
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully inkjet-printed multilayered graphene-based flexible electrodes for repeatable electrochemical response†
Twinkle Pandhi a,
Casey Cornwellb,
Kiyo Fujimotoa,
Pete Barnesa,
Jasmine Coxc,
Hui Xionga,
Paul H. Davisa,
Harish Subbaramanc,
Jessica E. Koehned and
David Estrada*ae
a,
Casey Cornwellb,
Kiyo Fujimotoa,
Pete Barnesa,
Jasmine Coxc,
Hui Xionga,
Paul H. Davisa,
Harish Subbaramanc,
Jessica E. Koehned and
David Estrada*ae
aMicron School of Materials Science and Engineering, Boise State University, Boise, ID 83725-2090, USA. E-mail: daveestrada@boisestate.edu
bDepartment of Chemistry, Northwest Nazarene University, Nampa, ID 83686, USA
cDepartment of Electrical and Computer Engineering, Boise State University, Boise, ID 83725-2075, USA
dNASA Ames Research Center, Moffett Field, CA 94035, USA
eCenter for Advanced Energy Studies, Boise State University, Boise, ID 83725-1012, USA
First published on 16th October 2020
Abstract
Graphene has proven to be useful in biosensing applications. However, one of the main hurdles with printed graphene-based electrodes is achieving repeatable electrochemical performance from one printed electrode to another. We have developed a consistent fabrication process to control the sheet resistance of inkjet-printed graphene electrodes, thereby accomplishing repeatable electrochemical performance. Herein, we investigated the electrochemical properties of multilayered graphene (MLG) electrodes fully inkjet-printed (IJP) on flexible Kapton substrates. The electrodes were fabricated by inkjet printing three materials – (1) a conductive silver ink for electrical contact, (2) an insulating dielectric ink, and (3) MLG ink as the sensing material. The selected materials and fabrication methods provided great control over the ink rheology and material deposition, which enabled stable and repeatable electrochemical response: bending tests revealed the electrochemical behavior of these sensors remained consistent over 1000 bend cycles. Due to the abundance of structural defects (e.g., edge defects) present in the exfoliated graphene platelets, cyclic voltammetry (CV) of the graphene electrodes showed good electron transfer (k = 1.125 × 10−2 cm s−1) with a detection limit (0.01 mM) for the ferric/ferrocyanide redox couple, [Fe(CN)6]−3/−4, which is comparable or superior to modified graphene or graphene oxide-based sensors. Additionally, the potentiometric response of the electrodes displayed good sensitivity over the pH range of 4–10. Moreover, a fully IJP three-electrode device (MLG, platinum, and Ag/AgCl) also showed quasi-reversibility compared to a single IJP MLG electrode device. These findings demonstrate significant promise for scalable fabrication of a flexible, low cost, and fully-IJP wearable sensor system needed for space, military, and commercial biosensing applications.
Introduction
Graphene has been used for many electrochemical applications such as in fuel cells, electric double-layer capacitors, and even in lithium-ion batteries.1–5 So far research has been conducted for graphene oxide electrodes, screen printed graphene electrodes, and IJP graphene electrodes modified with PEDOT-PSS or polyaniline, but fully IJP printed bare graphene-based electrodes with high stability, sensitivity, and repeatability have not been developed.2,6–18 Conventional fabrication processes for sensor development, such as vacuum deposition, photolithography, and epitaxial growth of electronic materials, tend to be complicated and expensive, often requiring lithographic patterning and high-temperature processing.19 As a result, additive electronics manufacturing techniques, such as inkjet printing (IJP), aerosol jet printing (AJP), and micro-dispense printing (MDP), are being explored as potential low-cost scalable fabrication methods for flexible sensor systems.20–24 Previous studies have demonstrated that inkjet printing, a drop-on-demand process, eliminates the need for the prefabricated masks or stencils required for lithographic and contact-printing processes.25–28 An inkjet-printed ion-selective single layer reduced graphene oxide-based sensor by Claussen et al. demonstrated a wide sensing range and low detection limits.29 However, such studies involved rigid substrates and high annealing temperatures that are not compatible with flexible substrates or included lithographic processes in the overall device design.Over the last two decades, carbon-based materials such as graphene or functionalized/doped graphene, glassy carbon, carbon nanotubes (CNTs), fullerenes, and boron-doped diamond have been investigated for their use as electrodes in electrochemical sensing due to their advantageous structural and electrical properties.13,18,30–36 As each carbon allotrope possesses a unique structure and surface chemistry, the electrochemical behavior of each is also unique. For electrochemical applications, highly ordered pyrolytic graphite, pseudographite, graphene, and orientated CNTs are excellent candidates due to their high conductivity, large surface area, and unique heterogeneous electron transfer rates.32,35,37
Graphene, a monolayer of sp2 hybridized carbon in a 2-dimensional hexagonal lattice structure, has received much attention in the research community due to its unique physical and chemical properties. The sp2 bonding between the carbon atoms in graphene creates three σ-bonds, which are responsible for its high mechanical strength and high in-plane thermal conductivity.38–42 Graphene's remarkable conductivity is associated with overlapping pz orbitals above and below the molecular plane, which creates a delocalized π – electron system to allow for free movement of electrons. These unique bonding characteristics give rise to a linear band structure with a zero-band gap near the K and K′ points, leading to graphene's high electrical conductivity.39,43 Moreover, graphene makes for an excellent candidate for electrochemical applications due to its high conductivity, large surface area, unique heterogeneous electron transfer rate, and low production cost. The edge plane and basal plane-defect sites of the highly ordered pyrolytic graphite greatly favors electrochemical activity.2,18 Three common techniques used to obtain graphene are exfoliation, chemical vapor deposition (CVD), and epitaxial growth. While these are widely used techniques, they are known to introduce defects to the graphene structure that are detrimental to electrical and thermal transport properties, while conversely improving chemical and electrochemical sensitivity.1,5,42,44–48
The method of fabricating electrochemical graphene sensors is vital in creating edge and basal plane defects to improve chemical sensitivity. Work such as Banerjee et al. reported ultrahigh electrochemical current densities for graphene edges embedded in dielectric nanopores.1 Yuan et al. further demonstrated that the electrochemical activity on the edge states of single-layer CVD grown graphene is higher than on the basal plane.49 Shang et al. showed that increased graphitic edge and basal plane defects in CVD grown multilayer graphene resulted in superior electron transfer kinetics compared to the edge state of highly ordered pyrolytic graphite.50 Fisher et al. used microwave plasma-enhanced chemical vapor deposition grown multilayered graphene petal nanosheets to develop a versatile glucose sensor on a silicon wafer with high sensitivity, selectivity, and stability.51 Furthermore, Tang et al. showed excellent electrocatalytic activity for reduced graphene oxide sheets synthesized by chemical exfoliation and cast onto a glassy carbon electrode.52 We chose a high yield, solvent assisted exfoliation method to synthesize multilayer graphene to retain desirable edge and basal-plane defects that promote electrochemical activity. Moreover, it has been previously shown that the annealing conditions, ink properties, and number of print passes impact the electrical and structural porosity of printed graphene microstructures.53 Such porosity in functionalized graphene electrodes has a significant impact on enhancing the electrochemical performance as well.54
In this report, the electrochemical performance of IJP MLG electrodes and fully IJP three-electrode sensors is investigated by measuring the cyclic voltammetry response of a ferric/ferrocyanide redox couple and by performing pH sensitivity studies. Additionally, the effect of electrode porosity is examined with a comparison between the electrochemical performance of MLG electrodes having different porosities as a result of the printing process. It is observed that the structure–property–processing correlations of fully additively manufactured graphene-based electrochemical electrodes are essential factors in improving consistency, repeatability, and uniformity of such fully printed sensor systems. Finally, the IJP MLG electrodes are shown to exhibit robust electrochemical performance over 1000 bend cycles, highlighting the attractive properties and behavior of IJP MLG electrodes for use in wearable electroanalysis. Advances such as this will further enable additive electronics manufacturing of flexible sensors for human performance monitoring in space, military, and commercial applications.
Results and discussion
Characterization of multilayer graphene (MLG) ink
The multilayer graphene ink (MLG) was formulated via solvent assisted exfoliation of bulk graphite powder using a process reported previously.53 In Fig. S1,† we see the characteristic of the individual graphene flakes with TEM (Fig. S1c†) and Raman spectroscopy (inset Fig. S1b†). Raman spectroscopy reveals the characteristic of D, G, and 2D peaks for graphene at 1350 cm−1, 1580 cm−1, and 2700 cm−1, respectively. The ratio of the D/G peak intensities (ID/IG) determines the graphene flakes' quality (defect/disorder). The ID/IG peak ratio of 0.24 is lower than previously reported values (0.33–0.7), suggesting the exfoliated flakes are of higher quality with fewer defects.55 TEM images show the graphene flakes vary in lateral size from ∼50–300 nm. To correlate the TEM and Raman data, the ID/IG peak ratio and 532 nm excitation wavelength was used in Cancado's general equation:56La (nm) = (2.4 × 10−10) 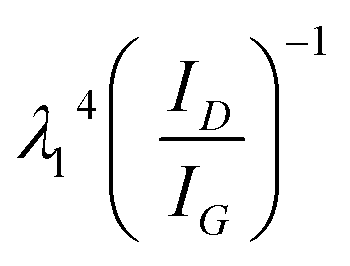 to extract the crystal size (La ≈ 102 nm) of the graphene flakes. AFM characterization of the flakes shows the thickness (tg) ranges from monolayer to flakes with an average thickness of tg = 16 nm ±15 nm (Fig. S1d†).
to extract the crystal size (La ≈ 102 nm) of the graphene flakes. AFM characterization of the flakes shows the thickness (tg) ranges from monolayer to flakes with an average thickness of tg = 16 nm ±15 nm (Fig. S1d†).
This unique process results in a highly concentrated ink compatible with a variety of print modalities.53 Specifically, the layered graphene/ethyl cellulose (EC) flakes were dispersed in a mixture of 85% cyclohexanone and 15% terpineol to yield a viscosity of ∼8 cP, as measured with a Wells-Brookfield Cone/Plate viscometer at an ink concentration of 3.5 mg mL−1. To prepare stable and homogeneous MLG ink, one of the most important parameters is to optimize the ink viscosity. Viscosities and concentrations in this range have shown to be compatible with the ultrasonic atomizer of an aerosol jet printer (AJP) and the Dimatix DMP 2850 materials inkjet printer (IJP).57 Viscosity higher than 10 cP results in the printer's clogging and too low of value (<5 cP) doesn't allow to form a droplet. Furthermore, the ink's viscosity is greatly affected by the concentration and dispersing agents.58 So, it is essential to carefully control the ink fabrication process as it can greatly affect the printability and quality of the printed patterns.
Microstructural and electrical characterization was performed on multilayered graphene (MLG) printed lines with silver contact pads in a transmission line measurement (TLM) structure with varying numbers of print passes (15–30, in increments of 5 passes) using a Dimatix inkjet printer (seen in Fig. 1a). To minimize interference due to excessive charging and fluorescence from the large surface roughness of Kapton HN substrates, the MLG and silver TLM structures for microstructural and electrical characterization were printed on glass instead. The tool platen temperature, nozzle diameter, and cartridge temperature were optimized to ensure that the dimensions and material deposition were adequate to obtain uniform structures. Scanning electron microscopy (SEM) of a line with 25 print passes of IJP multilayered graphene (Fig. 1b – left), demonstrated good uniformity of the printed layer. Transmission electron microscopy (TEM) (Fig. 1b – right) showed multilayer graphene flakes ranging in thickness from 5–20 nm layers. The surfactant ethyl cellulose (EC) stabilized the graphene flakes in the solution but required decomposition after printing to achieve optimal electrical conductivity. Thermal gravimetric analysis (TGA) was employed to analyze the thermal stability of the EC coated graphene flakes. Fig. 1c shows weight percent (black) and differential scanning calorimetry (blue) as a function of temperature. The decomposition peak of EC occurs around 250 °C, while the decomposition temperature of the solvents is seen around 350 °C. Using the TGA, the graphene printed structures were then annealed in two stages: first at 250 °C for 30 minutes to evaporate the surfactant, and subsequently at 350 °C for another 30 minutes to remove the remaining solvents, leading to enhanced electrical conductivity. The silver contacts were deposited via IJP on top of the graphene in a TLM structure using commercially available 40 wt% silver ink.
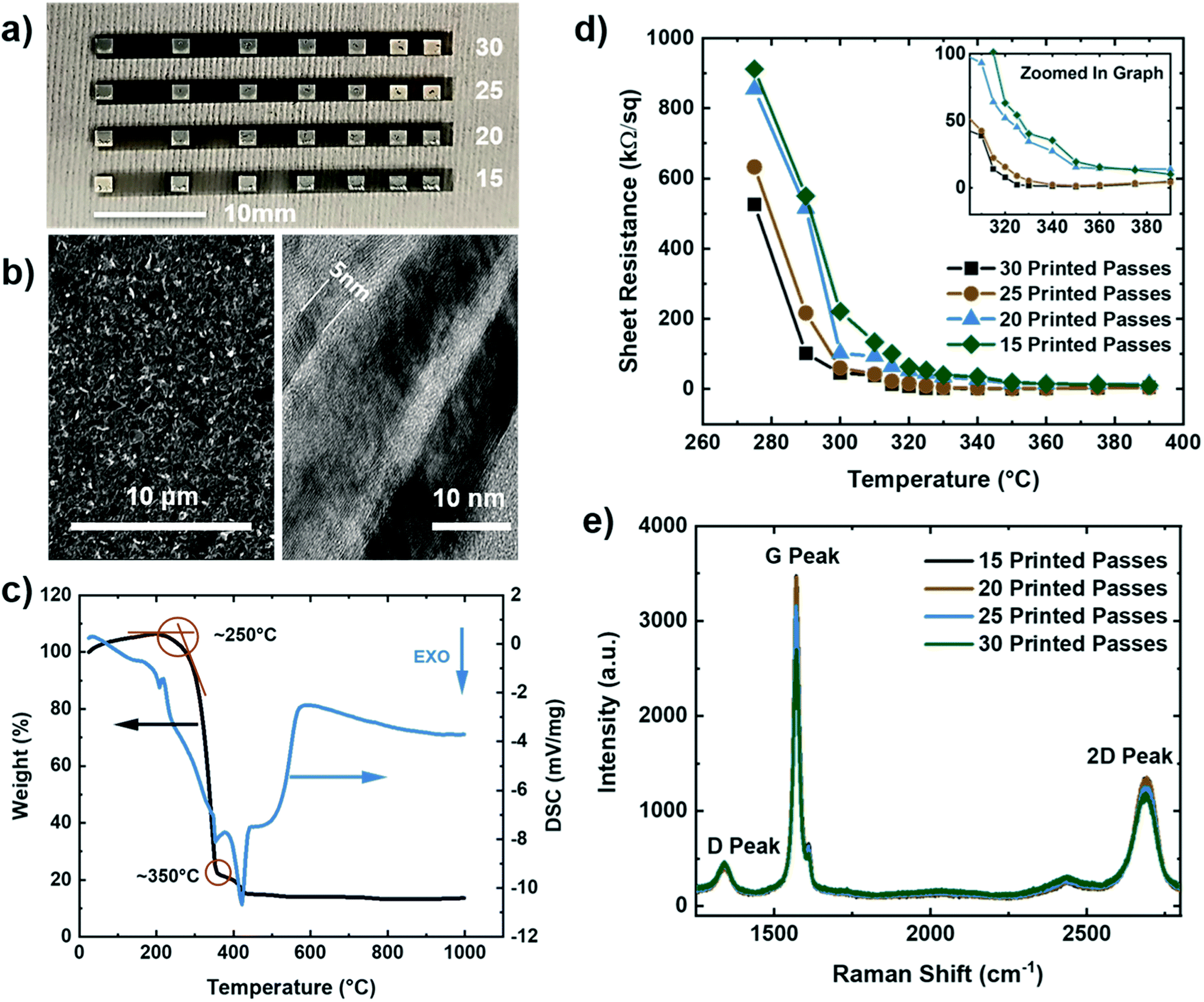 | ||
| Fig. 1 Inkjet printed graphene layer characterization. (a) Optical image of inkjet printed graphene (15–30 printed passes) structure on glass. (b) SEM image of the 25-pass printed line (left) and TEM image of the multilayer graphene flakes (right) on Kapton HN. (c) TGA and DSC data of the graphene/ethyl cellulose ink. (d) Sheet resistance vs. temperature for 15 to 30 printed passes on glass. (e) Typical Raman spectra (532 nm excitation) for 15 to 30 printed passes MLG films. | ||
To measure the electrical properties of the printed interconnects, a 2-point probe (Keithley 4200 SCS, Textronix, Beaverton, OR) measurements on the TLM structure were conducted. Fig. 1d shows the calculated sheet resistance as a function of annealing temperature for 15–30 print passes. Based on the TLM measurements, the lowest sheet resistance was calculated to be 0.89 kΩ sq−1, and 1.60 kΩ sq−1 for 30 and 25 print passes, respectively, at an annealing temperature of 350 °C. Moreover, Raman spectroscopy results are shown in Fig. 1e reveal the graphitic nature of our electrodes through the characteristic D, G, and 2D peaks for the IJP graphene layers at 1350 cm−1, 1580 cm−1, and 2700 cm−1, and I2D/IG peak ratios in the range of 0.38–0.43.
Electrochemical response of MLG electrodes
To investigate our IJP MLG electrode's electrochemical properties, the synthesized MLG was printed into 1 cm × 1 cm squares (15, 20, 25, and 30 printed passes) with silver contact pads (1 printed layer) and SU-8 (3 printed passes) as a passivation layer for the silver electrode. All layers were printed by IJP on 2 mil thick untreated Kapton HN film (Fig. 2a and b). Before printing MLG ink on Kapton, contact angle (CA) measurements of MLG inks on a Kapton HN substrate were performed to ensure wettability. In Fig. 2c, we present a low CA of 15.6°, suggesting good wettability of MLG ink on untreated Kapton HN substrates. An example of the flexible and fully printed MLG electrode with 25-layer print passes on Kapton HN is seen in Fig. 2d.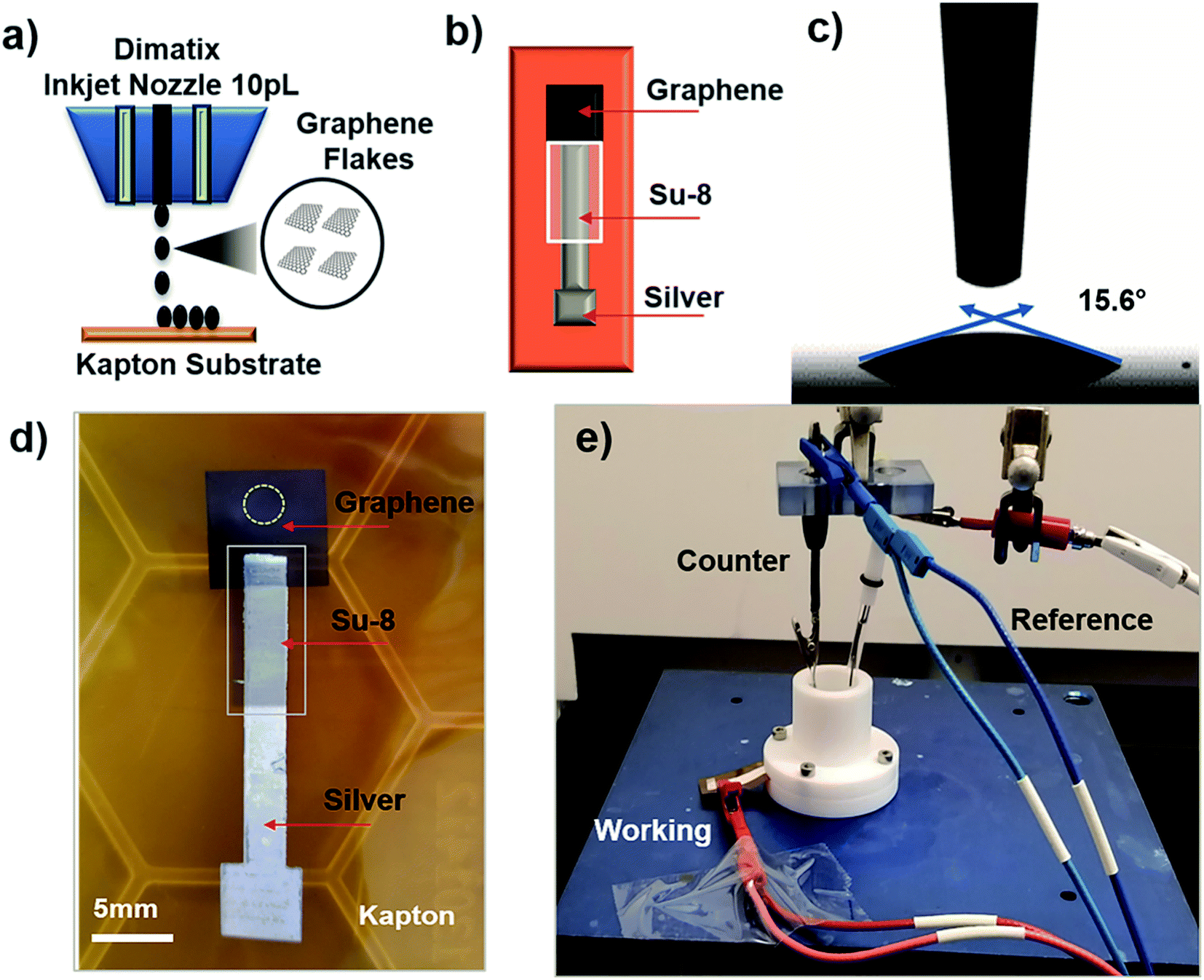 | ||
| Fig. 2 (a) Sketch of the Dimatix inkjet printer printing graphene on Kapton. (b) Design and layers of the graphene electrode. (c) Contact angle measurements of graphene ink on Kapton. (d) Optical image of the printed graphene electrode, where the yellow ring indicates the surface area of the electrode exposed to the electrolyte. (e) Photograph of the electrochemical experimental setup of the graphene electrode in aqueous ferricyanide redox couple with Ag/AgCl as the reference electrode and platinum wire as the counter electrode. | ||
The experimental setup to study the electrochemical response of printed MLG electrodes is shown in Fig. 2e, like the setups used by Munoz and Richter.59–61 Cyclic voltammetry (CV) measurements were carried out with a Bio-Logic VMP-300 potentiostat with scan rates from 10–100 mV s−1 in 1 mM K3[Fe(CN)6] with 1 M KCl as the supporting electrolyte, at room temperature for MLG working electrodes made by 15 to 30 printed passes (ESI Fig. S2a, b† and 3a, b). Ferro/ferri cyanide redox couple is often used in physiological experiments because of its sensitivity to relatively small surface changes. The iron is low spin and quickly reduce to the ferric/ferrocyanide Fe(CN)6]−3/−4.13 For this experiment, the electrochemical cell is comprised of an IJP MLG working electrode, a standard platinum wire counter electrode and a conventional aqueous Ag/AgCl/KCl (sat.) reference electrode with saturated (sat.) KCl solution from SYC Technologies. A ∼0.07 cm2 circular surface area of IJP MLG is defined by the size of the O-ring in the liquid cell. Studies were performed with the 25 and 30 printed pass electrodes due to their superior electrochemical performance. The observed electrochemical behavior was evaluated, as can be seen from the CV curves in Fig. 3, distinct redox peaks can be observed. The 25 printed passes of MLG electrode exhibits quasi-reversible CV characteristics with a peak to peak separation ∼60 mV at a scan rate of 10 mV s−1 in a 1 mM K3[Fe(CN)6] electrolyte. This peak to peak separation (i.e., near-ideal ΔEp of 59 mV for Nernstian reactions) is evidence of the fast electrode kinetics, while shifts in peak to peak separation with an increase in scan rate point to the electrodes' quasi-reversible nature.
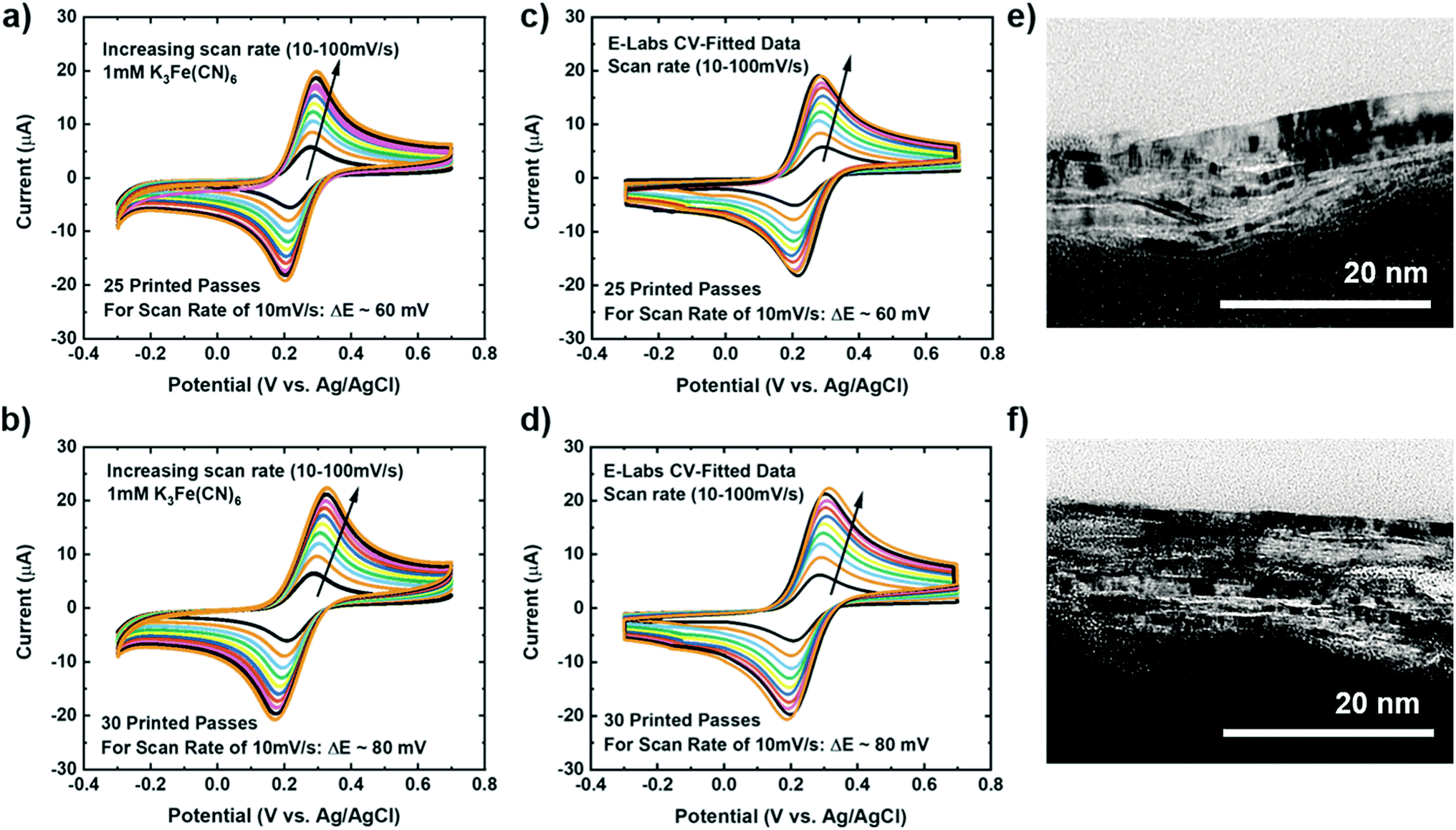 | ||
| Fig. 3 Graphene electrode cyclic voltammetry (CV) scan rate data for 1 mM K3[Fe(CN)6] in 1 M KCl as the supporting electrolyte with increasing scan rate 10–100 mV s−1 for (a) 25 printed passes of graphene and (b) 30 printed passes of graphene. E-labs CV-Sim fitted data for (c) 25 printed passes of graphene and (d) 30 printed passes of graphene. Cross-section TEM images of (e) 25 printed passes graphene and (f) 30 printed passes graphene. | ||
The kinetics (extracted dimensionless coefficient φ) of our IJP MLG electrode (25.14) is compared with that of other comparable graphene-based electrodes and inkjet-printed electrodes in Table 1. Electrodes shaded in pink shows functionally of a fully printed three electrode sensor system. Our electrode shows much better reversibility, stability, and repeatability on a flexible substrate than the other electrodes. Our data suggest that IJP MLG electrodes possess well-defined structures and electrochemical properties to support fast kinetics, comparable to results presented in the literature for MLG.51 Although 30 printed passes of MLG shows a higher current and lower sheet resistance than 25 printed passes, a higher peak to peak separation of 80 mV (Fig. 3b) is observed. It is hypothesized that while the additional printed passes for the (30 printed passes sample) electrode increase uniformity and decrease resistance, they also create denser packing of the graphene flakes, thereby reducing the porosity and slowing the redox reaction on the electrode surface.
| Electrode material | Fabrication technique | Substrate | Electrolyte | Redox couple | Scan rate (mV s−1) | Ψ, EpΔ (59–200) mV | Ref. |
|---|---|---|---|---|---|---|---|
| Multi-layered graphene | Inkjet-printed | Kapton (polyimide) | 1 M KCL | Fe(CN)63−/4− (1 mM) | 10 | 25.14 | This work |
| Reduced graphene oxide | Laser-scribed | PET | 1 M KCl | Fe(CN)63−/4− (1 mM) | 10 | 25.14 | 66 |
| COOH-terminated graphene nanoflakes | Coated | Boron doped dimond | 0.1 M KH2PO4 | Fe(CN)63−/4− (0.5 mM) | 50 | 2.54 | 96 |
| Reduced graphene oxide | Coated | Glassy carbon electrode | 0.1 M KCl | Fe(CN)63−/4− (5 mM) | 30 | 2.54 | 97 |
| Gold | Inkjet-printed | Kapton (polyimide) | 100 mM KCl | Fe(CN)63−/4− (1 mM) | 100 | 1.71 | 98 |
| Reduced graphene oxide | Screen-printed | Poly(vinyl chloride) | 0.1 M KCl | Fe(CN)63−/4− (5 mM) | 50 | 1.61 | 15 |
| Edge-oxidized graphene nanosheet | Inkjet-printed | Kapton (polyimide) | 0.1 M KNO3 | Fe(CN)63−/4− (2 mM) | 50 | 1.61 | 99 |
| Carbon nanotubes | Inkjet-printed | Kapton (polyimide) | 0.1 M KCl | FcMeOH (4 mM) | 100 | 1.29 | 100 |
| CNT | Inkjet-printed | PET | 0.1 M KNO3 | FcMeOH (2 mM) | 25 | 1.28 | 101 |
| Gold | Inkjet-printed | Paper | 3 M KCl | Fe(CN)63−/4− (3 mM) | 20 | 0.75 | 102 |
| Graphite | Screen printed | Ultra-flexible polyester materials | 0.1 M KCl | Fe(CN)63−/4− (1 mM) | 10 | 0.60 | 103 |
| Reduced graphene oxide | Inkjet-printed | FTO (fluorine-doped tin oxide) TEC-15 | 0.1 M TBAPF6 | Co(bpy)3(B(CN)4)3 (1 mM) | 50 | 0.60 | 104 |
| Graphene–PEDOT:PSS | Inkjet-printed | Screen-printed carbon electrode/non-conformal | 0.1 M KCL | Fe(CN)63−/4− (0.36 μM) | 100 | 0.33 | 105 |
| Functionalized graphene nanoribbons | Screen-printed | Polyethylene glycol terephthalate | 0.2 M PBS | Fe(CN)63−/4− (1 mM) | 100 | 0.20 | 106 |
| Graphene–polyaniline | Inkjet-printed | PET | 0.1 M KCl | Fe(CN)63−/4− (1 mM) | 100 | 0.20 | 107 |
| Graphite pencil | Drawn | Paper | 0.1 M KCl | Fe(CN)63−/4− (1 mM) | 100 | 0.10 | 108 |
| Graphene with platinum (laser annealed) | Inkjet mask-less lithography | Screen-printed carbon electrode/non-conformal substrate | 1 M PBS | Fe(CN)63−/4− (5 mM) | 10 | EpΔ > 300 (mV vs. Ag/AgCl) | 109 |
| Reduced graphene/polylactic acid | 3D-printed | — | 0.1 M KCl | Fe(CN)63−/4− (1 mM) | 100 | ∼500 (mV vs. Ag/AgCl) | 110 |
| Reduced graphene oxide | Inkjet-printed/laser sintered | Cellulose-based paper | 1 M KCl | Fe(CN)63−/4− | 10 | ∼0.7 (V vs. Ag/AgCl) | 111 |
| Reduced graphene oxide | Inkjet-printed | Poly(ethylene 2,6-naphthalate) PEN | 0.1 M KCl | Fe(CN)63−/4− (1 mM) | 10 | ∼400 to 5000 (mV vs. Ag/AgCl) | 112 |
| PEDOT:PSS | Inkjet-printed | Paper | 0.1 M PBS | FcMeOH (−) | 20 | ∼50 (mV vs. Ag/AgCl) | 113 |
| Multi-walled nanotubes with silver | Inkjet-printed | Paper | 0.5 M KCl | Fe(CN)63−/4− (3 mM) | 20 | ∼55 (mV vs. Ag/AgCl) | 114 |
| Graphite | Screen-printed | Chromatography paper | 0.1 M H2SO4 | Fe(CN)63−/4− (1 mM) | 100 | ∼56 (mV vs. Ag/AgCl) after 30 scans | 115 |
For further analysis, the cathodic and anodic peak currents (Ipc and Ipa, respectively) from the CV data were plotted versus the square root of the scan rate (V s(−1/2)) for both 25 and 30 printed passes (ESI Fig. S3a and b†). The cathodic (Ipc) and anodic (Ipa) peak currents for 25 and 30 printed passes graphene showed excellent linear regression, suggesting redox reaction controlled by diffusion. CV simulation was used to extract the diffusion coefficient (D) and the rate constant (k) from the experimental CV data (Fig. 3a and b). This simulation provided identical CV curves compared to the data for 25 and 30 printed passes, respectively, as seen in Fig. 3c and d. In order to calculate the active surface area, we accounted for surface roughness extracted from the AFM images of 25 printed passes and 30 printed passes (seen in ESI Fig. S4†). To compare our data, we also used Randles–Sevcik equation to calculate the electrochemically active surface area. All the steps for these calculations are presented in the supplementary active surface area calculations. The active surface area extracted from AFM images of ∼0.086 cm2 and ∼0.084 cm2 compared to the calculated electrochemically active surface area of ∼0.091 cm2, and ∼0.093 cm2 for 25 printed passes 30 printed passes respectively, are in good agreement with 0.7% error. Using the calculated electrochemically active surface area, the diffusion coefficients are D = 4.17 × 10−6 cm2 s−1 for 25 printed passes and D = 6.38 × 10−6 cm2 s−1 for 30 printed passes, respectively. The calculated diffusion coefficient values are comparable with the ferric/ferrocyanide electrolyte reported in Konopka et al.62 Furthermore, the average electron transfer rate constants, (25 printed passes) k = 1.125 × 10−2 cm s−1 with average double layer capacitance of 43.4 μF and (30 printed passes) k = 7.34 × 10−3 cm s−1 with average double layer capacitance of 45.5 μF, where α = 0.5 (shows symmetric free energy curve, where the influence of applied voltage at transition state is about mid-way between reactants and products).63
To compare our extracted rate constant (k), we used the dimensionless kinetic parameter equation to estimate the heterogenous standard rate constant (k0).64
| φ = (−0.6288 + 0.0021x)/(1 − 0.017x) |
where D is the diffusion coefficient of the redox mediator (potassium ferricyanide is about 4.17 × 10−6 cm2 s−1), v is the scan rate (10 mV s−1), F is the Faraday constant, R is the gas constant, and T is the temperature (25 °C). The k0 of MLG was calculated as 2.38 × 10−2 cm s−1 close to our extracted value of k = 1.125 × 10−2 cm s−1.
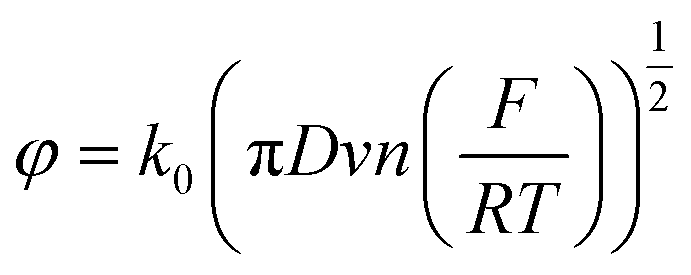
Furthermore, we conducted CV with ferrocene methanol (C11H12FeO) an outer sphere redox species, which is not sensitive to surface oxides and only depended on the density of states.65–67 Unlike [Fe(CN)6]−3/−4 which is an inner sphere redox system that is sensitive to changes on the surface. From the CV scans in Fig. S7† we see that the peak separation remines close to 65 mV at a scan rate of 10 mV s−1 similar suggesting the quasi-reversible electrode kinetics. Our MLG demonstrates good electrochemical response due to many edge sites available on the surface of the electrodes.
This suggests that 25 printed passes of MLG has slightly faster electron transfer kinetics than 30 printed passes, likely due to a higher surface roughness of 25 printed passes extracted from the AFM images in the (ESI Fig. S4a and b†). This electron transfer rate (k = 1.125 × 10−2 cm s−1) is close to or better than that of graphite, graphene oxide, mechanically exfoliated graphene, and screen-printed carbon or graphene or CNT electrode which range from 10−4 cm s−1 to 10−2 cm s−1 for the ferric/ferrocyanide redox reaction.5,68–73
Cross-sectional TEM was used to image the porosity of the printed MLG electrodes (25 printed passes and 30 printed passes) on untreated Kapton substrates and better understand the structure of IJP deposited MLG. From the cross-sectional TEM images seen in Fig. 3e and f, it is evident that the 25 printed passes (Fig. 3e) IJP MLG sheets are less dense than the 30 printed passes of MLG (Fig. 3f). It is seen that 25 printed passes of graphene exhibit a higher disordered stacking than the 30 printed pass case. A higher porosity between the stacked multilayers of graphene is observed in 25 printed passes of graphene than in the 30 printed passes. Surface porosity and packing morphology play a significant role in electrochemical performance, as established by Punckt et al.54 Moreover, using our CV data with varying scan rate (v), we can obtain the values of max current at v = 10 mV s−1 and v = 100 mV s−1 and calculate the porosity (P) according to the equation, 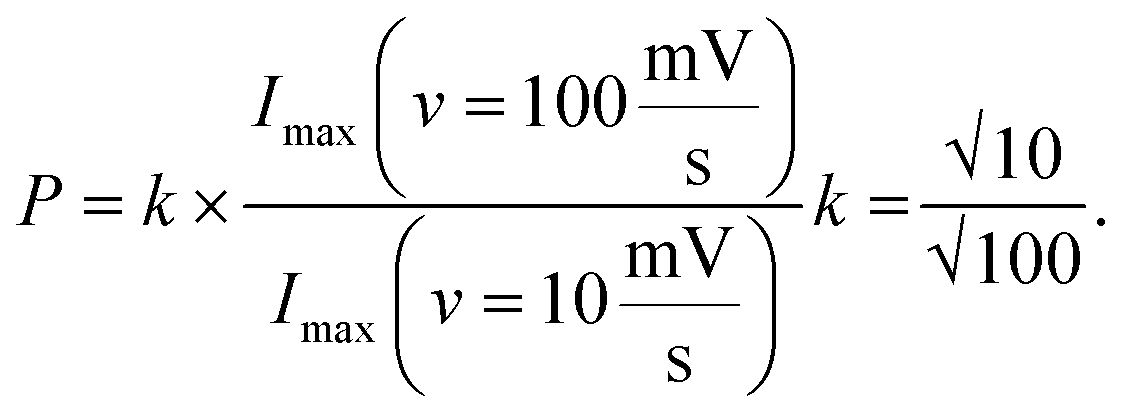 For an ideal planar electrode, P = 1 since then Imax ∼ (v)1/2. We find that for our IJP MLG electrodes, P = 1.17 for 25 printed passes and P = 1.06 for 30 printed passes. This suggests that 25 printed passes show more porosity than 30 printed passes, further supporting our hypothesis that enhanced electrocatalytic behavior is influenced by packing morphology in our printed graphene electrodes.74
For an ideal planar electrode, P = 1 since then Imax ∼ (v)1/2. We find that for our IJP MLG electrodes, P = 1.17 for 25 printed passes and P = 1.06 for 30 printed passes. This suggests that 25 printed passes show more porosity than 30 printed passes, further supporting our hypothesis that enhanced electrocatalytic behavior is influenced by packing morphology in our printed graphene electrodes.74
Stability of MLG electrodes
It is important that these electrodes are inherently stable in the electrolyte and can be reproduced via the printing methods, to enable biosensor design, optimization, and efficient collection of data in real-time.75 To investigate the stability of our MLG electrodes, CV measurements were carried out for a series of redox mediator couple dilutions between 1 mM to 5 mM K3[Fe(CN)6] with 1 M KCl as the supporting electrolyte. Scans were performed at room temperature for 25 (Fig. 4a–c) and 30 (Fig. 4d and e) printed passes with varying scan rate of 10 mV s−1 (Fig. 4a and d), 50 mV s−1 (Fig. 4b and e), and 100 mV s−1 (Fig. 4c and f). We observed that 25 printed passes showed a lower peak to peak separation for all three scan rates in each dilution as compared to 30 printed passes of MLG. Additionally, time-dependent effects were investigated by recording the CV curves at 100 mV s−1 every 5 min in the same electrolyte for up to 2 hours, similar to the study of Patel et al.76 These studies were carried out with 25 printed passes of MLG electrode in 1 mM ferric/ferrocyanide solution. Fig. 5a shows great stability with negligible change in the peak to peak potential separation, even after 16 hours in the electrolyte.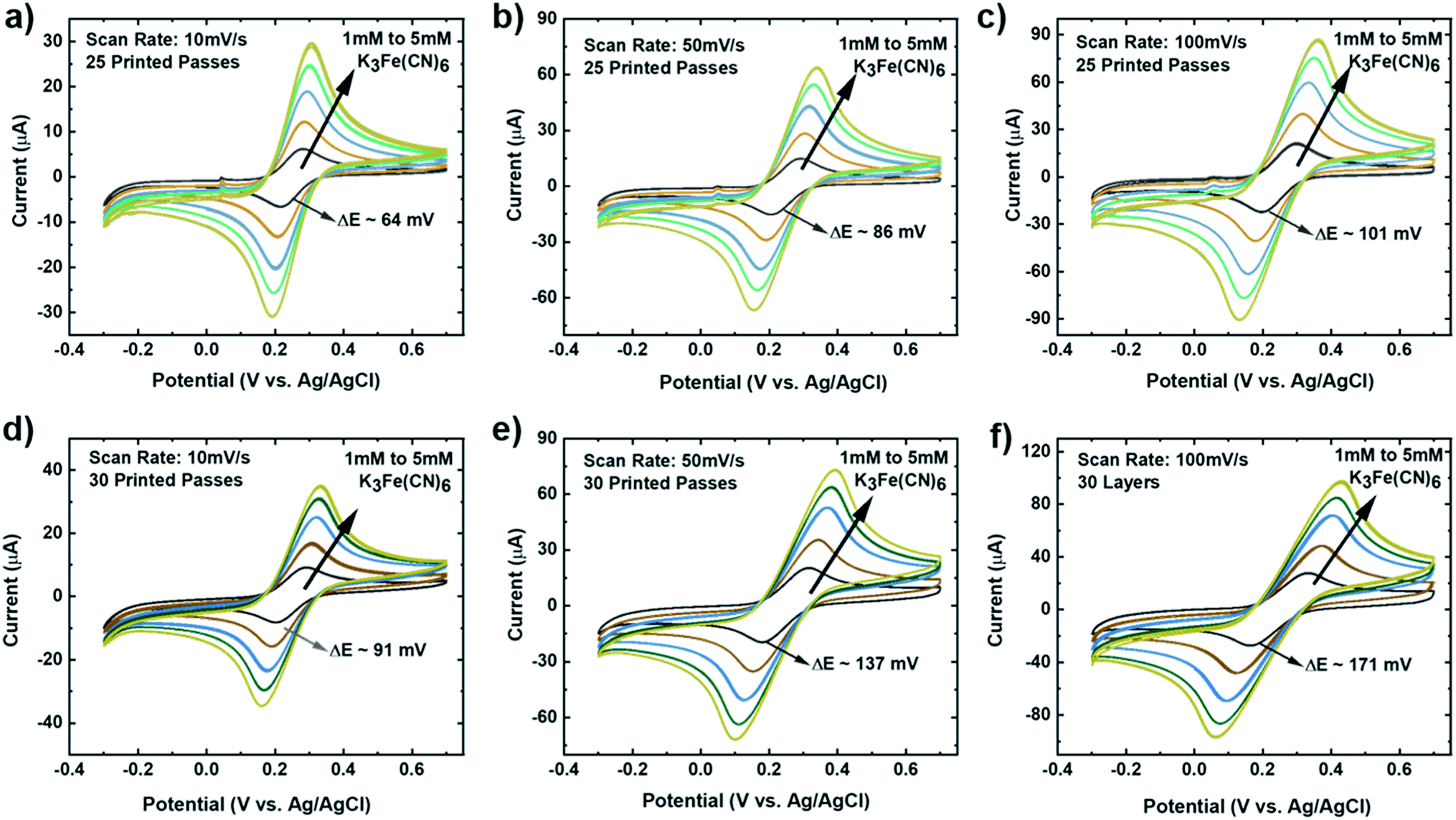 | ||
| Fig. 4 Graphene electrode CV dilution data for 1–5 mM K3[Fe(CN)6] in 1 M KCl as the supporting electrolyte for 25 printed passes at scan rates of (a) 10 mV s−1, (b) 50 mV s−1, and (c) 100 mV s−1 (d–f) are corresponding data for 30 printed passes, with scan rates of (d) 10 mV s−1, (e) 50 mV s−1, and (f) 100 mV s−1. | ||
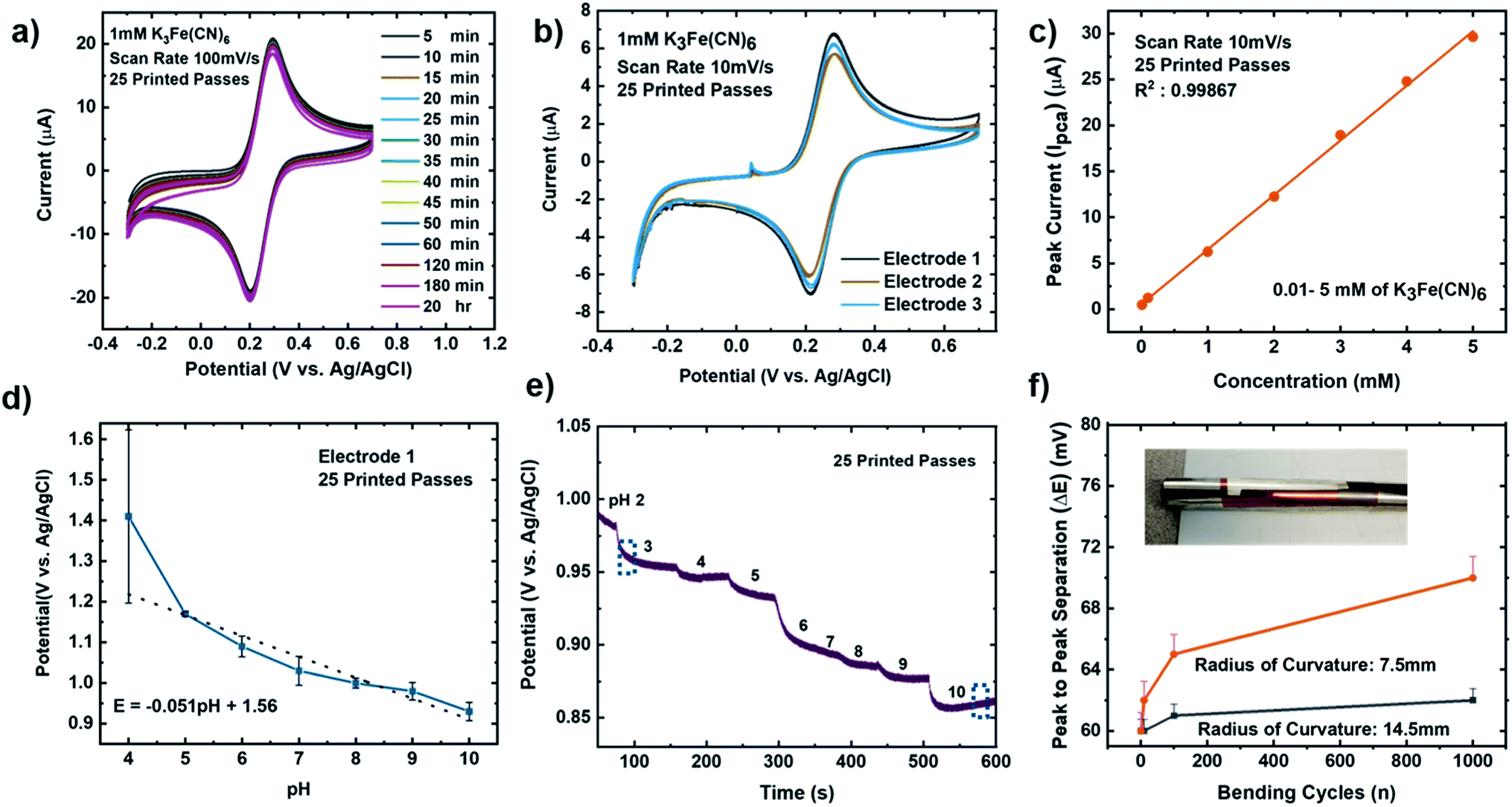 | ||
| Fig. 5 (a) Time-dependent CV scans for 25 IJP printed passes, 1 mM K3[Fe(CN)6] in 1 M KCl as the supporting electrolyte at a scan rate of 100 mV s−1. (b) CV repeatability data for 25 IJP printed passes (3 graphene electrodes), 1 mM dilution K3[Fe(CN)6] in 1 M KCl as the supporting electrolyte at a scan rate of 10 mV s−1. (c) CV dilution data showing peak current (Ipca) vs. concentration from 5 mM to 0.01 mM of K3[Fe(CN)6] in 1 M KCl as the supporting electrolyte for 25 IJP printed passes, one graphene electrode at scan rate from 10 mV s−1. (d) Static linear pH data vs. potential (potassium phosphate monobasic with sodium hydroxide commercial pH buffer solutions: 4–10 pH) using the 25 passes graphene printed electrode. Error bars represents the interelectrode standard variation in slope compared to the theoretical values based on the Nernst equation.93–95 (e) Time vs. potential data with changing pH from 2 to 10 for a single 25 printed passes graphene electrode. The error bar represents the standard deviation of potential across three independent samples. (f) Bending cycles (1, 10, 100, and 1000) conducted on the electrodes with radius of curvature either 7.5 mm (orange) or 14.5 mm (black) vs. peak to peak separation potential. | ||
The reproducibility of the MLG electrodes is demonstrated via the CV scans seen in Fig. 5b, showing a triplicate study with equivalent ink, printing, and other experimental and measurement conditions. The CV scans show consistent and reproducible results for all three electrodes, with a mean peak to peak potential separations of 64 ± 1 mV. To understand the sensing range of the MLG electrode, CV measurements were performed with varying concentrations of ferric/ferrocyanide solution from 10 mM to 1 μM. The bare MLG flexible electrode exhibits a broad sensing range from 10 mM down to 0.01 mM (Fig. 5c), which is comparable to the sensing range for non-flexible, modified graphene, reduced graphene oxide, or CNT electrodes shown in Table 1 and reported in the literature.8,9,11,77–84
MLG electrode pH sensitivity
The pH of a system is critical to chemical/biological/biochemical processes.78,80,85,86 It is also an essential factor for accurately determining the stability and sensitivity of a biosensor as biochemical reactions that take place on the sensor are highly dependent on pH. Potentiometric pH sensors can extract information about pH values by measuring the open circuit potentials.11 For this study, pH sensitivity experiments were conducted on bare MLG electrodes to observe the potentiometric response of the electrode as the pH was varied in the range of 1–10. First, static pH data were acquired using commercially available potassium phosphate monobasic with sodium hydroxide pH buffer solutions (pH 1, 4, 7, and 10) on bare 25 printed passes of MLG printed electrode. Solutions with pH values of 2, 3, 5, 6, 8, and 9 were then formulated by mixing the high pH standard solution with low pH solutions. The pH of the buffer solutions was confirmed using a glass-electrode benchtop pH meter (Mettler-Toledo, Columbus, OH) in a stirred solution. Chronopotentiometry measurements were performed with the printed MLG electrodes while varying the pH buffer solution.The open circuit potential values were captured for different pH solutions for a 120 second duration. Since the MLG electrode electrochemical process is reversible, the Nernst equation for pH calculation from open circuit potentials is as follows, 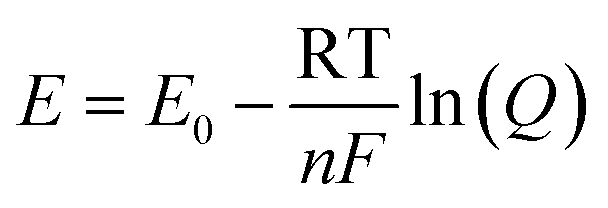 , where for an ideal electrode, E = E′ + 0.0591pH, with E the measured open circuit potential and E′ the standard potential, R the gas constant (8.314 J K−1 mol−1), T the absolute temperature (K), n the signed ionic charge and F is the Faraday constant (96
, where for an ideal electrode, E = E′ + 0.0591pH, with E the measured open circuit potential and E′ the standard potential, R the gas constant (8.314 J K−1 mol−1), T the absolute temperature (K), n the signed ionic charge and F is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 487.3415C mol−1).87 The equation of the fitted line (pH values 4–10) is as follows: E = 1.56 − 0.051pH, where the slope of 51 mV pH−1 is close to that of an ideal electrode (i.e., 59 mV pH−1) seen in Fig. 5d and the raw data is seen in the ESI Fig. S5.† To examine the reproducibility of the MLG electrodes, the pH experiment was conducted on three different 25 print pass electrodes fabricated with identical print conditions as described above. ESI Fig. S6† displays the potential (MLG vs. Ag/AgCl) versus pH data for all three electrodes. Additionally, from ESI Fig. S6,† it can be determined that the MLG electrodes are capable of providing a consistent response with potential (V vs. Ag/AgCl). Furthermore, we employed a response time experiment of our electrode with changing pH values. We started by measuring pH 2 buffer solution and added aliquots (100 μL to 1 mL) of pH 10 buffer solution to change the pH of the solution tested from 2 to 10 and recorded the change in the open circuit potential. The solution tested was stirred between measurements with a magnetic stirrer placed under the cell. Fig. 5e displays the change in potential with the pH of the solution. This suggests that the IJP of bare MLG on a Kapton substrate showed a significant response to the change in pH in the solution.
487.3415C mol−1).87 The equation of the fitted line (pH values 4–10) is as follows: E = 1.56 − 0.051pH, where the slope of 51 mV pH−1 is close to that of an ideal electrode (i.e., 59 mV pH−1) seen in Fig. 5d and the raw data is seen in the ESI Fig. S5.† To examine the reproducibility of the MLG electrodes, the pH experiment was conducted on three different 25 print pass electrodes fabricated with identical print conditions as described above. ESI Fig. S6† displays the potential (MLG vs. Ag/AgCl) versus pH data for all three electrodes. Additionally, from ESI Fig. S6,† it can be determined that the MLG electrodes are capable of providing a consistent response with potential (V vs. Ag/AgCl). Furthermore, we employed a response time experiment of our electrode with changing pH values. We started by measuring pH 2 buffer solution and added aliquots (100 μL to 1 mL) of pH 10 buffer solution to change the pH of the solution tested from 2 to 10 and recorded the change in the open circuit potential. The solution tested was stirred between measurements with a magnetic stirrer placed under the cell. Fig. 5e displays the change in potential with the pH of the solution. This suggests that the IJP of bare MLG on a Kapton substrate showed a significant response to the change in pH in the solution.
Using the separate solutions methods (SSM) with different interfering ions of K+, Na+, and NH+4 (pH 6) to estimate the potentiometric selectivity coefficients KpotIJ at different ion concentration (10−2 M), where K is the selectivity coefficient, I is the primary ion, and J is the interfering ion.88,89 Equation is as follows:
| KpotIJ = aI/(aJ)ZI/ZJ |
| Ions (J) | logKpotI,J |
KpotI,J |
|---|---|---|
| Na+ | −5.34 | 4.52 × 10−6 |
| K+ | −4.48 | 3.32 × 10−5 |
| NH4+ | −6.87 | 1.34 × 10−7 |
To investigate the flexibility of the IJP MLG electrodes, bending cycle testing (n = number of bending cycles) was performed on five IJP printed MLG electrodes fabricated with identical printing conditions (25 printed passes), and having similar resistance measurements. Bending cycles were performed with 7.5 mm and 14.5 mm radius of curvature tubes, and tests were performed from n = 1 to 1000, with CV measurements performed at n = 10 intervals, as shown in the images in Fig. 5f. Bending is expected to increase the resistance of the electrodes, which should increase the peak to peak separation. Fig. 5f, shows that the IJP MLG electrodes show a robust performance over this range of bend cycle testing. Additionally, bending cycles resulted in a ∼2% increase peak-to-peak separation with a 14.5 mm radius of curvature and an ∼10% increase in peak-to-peak separation with a 7.5 mm radius of curvature at n = 1000. A summary of flexible, graphene-based sensors is listed in Table 1. Electrodes shaded in pink shows functionally of a fully printed three electrode sensor system, however only our study show functionality or bare graphene electrodes with inkjet printed Pt counter electrodes. To the best of our knowledge, this is the first study amongst all other flexible, graphene-based sensors, to report on and demonstrate the stability of these types of sensors subjected to cyclic bending tests.
Fully printed three-electrode devices
The studies mentioned above provide insights into the electrochemical performance of individual working IJP MLG electrodes using conventional external reference and counter electrodes, as seen in Fig. 2e. Here we compare the electrochemical performance of fully flexible IJP three-electrode (working, counter, and reference) sensor systems to the individual IJP MLG electrode. Fully IJP flexible electrodes could enable large scale, roll-to-roll level production of such sensors. To fabricate the IJP three-electrode sensor system, custom made polyvinyl pyrrolidone capped Pt nanoparticle (PVP–PtNP) ink (seen inset of Fig. 6a) was prepared to print a counter electrode. The TEM image seen in Fig. 6a shows the PtNP ranging from 5–8 nm. To measure the electrical properties of the printed platinum lines, a 4-point probe (Keithley 4200 SCS, Textronix) measurements were conducted. Fig. 6b shows the calculated resistivity as a function of annealing temperature for 4 print passes. Based on the graph, the lowest resistivity was calculated to be 1.3 × 10−6 Ωm for 4 print passes at an annealing temperature of ∼425 °C which is ∼10× greater than bulk platinum (1.06 × 10−7 Ωm).91 Silver (NovaCentrix), Su8 (PriElex Microchem), NaClO, and a cocktail mixture of PVP (Butvar B-98) ink solutions were used to fabricate the Ag/AgCl/(Na+) reference electrode (see Methods section for further details). Fig. 6c displays the fully IJP three-electrode sensor system using MLG as the working electrode, Pt counter electrode and Ag/AgCl/(Na+) reference electrode. Su8 ink was used as a passivation layer for the MLG electrode, and Ag/AgCl/(Na+) electrode.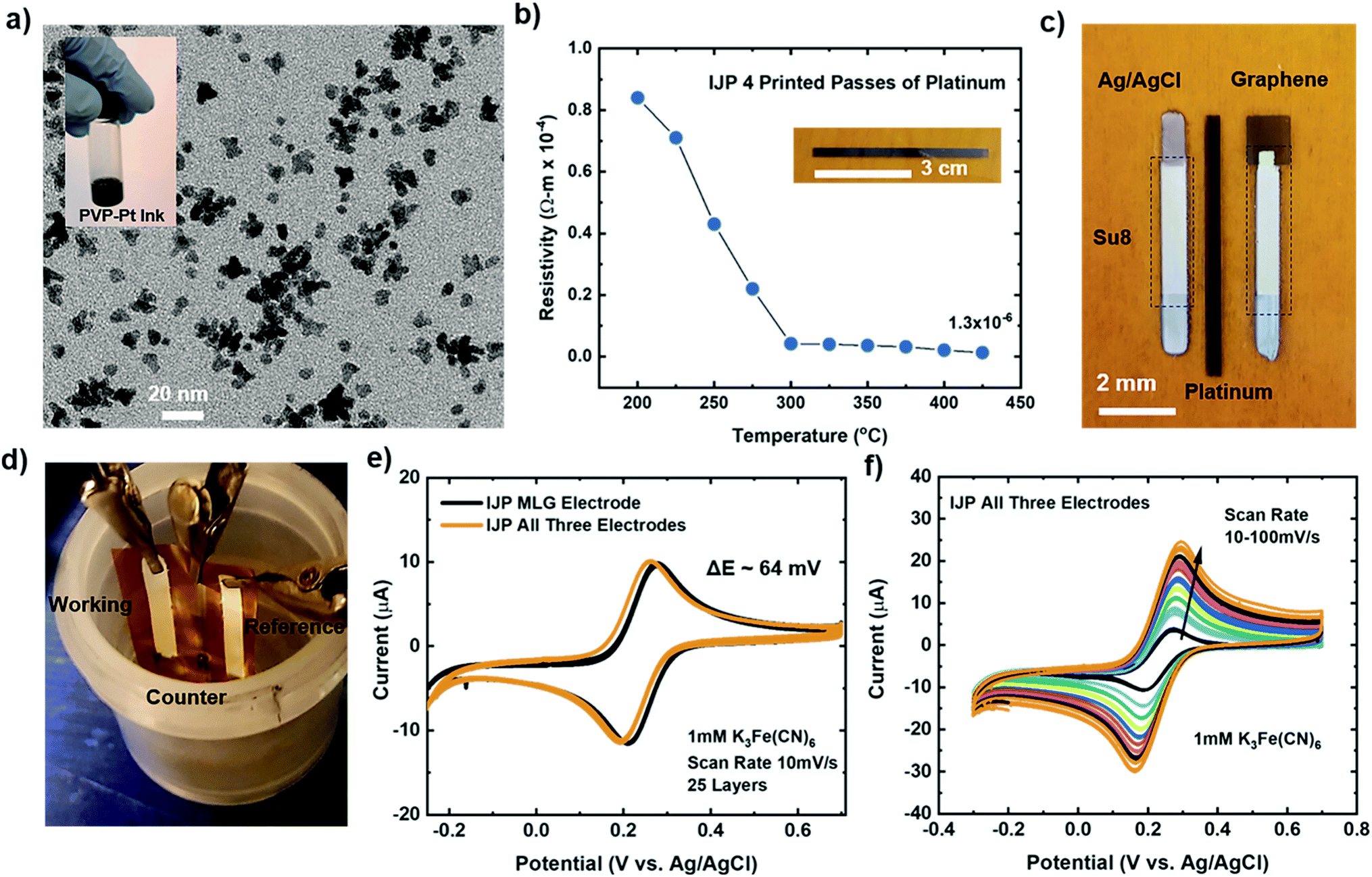 | ||
| Fig. 6 (a) TEM image of the platinum nanoparticles (PtNP) with an optical image of the platinum ink (inset) (b) resistivity vs. temperature for 4 printed passes of platinum lines on Kapton (inset). (c) Optical image of IJP all three electrodes (Ag/AgCl as the reference electrode, platinum as the counter electrode, and MLG as the working electrode). (d) Optical picture of the electrochemical setup for the fully printed three electrode sensors. (e) Cyclic voltammetry (CV) data of IJP MLG (black) compared to all three electrodes (orange) in 1 mM K3[Fe(CN)6] and 1 M KCl as the supporting electrolyte with scan rate 10 mV s−1 and (f) CV scans of all three printed electrodes with increasing scan rate from 10–100 mV s−1. | ||
The experimental setup shown in Fig. 6d was used to study and compare the electrochemical response of a fully printed three-electrode device to our individual IJP MLG electrode. CV measurements were carried out with a scan rate of 10 mV s−1 in 1 mM K3[Fe(CN)6] with 1 M KCl as the supporting electrolyte, for the individual IJP MLG electrode (25 printed passes) and the fully printed three-electrode device. Fig. 6e demonstrates that the response of the fully printed three-electrode devices is comparable to the individual IJP MLG electrodes, and that the three-electrode device exhibits excellent reversibility with a peak to peak separation of ∼64 mV. Furthermore, CV measurements were carried out with an increasing scan rate from 10–100 mV s−1 in 1 mM K3[Fe(CN)6], as seen in Fig. 6f. Again, CV simulation was used to extract the rate constant (k) from the experimental CV data seen in Fig. 6f. The electron rate transfer constant extracted for the fully IJP three-electrode sensors was determined to be k = 1.22 × 10−2 cm s−1 for a scan rate of 10 mV s−1. These CV measurements suggest that the fully printed three-electrode device shows fast electron transfer with this redox system, similar to the results presented for the individual IJP MLG electrodes.
Conclusion
In this study, the electrochemical behavior of fully inkjet printed multilayer graphene electrodes on flexible Kapton substrates was investigated. Cyclic voltammetry was used to analyze the electrochemical reversibility of a fully inkjet-printed MLG electrode and a fully inkjet-printed three-electrode device using the ferric/ferrocyanide [Fe(CN)6]−3/−4 redox couple as the analyte. It was confirmed that electrodes optimized at 25 printed passes (with adequate inert edge defects and surface porosity) showed quasi-reversibility with a low peak to peak potential separation of 60 mV and fast electron-transfer kinetics (k = 1.125 × 10−2 cm s−1). Moreover, it was verified that the printed MLG electrode was responsive to varying solution pH and displayed good electrochemical stability even after 1000 bending cycles (7.5 mm radius of curvature) with less than 10% change in peak to peak separation. Cross-sectional TEM images also revealed that the morphology of the printed graphene electrodes enhanced the electrochemical response and behavior of the printed electrodes. These studies indicate that fully IJP three-electrode sensors are a promising approach to fabricating flexible electrodes with an excellent electrochemical response comparable to those reported in the literature. These electrodes can be produced quickly, easily, and repeatedly, thus showing excellent potential for scalable manufacturing and flexible biosensing applications. The approach reported here enables a deeper understanding of how the combination of ink rheology and additive electronics manufacturing can enable the scalable manufacturing of flexible biosensors for space, military, and commercial applications.Methods section
Preparation and characterization of multilayered graphene ink
Similar to previous work, graphene flakes were obtained by solvent assisted exfoliation of 50 mg mL−1 graphite powder in a suspension of 2% ethyl cellulose (EC) in ethanol using a Qsonica (Q125) (Newtown, CT) probe tip sonicator for 90 minutes.20,53,57 To remove the larger graphite flakes, the dispersion was centrifuged (Heraeus™ Megafuge™ 8 Centrifuge TX-150 rotor) at 3402 RCF for 60 min and the supernatant was collected immediately. In a 1:2 volume ratio, the collected supernatant and 0.04 g mL−1 aqueous solution of NaCl (Sigma-Aldrich, >99.5%) were centrifuged for 30 min at 3402 RCF to facilitate the flocculation of graphene flakes. The resulting graphene/EC dispersion was dried overnight on a PTFE (Teflon) plate. To tailor the concentration and viscosity of ink to be compatible with the Dimatix IJP (Fujifilm, Santa Clara, CA), the dried graphene/EC paper was then dispersed by sonication for 30 min in a mixture of 85% cyclohexanone and 15% terpineol solution, followed by centrifugation at 3402 RCF for 15 min. The resulting ink concentration was 3.5 mg mL−1 with a viscosity of 8 cP (Wells-Brookfield Cone/Plate Middleboro, MA). The ink concentration was quantified by UV-vis absorption spectroscopy and Beer–Lamberts law at λ = 600 nm.
Fabrication of the MLG electrodes
The MLG was printed using a Dimatix inkjet printer. The tool platen was heated to 60 °C to ensure rapid drying of the ink once deposited on the Kapton HN (Dupont, Wilmington, DE) substrate. A 10 pL cartridge was used to print the MLG. The waveform, jetting voltage, and drop spacing were adjusted to achieve uniform droplets in volume and velocity of the MLG ink. Moreover, 4 nozzles were used to print, and the cartridge was at room temperature. After printing, any remaining ethyl cellulose and solvent was driven out of the lines with a 250 °C bake for 30 min followed by a ramp to a 350 °C bake for 45 min to maximize conductivity of the printed features. Next, NovaCentrix, Metalon (JS-B40G, Austin, TX) silver ink was used to print the contact pad connecting the printed MLG before sintering at 250 °C for 15 min. Lastly, PriElex Microchem (Westborough, MA) SU-8 ink was printed on top of the silver contact pad connecting the printed MLG. SU-8 acted as a passivation layer to isolate the silver while conducting electrochemical experiments.Fabrication of the platinum ink and platinum electrodes
98% sodium tetrahydroborate (Alfa Aesar), 99.999% hexachloroplatinic(IV) acid hydrate (40% platinum metals basis, BeanTown Chemical), 10 kDa polyvinylpyrrolidone (Alfa Aesar), 95–100.5% sodium hydroxide pellets (Macron), ethylene glycol (VWR), ≥99.5% carboxymethyl cellulose sodium salt, and 40 kDa MWCO Slide-A-Lyzer Dialysis flasks (Thermo Scientific) were purchased from commercial sources and used without further purification. A 0.58 M hexachloroplatinic acid (H2PtCl6) stock solution was prepared with the addition of 5 g of H2PtCl6 to 20 mL of nanopure (18 MΩ) water. Additionally, a 2.2 M stock solution of sodium tetrahydroborate (NaBH4) was prepared with the addition of 0.500 g of NaBH4 to 6 mL of nanopure (18 MΩ) water buffered to a pH of 12 with NaOH. Both solutions were used without further purification or dilution.A platinum nanoparticle ink containing ∼20 wt% of 5–8 nm of polyvinyl pyrrolidone capped Pt nanoparticles (PVP–PtNP) suspended in a water/ethylene glycol co-solvent mixture was prepared to be compatible with ink jet printing. The synthesis of PVP stabilized Pt nanoparticles was accomplished through wet chemical methods where 10 mL of stock H2PtCl6 solution was added to 1.5 L of nano-pure water containing 6 g of dissolved PVP. The H2PtCl6/H20/PVP solution was allowed to stir for two hours and was followed by the drop-wise addition of 6 mL of stock NaBH4 to form PVP–PtNP. The resulting suspension was allowed to stir vigorously for 24 hours, and this process was repeated until a total of 20 g of H2PtCl6 had been reduced or 6 L of PVP–PtNP suspension had been synthesized to form ∼8 g of PVP capped platinum nanoparticles.
The removal of excess capping agent and reaction by-products was performed through dialysis while utilizing 40 kDa MWCO Slide-A-Lyzer Dialysis flasks. As the dialysis process can be extremely time consuming, the PVP capped Pt nanoparticle suspension was dialyzed against a very high concentration solution of carboxymethyl cellulose in order to accelerate this process. A total of 6 L of PVP–PtNP was concentrated to 50 mL, which was followed by rotary evaporation to further concentrate the suspension to 20 mL. The viscosity of the PVP–PtNP suspension was tuned through the addition of ethylene glycol solution to ensure the ink rheology was compatible with inkjet printing.
The PVP–Pt was printed using a Dimatix inkjet printer. The tool platen was heated to 30 °C to ensure rapid drying of the ink once deposited on the Kapton HN (Dupont, Wilmington, DE) substrate. A 10 pL cartridge was used to print the PVP–Pt. The waveform, jetting voltage, and drop spacing were adjusted to achieve uniform droplets in volume and velocity of the PVP–Pt ink. Moreover, 2 nozzles were used to print, and the cartridge was at room temperature. After printing, any remaining surfactant and solvent was driven out of the lines with a 150 °C bake for 15 min followed by a ramp to a 400 °C bake for 45 min to maximize conductivity of the printed features.
Fabrication of Ag/AgCl electrodes
A good method to fabricate fully inkjet printed Ag/AgCl reference electrodes has been described by Moya et al.92 NovaCentrix, Metalon (JS-B40G, Austin, TX) silver ink was used to print the silver layers on Kapton substrate and then sintered at 250 °C for 30 min. Then, PriElex Microchem (Westborough, MA) SU-8 ink was printed on top of the silver layers for passivation and then sintered 250 °C for 50 min. For chlorination, diluted NaClO (5 v/v% purchased from Sigma-Aldrich) was printed (2 passes) on the exposed silver and then washed with deionized water. For the formulation of a protecting membrane, a cocktail mixture of PVB (Butvar B-98) (10 w%) in methanol (40%), xylene (30%), diacetone alcohol (15%), and 1-butanol (15%), all purchased from Sigma-Aldrich, was printed (5 passes) on top of the chlorinated area. Lastly, the electrode was left in a fume hood to dry overnight.Thermogravimetric analysis
To determine the annealing temperature of printed graphene features, thermogravimetric analysis (TGA) was employed. A Netzsch STA 449 F1 Jupiter (Burlington, MA) TGA instrument was used to measure the weight percent loss as a function of temperature (25 °C to 1000 °C) at a heating rate of 5 °C min−1 in air. TGA analysis revealed the decomposition peak of ethyl cellulose is around 250 °C, while the other volatile solvent components (cyclohexanone and terpineol) are driven off at 390 °C (seen in Fig. 1e).SEM, TEM and Raman imaging
A FEI Teneo (Hillsboro, OR), field emission SEM was used to image the printed films. Using the Dimatix inkjet printer, MLG and silver ink were used to print a transmission line measurement (TLM) structure with varying number of print passes from 15–30, with increments of 5 passes, on glass substrate (seen in Fig. 1a). The SEM image for 25-layer pass line of IJP multilayered graphene is shown in Fig. 1b to demonstrate its uniformity. TEM images were obtained using a JOEL JEM 2100 (Peabody, MA) system, with the particles characterized using ImageJ software. 25 and 30 printed passes of MLG were printed on untreated Kapton and the TEM samples were prepared by a FIB (focused ion beam) tool at the Center for Advanced Energy Studies. Lastly, Raman spectra were obtained using a Horiba LabRAM HR Evolution Raman microscope (Irvine, CA) with a 532 nm excitation wavelength. The spectra (1000–3000 cm−1) were collected at a relative laser power of 25% with a 100× objective and 30 s exposure time.Electrochemical setup
The electrochemical experiments were conducted using a customized 3D printed cell (Fig. 2e), and potentiostat (BioLogic VMP-300 instrument, Knoxville, TN) with EC-Lab for the software. The 3D printed cell allowed for the printed MLG (working electrode) to be placed on the bottom of the cell, with only 0.07 cm2 of the MLG exposed to the solution in the cell. Ag/AgCl was used as a reference electrode and with a platinum mesh as the working electrode. Room temperature cyclic voltammetry (CV) measurements were carried out in a fume hood with an increasing scan rate from 10–100 mV s−1, with dilutions ranging from 1 mM to 5 mM K3[Fe(CN)6] and 1 M KCl (both purchased from Sigma Aldrich) as the supporting electrolyte. The EC-lab software was used to extract the fitted CV data seen in Fig. 3c and d.Authors contributions
T. P. designed and printed the test structures, and together with C. C., P. B., and J. C. characterized the devices. J. K. and D. E. conceived of the experimental design, and together with H. S and C. X. supervised the experiments. K. F. synthesized the platinum (PVP–Pt) inks. All authors contributed to the development of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by NASA EPSCoR under Award #ID-80NSSC17M0029. The authors also thank Dr Yaqiao Wu at the Center for Advanced Energy Studies for his support on FIB and TEM experiments. D. E. also acknowledges career development support by Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grants #P20GM103408 and #P20GM109095.References
- S. Banerjee, J. Shim, J. Rivera, X. Jin, D. Estrada, V. Solovyeva, X. You, J. Pak, E. Pop, N. Aluru and R. Bashir, ACS Nano, 2013, 7, 834–843 CrossRef CAS.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, TrAC, Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS.
- R. A. W. Dryfe and P. R. Unwin, J. Electroanal. Chem., 2015, 753, 1–2 CrossRef CAS.
- R. A. W. Dryfe and P. R. Unwin, J. Electroanal. Chem., 2015, 753, 1–46, DOI:10.1021/nn202878f.
- L. Huang, Y. Huang, J. Liang, X. Wan and Y. Chen, Nano Res., 2011, 4, 675–684 CrossRef CAS.
- M. S. Mannoor, H. Tao, J. D. Clayton, A. Sengupta, D. L. Kaplan, R. R. Naik, N. Verma, F. G. Omenetto and M. C. McAlpine, Nat. Commun., 2012, 3, 763 CrossRef.
- N. Lei, P. Li, W. Xue and J. Xu, Meas. Sci. Technol., 2011, 22, 10, DOI:10.1088/0957-0233/22/10/107002.
- P. K. Ang, W. Chen, A. Thye, S. Wee and K. P. Loh, J. Am. Chem. Soc., 2008, 130(44), 14392–14393 CrossRef CAS.
- D. Dodoo-Arhin, R. C. T. Howe, G. Hu, Y. Zhang, P. Hiralal, A. Bello, G. Amaratunga and T. Hasan, Carbon, 2016, 105, 33–41 CrossRef CAS.
- B. Melai, P. Salvo, N. Calisi, L. Moni, A. Bonini, C. Paoletti, T. Lomonaco, V. Mollica, R. Fuoco and F. Di Francesco, in Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society, EMBS, 2016, vol. 2016, pp. 1898–1901 Search PubMed.
- Y. Xu, I. Hennig, D. Freyberg, A. James Strudwick, M. Georg Schwab, T. Weitz and K. Chih-Pei Cha, J. Power Sources, 2014, 248, 483–488 CrossRef CAS.
- M. Zhou, Y. Zhai and S. Dong, Anal. Chem., 2009, 81, 5603–5613 CrossRef CAS.
- E. P. Randviir, D. A. C. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- J. Ping, J. Wu, Y. Wang and Y. Ying, Biosens. Bioelectron., 2012, 34, 70–76 CrossRef CAS.
- F.-Y. Kong, S.-X. Gu, W.-W. Li, T.-T. Chen, Q. Xu and W. Wang, Biosens. Bioelectron., 2014, 56, 77–82 CrossRef CAS.
- C. Sriprachuabwong, C. Karuwan, A. Wisitsorrat, D. Phokharatkul, T. Lomas, P. Sritongkham and A. Tuantranont, J. Mater. Chem., 2012, 22, 5478–5485, 10.1039/c2jm14005e.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- Q. He, S. Wu, Z. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764 RSC.
- E. Jabari and E. Toyserkani, Carbon, 2015, 91, 321–329 CrossRef CAS.
- Z. Zhan, J. An, Y. Wei, V. T. Tran, H. Du, X. Niu, Y. Chen, Q. Pei, K. Schierle-Arndt, P. Yang, J.-U. Park, G. Shvets, R. S. Ruoff, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nanoscale, 2017, 9, 965–993 RSC.
- M. J. Renn, WHITEPAPER – Optomec, 2010, pp. 3–5 Search PubMed.
- R. G. Clinton, NASA's In Space Manufacturing Initiative and Additive Manufacturing Development for Rocket Engine Space Flight Hardware, 2016 Search PubMed.
- F. Torrisi, T. Hasan, W. Wu, Z. Sun, A. Lombardo, T. S. Kulmala, G.-W. W. Hsieh, S. Jung, F. Bonaccorso, P. J. Paul, D. Chu, A. C. Ferrari, H. Tang, D. Liu, Y. Zhao, X. Yang, J. Lu, F. Cui, P. A. Brooksby, A. K. Farquhar, H. M. Dykstra, M. R. Waterland, A. J. Downard, R. Zappacosta, M. Di Giulio, V. Ettorre, D. Bosco, C. Hadad, G. Siani, S. Di Bartolomeo, A. Cataldi, L. Cellini, A. Fontana, F. Torrisi, T. Hasan, W. Wu, Z. Sun, A. Lombardo, T. S. Kulmala, G.-W. W. Hsieh, S. Jung, F. Bonaccorso, P. J. Paul, D. Chu, A. C. Ferrari, A. Pattammattel, C. V. Kumar, E. del Corro, L. Kavan, M. Kalbac, O. Frank, T. A. Faculty, S. K. Bidasaria, I. P. Fulfillment, U. Mogera, R. Dhanya, R. Pujar, C. Narayana, G. U. Kulkarni, F. Irin, M. J. Hansen, R. Bari, D. Parviz, S. D. Metzler, S. K. Bhattacharia, M. J. Green, A. a. Green, M. C. Hersam, K. D. Papadimitriou, E. N. Skountzos, S. S. Gkermpoura, I. Polyzos, V. G. Mavrantzas, C. Galiotis, C. Tsitsilianis, H. H. Winter, R. J. T. Nicholl, H. J. Conley, N. V. Lavrik, I. Vlassiouk, Y. S. Puzyrev, V. P. Sreenivas, S. T. Pantelides, K. I. Bolotin, J. Yuan, A. Luna, W. Neri, C. Zakri, T. Schilling, A. Colin, P. Poulin, D. Du, P. Li, J. Ouyang, A. M. Dimiev, G. Ceriotti, A. Metzger, N. D. Kim, J. M. Tour, A. N. J. Rodgers, M. Velický and R. A. W. Dryfe, ACS Nano, 2012, 6, 2992–3006 CrossRef CAS.
- Z. P. Yin, Y. a. Huang, N. B. Bu, X. M. Wang and Y. L. Xiong, Chin. Sci. Bull., 2010, 55, 3383–3407 CrossRef.
- a Sridhar, T. Blaudeck and R. Baumann, Mater. Matters, 2009, 6, 1–8 Search PubMed.
- D. McManus, S. Vranic, F. Withers, V. Sanchez-Romaguera, M. Macucci, H. Yang, R. Sorrentino, K. Parvez, S. K. Son, G. Iannaccone, K. Kostarelos, G. Fiori and C. Casiraghi, Nat. Nanotechnol., 2017, 12, 343–350, DOI:10.1038/nnano.2016.281.
- R. Worsley, L. Pimpolari, D. McManus, N. Ge, R. Ionescu, J. A. Wittkopf, A. Alieva, G. Basso, M. MacUcci, G. Iannaccone, K. S. Novoselov, H. Holder, G. Fiori and C. Casiraghi, ACS Nano, 2019, 13(1), 54–60, DOI:10.1021/acsnano.8b06464.
- Q. He, S. R. Das, N. T. Garland, D. Jing, J. A. Hondred, A. A. Cargill, S. Ding, C. Karunakaran and J. C. Claussen, ACS Appl. Mater. Interfaces, 2017, 9(14), 12719–12727, DOI:10.1021/acsami.7b00092.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng, H. L. Poh, E. Lay, K. Chng, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, TrAC, Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS.
- J. L. R. C. Alkire and P. N. Bartlett, Electrochemistry of Carbon Electrodes, Urbana, 2015, vol. 16 Search PubMed.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, Chem. Soc. Rev., 2012, 41, 6944 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- H. Choi, H. Kim, S. Hwang, M. Kang, D.-W. Jung and M. Jeon, Scr. Mater., 2011, 64, 601–604 CrossRef CAS.
- H. Kabir, H. Zhu, J. May, K. Hamal, Y. Kan, T. Williams, E. Echeverria, D. N. McIlroy, D. Estrada, P. H. Davis, T. Pandhi, K. Yocham, K. Higginbotham, A. Clearfield and I. F. Cheng, Carbon, 2019, 144, 831–840, DOI:10.1016/j.carbon.2018.12.058.
- L. Wang, Z. Sofer and M. Pumera, ACS Nano, 2020, 14, 21–25 CrossRef CAS.
- F. Du, L. Zhu and L. Dai, in Biosensors Based on Nanomaterials and Nanodevices, 2017 Search PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- C. M. Lewandowski, N. Co-investigator and C. M. Lewandowski, Carbon Nanotube and Graphene Device Physics, 2015, vol. 1 Search PubMed.
- H. Y. Chen, S. Maiti and D. H. Son, ACS Nano, 2012, 6, 583–591 CrossRef CAS.
- D. Estrada, Z. Li, G.-M. Choi, S. N. Dunham, A. Serov, J. Lee, Y. Meng, F. Lian, N. C. Wang, A. Perez, R. T. Haasch, J.-M. Zuo, W. P. King, J. A. Rogers, D. G. Cahill and E. Pop, npj 2D Mater. Appl., 2019, 3, 10, DOI:10.1038/s41699-019-0092-8.
- R. Mas-Ballesté, C. Gómez-Navarro, J. Gómez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- K. L. Grosse, V. E. Dorgan, D. Estrada, J. D. Wood, I. Vlassiouk, G. Eres, J. W. Lyding, W. P. King and E. Pop, Appl. Phys. Lett., 2014, 105, 143109, DOI:10.1063/1.4896676.
- P. Yasaei, A. Fathizadeh, R. Hantehzadeh, A. K. Majee, A. El-Ghandour, D. Estrada, C. Foster, Z. Aksamija, F. Khalili-Araghi and A. Salehi-Khojin, Nano Lett., 2015, 15(7), 4532–4540, DOI:10.1021/acs.nanolett.5b01100.
- J. C. Koepke, J. D. Wood, D. Estrada, Z. Y. Ong, K. T. He, E. Pop and J. W. Lyding, ACS Nano, 2013, 7, 75–86 CrossRef CAS.
- B. Kumar, K. Min, M. Bashirzadeh, A. B. Farimani, M. H. Bae, D. Estrada, Y. D. Kim, P. Yasaei, Y. D. Park, E. Pop, N. R. Aluru and A. Salehi-Khojin, Nano Lett., 2013, 13, 1962–1968 CrossRef CAS.
- A. Salehi-Khojin, D. Estrada, K. Y. Lin, K. Ran, R. T. Haasch, J. M. Zuo, E. Pop and R. I. Masel, Appl. Phys. Lett., 2012, 9–12 Search PubMed.
- W. Yuan, Y. Zhou, Y. Li, C. Li, H. Peng, J. Zhang, Z. Liu, L. Dai and G. Shi, Sci. Rep., 2013, 3, 2248 CrossRef.
- N. G. Shang, P. Papakonstantinou, M. McMullan, M. Chu, A. Stamboulis, A. Potenza, S. S. Dhesi and H. Marchetto, Adv. Funct. Mater., 2008, 18(21), 3506–3514, DOI:10.1002/adfm.200800951.
- J. C. Claussen, A. Kumar, D. B. Jaroch, M. H. Khawaja, A. B. Hibbard, D. M. Porterfield and T. S. Fisher, Adv. Funct. Mater., 2012, 22(16), 3399–3405, DOI:10.1002/adfm.201200551.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19(17), 2782–2789, DOI:10.1002/adfm.200900377.
- T. Pandhi, E. Kreit, R. Aga, K. Fujimoto, M. T. Sharbati, S. Khademi, A. N. Chang, F. Xiong, J. Koehne, E. M. Heckman and D. Estrada, Sci. Rep., 2018, 8, 10842, DOI:10.1038/s41598-018-29195-y.
- C. Punckt, M. A. Pope and I. A. Aksay, J. Phys. Chem. C, 2013, 117(31), 16076–16086, DOI:10.1021/jp405142k.
- K. S. Rao, J. Senthilnathan, Y.-F. F. Liu and M. Yoshimura, Sci. Rep., 2014, 4, 4237 CrossRef.
- L. G. Caņado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, A. Jorio, L. N. Coelho, R. Magalhães-Paniago and M. A. Pimenta, Appl. Phys. Lett., 2006, 88(16), 163106, DOI:10.1063/1.2196057.
- E. B. Secor, P. L. Prabhumirashi, K. Puntambekar, M. L. Geier and M. C. Hersam, J. Phys. Chem. Lett., 2013, 4(8), 1347–1351 CrossRef CAS.
- L. Dybowska-Sarapuk, K. Kielbasinski, A. Arazna, K. Futera, A. Skalski, D. Janczak, M. Sloma and M. Jakubowska, Nanomaterials, 2018, 8(8), 602, DOI:10.3390/nano8080602.
- E. F. Silva, A. A. Tanaka, R. N. Fernandes, R. A. A. Munoz and I. S. da Silva, Microchem. J., 2020, 157, 105027 CrossRef CAS.
- L. A. J. Silva, W. P. da Silva, J. G. Giuliani, S. C. Canobre, C. D. Garcia, R. A. A. Munoz and E. M. Richter, Talanta, 2017, 165, 33–38 CrossRef CAS.
- D. N. Barreto, M. M. A. C. Ribeiro, J. T. C. Sudo, E. M. Richter, R. A. A. Muñoz and S. G. Silva, Talanta, 2020, 217, 120987 CrossRef CAS.
- S. J. Konopka and B. McDuffie, Anal. Chem., 1970, 42(14), 1741–1746, DOI:10.1021/ac50160a042.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, The Handbook of Graphene Electrochemistry, 2014 Search PubMed.
- A. S. Ambolikar, N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95(2), 197–206, DOI:10.1021/acs.jchemed.7b00361.
- K. Griffiths, C. Dale, J. Hedley, M. D. Kowal, R. B. Kaner and N. Keegan, Nanoscale, 2014, 6, 13613–13622, 10.1039/c4nr04221b.
- E. P. Randviir, D. A. C. Brownson, M. Gómez-Mingot, D. K. Kampouris, J. Iniesta and C. E. Banks, Nanoscale, 2012, 4, 6470–6480, 10.1039/c2nr31823g.
- J. Yang and S. Gunasekaran, Carbon, 2013, 51, 36–44 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Actuators, B, 2009, 138(2), 556–562, DOI:10.1016/j.snb.2009.01.044.
- M. V. Mirkin and A. J. Bard, Anal. Chem., 1992, 64(19), 2293–2302, DOI:10.1021/ac00043a020.
- H. He, Q. Xie, Y. Zhang and S. Yao, J. Biochem. Biophys. Methods, 2005, 62(3), 191–205, DOI:10.1016/j.jbbm.2004.11.004.
- S. Cinti, F. Arduini, M. Carbone, L. Sansone, I. Cacciotti, D. Moscone and G. Palleschi, Electroanalysis, 2015, 27, 2230–2238, DOI:10.1002/elan.201500168.
- S. H. Lee, H. Y. Fang, W. C. Chen, H. M. Lin and C. A. Chang, Anal. Bioanal. Chem., 2005, 383, 532–538, DOI:10.1007/s00216-005-0034-5.
- D. Menshykau and R. Compton, Electroanalysis, 2008, 20, 2387–2394, DOI:10.1002/elan.200804334.
- H. Dong, S. Kyung, H. Chang, K. Roh and J. Choi, Biosens. Bioelectron., 2012, 38, 184–188 CrossRef.
- A. N. Patel, M. G. Collignon, M. A. OConnell, W. O. Y. Hung, K. McKelvey, J. V. MacPherson and P. R. Unwin, J. Am. Chem. Soc., 2012, 134(49), 20117–20130, DOI:10.1021/ja308615h.
- N. O. Gomes, E. Carrilho, S. A. S. Machado and L. F. Sgobbi, Electrochim. Acta, 2020, 349, 136341 CrossRef CAS.
- P. Salvo, B. Melai, N. Calisi, C. Paoletti, F. Bellagambi, A. Kirchhain, M. G. Trivella, R. Fuoco and F. Di Francesco, Sens. Actuators, B, 2018, 256, 976–991 CrossRef CAS.
- W. Fu, C. Nef, O. Knopfmacher, A. Tarasov, M. Weiss, M. Calame and C. Schönenberger, Nano Lett., 2011, 11(9), 3597–3600, DOI:10.1021/nl201332c.
- D. Janczak, A. Peplowski, G. Wroblewski, L. Gorski, E. Zwierkowska and M. Jakubowska, J. Sens., 2017, 2017, 1–6 CrossRef.
- J. G. Giuliani, T. E. Benavidez, G. M. Duran, E. Vinogradova, A. Rios and C. D. Garcia, J. Electroanal. Chem., 2016, 765, 8–15 CrossRef CAS.
- W. R. de Araujo, C. M. R. Frasson, W. A. Ameku, J. R. Silva, L. Angnes and T. R. L. C. Paixão, Angew. Chem., Int. Ed., 2017, 56, 15113–15117 CrossRef CAS.
- L. García-Carmona, A. Martín, J. R. Sempionatto, J. R. Moreto, M. C. González, J. Wang and A. Escarpa, Anal. Chem., 2019, 91, 13883–13891 CrossRef.
- A. A. Dias, C. L. S. Chagas, H. D. A. Silva-Neto, E. O. Lobo-Junior, L. F. Sgobbi, W. R. De Araujo, T. R. L. C. Paixaõ and W. K. T. Coltro, ACS Appl. Mater. Interfaces, 2019, 11, 39484–39492 CrossRef CAS.
- C.-H. Chuang, H.-P. Wu, C. H. C.-H. Chen and P.-R. Wu, Proc. Int. Conf. Sens. Technol., ICST, 2011, vol. 5, pp. 1–13 Search PubMed.
- R. Rahimi, M. Ochoa, T. Parupudi, X. Zhao, I. K. Yazdi, M. R. Dokmeci, A. Tamayol, A. Khademhosseini and B. Ziaie, Sens. Actuators, B, 2016, 229, 609–617 CrossRef CAS.
- Y. Wen, X. Wang, P. Cai and X. Yu, Sens. Actuators, B, 2015, 216, 409–411 CrossRef CAS.
- R. P. Buck and E. Lindner, Pure Appl. Chem., 1994, 66(12), 2527–2536, DOI:10.1351/pac199466122527.
- E. Lindner and Y. Umezawa, Pure Appl. Chem., 2008, 80(1), 85–104 CAS.
- J. Ping, Y. Wang, J. Wu and Y. Ying, Electrochem. Commun., 2011, 13(12), 1529–1532, DOI:10.1016/j.elecom.2011.10.018.
- D. C. Giancoli, Phys. Educ., 2000, 35, 5, DOI:10.1088/0031-9120/35/5/705.
- A. Moya, R. Pol, A. Martínez-Cuadrado, R. Villa, G. Gabriel and M. Baeza, Anal. Chem., 2019, 91, 15539–15546 CrossRef CAS.
- G. Chen, S. Xiao, A. Lorke, J. Liu and P. Zhang, J. Electrochem. Soc., 2018, 165, 16, DOI:10.1149/2.0101816jes.
- D. De Venuto, S. Carrara, A. Cavallini and G. De Micheli, in Proceedings of the 12th International Symposium on Quality Electronic Design, ISQED, 2011, vol. 2011 Search PubMed.
- S. Carrara, M. D. Torre, A. Cavallini, D. De Venuto and G. De Micheli, in 2010 IEEE Biomedical Circuits and Systems Conference, BioCAS, 2010, vol. 2010 Search PubMed.
- M. M. Lounasvuori, M. Rosillo-Lopez, C. G. Salzmann, D. J. Caruana and K. B. Holt, Faraday Discuss., 2014, 172, 293–310, 10.1039/c4fd00034j.
- K. L. S. Castro, S. M. Oliveira, R. V. Curti, J. R. Araújo, L. M. Sassi, C. M. Almeida, E. H. M. Ferreira, B. S. Archanjo, M. F. Cabral, A. Kuznetsov, L. A. Sena, C. A. Achete and E. D’Elia, Int. J. Electrochem. Sci., 2018, 13, 71–87, DOI:10.20964/2018.01.02.
- G. C. Jensen, C. E. Krause, G. A. Sotzing and J. F. Rusling, Phys. Chem. Chem. Phys., 2011, 13, 4888–4894, 10.1039/c0cp01755h.
- B. Nagar, M. Jović, V. C. Bassetto, Y. Zhu, H. Pick, P. Gómez-Romero, A. Merkoçi, H. H. Girault and A. Lesch, ChemElectroChem, 2020, 7, 460, DOI:10.1002/celc.201901697.
- A. Lesch, F. Cortés-Salazar, V. Amstutz, P. Tacchini and H. H. Girault, Anal. Chem., 2015, 87(2), 1026–1033, DOI:10.1021/ac503748g.
- A. Lesch, M. Jović, M. Baudoz, Y. Zhu, P. Tacchini, F. Gumy and H. H. Girault, ECS Trans., 2017, 77(7), 73, DOI:10.1149/07707.0073ecst.
- A. Määttänen, U. Vanamo, P. Ihalainen, P. Pulkkinen, H. Tenhu, J. Bobacka and J. Peltonen, Sens. Actuators, B, 2013, 177, 153–162, DOI:10.1016/j.snb.2012.10.113.
- C. W. Foster, J. P. Metters, D. K. Kampouris and C. E. Banks, Electroanalysis, 2014, 26, 262–274, DOI:10.1002/elan.201300563.
- R. Martínez-Flores, E. J. Canto-Aguilar, G. Rodríguez-Gattorno, G. Oskam, D. Meneses-Rodríguez and M. A. Ruiz-Gómez, J. Electrochem. Soc., 2019, 166(5), H3279, DOI:10.1149/2.0381905jes.
- C. Sriprachuabwong, C. Karuwan, P. Ditsayut, T. Lomas, P. Sritongkham, A. Tuantranont, A. Wisitsorrat, D. Phokharatkul, T. Lomas, P. Sritongkham, A. Tuantranont, P. Ditsayut, T. Lomas, P. Sritongkham and A. Tuantranont, J. Mater. Chem., 2012, 22, 5478 RSC.
- F. Valentini, D. Romanazzo, M. Carbone and G. Palleschi, Electroanalysis, 2012, 24, 872–881, DOI:10.1002/elan.201100415.
- C. Bardpho, P. Rattanarat, W. Siangproh and O. Chailapakul, Talanta, 2016, 148, 673–679 CrossRef CAS.
- W. Li, D. Qian, Y. Li, N. Bao, H. Gu and C. Yu, J. Electroanal. Chem., 2016, 769, 72–79, DOI:10.1016/j.jelechem.2016.03.027.
- J. A. Hondred, J. C. Breger, N. J. Alves, S. A. Trammell, S. A. Walper, I. L. Medintz and J. C. Claussen, ACS Appl. Mater. Interfaces, 2018, 10(13), 11125–11134, DOI:10.1021/acsami.7b19763.
- C. L. Manzanares Palenzuela, F. Novotný, P. Krupička, Z. Sofer and M. Pumera, Anal. Chem., 2018, 90(9), 5753–5757, DOI:10.1021/acs.analchem.8b00083.
- S. R. Das, Q. Nian, A. A. Cargill, J. A. Hondred, S. Ding, M. Saei, G. J. Cheng and J. C. Claussen, Nanoscale, 2016, 8, 15870–15879, 10.1039/c6nr04310k.
- J. Kudr, L. Zhao, E. P. Nguyen, H. Arola, T. K. Nevanen, V. Adam, O. Zitka and A. Merkoçi, Biosens. Bioelectron., 2020, 156, 112109, DOI:10.1016/j.bios.2020.112109.
- E. Bihar, S. Wustoni, A. M. Pappa, K. N. Salama, D. Baran and S. Inal, npj Flexible Electron., 2018, 2, 30, DOI:10.1038/s41528-018-0044-y.
- H. Shamkhalichenar and J.-W. Choi, J. Electrochem. Soc., 2017, 164, 5, DOI:10.1149/2.0161705jes.
- P. J. Lamas-Ardisana, P. Casuso, I. Fernandez-Gauna, G. Martínez-Paredes, E. Jubete, L. Añorga, G. Cabañero and H. J. Grande, Electrochem. Commun., 2017, 75, 25–28, DOI:10.1016/j.elecom.2016.11.015.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04786d |
| This journal is © The Royal Society of Chemistry 2020 |