the small pore chabazite (CHA).11–16 The chabazite zeolite consists of double six ring (d6r) rings and cha composite building units (cages). The cha composite building units are connected through 8 ring openings making the channels of about 3.8 Å that run in three dimensions.17 One of the applications where the chabazite zeolite has shown to be useful is in the automotive industry where it acts as a catalyst support for the selective catalytic reduction (SCR) reaction in diesel vehicles due to its high stability and activity.14,18–20 Other studies have shown that chabazite can be useful in gas adsorption and separation, where it can be used for natural gas or flue gas purification and for carbon capture due to the size selectivity for gasses.7,21,22
Chabazite can be synthesized using organic structure directing agents (OSDAs) which are typically costly and require calcination of the zeolite to remove the organic template.23–27 Another way to obtain the CHA zeolite is through interzeolite transformation where only inorganic structure directing agents are used.13,28–31 CHA obtained by an interzeolite transformation of faujasite (FAU) has been reported, however, only limited research has been dedicated on the effect of the starting material on the size and morphology of the final CHA crystals.
In the application of zeolites, mass transfer and diffusion of molecules through zeolite channels are very important and size of both zeolite crystals and pores have an effect on the utilization of the zeolite.32 Higher utilization of the zeolite can be accomplished by introducing an additional porosity in the zeolite or by decreasing the size to reduce the mean diffusion path-length for molecules.33–35
Herein, we present the successful interzeolite transformation of micronsized FAU zeolite to nanosized CHA zeolite via alkali treatment. The composition, size, morphology, and adsorption characteristics of the produced nanosized CHA zeolites made from different FAU zeolites were studied and showed that the starting FAU zeolite has a large impact on the size, composition, and morphology of the final CHA zeolite product.
Experimental
Materials
Commercially available chemical were used as received. NaOH (99%, VWR chemicals), KOH (90%, flakes, Sigma Aldrich), water (double distilled), FAU(2.6) (CBV400, Zeolyst) zeolite, FAU(15) (CBV720, Zeolyst) zeolite, H3BO3 (99.8%, Sigma Aldrich), HF (40%, Fisher chemical), HCl (37%, VWR), HNO3 (65%, Carlo Erba) CO2 (99.9999, Air Liquide), CO (99.9999, Air Liquide).Synthesis
000 rpm, 20 min), washed with water until neutral pH of the wash water, and dried at 50 °C overnight. See Table S1 in ESI† for details on amounts.000 rpm for 20 min). The solid were washed with water by first dispersing the solid in 90 °C water followed by sonicated for 20 min and isolation by centrifugation (20000 rpm, 20 min). The washing procedure was repeated until neutral pH of the wash water. The solid was dried at 50 °C overnight. See Table S1 in ESI† for details on amounts.Characterization
X-ray powder diffraction was performed using a PANalytical X'Pert Pro diffractometer with CuKα monochromatized radiation (λ = 1.5418 Å). Nitrogen physisorption was performed on a Micromeretics 3 flex at liquid nitrogen temperature (77 K). The samples were degassed at 350 °C under vacuum prior to analysis. The surface area was determined by the Brunauer–Emmett–Teller (BET) method and micropore volume by the t-plot method. The total pore volume was determined from a single point at P/P0 = 0.97 on the adsorption branch. CO2 adsorption was performed on a Micromeretics ASAP 2020 at 273.15 K. The sample was degassed at 350 °C under vacuum prior to analysis. Scanning electron microscopy (SEM) was performed using a MIRA-LMH (TESCAN) fitted with a field emission gun using an accelerating voltage of 30.0 kV. The samples were dispersed on carbon tape and coated with Pt and Pd prior to analysis. Transmission electron microscopy (TEM) was performed on a FEI Tecnai T20 G2 microscope operating at 200 kV. The samples were dispersed on lacey carbon grid prior to analysis. Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES) was performed using a Varian ICP-OES 720-ES instrument. 50 mg of solid samples was dissolved in 0.5 mL aqua regia and 3 mL HF at 110 °C for 1 h in a PTFE beaker and cooled to rt. After that, boric acid (1.8 g) was added to the beaker along with 96.5 g of water and shaken overnight for homogenization. In situ infrared spectroscopy with CO2 as probe molecule was performed using a Nicolet 6700 IR spectrometer (Thermo Scientific, Villebon sur Yvette, France) equipped with a mercury cadmium telluride (MCT) detector and an extended KBr beam splitter. The spectra were recorded with 128 scans and a resolution of 4 cm−1. For analysis, the solid was pressed as a self-supported wafer (about 20 mg for a disc of 2.01 cm2). Prior to CO2 adsorption, the solid was activated by heating the pellet at 350 °C for 2 h with a 3 h heating ramp under vacuum (∼10−5 torr) which was then allowed to cool to room temperature. A spectrum with the activated pellet was used as a background and subtracted from the obtained spectra recorded at different concentrations of CO2.Additionally, in situ IR spectroscopy study using CO as a probe molecule was performed; the sample was activated at 350 °C for 2 h under vacuum (∼10−5 torr). The IR spectra under CO delivery were recorded at 77 K.
Results and discussion
Materials and characterization
Interzeolite transformation of micronsized FAU zeolites with an initial Si/Al ratios of 15 (FAU(15)) and 2.6 (FAU(2.6)) to CHA was performed under treatment at 95 °C for 4–7 days in solution with the following chemical composition: vSi/xK/yNa/zH2O ratio of v = 1, x = 0.76–1, y = 0–0.25, z = 30–40. The preparation of nanosized CHA zeolites is based on a modified version reported by Bourgogne et al.36 using a purely inorganic synthesis approach.In general, the choice of reactions conditions, silica/alumina source and Si/Al ratio strongly affects the size and morphology of resulting crystal.37 In this study, the Si/Al ratio of FAU (2.6 to 15 from Zeolyst) and the concentration of cations are varied to study the effect on the physicochemical properties of formed nanosized CHA materials under mild conditions (95 °C). In comparison, J. Kim and D. H. Kim previously produced CHA crystals of 300–500 nm by an interzeolite transformation of FAU (from Zeolyst) with Si/Al ratio of 6 at 140 °C.39 Moreover from previous studies on interzeolite transformation of FAU to CHA via an OSDA-free synthesis it has been only successfully shown for FAU with a Si/Al = 6 or below, while studies using higher Si/Al ratio FAU are reported only via synthesis with OSDA and higher temperature.31 T. Takata et al. has previously shown that it is possible to form nanosized CHA crystals from FAU with Si/Al = 15 and higher in the presence of a OSDA.37 Our study is exclusively focusing on the OSDA-free synthesis. Details of selected experiments are presented in Table S1, ESI.†
X-ray powder diffraction
The X-ray powder diffraction (XRPD) patterns for the starting materials FAU(15) and FAU(2.6) are shown in Fig. 1. The XRPD patterns of corresponding products (CHA(2.3) and CHA(1.9)) after 7 and 4 days interzeolite transformations, respectively, are also shown in Fig. 1. The diffraction patterns clearly show intensive peaks corresponding to the CHA type framework structure. Fig. 1 clearly indicate successful interzeolite transformations since no traces of FAU zeolite are observed. A minor impurity peak likely corresponding to GME type zeolite in sample CHA(1.9) is observed (Fig. 1D). The presence of GME peak and the combination of broad and narrow peaks indicates the presence of stacking fault in CHA(1.9) as described in literature.38,39 For CHA(2.3) there is no GME peak, however, there is a combination of broad and narrow peaks, which could indicate minor faulting or direction oriented growth.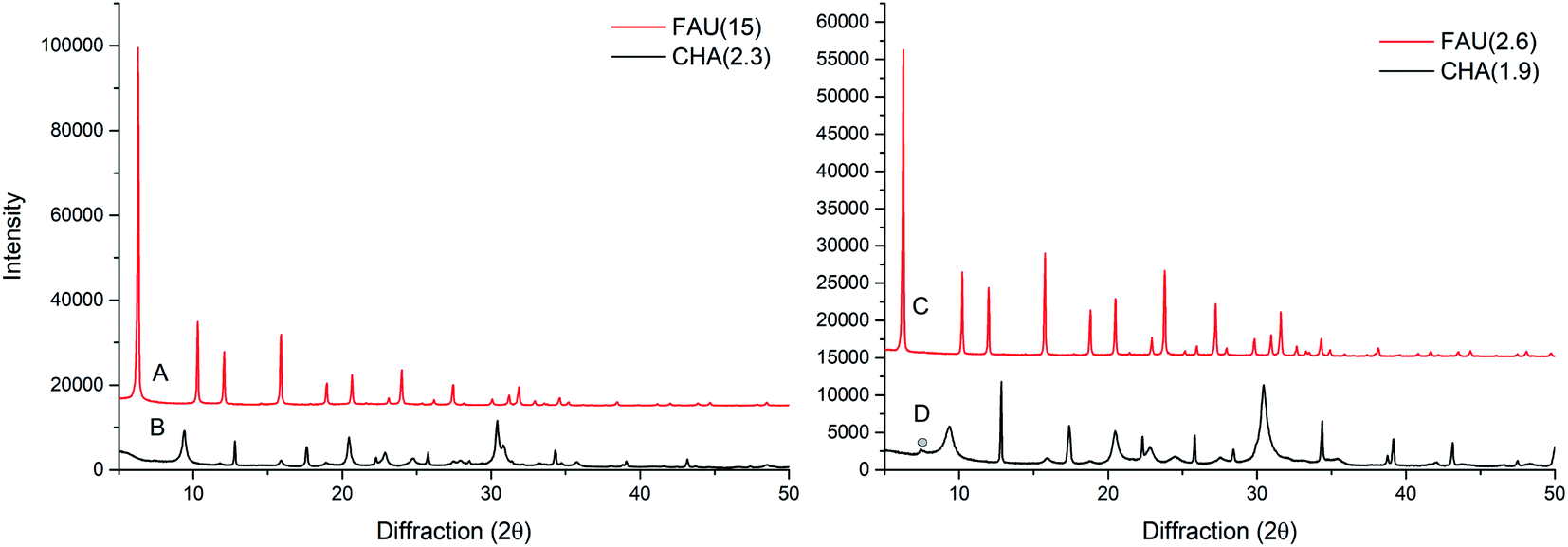 | ||
| Fig. 1 XRPD patterns of (A) FAU(15), (B) CHA(2.3), (C) FAU(2.6), and (D) CHA(1.9); trace of impurity marked with circle. | ||
Interzeolite transformation of micronsized FAU(15) to CHA (Table S1, entry 4†) results in CHA with a Si/Al ratio of 2.3 (CHA(2.3)), whereas the interzeolite transformation of low silica FAU(2.6) (Table S1, entry 1†) results in CHA with a Si/Al ratio of 1.9. During this interzeolite transformation, only a part of the Si is retained in the CHA product from the starting FAU, however, the Si/Al of the starting materials has an influence on the chemical composition of the product. As evidenced, a higher Si/Al ratio in the starting FAU material results in a higher Si/Al in the product CHA. Considering the relative amount of Si going into the CHA zeolite, less of the initial Si is retained in the CHA(2.3) from FAU(15) compared to CHA(1.9) from FAU(2.6). The remaining Si is likely present as unreacted species in the reaction medium, lowering the yield of the zeolite. The difference in composition between samples CHA(2.3) and CHA(1.9) could potentially affect the application of the materials, since a higher Si/Al normally results in a higher thermal stability,40 whereas a lower Si/Al has shown a stronger effect in selectivity for different gasses.22
Electron microscopy
The size and morphology of the zeolites were assessed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM image of FAU(15) (Fig. S1A†) shows that it consists of zeolite crystals of varying sizes (200–1000 nm) with a little rough surface. After interzeolite transformation to the product CHA(2.3) the crystals appearance changed. CHA(2.3) consists of agglomerates (∼500 nm in size) of smaller zeolite crystals of 50–100 nm (Fig. S1B and S2†). The shaped of the agglomerated indicates some oriented growth of the nano-sized crystallites. The mechanism for interzeolite transformation has been suggested to proceed through a dissolution of the zeolite crystals that breaks up into smaller building units which then are used to construct new zeolite form.41–44 These nanoparts are then directed by the K+ to form CHA rather than other zeolite structures which are directed by different cations, as described by van Tendeloo et al.30 It is speculated if this is the case and that the mesopores in the FAU(15) could facilitate a dissolution of the crystal into smaller crystals which would result in the formation of smaller CHA zeolite crystals. It is unknown whether the zeolites dissolves and recrystallize in the solution or on the surface of the dissolving FAU zeolites. For the interzeolite transformation of FAU(2.6), the parent zeolite consists of both larger zeolite crystals of 200–1000 nm but also has smaller crystals of 100–150 nm (Fig. S1C†). Based on SEM analysis, the CHA(1.9) crystals compared to CHA(2.3) is much different (Fig. S1D and S3†). The CHA(1.9) zeolite contains more plate/disc-like structured particles consisting of smaller zeolite crystals with its rough looking surface, however, it is not possible to see individual small zeolite crystals making up the plate. It is possible that the presence of small FAU crystals in the FAU(2.6) zeolite aids the synthesis of smaller plate-like CHA zeolite crystals. The plate-like morphology is a typical shape of faulted CHA, which is consistent with the observations in the XRD pattern.39 For both CHA(1.9) and CHA(2.3) there is some oriented growth of the zeolite. It is speculated if this could arise from faulting, when purely inorganic SDAs are used rather than when OSDAs are used for synthesis.Fig. 2 shows the TEM images of both CHA(1.9) and CHA(2.3) samples. The CHA(1.9) contains crystals with a size of 300–500 nm (Fig. 2A) consisting of densely packed smaller crystallites. Fig. S4† shows lower magnification TEM images of CHA(1.9) which indicates the plate-like morphology with smaller crystallites growing from the plate. This is consistent with the SEM observations made for the CHA(1.9) sample. The CHA(2.3) sample contains very small crystallites in discrete form (Fig. 2B). The size of the individual zeolite crystals is about 50 nm, and the crystalline fringes are clearly observed in the TEM images. Fig. S5† shows TEM images of agglomerates of CHA(2.3). Here, it appears that smaller zeolite crystals are overlapping, which is consistent with the agglomerates observed in SEM, however, it is difficult to see any direction oriented growth. The Si/Al ratio of the starting FAU materials and the concentration of both cations and hydroxide are different for the synthesis of two CHA zeolites. Those are the main factors that affect the morphology and the size of the final product. Based on those two samples the larger Si/Al ratio in FAU results in the smallest crystal size (FAU(15) to CHA(2.3) gives crystals of 50–100 nm). This correspond well with the previous studies where Si/Al = 15 and higher (and using a OSDA at 125–170 °C) also results in nanosized crystals.37 However, J. Kim and D. H. Kim39 showed a Si/Al = 6 (OSDA-free at 140 °C) results in larger crystal size similar to the once obtained here for CHA(1.9) produced from FAU(2.6). By studying different Si/Al ratios and silica/alumina sources, T. Takata concluded that the starting reaction mixture with common composite building unit and suitable Si/Al ratio causes abundant and uniform nucleation, resulting in the formation of nanosized CHA zeolites.37
 | ||
| Fig. 2 TEM images of samples CHA(1.9) (A) and CHA(2.3) (B). | ||
Porosity and CO2 capacity
The porosity of CHA(2.3) and CHA(1.9) zeolite samples was assessed by nitrogen physisorption measurements. The N2 physisorption isotherm of FAU(2.6), FAU(15), CHA(1.9), and CHA(2.3) are shown in Fig. S6–S9.† The N2 isotherm for CHA(2.3) reveals the presence of both micro- and mesoporosity from the uptake at low pressure (<0.01 P/P0) and from the uptake of N2 at higher pressure, respectively (Fig. S9†). The calculated porosity for FAU and CHA zeolites are listed in Table S3.† CHA(2.3) exhibits a high BET surface area of 251 m2 g−1 compared to other CHA zeolites in the K-form obtained from interzeolite conversion.45–47 The total pore volume of 0.39 cm3 g−1 has only a limited contribution from the micropores with 0.04 cm3 g−1, hence the total porosity of the material is primarily mesopores. The hysteresis loop at high P/P0 for nanosized CHA(2.3) is due the presence of textural (interparticle) porosity, which is typical for nanosized zeolites.34 On the other hand CHA(1.9) has significantly lower surface area of 43 m2 g−1 and a total pore volume of 0.07 cm3 g−1 of which 0.004 cm3 g−1 is the micropore volume. This is typically seen for the K form of the CHA zeolite obtained from the transformation in the potassium form.45–47 CHA(1.9) is almost exclusively in the K form determined by ICP analysis (93% K and 6% Na relative to Al) which is the reason for this low porosity. It's reported that the K+ extra-framework cations sit in the channels of CHA with low Si/Al ratio and limits the access of N2 to the channels, thus, lowering the surface area determined by N2 physisorption at 77 K.12,45 CHA(1.9) does not show the same interparticle porosity which suggest that smaller crystals are grown together and are not accessible for the N2 molecules at 77 K. Considering the higher micropore volume for CHA(2.3) compared to CHA(1.9), it could be a result of the higher mesoporosity which provides access to more pore mouth openings of the CHA zeolite. The interparticle mesoporosity of CHA(2.3) and the lack of this in CHA(1.9) is consistent with the SEM and TEM observations for the two samples. This illustrates the large effect that the starting zeolite has on the porosity of the obtained CHA with the agglomerates of discrete nanosized CHA zeolite crystals (CHA(2.3)) rather than the thin plates of intergrown CHA zeolite (CHA(1.9)). Changing the cation in CHA greatly affects the adsorption properties of CHA. This was nicely demonstrated by Shang et al.,12 who showed that changing to a smaller cation than K+ increases the uptake of N2. However, both Si/Al, type of cation and degree of ion-exchange affects the adsorption properties of CHA.One of the applications of CHA is the selective capture of CO27,21,22 hence, the capacity of CO2 was measured to evaluate the potential use of the newly synthesized materials. The accessibility of CO2 and not N2 in the K-CHA zeolite has been shown before and this is related to the different polarizability of the two gas molecules with CO2 being much more polarizable.45 CHA(2.3) shows a large uptake of CO2 at low relative pressure and then levels off at higher relative pressure (Fig. 3). Interestingly, the difference of the two CHA zeolites with either interparticle porosity (CHA(2.3)) or intergrown CHA (CHA(1.9)) does not have a large impact on the adsorption profile and capacity of CO2. At P/P0 = 0.030, the CO2 capacity of CHA(2.3) is 4.26 mmol g−1, which is similar to the CHA(1.9) of 4.25 mmol g−1. The CO2 capacity is close to other similar reported materials7,12 (see reference points in Fig. 3), however, it is hard to make a profound comparison, since both Si/Al ratio together with the location of cations affect the capacity. Additionally, the amount and location of cations also affect the selectivity of CHA zeolite for different gasses.7
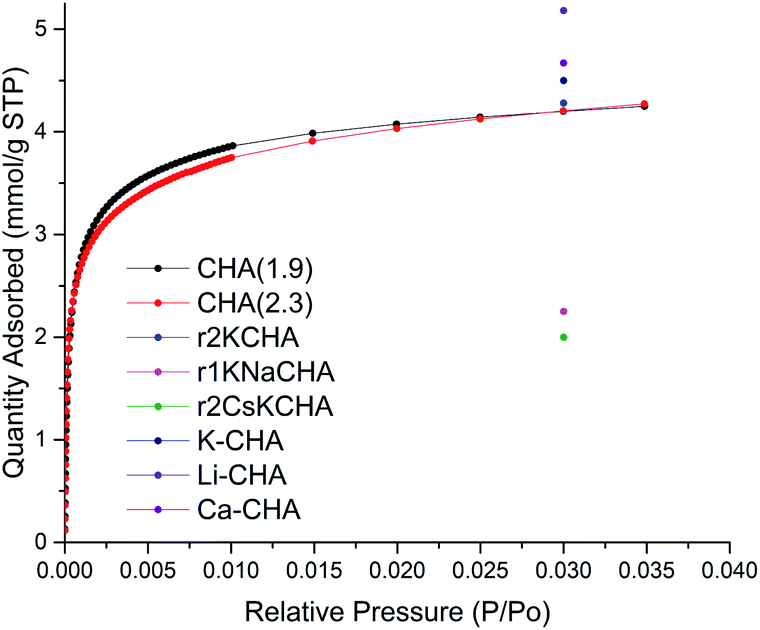 | ||
| Fig. 3 CO2 adsorption of samples CHA(2.3) (red) and CHA(1.9) (black) at 273.15 K. CO2 capacity from Zhang et al.7 (K-CHA, Li-CHA, and Ca-CHA) and Shang et al.12 (r1KNaCHA, r2KCHA, and r2CsKCHA) reported as single points at relative pressure of 0.030 (104 kPa). | ||
FT-IR
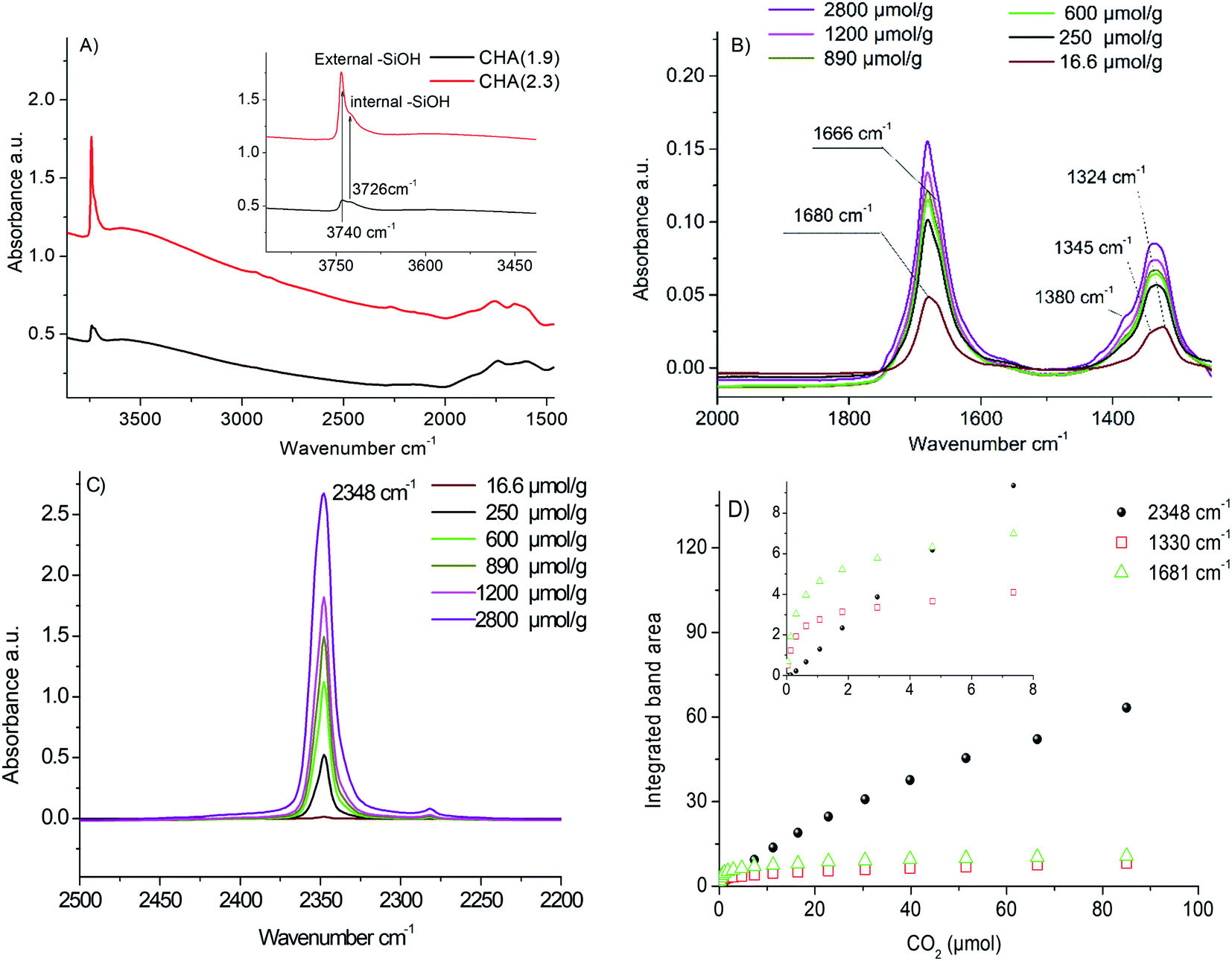 | ||
| Fig. 4 IR spectra of CHA(1.9) and CHA(2.3) after activation at 350 °C under vacuum in the region 1500–4000 cm−1; the spectra were normalized to 20 mg (A). IR spectra under pulsed CO2 adsorption on CHA(2.3) in the regions of 2000–1250 cm−1 (B), 2500–2200 cm−1 (C) and integrated area of bands in FTIR spectra corresponding to chemisorbed CO2 at 1330 cm−1 and 1681 cm−1 and physisorbed CO2 at 2348 cm−1 (D). | ||
Fig. 4B and C reports the difference spectra obtained at adsorption of a specific dose of CO2 after subtraction of the background (the IR spectrum recorded after activation of the samples). After the first CO2 dose delivered, two broad bands are observed at 1324 cm−1 and 1680 cm−1. After several doses, two additional distinct bands at 1665 cm−1 and 1345 cm−1 appeared. The bands at 1324 cm−1 and 1680 cm−1 are attributed to carbonate species formed during CO2 adsorption (Fig. 4B). As shown in Fig. 4D, these bands increase by increasing CO2 doses to reach saturation at 40–60 μmol g−1 CO2 (∼1 μmol CO2 for 20 mg sample). It is already reported in the literature that the double degenerated ν3 vibration of the carbonates is usually split into two bands.54 Busca et al.55 used the shift between these two bands to identify the carbonate species. In our case, the Δν3 for 1680–1324 cm−1 and 1665–1354 cm−1 is higher than 300 cm−1, which corresponds to bidentate carbonates. Such a result is in a good agreement with the low Si/Al ratio of the samples, which gives rise to strong basic zeolite sites. In addition, a new band assigned to physisorbed CO2 appears at 2348 cm−1 due to the polarization of CO2 by the compensating cations present in the CHA crystals.48 The intensity of the band at 2348 cm−1 increases with an increase of the CO2 pressure (Fig. 4C). Moreover, a new shoulder at 1380 cm−1 appeared. This shoulder is also attributed to physisorbed CO2 molecules.48 At high loading, more than 3000 μmol g−1 of CO2, a shift of the silanol bands at 3740–3725 cm−1 is observed, which is due to the perturbation of silanol sites by CO2 molecules (see Fig. S10†).
The evolution of the integrated band area of the carbonates at 1325 cm−1 and 1681 cm−1 and the polarized CO2 at 2348 cm−1 as a function of the amount of CO2 introduced in the cell is depicted in Fig. 4D. Three distinct phenomena are observed (i) first at a low CO2 loading, CO2 is interacting with strong basic sites of zeolite leading to the formation of carbonates. This is traduced by the rising of the bands in the region 1300–1400 cm−1 and 1600–1700 cm−1; (ii) after introduction of 4–5 μmol of CO2, the strongest basic sites of zeolite are completely neutralized with carbonates. No more carbonates are formed; the IR bands at 1325 cm−1 and 1681 cm−1 corresponding to carbonates reached saturation that correspond to 200 μmol CO2 per g of zeolite. Only the band at 2348 cm−1 related to the interaction of CO2 with the extra-framework cations continues to increase linearly with the amount of CO2 introduced; (iii) at a higher amount of CO2, above 3500–4000 μmol g−1 of zeolite, after a complete consumption of the cationic sites, a shift of the silanol bands in the region 3700–3750 cm−1 is observed. This is due to the perturbation of the external silanol sites by CO2. This amount is in a good agreement with the amount of extra-framework cations as determined by ICP (about 3600 μmol of potassium per g of zeolite).
The amount of carbonates formed on the two samples is estimated based on the integrated areas of the bands at 1325 cm−1 and 1681 cm−1. Since the absorption coefficient is unknown, we calculated the height ratio of the two bands (A1325 CHA(1.9)/A1325 CHA(2.3) = 1.56 and A1681 CHA(1.9)/A1681 CHA(2.3) = 1.6). This means that the amount of strong basic sites of CHA(1.9) compared to CHA(2.3) is 1.6 times higher. For physisorbed species, the integrated band area is almost the same for the two samples with a ratio equal to 0.9.
The desorption properties of CHA(2.3) was tested by desorption of CO2 until a pressure of 10−5 torr. Fig. S14† shows the IR bands for the chemisorbed and physisorbed species at 2800 μmol g−1 and at 10−5 torr. This clearly shows that the physisorbed species are absent after desorption whereas the chemisorbed species are much more strongly bound at present and still are present at 10−5 torr in similar quantities. Thus, the physisorbed species can reversibly be adsorbed and desorbed to CHA(2.3). A similar trend is observed for CHA(1.9) (not shown).
Conclusions
The morphology, size and porosity of the CHA zeolite was found to be highly dependent by the type of the initial FAU zeolite samples. Using appropriate conditions, the transformation from two FAU-type zeolites to CHA-type zeolites was accomplished by alkali treatment without the use of an organic structure directing agent. The CHA(2.3) sample consisted of 50–100 nm discrete zeolite crystals while the CHA(1.9) sample consisted of big agglomerated crystals. Both zeolite samples CHA(1.9) and CHA(2.3) showed an extremely high CO2 adsorption capacity. The amount of chemisorbed CO2 species on the CHA zeolite is strongly related to the Si/Al ratio of the material. The sample CHA(1.9) with low Si/Al ratio showed higher amount of carbonate species which is proportional to the amount of compensating cations in zeolite structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Farnoosh Goodarzi is thanked for doing TEM analysis on the zeolites. Engineer Valérie Ruaux and Technician Marie Desmurs is thanked for their help with obtaining IR, ICP, N2 physisorption and CO2 adsorption data. The authors are grateful for funding from Haldor Topsøe A/S, Independent Research Fund Denmark (grant no. 6111-00237), and Villum fonden (grant no. 13158).Notes and references
- T. Maesen, in Introduction to Zeolite Science and Practice, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, Amsterdam, 2007, vol. 168, pp. 1–12 Search PubMed.
- C. Liu, J. Liu, S. Yang, C. Cao and W. Song, ChemCatChem, 2016, 8, 1279–1282 CrossRef CAS.
- M. Choi, Z. Wu and E. Iglesia, J. Am. Chem. Soc., 2010, 132, 9129–9137 CrossRef CAS.
- D. Fraenkel, M. Cherniavsky, B. Ittah and M. Levy, J. Catal., 1986, 101, 273–283 CrossRef CAS.
- F. Goodarzi, R. P. Thumbayil, K. Enemark-Rasmussen, J. Mielby, T. T. M. Nguyen, P. Beato, F. Joensen and S. Kegnæs, ChemCatChem, 2020, 12, 1519–1526 CrossRef CAS.
- F. Goodarzi, L. Kang, F. R. Wang, F. Joensen, S. Kegnæs and J. Mielby, ChemCatChem, 2018, 10, 1566–1570 CrossRef CAS.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478–487 CrossRef CAS.
- Y. Zheng, N. Hu, H. Wang, N. Bu, F. Zhang and R. Zhou, J. Membr. Sci., 2015, 475, 303–310 CrossRef CAS.
- S. Imasaka, M. Itakura, K. Yano, S. Fujita, M. Okada, Y. Hasegawa, C. Abe, S. Araki and H. Yamamoto, Sep. Purif. Technol., 2018, 199, 298–303 CrossRef CAS.
- International Zeolite Association Structure Commission, Database Zeolite Struct., 2017 Search PubMed.
- X. Li, H. Kita, H. Zhu, Z. Zhang, K. Tanaka and K. Okamoto, Microporous Mesoporous Mater., 2011, 143, 270–276 CrossRef CAS.
- J. Shang, G. Li, R. Singh, P. Xiao, J. Z. Liu and P. A. Webley, J. Phys. Chem. C, 2013, 117, 12841–12847 CrossRef CAS.
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- X. Wang, Q. Wu, C. Chen, S. Pan, W. Zhang, X. Meng, S. Maurer, M. Feyen, U. Müller and F. S. Xiao, Chem. Commun., 2015, 51, 16920–16923 RSC.
- Y. Shan, X. Shi, J. Du, Z. Yan, Y. Yu and H. He, Ind. Eng. Chem. Res., 2019, 58, 5397–5403 CrossRef CAS.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Amsterdam, 6th edn, 2007 Search PubMed.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, 800–802 RSC.
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. F. Peden, J. Catal., 2012, 287, 203–209 CrossRef CAS.
- D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- T. D. Pham, Q. Liu and R. F. Lobo, Langmuir, 2013, 29, 832–839 CrossRef CAS.
- J. Shang, G. Li, R. Singh, Q. Gu, K. M. Nairn, T. J. Bastow, N. Medhekar, C. M. Doherty, A. J. Hill, J. Z. Liu and P. A. Webley, J. Am. Chem. Soc., 2012, 134, 19246–19253 CrossRef CAS.
- M. Moliner, J. E. Gabay, C. E. Kliewer, R. T. Carr, J. Guzman, G. L. Casty, P. Serna and A. Corma, J. Am. Chem. Soc., 2016, 138, 15743–15750 CrossRef CAS.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441–448 CrossRef CAS.
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CrossRef CAS.
- M. Itakura, I. Goto, A. Takahashi, T. Fujitani, Y. Ide, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2011, 144, 91–96 CrossRef CAS.
- S. I. Zones, US Pat. US4544538, 1985.
- S. Miyagawa, K. Miyake, Y. Hirota, N. Nishiyama, M. Miyamoto, Y. Oumi and S. Tanaka, Microporous Mesoporous Mater., 2019, 278, 219–224 CrossRef CAS.
- H. Robson and K. P. Lillerud, Verified Syntheses of Zeolitic Materials, Elsevier, Amsterdam, 2nd rev., 2001 Search PubMed.
- L. van Tendeloo, E. Gobechiya, E. Breynaert, J. A. Martens and C. E. A. Kirschhock, Chem. Commun., 2013, 49, 11737–11739 RSC.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS.
- J. O. Abildstrøm, M. Kegnæs, G. Hytoft, J. Mielby and S. Kegnæs, Microporous Mesoporous Mater., 2016, 225, 232–237 CrossRef.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- H. Awala, J.-P. Gilson, R. Retoux, P. Boullay, J.-M. Goupil, V. Valtchev and S. Mintova, Nat. Mater., 2015, 14, 447–451 CrossRef CAS.
- J. van den Bergh, J. Gascon and F. Kapteijn, in Zeolites and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, vol. 1, pp. 361–387 Search PubMed.
- M. Bourgogne, J.-L. Guth and R. Way, US Pat. 4503024, 1985.
- T. Takata, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2016, 225, 524–533 CrossRef CAS.
- K. P. Lillerud, R. Szostak and A. Long, J. Chem. Soc., Faraday Trans., 1994, 90, 1547–1551 RSC.
- J. Kim and D. H. Kim, Microporous Mesoporous Mater., 2018, 256, 266–274 CrossRef CAS.
- L. B. McCusker and C. Baerlocher, in Introduction to Zeolite Science and Practice, eds. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, Amsterdam, 2007, vol. 168, pp. 13–37 Search PubMed.
- C. Li, M. Moliner and A. Corma, Angew. Chem., Int. Ed., 2018, 57, 15330–15353 CrossRef CAS.
- A. Yashiki, K. Honda, A. Fujimoto, S. Shibata, Y. Ide, M. Sadakane and T. Sano, J. Cryst. Growth, 2011, 325, 96–100 CrossRef CAS.
- I. Goto, M. Itakura, S. Shibata, K. Honda, Y. Ide, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2012, 158, 117–122 CrossRef CAS.
- H. Jon, N. Ikawa, Y. Oumi and T. Sano, Chem. Mater., 2008, 20, 4135–4141 CrossRef CAS.
- F. N. Ridha, Y. Yang and P. A. Webley, Microporous Mesoporous Mater., 2009, 117, 497–507 CrossRef CAS.
- K. H. Rasmussen, J. Mielby and S. Kegnaes, ChemCatChem, 2018, 10, 4380–4385 CrossRef CAS.
- R. Nedyalkova, C. Montreuil, C. Lambert and L. Olsson, Top. Catal., 2013, 56, 550–557 CrossRef CAS.
- A. Villarreal, P. Castillo-Villalón and J. Ramírez, J. Mex. Chem. Soc., 2017, 61, 102–108 CAS.
- T. Montanari and G. Busca, Vib. Spectrosc., 2008, 46, 45–51 CrossRef CAS.
- E. Gallei and G. Stumpf, J. Colloid Interface Sci., 1976, 55, 415–420 CrossRef CAS.
- R. W. Stevens, R. V. Siriwardane and J. Logan, Energy Fuels, 2008, 22, 3070–3079 CrossRef CAS.
- J. C. M. Pires, F. G. Martins, M. C. M. Alvim-Ferraz and M. Simões, Chem. Eng. Res. Des., 2011, 89, 1446–1460 CrossRef CAS.
- S. M. Maier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 8005–8013 CrossRef CAS.
- J. Fujita, A. E. Martell and K. Nakamoto, J. Chem. Phys., 1962, 36, 339–345 CrossRef CAS.
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 CrossRef CAS.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS.
- A. Zecchina and C. O. Areán, Chem. Soc. Rev., 1996, 25, 187–197 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04937a |
| This journal is © The Royal Society of Chemistry 2020 |