DOI:
10.1039/D0RA04885B
(Paper)
RSC Adv., 2020,
10, 32259-32264
Dynamic instability of lithiated phosphorene
Received
3rd June 2020
, Accepted 20th August 2020
First published on 1st September 2020
Abstract
Li-ion batteries are widely used energy storage units. Although phosphorene delivers a high Li capacity, the transition capacity between the intercalation reaction and the conversion reaction is still not clear. We investigate the structural and electronic properties of Li intercalated phosphorene and graphene/phosphorene/graphene sandwiches by first-principles calculations. The competition to obtain charge from Li between C and P reduces charge depletion on the interlayer P–P bonds, improving stability. Importantly, the sandwiches show higher transition capacities than freestanding phosphorene, confirmed by ab initio molecular dynamics simulations. The trilayer structures show better structural reversibility than the monolayers.
1 Introduction
Two-dimensional materials have recently drawn much attention after discovery of the unusual electronic properties of graphene.1 Silicene,2 germanene,3 phosphorene,4 and antimonene5 are monolayer Si, Ge, P, and Sb in buckled structures (sp2–sp3 hybridization) as compared to the planar structure of graphene (sp2 hybridization).6 Phosphorene can be prepared by mechanical and liquid exfoliation,7 whereas silicene and germanene can only be deposited on substrates.8,9
Phosphorene is considered to have potential applications in electronic devices (field-effect transistors),10–12 sensing devices (gas, ion and biology sensors),13,14 and energy storage devices (batteries and supercapacitors)15 based on recent advances in material synthesis.16,17 Li-ion and Na-ion batteries are widely used energy storage units because of high energy density and lack of memory effect. The Na capacity of graphene/phosphorene/graphene sandwiches is measured to be 2440 mA h g−1,18 which is close to the theoretical value of bulk P (2596 mA h g−1) based on the conversion reaction. However, only 20% capacities of the sandwich is retained for the 10C cycle as compared to the 0.02C cycle. Although bulk P has a much higher specific capacity than the commercial graphite anode (372 mA h g−1), the huge volume change of 491% leads to disintegration of the electrode material and capacity loss.19,20 Two-dimensional materials usually have much lower diffusion energy barriers than bulk materials because of flat potential surface,21,22 which is important to prevent capacity loss for high-rate cycles. Since intercalation reactions on two-dimensional materials do not affect structural stability and conversion reactions destroy the structures, the high-rate performance thus can be improved by enhancing the transition capacity between intercalation and conversion reactions.23
On the other hand, phosphorene is thermodynamically calculated to deliver a Li/Na capacity of 433 mA h g−1 based on the intercalation reaction, showing a ratio of Li/Na to P to be 0.5.24,25 Phosphorene/graphene heterostructures improve the theoretical Li capacity to 485 mA h g−1 with a low diffusion energy barrier of 0.12 eV.26 The structure of bulk P is calculated to be stable for the ratio of Na to P to be no more than 0.25 based on the intercalation reaction without considering temperature effect,27 being much lower than for phosphorene (Li0.5P)24,25 and for the fully charged electrode (Li3P). In addition, some optimized structures of decorated phosphorene are demonstrated to be unstable by ab initio molecular dynamics simulation at room temperature.28 The real capacities based on intercalation reactions for phosphorene-based electrode materials thus may be lower than the calculated values at 0 K. Since integration of electrode materials is important to the performance of high-rate charge/discharge, we will investigate the structural and electronic properties of Li intercalated multilayer phosphorene unsandwiched and sandwiched by graphene using first-principles calculations.
2 Computational method
All the calculations are carried out in the framework of density functional theory using the projector augmented wave method, implemented in Vienna Ab initio Simulation Package.29 The exchange-correlation potential is based on generalized gradient approximation proposed by Perdew, Burke and Ernzerhof.30 The van der Waals type interlayer interaction for multilayer phosphorene is taken into account by the DFT-D3 approach,31 which has been demonstrated to successfully reproduce experimental interlayer distance for two-dimensional material heterostructures as compared to van der Waals functionals in spite of different charge density distribution at the interface.32 The energy cutoff for the plane wave basis is set to 400 eV. The total energies converge to 10−6 eV in the self-consistent solution of Kohn–Sham equations. The residual forces on the atoms have declined to less than 0.01 eV Å−1. The graphene/phosphorene/graphene sandwiches are built by 4 × 2 × 1 and 3 × 2 × 1 and supercells of graphene in a rectangular unit cell and phosphorene, respectively. Double-sized supercells containing 352, 448, and 544 atoms (excluding Li atoms) for monolayer, bilayer, and trilayer phosphorene, respectively, are used for ab initio molecular dynamics simulations at 300 K with a time step of 1 and 3 fs. The crystal orbital Hamiltonian population calculations are performed by LOBSTER code.33
3 Results and discussion
Table 1 lists the optimized lattice constants and interlayer distance for the monolayer, bilayer, and trilayer phosphorene and the bulk. The calculated lattice constant a/b expands/shrinks with increasing number of layers and agrees with the experimental value. The interlayer distance decreases with increasing thickness and is smaller than the previous theoretical results (3.612 Å) because of different types of van der Waals functionals.26 The interlayer binding energy for the bulk turns out to be 0.099 eV per atom as compared to 0.012 eV per atom excluding van der Waals correction, leading to a smaller distance of 3.132 Å to 3.432 Å.
Table 1 Structural properties of monolayer, bilayer, and trilayer phosphorene and the bulk as compared to the experimental results, including the lattice constants a, b, and c and the interlayer distance d (in Å)
| |
Monolayer |
Bilayer |
Trilayer |
Bulk |
Exp.34 |
| a |
3.310 |
3.313 |
3.317 |
3.357 |
3.34 |
| b |
4.550 |
4.506 |
4.456 |
4.232 |
4.49 |
| c |
— |
— |
— |
10.59 |
10.81 |
| d |
— |
3.197 |
3.177 |
3.132 |
— |
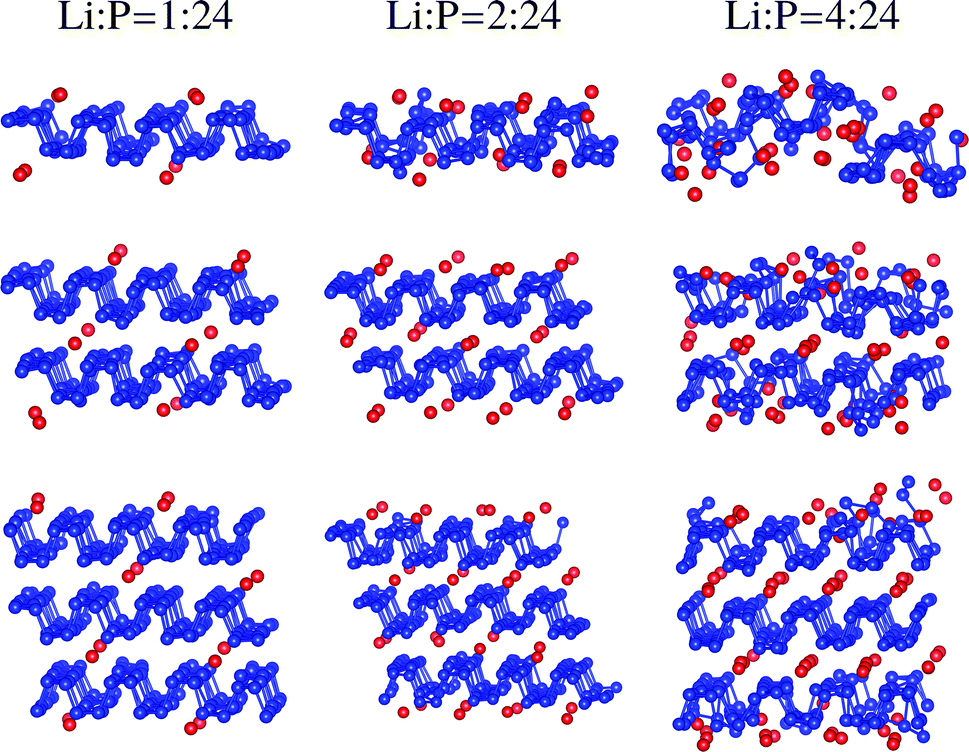
Total energies of different locations for Li intercalation have been compared to search the ground states. Uniform distribution is energetically preferred to clustering because of the repulsion between the Li atoms. Fig. 1 illustrates the structures for Li intercalated multilayer phosphorene after 3 ps ab initio molecular dynamics simulations at 300 K. The Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio is calculated by the Li atoms on (at) the surface (interface) and the P atoms in each layer to be independent of the number of layers. The structure is distorted at the ratio of 2:24 for the monolayer. The maximal Li capacity based on the intercalation reaction requiring stability (instead of full capacity) turns out to be less than 144 mA h g−1. Although some previous work reports stable structures and higher capacities for multilayer phosphorene calculated by structural optimization at 0 K, the structure may not be stable at room temperature confirmed by molecular dynamics simulations leading to lower capacities.16,24,35 The structure is completely destroyed at the ratio of 4:24. In the case of bilayer and trilayer, the structures are almost not distorted for the ratio of 2:24, confirmed by the average formation energy of Li intercalation (0.58 eV and 0.64 eV, respectively, as compared to 0.46 eV for the monolayer). The surface sheets are broken for the ratio of 4:24, whereas the center sheet is still stable for the trilayer further confirmed by the molecular dynamics simulations for 6 ps. The capacities are calculated to be 108 mA h g−1 and 96 mA h g−1 for the bilayer and the trilayer. The capacity converges to 72 mA h g−1 for bulk P, being lower than the value of 216 mA h g−1 obtained by the thermodynamic calculations at 0 K.27 High quality phosphorene nanoribbons have been successfully prepared with typical widths of 4–50 nm and lengths of up to 75 μm recently.16 Since the number of surface sites for Li adsorption is much more than the edge sites, our results obtained by molecular dynamics can also predict the stability and capacities based on the intercalation reaction for phosphorene nanoribbons. In addition, other kinds of phosphorene nanostructures such as nanorods and nanoneedles have also drawn much attention,36 which may have higher capacities than the bulk.
:P ratio is calculated by the Li atoms on (at) the surface (interface) and the P atoms in each layer to be independent of the number of layers. The structure is distorted at the ratio of 2:24 for the monolayer. The maximal Li capacity based on the intercalation reaction requiring stability (instead of full capacity) turns out to be less than 144 mA h g−1. Although some previous work reports stable structures and higher capacities for multilayer phosphorene calculated by structural optimization at 0 K, the structure may not be stable at room temperature confirmed by molecular dynamics simulations leading to lower capacities.16,24,35 The structure is completely destroyed at the ratio of 4:24. In the case of bilayer and trilayer, the structures are almost not distorted for the ratio of 2:24, confirmed by the average formation energy of Li intercalation (0.58 eV and 0.64 eV, respectively, as compared to 0.46 eV for the monolayer). The surface sheets are broken for the ratio of 4:24, whereas the center sheet is still stable for the trilayer further confirmed by the molecular dynamics simulations for 6 ps. The capacities are calculated to be 108 mA h g−1 and 96 mA h g−1 for the bilayer and the trilayer. The capacity converges to 72 mA h g−1 for bulk P, being lower than the value of 216 mA h g−1 obtained by the thermodynamic calculations at 0 K.27 High quality phosphorene nanoribbons have been successfully prepared with typical widths of 4–50 nm and lengths of up to 75 μm recently.16 Since the number of surface sites for Li adsorption is much more than the edge sites, our results obtained by molecular dynamics can also predict the stability and capacities based on the intercalation reaction for phosphorene nanoribbons. In addition, other kinds of phosphorene nanostructures such as nanorods and nanoneedles have also drawn much attention,36 which may have higher capacities than the bulk.
 |
| | Fig. 1 Snapshots of the structures for Li intercalated multilayer phosphorene after 3 ps ab initio molecular dynamics simulations at 300 K with a step of 1 fs. The P and Li atoms are labeled by blue and red. | |
Since stability of two-dimensional materials can be improved by formation of hybrids with graphene,18 the structures of Li intercalated multilayer phosphorene sandwiched by graphene after 3 ps molecular dynamics simulations are shown in Fig. 2. The structures are stable for the Li:P ratio of 4:24, which is higher than in the graphene-free cases. The Li capacities are calculated to be 142–144 mA h g−1 for the monolayer, bilayer and trilayer including the mass of C. The corresponding values are 289, 216, and 192 mA h g−1 excluding the mass of C. In addition, lithiation only expands the thickness of the sandwiches by less than 15%, as compared to the volume change of 491% (sodiation) for bulk P.19 High-rate charge/discharge may be achieved below these capacities, since the layered structures show low diffusion energy barriers.26 Moreover, the sandwich structures may obstruct reaction of phosphorene with oxygen and florine affecting applications35,37 because of high energy barriers for diffusion through graphene. The surface sheets are broken for the ratio of 6:24. Furthermore, the lithiation process changes to the conversion step for higher Li concentration due to complete destruction of the structures of multilayer phosphorene.
 |
| | Fig. 2 Snapshots of the structures for Li intercalated multilayer sandwiches after 3 ps ab initio molecular dynamics simulations at 300 K with a step of 1 fs. The P, C, and Li atoms are labeled by blue, black, and red. | |
Charge density difference for Li intercalated trilayer phosphorene and the sandwich is shown in Fig. 3. The charge density difference is defined as
| | |
Δρ = ρfull − ρsandwich − ρLi
| (1) |
where
ρfull,
ρsandwich, and
ρLi refer to the charge density for the full system, the sandwich without Li, and the Li atoms, respectively. The charge redistribution on the surface is stronger than at the interface for phosphorene, which leads to a shorter Li–P bond length on the surface than at the interface as listed in
Table 2. The charge depletion on the interlayer P–P bonds occurs with increasing Li concentration, weakening interaction and thus lengthening the bonds. Graphene obtains partial charge from the Li atoms in the sandwich, reducing the charge transfer to the P atoms on the surface. The competition to obtain charge from Li suppresses the charge depletion on the interlayer P–P bonds, improving stability. The Li–P bond on the surface is lengthened for higher Li concentration, reflecting stronger interaction between Li and C than between Li and P.
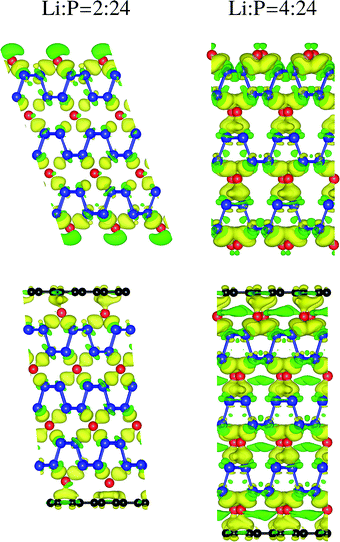 |
| | Fig. 3 Charge density difference for Li intercalated trilayer phosphorene and the sandwich. The yellow/green color represents charge accumulation/depletion. The isosurface value is 1.8 × 10−3 electrons per bohr3. The P, C, and Li atoms are labeled by blue, black, and red. | |
Table 2 Structural properties of trilayer phosphorene and the sandwich, including the distance of the Li–P and P–P bonds on the surface (bsLi–P and bsP–P) and at the center (bcLi–P and bcP–P) (in Å), respectively
| |
bsLi–P |
bcLi–P |
bsP–P |
bcP–P |
| Phosphorene |
| Li:P = 2:24 |
2.41 |
2.51 |
2.27 |
2.26 |
| Li:P = 4:24 |
2.40 |
2.44 |
2.31 |
2.32 |
|
| Sandwich |
| Li:P = 2:24 |
2.43 |
2.47 |
2.25 |
2.24 |
| Li:P = 4:24 |
2.45 |
2.42 |
2.30 |
2.32 |
The density of states (DOS) of the P 3p states for trilayer phosphorene and the sandwich with the Li:P ratio of 2:24 and the crystal orbital Hamiltonian populations (COHP) of the interlayer P–P bonds in the surface and center sheets are shown in Fig. 4. The P atoms in the surface and center sheets show similar DOS and –COHP in profile. The P atom in the surface sheet shows more states located at the Fermi level and consequently the P–P bond has a more negative –COHP. In addition, the –COHP for the P–P bond in the center sheet shows a higher integral value (–ICOHP) than that in the surface sheet (Table 3), reflecting better stability. The –ICOHP decreases by more than 50% from the Li:P ratio of 1:24 to 2:24 for phosphorene, whereas a reduction of 2% to 2:24 and 13% to 4:24 is found for the sandwich.
 |
| | Fig. 4 Density of states (DOS) of the P 3p states for trilayer phosphorene and the sandwich with the Li:P ratio of 2:24 and crystal orbital Hamiltonian populations (COHP) of the interlayer P–P bonds in the surface and center sheets. | |
Table 3 Integral of crystal orbital Hamiltonian populations (–ICOHP) of the interlayer P–P bonds in the surface and center sheets for trilayer phosphorene and the sandwich
| |
Phosphorene |
Sandwich |
| Surface |
Center |
Surface |
Center |
| Li:P = 1:24 |
4.17 |
4.46 |
4.56 |
4.66 |
| Li:P = 2:24 |
1.98 |
2.17 |
4.46 |
4.63 |
| Li:P = 4:24 |
— |
— |
3.96 |
3.97 |
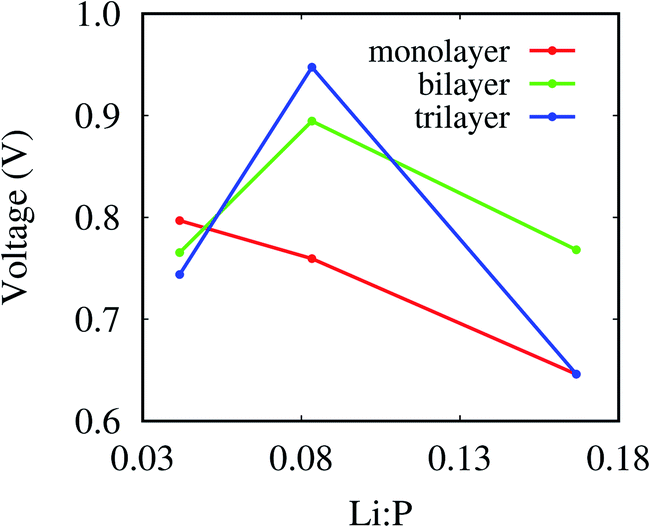
Fig. 5 depicts the voltage for the sandwiches defined as
| |
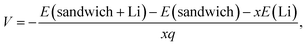 | (2) |
where
E,
x, and
q refer to the total energy, Li concentration, and charge on Li, respectively. The voltage for the monolayer decreases from 0.80 V to 0.65 V with increasing Li concentration as compared to the highest value of 1.01 V for phosphorene/graphene heterostructure.
26 Li intercalation at the interface leads to rotation of the
c axis to the armchair and the zigzag directions as shown in
Fig. 3, which is also reported in
ref. 27. In the case of the Li
:
P ratio of 2
:
24, the angle turns out to be 6.1°/0.9°, 0.8°/8.2° and 5.5°/7.8° to the armchair/zigzag direction for the monolayer, bilayer, and trilayer, respectively. A large rotation of 10.7° to the armchair direction takes place for the bilayer at the Li
:
P ratio of 4
:
24. The fluctuation of the voltage may be related to the distortion of the lattice.
 |
| | Fig. 5 Voltages for the sandwiches as function of the Li:P ratio calculated by the Li atoms at the interface and the P atoms in each layer. | |
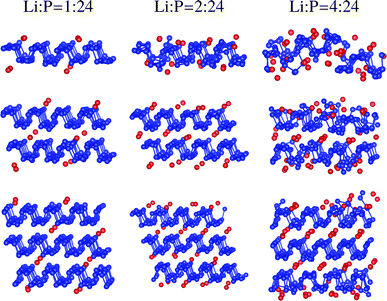
Structural reversibility of electrodes is important to prevent capacity loss during long-term cycles. Fig. 6 illustrates the structures of multilayer phosphorene and the sandwiches after molecular dynamics simulations starting from the configurations for the Li:P ratio of 4:24 and 6:24, respectively. The monolayer structures cannot be recovered after 3 ps because of high surface energy, which is confirmed by the results for up to 6 ps. Only slight surface distortion is retained for trilayer phosphorene after 1 ps, reflecting better reversibility. However, the surface reconstruction is hardly removed after 3 ps, which may lead to disintegrity of electrodes after many cycles. The structure is completely recovered without surface reconstruction for the sandwich after less than 1 ps.
 |
| | Fig. 6 Snapshots of the structures for multilayer phosphorene and the sandwiches after 1 and 3 ps ab initio molecular dynamics simulations at 300 K with a step of 3 fs starting from the configurations for the Li:P ratio of 4:24 and 6:24, respectively. The P and C atoms are labeled by blue and black. | |
4 Conclusions
Structural and electronic properties of Li intercalated multilayer phosphorene unsandwiched and sandwiched by graphene have been studied by first-principles calculations. Li intercalation has been demonstrated to donate charge to the P atom, weakening the interlayer P–P bond strength. Graphene obtains charge from the Li atoms on the surface of the sandwich, reducing charge depletion on the interlayer P–P bonds and thus improving structural stability. Consequently, the sandwiches achieve Li capacities of 142–144 mA h g−1 for the monolayer, bilayer, and trilayer without destroying structures in comparison to 144, 108, 96 mA h g−1 for freestanding multilayer phosphorene, respectively, which improves performance for high-rate charge/discharge. Structural reversibility of multilayer phosphorene is enhanced with increasing number of layers and can be improved by graphene sandwiching.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Science and Technology Foundation of Shenzhen (JCYJ20170818142419086), the Opening Project of Shanghai Key Laboratory of Special Artificial Microstructure Materials and Technology, the National Natural Science Foundation of China (Grant No. 41776142).
References
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, The Electronic Properties of Graphene, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- A. Molle, C. Grazianetti, L. Tao, D. Taneja, M. H. Alam and D. Akinwande, Silicene, Silicene Derivatives, and Their Device Applications, Chem. Soc. Rev., 2018, 47, 6370–6387 RSC.
- N. Liu, G. Bo, Y. Liu, X. Xu, Y. Du and S. X. Dou, Recent Progress on Germanene and Functionalized Germanene: Preparation, Characterizations, Applications, and Challenges, Small, 2019, 15, 1805147 CrossRef PubMed.
- J. L. Zhang, C. Han, Z. Hu, L. Wang, L. Liu, A. T. S. Wee and W. Chen, 2D Phosphorene: Epitaxial Growth and Interface Engineering for Electronic Devices, Adv. Mater., 2018, 30, 1802207 CrossRef PubMed.
- X. Wang, J. Song and J. Qu, Antimonene: From Experimental Preparation to Practical Application, Angew. Chem., Int. Ed., 2019, 58, 1574–1584 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric Field Effect in Atomically Thin Carbon Films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. C. Dhanabalan, J. S. Ponraj, Z. Guo, S. Li, Q. Bao and H. Zhang, Emerging Trends in Phosphorene Fabrication towards Next Generation Devices, Adv. Sci., 2017, 4, 1600305 CrossRef PubMed.
- L. Feng, K. Yabuoshi, Y. Sugimoto, J. Onoda, M. Fukuda and T. Ozaki, Structural Identification of Silicene on the Ag(111) Surface by Atomic Force Microscopy, Phys. Rev. B, 2018, 98, 195311 CrossRef CAS.
- J. Yuhara, H. Shimazu, K. Ito, A. Ohta, M. Araidai, M. Kurosawa, M. Nakatake and G. Le Lay, Germanene Epitaxial Growth by Segregation through Ag(111) Thin Films on Ge(111), ACS Nano, 2018, 12, 11632–11637 CrossRef CAS PubMed.
- F. Liu, J. Wang and H. Guo, Impact of Edge States on Device Performance of Phosphorene Heterojunction Tunneling Field Effect Transistors, Nanoscale, 2016, 8, 18180–18186 RSC.
- K. D. Pham, N. N. Hieu, H. V. Phuc, I. A. Fedorov, C. A. Duque, B. Amin and C. V. Nguyen, Layered Graphene/GaS van der Waals Heterostructure: Controlling the Electronic Properties and Schottky Barrier by Vertical Strain, Appl. Phys. Lett., 2018, 113, 171605 CrossRef.
- H. V. Phuc, V. V. Ilyasov, N. N. Hieu and C. V. Nguyen, Electric-Field Tunable Electronic Properties and Schottky Contact of Graphene/Phosphorene Heterostructure, Vacuum, 2018, 149, 231–237 CrossRef CAS.
- A. Yang, D. Wang, X. Wang, D. Zhang, N. Koratkar and M. Rong, Recent Advances in Phosphorene as a Sensing Material, Nano Today, 2018, 20, 13–32 CrossRef CAS.
- X. Liu, T. Ma, N. Pinna and J. Zhang, Two-Dimensional Nanostructured Materials for Gas Sensing, Adv. Funct. Mater., 2017, 1702168 CrossRef.
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Applications of Phosphorene and Black Phosphorus in Energy Conversion and Storage Devices, Adv. Energy Mater., 2018, 8, 1702093 CrossRef.
- M. C. Watts, L. Picco, F. S. Russell-Pavier, P. L. Cullen, T. S. Miller, S. P. Bartuś, O. D. Payton, N. T. Skipper, V. Tileli and C. A. Howard, Production of Phosphorene Nanoribbons, Nature, 2019, 568, 216–220 CrossRef CAS PubMed.
- M. Akhtar, G. Anderson, R. Zhao, A. Alruqi, J. E. Mroczkowska, G. Sumanasekera and J. B. Jasinski, Recent advances in Synthesis, Properties, and Applications of Phosphorene, npj 2D Mater. Appl., 2017, 1, 5 CrossRef.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, A Phosphorene–Graphene Hybrid Material as a High-Capacity Anode for Sodium-Ion Batteries, Nat. Nanotechnol., 2015, 10, 980 CrossRef CAS PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, High Capacity and Rate Capability of Amorphous Phosphorus for Sodium Ion Batteries, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- X.-L. Wu, Y.-G. Guo and L.-J. Wan, Rational Design of Anode Materials Based on Group IVA Elements (Si, Ge, and Sn) for Lithium-Ion Batteries, Chem.–Asian J., 2013, 8, 1948–1958 CrossRef CAS PubMed.
- G. A. Tritsaris, E. Kaxiras, S. Meng and E. Wang, Adsorption and Diffusion of Lithium on Layered Silicon for Li-Ion Storage, Nano Lett., 2013, 13, 2258–2263 CrossRef CAS PubMed.
- B. Peng, F. Cheng, Z. Tao and J. Chen, Lithium Transport at Silicon Thin Film: Barrier for High-Rate Capability Anode, J. Chem. Phys., 2010, 133, 034701 CrossRef PubMed.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, Metal Oxides and Oxysalts as Anode Materials for Li Ion Batteries, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- S. Zhao, W. Kang and J. Xue, The Potential Application of Phosphorene as an Anode Material in Li-Ion Batteries, J. Mater. Chem. A, 2014, 2, 19046–19052 RSC.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phosphorene as an Anode Material for Na-Ion Batteries: A First-Principles Study, Phys. Chem. Chem. Phys., 2015, 17, 13921–13928 RSC.
- G.-C. Guo, D. Wang, X.-L. Wei, Q. Zhang, H. Liu, W.-M. Lau and L.-M. Liu, First-Principles Study of Phosphorene and Graphene Heterostructure as Anode Materials for Rechargeable Li Batteries, J. Phys. Chem. Lett., 2015, 6, 5002–5008 CrossRef CAS PubMed.
- K. P. S. S. Hembram, H. Jung, B. C. Yeo, S. J. Pai, S. Kim, K.-R. Lee and S. S. Han, Unraveling the Atomistic Sodiation Mechanism of Black Phosphorus for Sodium Ion Batteries by First-Principles Calculations, J. Phys. Chem. C, 2015, 119, 15041–15046 CrossRef CAS.
- O. I. Malyi, K. V. Sopiha, C. Draxl and C. Persson, Stability and Electronic Properties of Phosphorene Oxides: From 0-Dimensional to Amorphous 2-Dimensional Structures, Nanoscale, 2017, 9, 2428–2435 RSC.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Zhu and U. Schwingenschlögl, Silicene on MoS2: Role of the van der Waals Interaction, 2D Mater, 2015, 2, 045004 CrossRef.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- A. Brown and S. Rundqvist, Refinement of the Crystal Structure of Black Phosphorus, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS.
- J. Zhu, A. N. Gandi and M. Gu, Phosphorene as Cathode for Metal-Ion Batteries: Importance of F Decoration, Materials Today Energy, 2018, 10, 141–145 CrossRef.
- A. Kumar, Controlled Nanostructures and Simultaneous Passivation of Black Phosphorus (phosphorene) with Nafion, J. Mater. Res., 2020, 35, 141–152 CrossRef CAS.
- A. Ziletti, A. Carvalho, D. K. Campbell, D. F. Coker and A. H. Castro Neto, Oxygen Defects in Phosphorene, Phys. Rev. Lett., 2015, 114, 046801 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Hongchun Yuana,
Yingli Chang*a,
Mu Gu
a,
Hongchun Yuana,
Yingli Chang*a,
Mu Gu