
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous mesostructured zirconia with high (hydro)thermal stability†
Bénédicte Lebeauab,
Issam Naboulsic,
Laure Michelinab,
Claire Marichalab,
Séverinne Rigoletab,
Cédric Carteretd,
Sylvette Brunete,
Magali Bonne ab and
Jean-Luc Blin*c
ab and
Jean-Luc Blin*c
aUniversité de Haute Alsace (UHA), CNRS, IS2M UMR 7361, F-68100 Mulhouse, France
bUniversité de Strasbourg, 67000 Strasbourg, France
cUniversité de Lorraine, CNRS, L2CM 7053, Faculté des Sciences et Technologies, BP 70239, F-54506 Vandoeuvre-lès-Nancy Cedex, France. E-mail: Jean-Luc.Blin@univ-lorraine.fr; Tel: +33 3 83 68 43 70
dUniversité de Lorraine, CNRS, LCPME, F-54000 Nancy, France
eUniversité de Poitiers, CNRS, IC2MP, UMR 7285, 86073 Poitiers Cedex 9, France
First published on 13th July 2020
Abstract
Here, combining the evaporation-induced self-assembly (EISA) method and the liquid crystal templating pathway, mesostructured amorphous zirconium oxides have been prepared by a soft templating method without addition of any heteroelement to stabilize the mesopore framework. The recovered materials have been characterized by SAXS measurements, nitrogen adsorption–desorption analysis and X-ray diffraction (XRD). The obtained mesostructured zirconia exhibits a high thermal stability. An in situ XRD study performed as a function of temperature shows that the amorphous ZrO2, obtained after removal of the pore templating agent (pluronic P123), begins to crystallize in air from 420 °C. Amorphous mesostructured ZrO2 also presents a high hydrothermal stability; these materials are not degraded after 72 hours in boiling water.
1. Introduction
Zirconium oxides have applications in many domains, for example they are extensively used in the field of solid oxide fuel cells, microelectronics, and they are known to be good ion exchangers;1 thanks to their excellent thermal stability, they are used as refractory materials,2,3 their interesting optical properties4,5 also make them excellent luminescent nanoprobes. Another application of these oxides deals with heterogeneous catalysis either as catalysts or as support.6–10 As catalyst, because of the presence of Lewis and Brønsted acid sites, zirconia is mainly used in its sulfated formed (SO4/ZrO2).11,12 For example, sulfated ZrO2 exhibits high activity for alkane isomerization.12 ZrO2 has also been used as a support for hydrodesulfurization (HDS) of gazole.13–18 It has been shown that, for the same Mo content per nm2 of support, MoS2 supported on zirconia has an activity three times higher than its analog supported on γ-alumina.17 Hamon et al.18 have observed that the stacking of MoS2 is less important on ZrO2 than on Al2O3 but a gain in activity is observed when ZrO2 is used as support. This was due to the fact that the NiMoS phase is more easily formed on zirconia than on alumina. In another study, Li et al.16 have shown that the catalyst promoted by Ni is more active in HDS, when it is supported on ZrO2 compared to Al2O3. Therefore, ZrO2 shows promising properties as support for the dispersion of the MoS2 slabs13 to design efficient hydrotreatment catalysts. But zirconium oxides generally have surface areas of 50 m2 g−1 or less, which is rather low compared with conventional supports such as SiO2, γ-Al2O3 (≈250 m2 g−1). Increasing the surface area of this support could potentially enhance the catalytic activity. One method to increase the specific surface area of ZrO2 consists in developing the preparation of templated-mesostructured zirconia in a similar way than what is done for silica materials. In 1995, Hudson et al.19 reported for the first time the synthesis of porous zirconia, using alkyltrimethylammonium halide as surfactant and zirconyl chloride as zirconium source. The authors proposed a scaffolding mechanism instead of a templating one to explain the formation of the mesoporous molecular sieve. In a first step, the template molecules are incorporated within the hydrous zirconium oxide and some water molecules are retained. Then in a second step, the mixture is slowly heated at a temperature that does not exceed 300 °C, and some water molecules are expulsed, which results in shrinkage of the structure. The trapped surfactant acts as filler, which avoids the structure collapse, and the continuation of the polymerization of zirconium chloride contributes to stabilize the structure. Finally, the porous framework is obtained after removal of the template by calcination at higher temperature (450 °C). Depending on the surfactant/zirconium ratio (varying from 0.082 to 0.341), mesostructured zirconia exhibit surface areas ranging from 238 to 329 m2 g−1. Since this date, using amphoteric, anionic, or neutral templates and, depending on the synthesis pathway, zirconyl chloride or zirconium propoxide as zirconium precursors, hexagonal, cubic or disordered mesoporous zirconia were successfully obtained.20–25 However, when obtained from the soft templating method, the main difficulty inherent in the synthesis of mesostructured ZrO2, is associated with the control of the reactivity of the zirconia source. To overcome this problem, the use of complexing agents makes the precursor less reactive and allows controlling its interaction with the surfactant.26 Moreover, as it is the case for most of other non-siliceous mesostructured oxides, it is very difficult to preserve the structure after the surfactant removal, as the structure generally collapses after the pores are freed from the surfactant. To enhance the stability, some phosphate or sulfate anions, which delay the crystallization of the amorphous ZrO2 into tetragonal or monoclinic form, can be incorporated to the framework.26–28 For example, Sachtler et al.26 prepared mesoporous zirconium oxides using zirconium isopropoxide Zr(iOpr)4 as zirconia source and hexadecylamine as a structuring agent via the sol–gel process. Acetylacetone, which is a bidentate chelating agent, is used to control the reactivity of the zirconia source. The materials obtained after removal of the surfactant by extraction in an alcoholic medium (ethanol) at 80 °C have a specific surface area of 347 m2 g−1 and a pore diameter of approximately 18.5 nm. However, the mesostructure collapses after calcination at 300 °C. The authors have shown that the treatment of the material with sulfate ions before the calcination stage preserves the mesostructure stable up to 700 °C, and a mixed tetragonal/monoclinic phase is obtained. In this study, we report the preparation and characterization of pure amorphous mesostructured zirconia with high thermal stability. Our methodology combined the evaporation induced self-assembly method (EISA)29 with the liquid crystal templating pathway.30–32 Pluronic P123, an amphiphilic triblock copolymer, is used as pore templating agent since such block copolymer is capable to impart larger pores, it is industrially available, hazard-free and easy to remove from the oxide framework. Most of zirconium(IV) alkoxides have a high cost, are toxic and present a fast hydrolysis rate, making difficult the control of homogeneity during the experimental processes. The fact that zirconium n-propoxide is diluted in propanol (70 wt% in propanol), its reactivity can be better controlled, so it has been selected as zirconia source.2. Materials and methods
The triblock copolymer pluronic P123 (EO)20(PO)70(EO)20 (EO = ethylene oxide, PO = propylene oxide) and the zirconium n-propoxide (70 wt% in propanol) were purchased from Sigma-Aldrich.2.1 Materials preparation
Amorphous mesostructured ZrO2 synthesis: first, x g of P123 are dissolved under stirring in a mixture of 20 g of ethanol and 2 g of a 15 M nitric acid solution (HNO3). Then, y g of zirconia precursor, zirconium n-propoxide (Zr(Opr)4) are added to this mixture. Finally, 2 g of distilled water are added. x and y varied from 1 to 3 g and from 1.15 to 4.65 g, respectively. These values correspond to a variation of the P123 concentration in the acidic solution from 33 to 60 wt% and of the P123/Zr(Opr)4 molar ratio from 0.012 to 0.092, respectively. Afterward, the mixture is evaporated using a rotary evaporator to remove the solvent (ethanol, propanol). The obtained hybrid mesophase is then dried for 12 hours at 40 °C and placed under an ammonia atmosphere (≈0.5 atm) for at least 12 hours. Finally, zirconia material is recovered after Soxhlet extraction with ethanol for 12 hours, to remove the P123 surfactant, and air drying.2.2 Characterization
SAXS experiments were performed on a SAXSess mc2 instrument (Anton Paar), using a line collimation system. This instrument is attached to a ID 3003 laboratory X-ray generator (General Electric) equipped with a sealed X-ray tube (PANalytical, λCu Kα = 0.1542 nm) operating at 40 kV and 50 mA. A multilayer mirror and a block collimator provide a monochromatic primary beam. A translucent beam stop allows the measurement of an attenuated primary beam at q = 0. Mesoporous materials or hybrid mesophase were put between two sheets of Kapton® placed in a powder cell before being introduced inside the evacuated chamber. All data were corrected for the background scattering from the Kapton® and for slit-smearing effects by a desmearing procedure from SAXSQuant software using the Lake method. Powder X-ray diffraction patterns were recorded using a PANalytical X'Pert PRO diffractometer equipped with a Cu X-ray tube (λCu Kα = 0.1542 nm) operating at 45 kV and 40 mA, and a X'Celerator detector. Fixed divergence slit (1/16°), mask (10 mm) and antiscatter slit (1/8°) were used at primary beam for the current analysis. The high temperature XRD patterns were recorded under dry air using a PANalytical X'Pert PRO diffractometer equipped with a Cu X-ray tube (λCu Kα1 = 0.15406 nm) operating at 45 kV and 35 mA, a X'Celerator detector and a high temperature oven chamber (HKT 1200 from Anton Paar). Fixed divergence slit (1°), mask (10 mm) and antiscatter slit (2°) were used at primary beam for the current analysis.N2 adsorption–desorption isotherms were recorded on a Micromeritics TRISTAR 3000 sorptometer at −196 °C. The specific surface area was obtained by using the BET model whereas the pore diameter and the pore size distribution were determined by the BJH (Barrett, Joyner, Halenda) method applied to the adsorption branch.33
The infrared spectra were recorded on a Fourier transform infrared spectrometer (Nicolet 8700), equipped with a KBr beam splitter and a DTGS detector. The spectra in diffuse reflectance (DRIFTS) mode were collected using a Harrick Praying Mantis™ diffuse reflection accessory and a HVC-DRP reaction chamber. To perform the analysis, the sample powder was first diluted in a KBr matrix (10 wt%). Then, the sample was kept inside the evacuated chamber (10−5 Torr). Reflectances Rs of the sample and Rr of pure KBr, used as a non-absorbing reference powder, were measured under the same conditions. The sample reflectance is defined as R = Rs/Rr. The spectra are shown in pseudo-absorbance (−log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) R) mode.
R) mode.
Raman Scattering Spectra were collected on a Jobin-Yvon T64000 spectrometer equipped with an optical microscope in confocal mode. The excitation beam (514.5 nm) was focused using a long-frontal ×50 objective (numerical aperture 0.5) on an area of about 3 μm2. The laser power on the sample was approximately 10 mW. The spectral resolution was 3 cm−1, with a wavenumber precision better than 1 cm−1.
1H decoupled 13C MAS NMR and 1H–13C Cross Polarization Magic Angle Spinning (CPMAS) NMR spectra have been recorded at room temperature on a Bruker Avance II 400WB spectrometer (B0 = 9.4 T) operating at 100.2 MHz. Samples were packed in a 4 mm diameter cylindrical zirconia rotor and spun at a spinning frequency of 12 kHz. 1H–13C CPMAS NMR experiments were performed with a proton π/2-pulse duration of 5.2 μs, a contact time of 1 ms, and a recycle delay of 5 s. 1H decoupled 13C MAS NMR spectra were recorded with a carbon π/4-pulse duration of 2.6 μs, a recycle delay of 60 s and a 1H high-power decoupling of 61 kHz. 1H MAS NMR experiments were performed with a proton π/2-pulse duration of 2.4 μs and a recycle delay of 5 s. 1H and 13C chemical shifts are relative to tetramethylsilane (TMS). The samples were analyzed by 13C and 1H solid state NMR before ethanol washing and after dehydration at 70 °C in an oven during 24 h.
2.3 Hydrothermal stability
2.5 g of amorphous mesostructured ZrO2 are placed under refluxed at 120 °C in 50 mL of distilled water. The mixture is stirred continuously for 7 days. Aliquots are taken at regular time intervals, between 15 minutes and 7 days. They were recovered by filtration and dried in air at room temperature.3. Results and discussion
3.1 Effect of the synthesis parameters on the mesostructured ZrO2 formation
In water, P123 forms liquid crystals for concentrations between 20 and 65 wt%.34,35 The latter is composed of a cubic phase (I1) and of a hexagonal one (H1) for P123 contents from 22 to 37 wt% and from 40 to 65 wt%, respectively. In an acidic solution, these limits are not modified.36 To explore the overall range of concentrations of the liquid crystal domain, materials have been prepared with a P123 concentration in the acidic solution of 33 (I1), 50 (H1) and 60 wt% (H1). The P123/Zr(Opr)4 molar ratio, noted R, has been varied from 0.012 to 0.092, depending on the P123 concentration. The large amount of ethanol prevents from the formation of a gel and controls the reactivity of the zirconia source. Materials have been prepared in the presence of nitric acid. Indeed, Ward et al.37 have investigated in detail the effect of nitric acid on the hydrolysis–condensation rate of zirconium n-propoxide. They have shown that the presence of nitric acid increases the rate of hydrolysis and decreases the speed of the condensation reaction. From the SAXS patterns reported in Fig. 1, it can be seen that, whatever the P123 surfactant concentration and the P123/Zr(Opr)4 molar ratio, neither a hexagonal or a cubic mesostructure is obtained. At the best, a broad reflection (Fig. 1B and C) is observed for materials prepared with a surfactant concentration belonging to the H1 domain (50 or 60 wt% of P123 in the acidic solution) for R values lower than 0.048. The presence of a single reflection indicates the formation of a disordered mesostructure. In this case, the mesoporous molecular sieve exhibits a wormhole like channel system, analogous to MSU-type materials.38 This reflection is an indication of the sum of the wall thickness and the distance between two mesopores.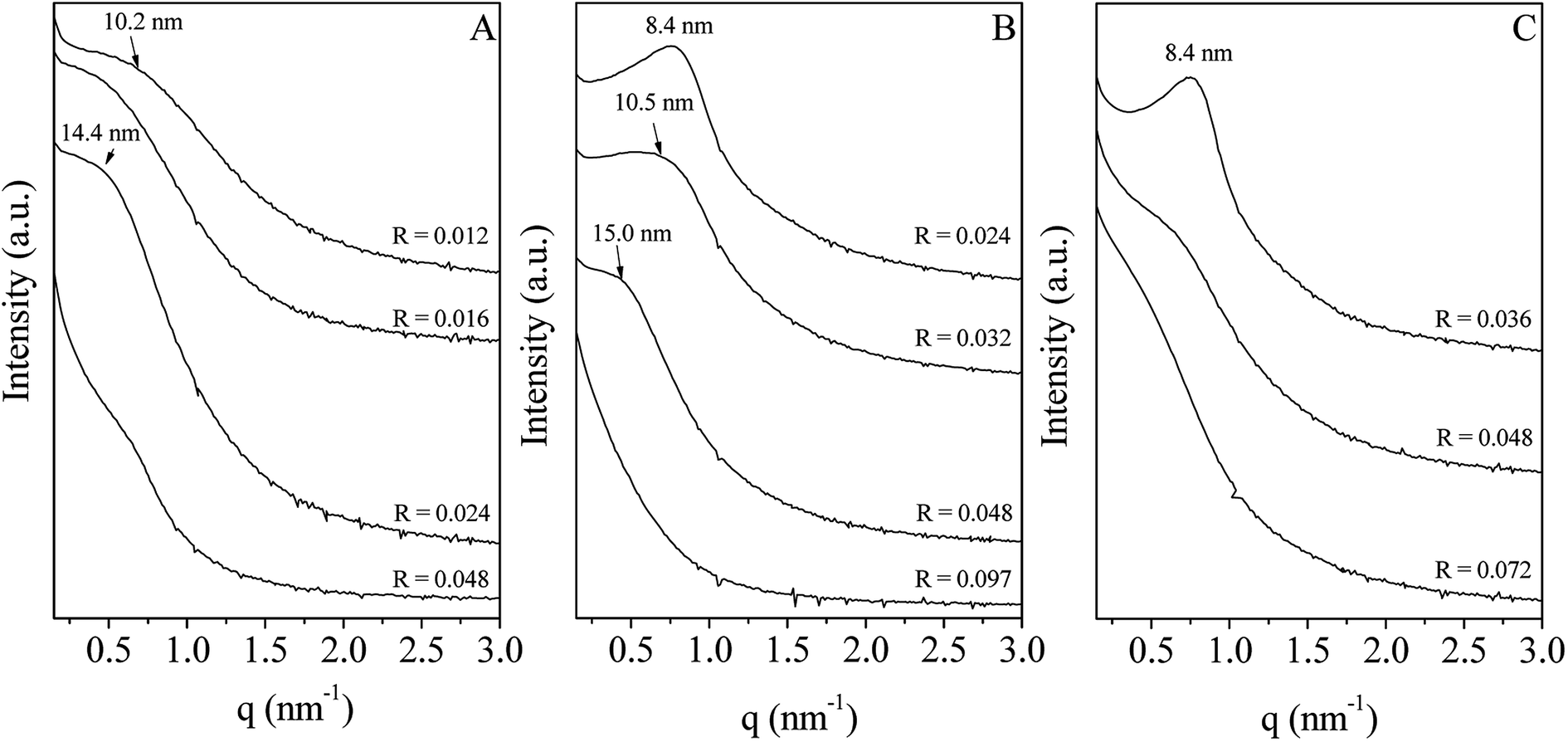 | ||
| Fig. 1 Evolution of the SAXS pattern as a function of the P123/Zr(Opr)4 molar ratio (R). The P123 concentration in the acidic solution is fixed to 33 (A), 50 (B) and 60 wt% (C). | ||
For higher P123/Zr(Opr)4 ratios or for zirconia obtained with a P123 concentration of 33 wt%, either a not well defined bump or no reflection line is detected (Fig. 1), meaning that the materials adopt a completely random pore arrangement.
Nitrogen adsorption–desorption isotherm of ZrO2 prepared with a P123 concentration of 33 wt% and a P123/Zr(Opr)4 molar ratio of 0.012 is intermediate between type I and type IV, meaning that the materials exhibit a supermicroporosity (Fig. 2A).39 This is also reflected by the mesopore size distribution, which presents no maximum in the mesopore range (inset of Fig. 2A). If the R value is increased, the isotherm becomes type IV, characteristic of mesoporous materials according to the IUPAC classification. Increasing the P123 content to 50 and 60 wt% in the acidic solution, all the materials show a type IV isotherm with a H2 hysteresis loop, encountered for materials having a wormhole-like structure, except for R = 0.097 for which the hysteresis is rather H3 meaning that the interparticular porosity predominates (Fig. 2B and C). Looking at the mesopore size distribution, it appears that by decreasing R, dV/dD gradually increases and the distribution becomes narrower (inset of Fig. 2B and C), which indicates a better uniformity in the mesopore diameter when the amount of zirconia precursor is increased. Whatever the considered P123 concentration in the acidic solution, the specific surface area is rather high and in a general way we can note a slight decrease as a function of the P123/Zr(Opr)4 molar ratio (Table 1). For example, when ZrO2 materials are prepared with a P123 content of 60 wt%, BET specific surface area decreases from 330 to 230 m2 g−1 if R is increased from 0.036 to 0.072. In all cases, taking into account the error on the measurement (5%), the mesopores volume is in the range of 0.20–0.40 cm3 gSTP−1.
 | ||
| Fig. 2 Variation of the nitrogen adsorption–desorption isotherms with the corresponding mesopores size distribution (inset) as a function of the P123/Zr(Opr)4 molar ratio (R). The P123 concentration in the acidic solution is fixed to 33 (A), 50 (B) and 60 wt% (C). | ||
| [P123] in acidic solution (wt%) | P123/Zr(Opr)4 molar ratio | SBET (m2 g−1) | Vp (cm3 gSTP−1) | ∅ (nm) |
|---|---|---|---|---|
| 33 | 0.012 | 345 | 0.10 | — |
| 0.016 | 337 | 0.20 | 2.6 | |
| 0.024 | 335 | 0.40 | 5.7 | |
| 0.048 | 295 | 0.30 | 4.0 | |
| 50 | 0.024 | 345 | 0.30 | 4.0 |
| 0.032 | 375 | 0.35 | 5.3 | |
| 0.048 | 385 | 0.45 | 6.5 | |
| 0.097 | 320 | 0.30 | — | |
| 60 | 0.036 | 330 | 0.30 | 4.2 |
| 0.048 | 293 | 0.17 | 4.4 | |
| 0.072 | 230 | 0.17 | 5.0 |
To find a possible explanation for the absence of any ordered mesostructure formation, we have to consider the different parameters which can affect the reconstitution of the liquid crystals during the evaporation of the solvent, and act as mold for the material structure. First, to control the reactivity of the zirconia source the preparation of ZrO2 is performed in a strong acidic solution, (HNO3 = 15 mol L−1), thus a large amount of NO3− is present in the synthesis medium. Due to its position in the Hofmeister series, this ion is known to have a kosmotropic effect.40 This means that this ion lowers the demixing temperature of the system by decreasing the solubility of the surfactant (salting out effect).40–42 Deyerle et al.43 have investigated the effect of a series of Hofmeister anions on the phase behavior of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymers and they have evidenced that poorly hydrated anions such as NO3− lower the interfacial tension and bind to the hydrophobic moieties of the polymer. Second, the precursor solution contains 30 wt% of propanol and even if it is removed during the evaporation step, propanol will, as other auxiliaries such as oil, modify the surfactant behavior in solution.44 Indeed, in the literature, it is reported that alcohols can act either as co-solvent or as co-surfactant, depending on their chain length.45 The tendency to act as a co-surfactant increases with the length of their carbon chain. Alcohols can also provoke salting in and salting out effects. For example, in a paper dealing with the influence of alcohol addition on the gelation in aqueous solution of pluronic F127 [(EO)100(PPO)65(EO)100], Kwon et al. have reported46 that ethanol and methanol increase the hard gel temperature and the critical micellar temperature, while butanol favors the aggregation of the polymer. Bharatiya et al.47 have also shown that in the presence of methanol, ethanol and propanol, the cloud point and the critical micellar temperature of pluronic P123 increase with the alcohol concentration. They conclude that these alcohols are good solvents for both PEO and PPO chains. If the carbon chain is longer than 3, alcohol replace water molecules in the PPO core and they induce a micellar growth in the P123 solution.47 Alany et al.48 have investigated the effect of alcohol incorporation on the phase behavior of a mixture of sorbian monolaurate and polyoxyethylene 20 sorbian mono-oleate surfactants. The authors conclude that the formation of liquid crystal is inhibited by the presence of low molecular weight alcohols (C3 and C4). These additives disturb the long-range organization of the surfactants mixed system. This phenomenon was also observed by Aramaki et al. for the octaethylene glycol dodecyl ether [C12(EO)8] system.49 The investigation by SAXS of the H1 phase of the water/propanol/C12(EO)8 system reveals that the hexagonal phase turns into an isotropic phase when increasing the volume fraction of propanol in the mixture. This phase transition is attributed to the penetration of the propanol molecules into the palisade layer of the cylinders.
For the synthesis of mesostructured silica materials, different groups have taken benefit of alcohol to tune the symmetry of the mesostructured materials or to induce phase transitions during the synthesis by modifying surface curvature of the micelles.50–52 For example, by increasing the amount of alcohol such as ethanol, propanol and methanol in the synthesis mixture of mesoporous materials prepared from the CTMABr-surfactant based system, Cool et al.52 have observed the following transition mesophase sequence: hexagonal → cubic→ lamellar → radially arranged hexagonal closed packed mesophase. To explain the phase transition, the authors consider a variation of the surfactant packing parameter. They claim that at the lower concentration, alcohol molecules penetrate into the surfactant micelles and act as a co-surfactant. Fang et al.53 have prepared large pore cubic Im3m mesostructured silica using F127 as template and tetraethylorthosilicate (TEOS) as silica precursor in the presence of butanol, which plays the role of a co-surfactant.
In a previous study dealing with the effect of alcohol on the phase behavior on a nonionic fluorinated surfactant [C8F17C2H4(OC2H4)9OH, labeled as R7F(EO)9], we have demonstrated that isopropanol acts as a co-solvent.54 We have also shown that, upon the addition of isopropanol, the range of surfactant composition belonging to H1 is progressively reduced and the hexagonal liquid crystal phase is completely melted when the isopropanol concentration reaches 7 wt%. This liquid crystal phase disappears in aid of less ordered ones. Preparing mesoporous silica from the R7F(EO)9 system in the presence of isopropanol according to the cooperative templating mechanism, the mesopore ordering is lost when the alcohol concentration reaches 5 wt%,54 since isopropanol acts as a micelles breaker.
Here, the combined effect of NO3− and propanol on pluronic P123 behavior in solution likely prevents the complete reconstitution of the hexagonal liquid crystal phase during the evaporation step, leading to the formation of a non-well ordered mesophase and as a consequence, at the best, a wormhole arrangement of the mesopores is recovered. Moreover, when the value of R is increased, the amount of zirconia source is not enough to cover all of the cylinders of the H1 phase and a random mesopore arrangement is obtained. Considering the SAXS and nitrogen adsorption–desorption results, in all following studies, the P123 concentration in the acidic solution and the P123/Zr(Opr)4 molar ratio have been fixed to 50 wt% and 0.024, respectively. The zirconia material synthesized under these conditions presents a wormhole-like mesostructure with a specific surface area of 345 m2 g−1 and a narrow mesopore size distribution centered around 4.0 nm.
3.2 Characterization of the hybrid mesophase and efficiency of surfactant removal
In accordance with the results reported above and to perform this study, the P123/Zr(Opr)4 molar ratio molar ratio is fixed to 0.024 and the surfactant concentration in the acidic solution is equal to 50 wt%.The SAXS pattern of the hybrid mesophase, i.e. before surfactant removal, shows a reflection at 9.8 nm (Fig. 3A(a)), confirming that no ordered mesostructure is recovered after the evaporation step and the treatment under NH3 atmosphere. After the template removal by ethanol extraction during 12 hours, no significant change is noted, except a shift of the position of the broad peak (Fig. 3A(b)) due to the contraction of the mesopore network (≈15%). To check the efficiency of the template extraction, Infrared and Raman analyses have been performed. The Infrared and Raman spectra of ZrO2 materials (mesophase and after extraction) were recorded between, 2000–4000 cm−1 and 2700–3400 cm−1, respectively (Fig. 3B and C). The infrared spectrum of the hybrid mesophase shows intense bands located at 2875, 2915 and 2971 cm−1 (Fig. 3B(a)), which are attributed to the CH2/CH3 stretchings of the P123 alkyl chains. The broad band in the 3200–3700 cm−1 range is characteristic of the O–H groups stretching, while the band at 3190 cm−1 corresponds to the N–H stretching of NH4(NO3). NH4(NO3) was formed upon NH3 treatment from residual NO3− coming from the nitric acid. Indeed, NO3− can strongly bond the amorphous zirconium hydroxyoxide network.55 Moreover, the TG curve recorded on the as-made mesostructured ZrO2 shows a brutal mass loss of 52% associated to a narrow and intense exothermic peak observed on the heat flow curve at 181 °C (Fig. S1†), that corresponds to the spontaneous thermal decomposition of the P123 promoted by the highly exothermic decomposition of the NH4NO3.56 It is noteworthy that, in the same condition, the decomposition of P123 occluded in SiO2 occurs at about the same temperature but in a broader temperature range with less intense exothermic peak.57 After ethanol extraction, the bands assigned to the stretching mode of the C–H bond of the P123 molecules (2800–3000 cm−1) are not observed anymore (Fig. 3B(b)). This indicates that the surfactant is not present in the pores. It is noteworthy that NH4NO3 was also removed with EtOH washing. The Raman spectra depicted in Fig. 3C confirm the results obtained by infrared spectroscopy. The intensities of the bands located at 2877, 2936 and 2990 cm−1, which are attributed to the alkyl chains of P123, are strongly reduced and almost negligible after ethanol extraction. We should note that the shoulders observed for the bands at 2936 and 2990 cm−1 indicate the presence of 2 others bands located at about 2978 and 2920 cm−1. That seem to reveal the presence of residual propanol and/or n-propoxy groups (vide infra). The weak and broad band at 3085 cm−1 corresponding to the N–H of NH4(NO3) is also strongly reduced after ethanol washing. These results indicate that ethanol extraction during 12 hours is an effective method to almost completely remove the surfactant located in the mesopores. This also suggests that weak interactions between P123 and the hydrolyzed zirconia precursor exist. To get informations about the potential interfacial contact between P123 triblock copolymer and the zirconia surface, 1H and 13C NMR experiments have been performed on the hybrid mesophase.
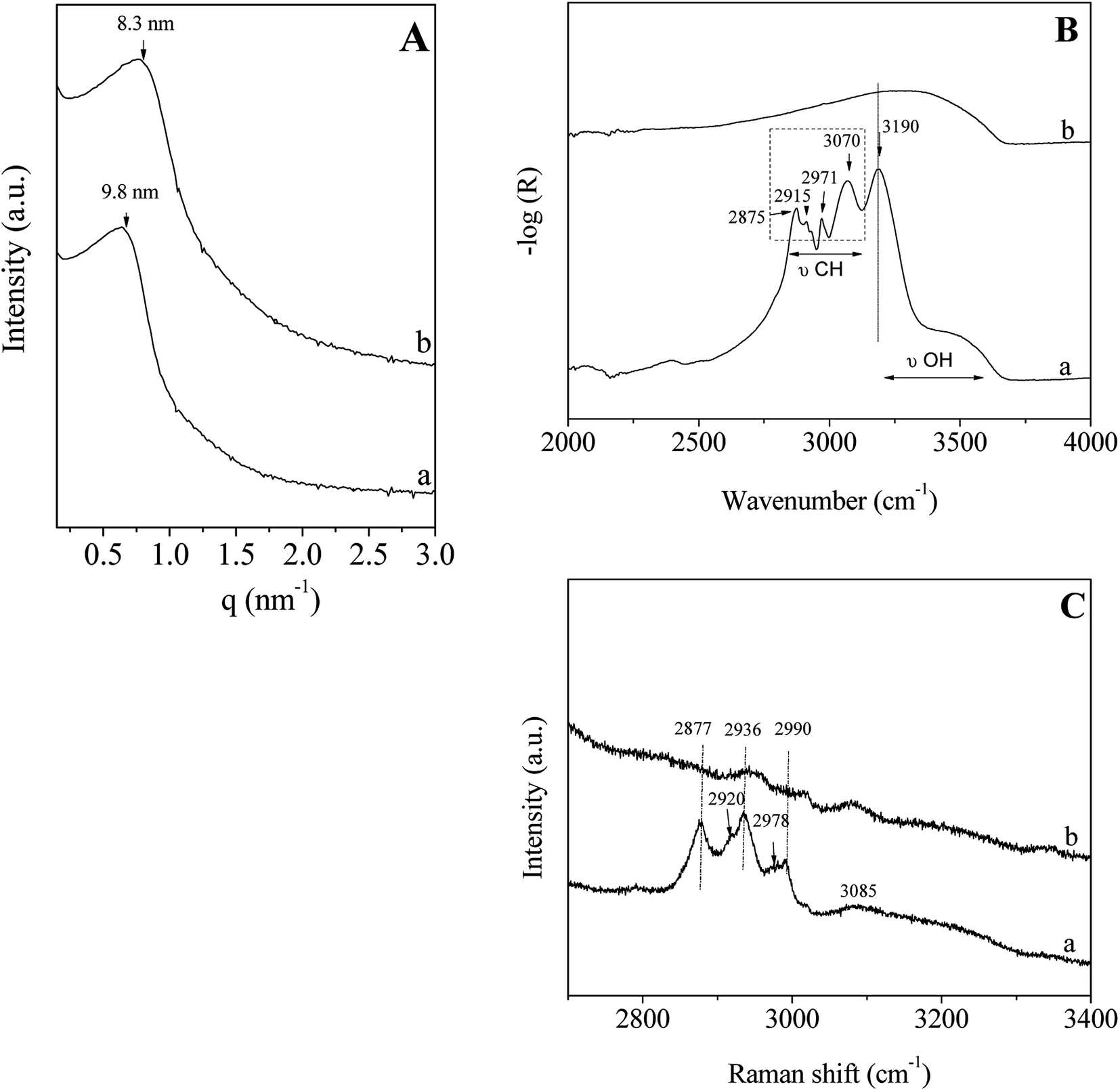 | ||
| Fig. 3 SAXS pattern (A); infrared (B) and Raman (C) spectra of the hybrid mesophase (a) and of the ZrO2 material obtained after ethanol extraction (b). | ||
13C MAS NMR spectrum of as-synthesized hybrid mesophase, shown in Fig. 4A, exhibits four resonances at 75.9, 73.8, 70.3 and 18.1 ppm, characteristic of P123. The first two resonances correspond to PPO moieties, the resonance at 70.3 ppm is assigned to PEO species while the CH3 belonging to PPO units of the P123 is detected at 18.1 ppm.55,56 All resonances are narrow, indicating that P123 alkyl chains are mobile. That suggests weak interactions with zirconia surface, which is consistent with the quasi total removal of P123 under Soxhlet ethanol extraction. 1H–13C CPMAS NMR spectrum (see Fig. S2A†) of as-synthesized hybrid mesophase shows that the previous resonances are broadened. It is worthy to note that CPMAS experiment emphasizes the rigid part of the micelles whereas the mobile part of the P123 molecules might be underestimated because motion prevents the magnetization transfer thanks to dipolar interaction from 1H to 13C. It is also worth noting that the resonance at 70.3 ppm appears asymmetric. This broadening can indicate the presence of several conformations of PEO units or a decrease in mobility.58 Moreover, an additional resonance, barely observable on the 13C MAS NMR spectrum, is clearly detected at 65.7 ppm thanks to the CPMAS NMR experiment. This resonance could correspond either to propanol or to propoxy groups coming from zirconium n-propoxide (Zr-Opr) that was not fully hydrolyzed during the synthesis, or to a mixture of both. The presence of residual propanol and/or Zr-Opr could explain the 2 weak resonances observed in Raman at 2978 and 2920 cm−1 corresponding to C–H stretching bands. Nevertheless, the amount of residual propanol or Zr-Opr is weak according the 13C MAS NMR spectrum that provides quantitative information.
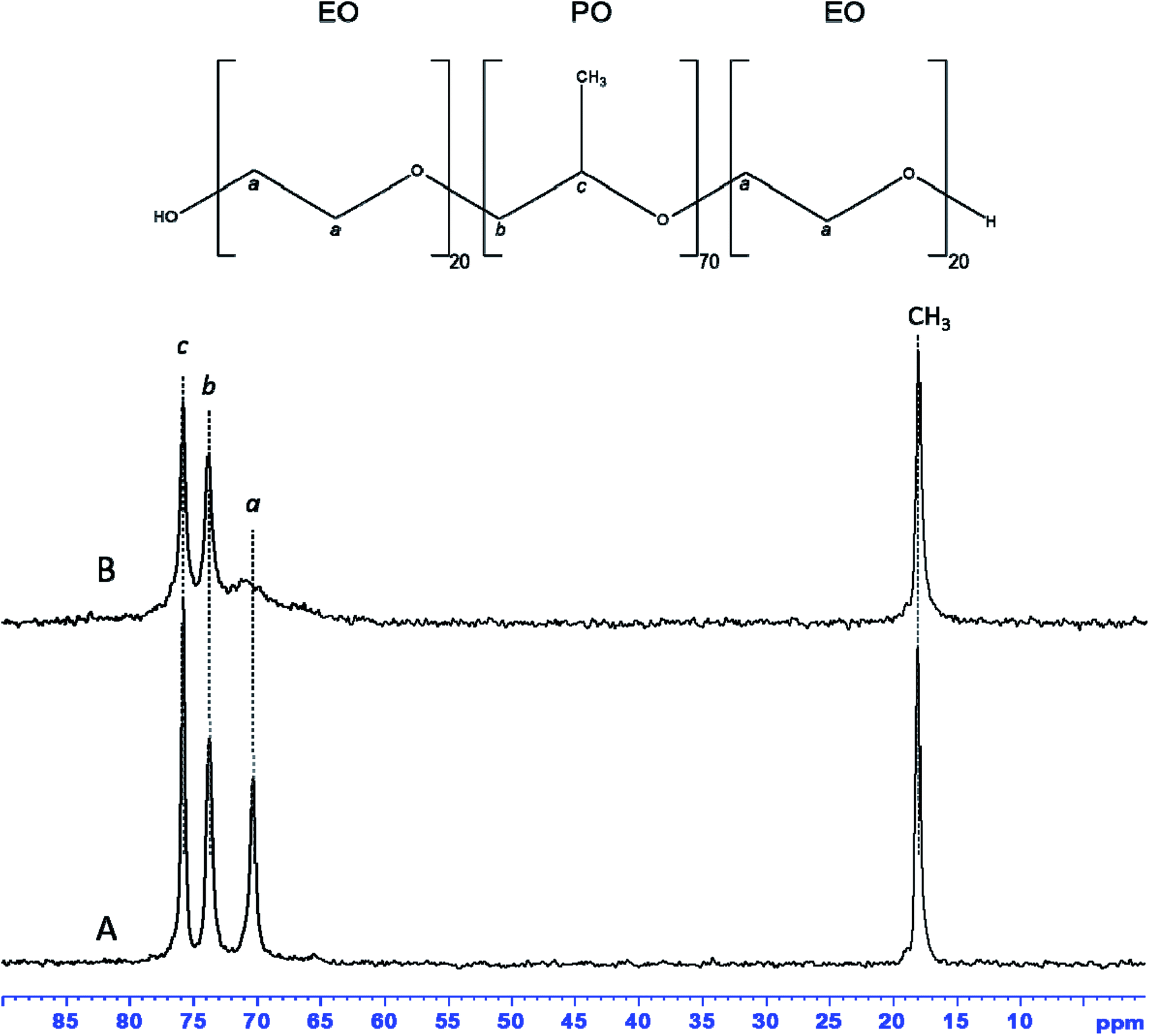 | ||
| Fig. 4 13C MAS NMR spectra with 1H decoupling of as-synthesized (A) and dehydrated at 70 °C (B) ZrO2 hybrid mesophase. | ||
The additional resonances, corresponding to propanol or Zr-Opr, expected around 25.8–27.1 and 10.2–10.4 ppm, are not detected on the 13C CPMAS NMR spectrum. In order to get some insight, dehydration at 70 °C of the hybrid mesophase was realized. After dehydration, all resonances detected on the 13C MAS NMR spectrum (compare Fig. 4A and B) are slightly broadened except the resonance assigned to the CH2 of PEO species that is drastically broadened by the water removal as already reported in the literature.58 Indeed, dehydration of PEO moieties obviously reduces their mobility. This again suggests low interaction with the zirconia walls, by comparison with the observation of Dragoi et al.58 who reported that a simple dehydration treatment at 70 °C during 26 hours was not sufficient to retract PEO chains from inside amorphous silica walls. The 1H–13C CPMAS NMR spectrum of the dehydrated sample (Fig. S2B†) clearly evidences a resonance at 23.9 ppm while the resonance corresponding to the CH3 groups of propanol or Zr-Opr may not be resolved from the one accounting for the methyl groups of P123. The presence of propanol in the hybrid mesophase supports our hypothesis of the lack of organization of the mesopore network. Indeed, propanol prevents from the formation of the hexagonal H1 phase and/or the uncompleted condensation of the precursor can also have a negative effect on the formation of an ordered mesostructure. However, resonances at 170.6 and 180.6 ppm are also detected on the 1H–13C CPMAS NMR spectrum of the dehydrated sample (Fig. S2B†), suggesting the presence of carbonyl groups. This is consistent with Raman analysis of as-synthesized ZrO2 hybrid mesophase that showed the presence of two bands at 1653–1687 cm−1, characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of carboxylic acids (Fig. S3†).59 This suggests a small degradation of PEO moieties of P123 due to acid induced hydrolysis as demonstrated by Yang et al.60 and Mesa et al.61 Indeed, the formation of formic acid (OCHOCH2–) due to polyoxides oxidation products was reported by Bérubé et al.57 Acid can also cleave the C–O–C bond leading to small PEO segments with terminal CH2–OH groups that are expected around 66 ppm. Consequently, because the 13C chemical shifts of propanol, Zr-Opr and terminal CH2–OH are very close, it is not possible to discriminate between those three hypotheses that may also coexist.
O bond of carboxylic acids (Fig. S3†).59 This suggests a small degradation of PEO moieties of P123 due to acid induced hydrolysis as demonstrated by Yang et al.60 and Mesa et al.61 Indeed, the formation of formic acid (OCHOCH2–) due to polyoxides oxidation products was reported by Bérubé et al.57 Acid can also cleave the C–O–C bond leading to small PEO segments with terminal CH2–OH groups that are expected around 66 ppm. Consequently, because the 13C chemical shifts of propanol, Zr-Opr and terminal CH2–OH are very close, it is not possible to discriminate between those three hypotheses that may also coexist.
1H MAS NMR experiment can also provide some interesting information regarding interaction with water. 1H MAS NMR spectra of as-synthesized and dehydrated hybrid mesophases are displayed Fig. S4.† At least 8 resonances are detected on the as-synthesized hybrid mesophase (Fig. S4A†). The splitted resonance in the 0.9–1.5 ppm range is assigned to CH3–P123 and to both CH3 and CH2 of propanol and/or Zr-Opr. Four resonances detected between 3 and 4 ppm correspond to CH2–O(H/Zr) group of propanol and/or Zr-OPr and CH–PPO, CH2–PPO and CH2–PEO groups of P123. CH2–OH moieties from PEO degradation are expected in the same range (at 3.45 ppm according to Mesa et al.61) and cannot be resolved. All resonances are narrow in agreement with the mobility of P123 molecules. Two additional resonances are detected at 7.4 and 6.3 ppm. The comparison with 1H MAS NMR spectrum of the dehydrated sample (Fig. S4B†) suggests that they are assigned to ZrOH groups and to H2O in interaction through hydrogen bonds with ZrOH, respectively. It can be noticed that the presence of ZrOH species was also evidenced by IR spectroscopy. The resonance at 7.4 ppm could also correspond to the proton of ammonium nitrate.62 It is also interesting to note that all resonances corresponding to P123 are broadened upon dehydration especially the one at 3.8 ppm corresponding, according to the literature, to the 1H of CH2–PEO species. This is consistent with 13C NMR results.
3.3 Thermal stability of the mesostructured ZrO2
The mesostructured ZrO2 has been synthesized from 50 wt% of P123 in the acidic solution with a P123/Zr(Opr)4 molar ratio of 0.024.XRD and Raman analysis show that the recovered mesostructured ZrO2 after ethanol extraction is amorphous. Only a broad vibration at around 500 cm−1 is observed on the Raman spectrum (Fig. S5†), which confirms the amorphous nature of the recovered zirconia.63 A XRD study as a function of temperature (HT-XRD) was carried out to determine the thermal stability of the mesostructured ZrO2. XRD patterns were recorded in the 2θ 27–33° range where the main peaks characteristic of the tetragonal and monoclinic ZrO2 phases are expected. Under dry air atmosphere, the crystallization of ZrO2 begins at 420 °C (Fig. 5). Peak located at 2θ = 30.37° indicates that the tetragonal form of zirconia is first obtained. Using the Scherrer law [D = 0.9λ/(wcosθ), w is the width at half maximum of the (101) peak], the size of the tetragonal particles is estimated between 5 and 10 nm at 440 °C. When the temperature reaches 700 °C (Fig. 5), the presence of very low intense and broad peaks at 2θ = 28.36 and 31.16° indicates that the monoclinic structure appears. It is noteworthy that for monoclinic and tetragonal phases, the peak positions are slightly shifted to the ones observed at room temperature because of the dilatation of the material and/or of the aluminum sample holder at high temperatures. Upon heating at 900 °C, the tetragonal ZrO2 is still detected (Fig. 5). After cooling at 25 °C, the monoclinic form of ZrO2 predominates. According to Davis et al.,64 the phase transformation during the cooling step can be attributed to anionic vacancies present at the zirconia surface. Indeed, these authors have investigated the crystallization and the phase transformation process of amorphous zirconia via in situ high temperature X-ray diffraction. They have shown that, during heating, amorphous ZrO2 crystallizes into the tetragonal structure at around 450 °C. Tetragonal zirconia is stable at temperature below 800 °C. Surface defects are generated at this temperature. These defects adsorb oxygen on cooling below 300 °C, which initiates the tetragonal to monoclinic phase transition. However, if the surface is covered with sulfates these defects do not have a pronounced effect on the phase transformation. Thus, the authors conclude that oxygen vacancies present on the surface are responsible for the low-temperature tetragonal to monoclinic transition on cooling.64
 | ||
| Fig. 5 XRD patterns of ZrO2 material obtained after ethanol extraction recorded in the 2θ 28–33° range at various temperatures from 25 °C to 900 °C and after cooling down at 25 °C. | ||
It is noteworthy that, when calcining the sample in a furnace at 440 °C under synthetic air, only amorphous ZrO2 is detected by XRD and Raman spectroscopy. The crystallization begins upon calcination at 480 °C. At this temperature, the tetragonal form is obtained and the mesostructure is preserved (Fig. S6A†). The recovered material exhibits a type IV nitrogen adsorption–desorption isotherm, characteristic of mesoporous materials according to the IUPAC classification (Fig. S6B†). The mesopore size distribution is quite narrow and centered around 3.6 nm (Fig. S6C†). The specific surface area and the mesopore volume are equal to 157 m2 g−1 and 0.16 cm3 g−1, respectively. The delay in the crystallization temperature between the HT-XRD and the calcination in a furnace can be due to the sample preparation. Indeed, for XRD HT experiments, ZrO2 powder was compacted contrary for XRD RT experiments.
3.4 Hydrothermal stability of the mesostructured ZrO2
To develop the application of mesostructured ZrO2, in particular in catalysis, the hydrothermal stability is a crucial parameter that should be considered. Thus, we have investigated it in boiling water. The starting ZrO2 has been obtained from a 50 wt% of P123 in the acidic solution and a 0.0124 P123/Zr(Opr)4 molar ratio. Before the immersion in boiling water, the surfactant has been removed by ethanol extraction during 12 hours. The wormhole-like mesostructure is still detected by SAXS even after 7 days in boiling water (Fig. 6). However, when the treatment is prolonged, the maximum of the broad line is shifted towards lower q value and its intensity decreases in the meantime. The Bragg distance varies from 8.2 to 7.7 nm when the immersion time is changed from 0.5 hour to 7 days. This evolution suggests that the mesostructure begins to be damaged when the treatment in boiling water is prolonged. Whatever the duration, the nitrogen adsorption–desorption isotherm is characteristic of mesoporous materials (Fig. 7). However, from 24 hours of presence in boiling water, at high relative pressure, the adsorbed volume of nitrogen increases instead of reaching a plateau (Fig. 7). This indicates the presence of an interparticular porosity. The condensation of nitrogen in these large mesopores occurs very likely, in addition to the adsorption inside the channels. This phenomenon is accentuated with the duration of stay in boiling water (Fig. 7). As a function of the immersion time, the maximum of the mesopore size distribution remains around 5.0 nm (inset of Fig. 7) and the pore size distribution becomes a bit broader, this reflects the beginning of the mesostructure collapse, evidenced by the SAXS analysis.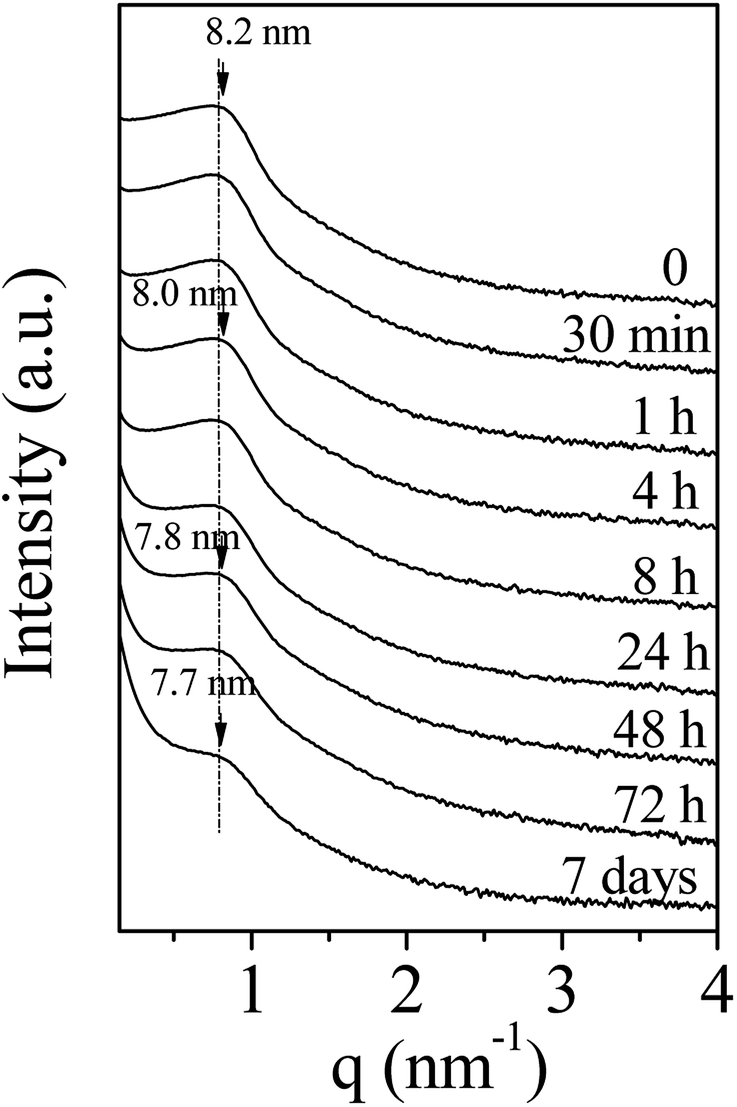 | ||
| Fig. 6 Evolution of the SAXS pattern of amorphous mesostructured ZrO2 with the immersion time into boiling water. | ||
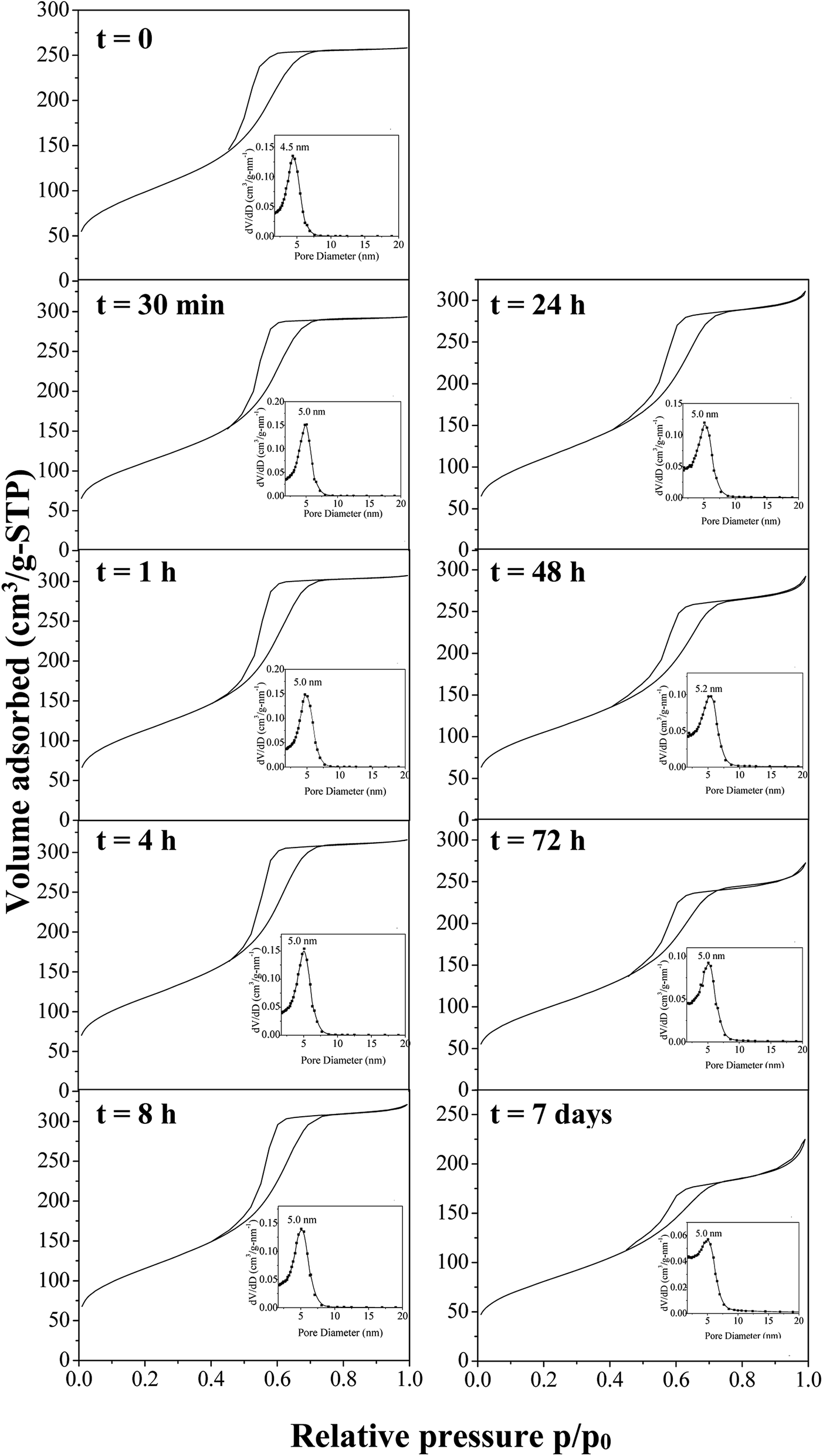 | ||
| Fig. 7 Nitrogen adsorption–desorption isotherms with the corresponding mesopores size distribution after time of immersion into boiling water of amorphous mesostructured ZrO2. | ||
The specific surface area and the pore volume follow the same trend as a function of the immersion time in boiling water (Fig. 8). First, from 0 to 4 hours, their values increase from 360 to 418 m2 g−1 and from 0.35 to 0.45 cm3 g−1, respectively. Since infrared and Raman analyses have shown that almost all the P123 molecules are removed after ethanol extraction, this variation cannot be attributed to surfactant removal but should rather be related to the dissolution of NH4(NO3), which is formed during the synthesis of the amorphous ZrO2. The presence of this salt has been evidenced by FTIR and Raman analyses. After this period, both parameters progressively decrease to reach after 7 days 290 m2 g−1 (i.e. ≈70% of the maximum value) and 0.28 cm3 g−1, respectively; reflecting the progressive alteration of the mesostructure upon treatment in boiling water. If mesostructured zirconia is calcined at 440 °C before their immersion in boiling water, the same evolution is noted, except the rise in the curves depicting the variations of the specific surface area and the pore volume for times less than 4 hours (Fig. S7†). This strengthens the fact that this phenomenon arises from the dissolution of NH4(NO3). The progressively collapse of the mesostructure can be attributed to dissolution–reprecipitation processes, as observed for mesostructured silica materials.65,66 Indeed, in the case of SiO2, during the treatment with boiling water, the water adsorbed onto the silanol groups causes the hydrolysis of nearby Si–O–Si bonds to yield more or less branched (Si–O–)n chains, resulting in the gradual collapse of the ordered mesoporous structure.65,66 We can assume that the OH groups present at the surface of the amorphous mesostructured ZrO2 generate a similar behavior.
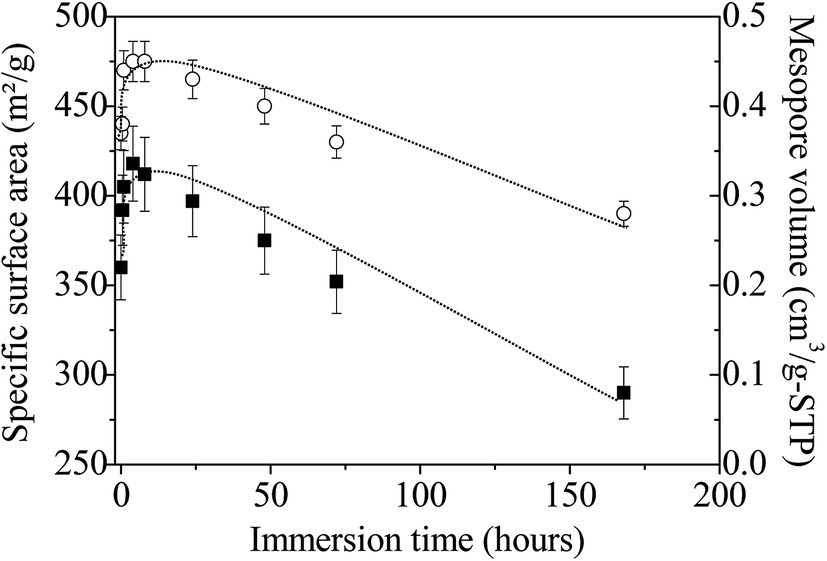 | ||
| Fig. 8 Evolution of the specific surface area (■) and pore volume (◯) of amorphous mesostructured ZrO2 as a function of the immersion time. Lines are just a guide for the eyes. | ||
4. Conclusions
Mesostructured amorphous ZrO2 has been synthesized by a surfactant templating process that combined the evaporation-induced self-assembly method and the liquid crystal templating pathway in an alcoholic acidic media. Nitric acid (HNO3) is used to control the reactivity of zirconium propoxide, used as zirconia precursor. However, the combined effect of NO3− and propanol on pluronic P123 behavior in solution likely prevents the complete reconstitution of the hexagonal liquid crystal phase during the evaporation step and, as a result, no ordered mesostructure can be obtain. Under the optimal conditions, which correspond to a 50 wt% of P123 concentration in the acidic solution and a P123/Zr(Opr)4 molar ratio of 0.024, a wormhole-like arrangement of the mesopores is recovered. The efficiency of the surfactant removal by ethanol extraction during 12 hours has been demonstrated by infrared and Raman analyses. This suggests weak interactions between the P123 pore templating agent and the hydrolyzed zirconia precursor in agreement with 13C and 1H MAS NMR results. These experiments also reveal the presence of propanol and/or unhydrolyzed propoxy group bonded to zirconium that may prevent the formation of an ordered mesostructure. However, 13C CPMAS NMR experiments suggest also the possible degradation of few PEO moieties of P123 upon acid induced hydrolysis.An in situ XRD temperature-dependent shows that the amorphous ZrO2 begins to crystallize in air from 420 °C into tetragonal zirconia. The monoclinic phase appears at 700 °C, and at 900 °C there is coexistence of the tetragonal and monoclinic structures. However, the material is amorphous when calcined at 440 °C under air in a furnace. Under these conditions, the crystallization begins at 480 °C without collapse of the mesostructure. The hydrothermal stability of the amorphous ZrO2 has also been investigated. The results obtained by SAXS and nitrogen adsorption desorption analyses show that these materials resist more than 72 hours in boiling water.
Conflicts of interest
There are no conflicts to declare.References
- Y. Inoue and H. Yamazaki, Bull. Chem. Soc. Jpn., 1987, 60, 891 CrossRef CAS.
- S. Jaenicke, G. K. Chuah, V. Raju and Y. T. Nie, Catal. Surv. Asia, 2008, 12, 153 CrossRef CAS.
- R. Srinivasan and B. H. Davis, in Materials Synthesis and Characterization, ed. D. L. Perry, Springer Science+Business Media, New York, 1997, pp. 147–188 Search PubMed.
- C. Zhang, Langmuir, 2009, 25, 7078 CrossRef CAS PubMed.
- C. Lin, C. Zhang and J. Lin, J. Phys. Chem. C, 2007, 111, 3300 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017 RSC.
- H. Xian, Q. Wang, G. Yu, H. Wang, Y. Li, Y. Wang and T. Li, Appl. Catal., A, 2019, 581, 116 CrossRef CAS.
- H. Jahangiri, A. Osatiashtiani, J. A. Bennett, M. A. Isaacs, S. Gu, A. F. Leed and K. Wilson, Catal. Sci. Technol., 2018, 8, 1134 RSC.
- Q. Cai, J. A. Lopez-Ruiz, A. R. Cooper, J. G. Wang, K. O. Albrecht and D. Mei, ACS Catal., 2018, 8, 488 CrossRef CAS.
- G. X. Yan, A. Wang, I. E. Wachs and J. Baltrusaitis, Appl. Catal., A, 2019, 572, 210 CrossRef CAS.
- V. Adeeva, H. Y. Liu, B. Q. Xu and W. M. H. Sachtler, Top. Catal., 1998, 6, 61 CrossRef CAS.
- M. Breysse, P. Afanasiev, C. Geantet and M. Vrinat, Catal. Today, 2003, 86, 5 CrossRef CAS.
- J. Mazurelle, C. Lamonier, C. Lancelot, E. Payen, C. Pichon and D. Guillaume, Catal. Today, 2008, 130, 41 CrossRef CAS.
- E. O. Orozco and M. Vrinat, Appl. Catal., A, 1998, 170, 195 CrossRef.
- Y. Ji, P. Afanasiev, M. Vrinat, W. Li and C. Li, Appl. Catal., A, 2004, 257, 157 CrossRef CAS.
- M. Breysse, J. L. Portefaix and M. Vrinat, Catal. Today, 1991, 10, 489 CrossRef CAS.
- D. Hamon, M. Vrinat, M. Breysse, B. Durand, F. Beauchesne and T. des Courieres, Bull. Soc. Chim. Belg., 1991, 100, 933 CrossRef CAS.
- J. A. Knowles and M. J. Hudson, Chem. Commun., 1995, 20, 2083 RSC.
- S. Reddy and A. Sayari, Catal. Lett., 1996, 38, 219 CrossRef.
- A. Kim, P. Bruinsma, Y. Chen, L. Q. Wang and J. Liu, Chem. Commun., 1997, 2, 161 RSC.
- G. Pacheco, E. Zhao, A. Garcia, A. Sklyarov and J. J. Fripiat, J. Mater. Chem., 1998, 8, 219 RSC.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. Stucky, Chem. Mater., 1999, 11, 2813 CrossRef CAS.
- J. L. Blin, R. Flamant and B. Su, Int. J. Inorg. Mater., 2001, 3, 959 CrossRef CAS.
- B. Tian, H. Yang, X. Liu, S. Xie, C. Yu, J. Fan, B. Tu and D. Zhao, Chem. Commun., 2002, 17, 1824 RSC.
- Y. Y. Huang, T. J. McCarthy and W. M. H. Sachtler, Appl. Catal., A, 1996, 148, 135 CrossRef CAS.
- U. Ciesla, S. Schacht, G. D. Stucky, K. K. Unger and F. Schiith, Angew. Chem., Int. Ed. Engl., 1996, 35, 541 CrossRef CAS.
- Y. Chen, L. Y. Jang and S. Cheng, J. Phys. Chem. B, 2006, 110, 11761 CrossRef PubMed.
- T. Kimura, Chem. Rec., 2016, 16, 445 CrossRef CAS PubMed.
- G. S. Attard, J. C. Glyde and C. G. Göltner, Nature, 1995, 378, 366 CrossRef CAS.
- S. A. El-Safty, Y. Kiyozumi, T. Hanaoka and F. Mizukami, J. Phys. Chem. C, 2008, 112, 5476 CrossRef CAS.
- K. Zimny, J. L. Blin and M. J. Stébé, J. Phys. Chem. C, 2009, 113, 11285 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CAS.
- S. S. Soni, G. Brotons, M. Bellour, T. Narayanan and A. Gibaud, J. Phys. Chem. B, 2006, 110, 15157 CrossRef CAS PubMed.
- K. Assaker, I. Naboulsi, M. J. Stébé, M. Emo and J. L. Blin, J. Colloid Interface Sci., 2015, 446, 170 CrossRef CAS PubMed.
- K. Zimny, C. Carteret, M. J. Stébé and J. L. Blin, J. Phys. Chem. C, 2012, 116, 6585 CrossRef CAS.
- D. A. Ward and E. I. Ko, Chem. Mater., 1993, 5, 956 CrossRef CAS.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242 CrossRef PubMed.
- M. M. Dubinin, in Progress in Surface and Membrane Science, ed. D. A. Cadenhead, Academic Press, New York, 1975, vol. 9, p. 1 Search PubMed.
- H. Schott, J. Colloid Interface Sci., 1997, 189, 117 CrossRef CAS.
- R. Zangi, J. Phys. Chem. B, 2010, 114, 643 CrossRef CAS PubMed.
- B. Bharatiya, G. Ghosh, P. Bahadur and J. Mata, J. Dispersion Sci. Technol., 2008, 29, 696 CrossRef CAS.
- B. A. Deyerle and Y. Zhang, Langmuir, 2011, 27, 9203 CrossRef CAS PubMed.
- Nonionic Surfactant Physical Chemistry, ed. M. J. Schick, Surfactant Science Series, M. Dekker, New York, 1987, vol. 23 Search PubMed.
- M. Tomsic, M. Bester-Rogac, A. Jamnik, W. Kunz, D. Touraud, A. Bergmann and O. Glatter, J. Colloid Interface Sci., 2006, 294, 194 CrossRef CAS PubMed.
- K. W. Kwon, M. J. Park, J. Hwang and K. Char, Polym. J., 2001, 33, 404 CrossRef CAS.
- B. Bharatiya, C. Guo, J. H. Ma, P. A. Hassan and P. Bahadur, Eur. Polym. J., 2007, 43, 1183 CrossRef.
- R. G. Alany, T. Rades, S. Agatonovic-Kustrin, N. M. Davies and I. G. Tucker, Int. J. Pharm., 2000, 196, 141 CrossRef CAS PubMed.
- K. Aramaki, U. Olsson, Y. Yamaguchi and H. Kunieda, Langmuir, 1999, 15, 6226 CrossRef CAS.
- H. M. Kao, C. C. Cheng, C. C. Ting and L. Y. Hwang, J. Mater. Chem., 2005, 15, 2989 RSC.
- T. W. Kim, F. Kleitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601 CrossRef CAS PubMed.
- S. Liu, P. Cool, O. Collart, P. Van Der Voort, E. F. Vansant, O. I. Lebedev, G. Van Tendeloo and M. Jiang, J. Phys. Chem. B, 2003, 107, 10405 CrossRef CAS.
- H. Fang, L. Zhang, W. H. Shi and T. L. Wan, J. Non-Cryst. Solids, 2006, 352, 2279 CrossRef CAS.
- J. L. Blin, N. Du and M. J. Stébé, J. Colloid Interface Sci., 2012, 373, 34 CrossRef CAS PubMed.
- A. Ayral, T. Assih, M. Abenoza, J. Phalippou, E. Lecomte and A. Dauger, J. Mater. Sci., 1990, 25, 1268 CrossRef CAS.
- B. B. Tatykaev, M. M. Burkitbayev, B. M. Uralbekov and F. Kh. Urakaev, Acta Phys. Pol., A, 2014, 126, 1044 CrossRef CAS.
- F. Bérubé and S. Kaliaguine, Microporous Mesoporous Mater., 2008, 115, 469 CrossRef.
- B. Dragoi, G. Laurent, S. Casale, T. Benamor, B. Lebeau, C. Boissière, F. Ribot, M. Selmane, P. Schmidt, D. Kreher and A. Davidson, J. Sol-Gel Sci. Technol., 2019, 91, 552 CrossRef CAS.
- T. Lana-Villarreal, J. M. Pérez and R. Gómez, C. R. Chim., 2006, 9, 806 CrossRef CAS.
- B. Yang, C. Guo, S. Chen, J. Ma, J. Wang, X. Liang, L. Zheng and H. Liu, J. Phys. Chem. B, 2006, 110, 23068 CrossRef CAS PubMed.
- M. Mesa, L. Sierra, J. Patarin and J. L. Guth, Solid State Sci., 2005, 7, 990 CrossRef CAS.
- S. Sivadevi, S. Selvasekarapandian, S. Karthikeyan, N. Vijaya, F. Kingslin Mary Genova, C. Sanjeeviraja, H. Nithya and I. J. Kawamura, International Journal of Electroactive Materials, 2013, 1, 64 Search PubMed.
- C. Schild, A. Wokaun, R. A. Koppel and A. Baiker, J. Catal., 1991, 130, 657 CrossRef CAS.
- R. Srinivasan and B. H. Davis, J. Am. Ceram. Soc., 1992, 75, 1217 CrossRef CAS.
- A. Léonard, J. L. Blin and B. L. Su, Colloids Surf., A, 2004, 241, 87 CrossRef.
- F. Michaux, C. Carteret, M. J. Stébé and J. L. Blin, Microporous Mesoporous Mater., 2008, 116, 308 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TG and heat-flow curves of the hybrid mesophase and EtOH-extracted mesostructured ZrO2 (S1). 1H–13C CPMAS NMR spectra of as-synthesized (A) and dehydrated at 70 °C (B) ZrO2 hybrid mesophase (S2). Raman spectra of pluronic P123 (A) and ZrO2 hybrid mesophase (B) (S3). 1H MAS NMR spectra of as-synthesized (A) and dehydrated at 70 °C (B) ZrO2 hybrid mesophase (S4). Raman spectrum of amorphous mesostructured ZrO2 recovered after surfactant extraction (S5). SAXS pattern (A), nitrogen adsorption–desorption isotherm (B) and mesopores size distribution (C) of ZrO2 after calcination at 480 °C under air atmosphere in a furnace (S6). Evolution as a function of the immersion time of the specific surface area (■) and pore volume (○) of amorphous ZrO2 after calcination at 440 °C under air atmosphere in a furnace. Lines are just a guide for the eyes (S7). See DOI: 10.1039/d0ra04824k |
| This journal is © The Royal Society of Chemistry 2020 |