
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium/graphene oxide nanocomposites with carbon nanotubes and/or magnetite for the reduction of nitrophenolic compounds†
L. K. Parrott and
E. Erasmus *
*
Department of Chemistry, University of the Free State, PO Box 339, Bloemfontein 9300, South Africa. E-mail: Erasmus@ufs.ac.za; Fax: +27 51 4017295; Tel: +27 51 4019656
First published on 4th September 2020
Abstract
Graphene oxide (GO) was synthesised via the oxidation of graphite and was characterised using ATR FTIR, PXRD, SEM, TEM and TGA. These techniques confirmed the presence of characteristic oxygen-containing functional groups and the resulting increase in interlayer spacing in the nanostructure. GO is used as the support to form nanocomposites composed of combinations of the following: iron oxide nanoparticles (Fe3O4), carbon nanotubes (CNT) and palladium nanoparticles (Pd). The four final nanocomposites formed are: Pd/GO, Pd/Fe3O4/GO, Pd/CNT/GO, and Pd/CNT/Fe3O4/GO. Key intermediates were analysed using ATR FTIR for the confirmation of the modification. Additionally, all composites and their precursors underwent electron microscopic analysis to visually assess composite morphologies and the size distribution of deposited nanoparticles. The Fe3O4 and Pd nanoparticles were indistinguishable from each other in their spherical shape and particle diameters, which were no bigger than 32 nm. From the TGA, incorporation of Fe3O4, CNT and finally Pd into the nanocomposites increased total thermal stability in terms of mass percentage lost over the temperature programme. GO showed significant decomposition, with all nanocomposites remaining relatively stable up to 120 °C. ICP OES results showed total Pd content by mass percentage for each final composite, varied from 7.9% to 9.1% mass Pd/collective mass. XPS confirmed the expected elemental compositions of composites according to their structures and the Pd0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PdII ratios are obtained. The nanocomposites were tested for the catalytic reduction of nitrophenols. Pd/CNT/Fe3O4/GO gave the highest TOF′ for the reduction of 4-NP and 2-NP. For the reduction of 3-NP, Pd/GO showed the highest TOF′. Nitrophenol's pKa and catalyst TOF′ correlated in a direct proportional relationship for Pd/GO and Pd/Fe3O4/GO. It was found that Pd0 surpassed PdII in catalytic activity. Reduction of PdII to Pd0 took place during the first catalytic cycle.
:PdII ratios are obtained. The nanocomposites were tested for the catalytic reduction of nitrophenols. Pd/CNT/Fe3O4/GO gave the highest TOF′ for the reduction of 4-NP and 2-NP. For the reduction of 3-NP, Pd/GO showed the highest TOF′. Nitrophenol's pKa and catalyst TOF′ correlated in a direct proportional relationship for Pd/GO and Pd/Fe3O4/GO. It was found that Pd0 surpassed PdII in catalytic activity. Reduction of PdII to Pd0 took place during the first catalytic cycle.
Introduction
Creating efficient and robust catalysts is becoming increasingly important for the human race to keep up with its own energy, food, chemical and environmental needs.1 Heterogeneous catalysis in particular, as opposed to homogeneous catalysis, is responsible for the more energy-demanding processes. The advantages that make heterogeneous catalysis particularly suitable to the chemical and energy industries include the ease with which the catalyst can be separated from the reaction mixture.1,2 There are, however, certain drawbacks, many heterogeneous catalysts have inferior selectivity and activity.2,3 The selectivity problem is due to the difficulty with which catalyst properties (e.g. steric and electronic properties) can be fine-tuned for optimisation. One modern answer to the problem of activity is the use of nanocatalysts.With decreasing particle size there is an increase in the surface area-to-volume ratio that results in higher catalytic activity for nanocatalysts, since more active sites are exposed to the reaction mixture, as opposed to being trapped in the bulk material.2 With atoms on the surface of the nanoparticle being surrounded by fewer neighbouring atoms, especially at edges, they also have decreased coordination that contribute to their amplified reactivity.4
Due to the unique structures, large specific surface area, exotic properties, 2D nanomaterials are becoming an important platform to design single site catalysts for various reactions,5–15 and provide a good support for nanoparticles. One such material is graphene oxide (GO), which is a two-dimensional single-layer sheet of oxygen-containing graphite, consisting of carbon atoms arranged in a hexagonal lattice. GO displays unique properties that have made it a valued material, such as high surface area, ease of preparation, its ability to be tuned for solubility in a plethora of solvents, its alterability in terms of its electrical and optical properties and compatibility for the formation of composites due to its oxygen functional groups.16,17
Graphene oxide's surface area, transparency and flexibility in particular are attractive properties in many fields of use, including but not limited to catalysis, electronics, sensing, materials, adsorbents, energy storage and biomedical applications.18,19 Two of GO's greatest virtues, compared to graphene, are its solubility in water and capacity to be readily functionalised. Both advantages have special relevance to catalysis in particular. In the case of the latter, functional groups (on both sides of a sheet) and other defects in the carbon network can be home to active sites themselves or nucleation sites for catalytically active nanoparticles (NPs) such that GO can act as a support and possibly as a synergistic co-catalyst.16,20 The attachment of catalytically active compounds to GO can occur by covalent bonds but also hydrogen bonding, hydrophobic interactions, π–π* stacking and electrostatic interactions.16
Palladium metal is more affordable than its congener platinum and is one of the noble metals very prevalent in catalytic applications today.21 The metal does, however, tend to aggregate when unsupported, precipitating out of solution. This compromises activity due to the subsequently impoverished surface area.22,23 In response to this problem stabilizers or supports are used to distribute palladium nanoparticles more uniformly and to maintain integrity. Common stabilisers for palladium nanoparticle (Pd NPs) include metal oxides, dendrimers, polymeric compounds and carbonaceous material.24
Carbon nanotubes (CNT) is an allotrope of carbon discovered in 1991, preceding the discovery of graphene.25,26 It is a nanostructure that can be described as graphene rolled up and sealed to form a 1D cylindrical shape with a high aspect ratio.25–27 Carbon nanotubes have properties that make them suitable for many applications in a variety of different fields, such as sensors, electrode components, in capacitors, as adsorbents and of course as catalyst supports.25,28 The ultrasonication of GO and CNTs forms relatively orderly self-aligned frameworks,28–30 which might allow for even more ease of access to the supported catalytically active species. The basal planes of GO sheets have some conjugated rings of carbon that exert π–π as well as hydrophobic attraction to the surfaces of the CNTs.29,31 Further, the combination of GO and CNT improves the hydrophilic–hydrophobic property of the nanocomposite, making it good support in aqueous medium.32
Another useful additive to catalytic composites is magnetite (Fe3O4) particles. Magnetite (Fe3O4) NPs are associated with paramagnetism, biocompatibility and low toxicity, this renders them easy to work with in a number of applications such as drug delivery, medical treatment and in biosensors.33 It is the magnetic quality of magnetite in particular that makes it valuable as part of a catalytic support since, application of an external magnetic field can readily separate the catalyst from the reaction mixture to be recycled for further use.
Nitroaromatic compounds are very prevalent in industrial and agricultural use today.34,35 Their basic chemical structure comprise of at least one nitro group bound to an aromatic ring, which affords unique properties useful in the manufacturing of explosives, pesticides, agrochemicals, dyes, paper and pharmaceuticals.24,34,36 However, the same qualities that can be exploited for productive use have been observed to be potentially harmful to living things by toxicity and mutagenicity.34,36 Environmental contamination, especially of water and soil, is an unfortunate consequence of prevalent and unskilful handling and storage of nitrophenols.24,35 Subsequently many approaches have been investigated for the removal of such pollutants including adsorption, degradation, oxidation, electrocoagulation and electrochemical treatment.37 One approach is to convert the nitrophenol to a more benign and/or usable chemical, for instance the nitro functionality on 4-nitrophenol (4-NP) can be reduced to an amine functionality, producing 4-aminophenol (4-AP).37 Aminophenol compounds are relevant to a number of applications: it has pharmaceutical value as an intermediate for the production of analgesic and antipyretic drugs, it has photographic and corrosion prevention value, as well as applications in dyes, surfactants, polymers, agriculture etc.5,37,38
In this study we report on the preparation of GO, and using it as the base to support the Pd NPs as the active catalyst as well as the other modifiers namely CNT and/or Fe3O4. It is expected that the CNT used in this study will act as spacers between layers of GO, playing a role to prevent unwanted stacking.28,31 In turn GO acts as a dispersing agent for the CNTs.25 The effect of the combination of CNTs with graphene oxide (nanocomposite) on factors like catalyst loading and surface area can be investigated. The magnetite will render the nanocomposite magnetic for ease of separation from the reaction mixture. Three different nitrophenols, namely 4-, 3- and 2-nitrophenol, will be reduced by means of sodium borohydride (NaBH4) as reductant and hydride source, accompanied by the Pd-supported GO nanocomposites, which mediates charge transfer.5,24,38
Experimental
Materials and spectroscopic characterisation
All reagents (solid and liquid) were reagent grade, purchased from Sigma-Aldrich and used without further purification. Distilled water was used throughout. Separation of NPs out of solution was conducted using an external magnet where applicable, otherwise centrifugation was employed using a Beckman Coulter, Allegra™ 25R Centrifuge. The dialysis tubes used had 12000 Da pores. To make homogeneous dispersions Qsonica Sonicators probe ultrasonic processor (500 W, 20 KHz) with a 1.4 cm tip diameter was used as necessary. Powder X-ray diffraction (PXRD) measurements were collected using a multipurpose X-ray diffractometer D8-Advance from Bruker operated in a continuous θ–θ scan in locked coupled mode with Cu-Kα radiation. Measurements ran within a range in 2θ defined by the user with a typical step size of 0.034° in 2θ. A position sensitive detector, Lyn-Eye, was used to record diffraction data at a typical speed of 0.5 s per step which is equivalent to an effective time of 92 s per step for a scintillation counter. To obtain Transmission Electron Microscopy (TEM) images a Philips (FEI) CM100 equipped with a Megaview III digital camera was used. Resulting images were analysed utilising Soft Imaging System (analySIS) software. For Scanning Electron Microscopy (SEM) a JEOL-JSM7800 Field Emission Scanning Electron Microscope was employed. A PHI 5000 Versaprobe system equipped with a monochromatic Al Kα X-ray source, was used to measure X-ray photoelectron spectroscopic (XPS). Operating conditions and settings are similar as reported in previous work from this lab.39–43 A Shimadzu ICPS-7510 inductively coupled plasma-optical emission spectrometry (ICP-OES) with a radial-sequential plasma spectrometer was used for wet chemical analysis of Pd-containing samples. Thermo-Gravimetric Analysis (TGA) was performed on a Mettler-Toledo TGA/SDTA851e under nitrogen atmosphere. The temperature program for TGA analysis involved a pre-emptive isothermal step in which the temperature was held constant at 25 °C for 10 minutes, followed by heating from 25 °C to 800 °C at a rate of 10 °C min−1 and cooling to 25 °C again to initiate the next analysis. TGA data were analysed utilising Mettler Stare Evaluation software. UV/Vis spectra for catalysis were obtained from a Shimadzu UV-1800 PC UV-Visible spectrophotometer. Spectra were analysed with the UVProbe Version 2.20 software. Attenuated Total Reflection Fourier Transformed Infrared (ATR FTIR) spectra were obtained from a Nicolet IS50 ATR Fourier transform spectrometer. Transmittance was measured on solid phase samples only (not dispersions).
Synthesis
:Fe3O4 ratio was 2:1. This was followed by stirring at 60 °C for 1 h. The product was separated by the application of an external magnet, washed with distilled water three times and dried in vacuum overnight at 60 °C to give a final mass of 170 mg.:CNT (3:1) by mass. This mixture was ultrasonicated for 1 h followed directly by the deposition of Pd NPs as described in the next section.:Pd (4:1), see Table 1). This mixture was stirred at 60 °C for 1 h. The resulting composite was washed by centrifugation with water (×3) and then acetone. The product was dried overnight in vacuum at 60 °C.
| GO-precursor | Precursor mass (mg) | Pd(OAc)2 mass (mg) | Ethanol (mL) | Product | Product mass (mg) |
|---|---|---|---|---|---|
| GO | 50.1 | 12.8 | 50 | Pd/GO, 5 | 50.4 |
| GO–Fe3O4 | 60.2 | 15.0 | 60 | Pd/Fe3O4/GO, 6 | 55.0 |
| GO–CNT | 60.0 | 15.4 | 60 | Pd/CNT/GO, 7 | 72.4 |
| GO–Fe3O4–CNT | 26.3 | 6.7 | 27 | Pd/CNT/Fe3O4/GO, 8 | 28.7 |
Catalysis
Reaction procedure. A stock solution of 4-nitrophenol (4-NP) was prepared (40.36 mg, 0.250 L, 161.44 ppm). A 5 mL aliquot of the 4-NP (5.803 × 10−6 mol) solution was added to 19.0 mg NaBH4 (5.022 × 10−4 mol) in addition to enough distilled water to give a final volume of 80 mL. Intermittent UV/Vis measurements were taken to ensure no reaction occurred without the addition of a catalyst. Following this, an appropriate amount of catalyst (see Table 2) was dispersed in 5 mL distilled water by ultrasonication and added to the reaction mixture, thus initiating the reaction. UV/Vis measurements were taken in the wavelength range of 200–500 nm to follow the progression of the reaction until completion. Afterwards the catalyst was separated from the reaction mixture, it was washed with water and dried. The catalytic reaction was repeated with the spent catalyst in an identical manner as described for the first cycle. See Table 3 for the mass of the spent catalyst and NaBH4 used in the second catalytic cycle.
| Catalyst | Catalyst mass (mg) | NaBH4 mass (mg) |
|---|---|---|
| Pd/GO, 5 | 0.45 | 19.00 |
| Pd/Fe3O4/GO, 6 | 0.75 | 19.08 |
| Pd/CNT/GO, 7 | 0.32 | 19.06 |
| Pd/CNT/Fe3O4/GO, 8 | 0.35 | 19.23 |
| Catalyst | Catalyst mass (mg) | NaBH4 mass (mg) |
|---|---|---|
| Pd/GO, 5 | 0.40 | 19.16 |
| Pd/Fe3O4/GO, 6 | 0.59 | 19.06 |
| Pd/CNT/GO, 7 | 0.70 | 19.14 |
| Pd/CNT/Fe3O4/GO, 8 | 1.18 | 18.93 |
Calibration curve for 4-NP and NaBH4. The reaction mixture was set up as it would be for a typical catalytic reaction as stipulated above, involving 5 mL 4-NP, 19 mg NaBH4 and enough water to give a final volume of 80 mL. Aliquots of 1 mL 4-NP stock solution were added to the reaction mixture and UV measurements taken in between each addition. The peak absorbance was found to be at 414 nm.
Reaction procedure. A stock solution of 3-nitrophenol (3-NP) was prepared (40.02 mg 3-NP, 0.250 L H2O, 160.08 ppm). A 15 mL aliquot was added to 57.5 mg NaBH4 in addition to enough distilled water to give a final volume of 85 mL. Intermittent UV/Vis measurements were taken to ensure no reaction occurred without the addition of a catalyst. Following this an appropriate amount of catalyst (see Table 4) was dispersed in 5 mL distilled water by ultrasonication and the appropriate amount added to the reaction mixture, thus initiating the reaction. UV/Vis measurements were taken in the wavelength range of 300–500 nm to follow the progression of the reaction until completion.
| Catalyst | Catalyst mass (mg) | NaBH4 mass (mg) |
|---|---|---|
| Pd/GO, 5 | 0.11 | 57.46 |
| Pd/Fe3O4/GO, 6 | 0.09 | 57.47 |
| Pd/CNT/GO, 7 | 1.16 | 57.53 |
| Pd/CNT/Fe3O4/GO, 8 | 1.24 | 57.48 |
Calibration curve for 3-NP and NaBH4. The reaction mixture was set up as it would be for a typical catalytic reaction as stipulated above, involving 58 mg NaBH4 and enough distilled water to give a final volume of 85 mL. Aliquots of 2 mL 3-NP stock solution were added to the reaction mixture and UV measurements taken in between each addition. The peak absorbance was found to be at 390 nm.
Reaction procedure. A stock solution of 2-nitrophenol (2-NP) was prepared (40.34 mg, 0.250 mL, 161.36 ppm). A 15 mL aliquot was added to 57.5 mg NaBH4, followed by the addition of enough water to give a final volume of 90 mL. Intermittent UV/Vis measurements were taken to ensure no reaction occurred without the addition of a catalyst. Following this an appropriate amount of catalyst (see Table 5) was dispersed in 5 mL distilled water by ultrasonication and the appropriate amount added to the reaction mixture, thus initiating the reaction. UV/Vis measurements were taken in the wavelength range of 300–550 nm to follow the progression of the reaction until completion.
| Catalyst | Catalyst mass (mg) | NaBH4 mass (mg) |
|---|---|---|
| Pd/GO, 5 | 0.54 | 57.05 |
| Pd/Fe3O4/GO, 6 | 0.47 | 57.41 |
| Pd/CNT/GO, 7 | 0.20 | 57.49 |
| Pd/CNT/Fe3O4/GO, 8 | 0.05 | 57.52 |
Calibration curve for 2-NP. The reaction mixture was set up as it would be for a typical catalytic reaction as stipulated above, involving 58 mg NaBH4 and 75 mL distilled. Aliquots of 2 mL 2-NP stock solution were added to the reaction mixture and UV measurements taken in between each addition. The peak absorbance was found to be at 414 nm.
Results and discussion
To prepare graphene oxide (GO), graphite was oxidised using a modified Hummer's method.44 This process uses H2SO4 and NaNO3 for the intercalation of graphite with oxygen. KMnO4 is employed as an oxidant of the acid-intercalated graphite to graphite oxide.17 The downside to this approach is the use of NaNO3, which leads to the release of toxic NO2/N2O4 gases, exposing Na+ and NO3− ions to waste water. This procedure allows the intercalation of oxygen functionalities and water between individual carbon sheets. Upon ultrasonication graphene oxide (GO) was obtained and is used as the basis, to support the other nanoparticles during the preparation of the nanocomposites.FeCl2·4H2O and FeCl3 were used as sources of FeII and FeIII respectively, which were combined in a ratio of 1:1.5 under nitrogen atmosphere to produce Fe3O4 NPs. These were characterised before being supported on GO to render the composite magnetic.19
Using GO in combination with Fe3O4 NPs and CNT (purchased), followed by the depositing of Pd NPs, the nanocomposites were made according to Scheme 1.
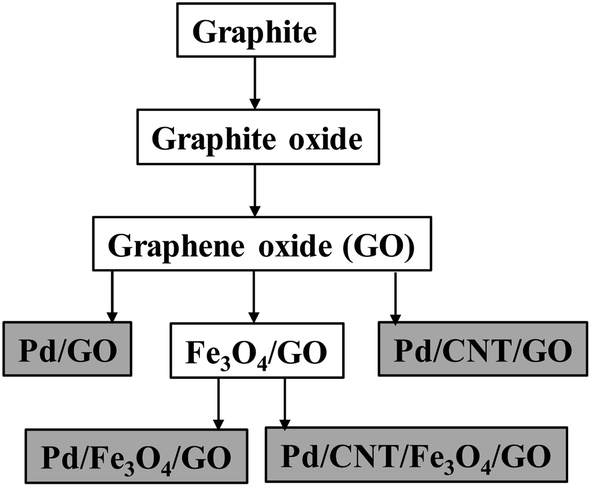 | ||
| Scheme 1 A schematic representation of routes for nanocomposite preparation, showing the final composites in grey blocks. For the CNT and Pd NPs, both are combined with the preceding composite in a single step. | ||
Four final Pd-containing nanocomposites were produced in total. The GO and Fe3O4,/GO, are the key intermediates onto which Pd NPs were deposited using ethanol as solvent and mild reductant at 60 °C. Pd(OAc)2 served as the Pd-source.20 CNT and the Pd NPs were deposited onto the composite together in the same reaction step.29
Attenuated Total Reflectance Fourier Transformed Infrared (ATR FTIR) analysis of key intermediates were conducted, to ensure successful combination of components, since it is easily accessible and gives immediate insight into the chemical composition of samples (seen in Fig. S1–S3 in the ESI†).
Graphite consists of carbon–carbon bonds, which does not give any significant transmission bands. It does, however, show a curvature in the baseline, that is typical for carbon compounds and is therefore also expected (and obtained) for CNT-containing composites.45,46 The lack of strong transmission bands for graphite is contrasted with the multitude of bands appearing for GO, which is indicative of the oxygen moieties that is now included in the structure. Most noticeable for GO is the strong, broad band around 3000–3600 cm−1, representative of O–H stretching frequencies, including that of water.47 The band at 1605 cm−1 is also an indicator of –OH functionalities. The three other bands observed at 1714 cm−1, 1395 cm−1 and 1030 cm−1, correspond with literature as typical for an FTIR spectrum of GO, representing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH and C–O stretching frequencies, respectively.48
O, C–OH and C–O stretching frequencies, respectively.48
The ATR FTIR spectra of Fe3O4 NPs and the Fe3O4/GO nanocomposites show a prominent shoulder band at 620 cm−1, which is not present in the ATR FTIR spectra of GO. This shoulder forms part of the characteristic band of iron oxide nanoparticles around 570–580 cm−1.49,50 This affirms that GO was successfully combined with Fe3O4 to produce the Fe3O4/GO nanocomposite.
As expected, there is a curve in the baseline of the FTIR spectra for CNT and the Pd/CNT/GO nanocomposite. The addition of CNT to GO causes considerable flattening of GO's bands, but some characteristic bands are still identifiable.
Fig. 1 depicts the Powder X-Ray Diffraction (PXRD) spectra of graphite (above) and graphene oxide (below) respectively. Graphite's intense, sharp characteristic peak at ca. 26.4° (002) is present as well as a smaller peak at ca. 54.6° (004). The additional peak at 19.0° is attributed to an impurity in the as-purchased graphite. Graphite's 26.4° peak shifts to 12.6° in the PXRD spectra of graphene oxide, indicating an increase in interlayer spacing due to the intercalation of oxygen moieties.51 The diffraction peak of the GO is broader than the diffraction peak of the graphite due to the significant increase in spacing between the sheets, because of the oxygen functional groups separating the sheets. At higher angles more peaks appear that do not correspond to a typical graphene oxide spectrum, this is presumably also be due to the presence of impurities not removed by dialysis.
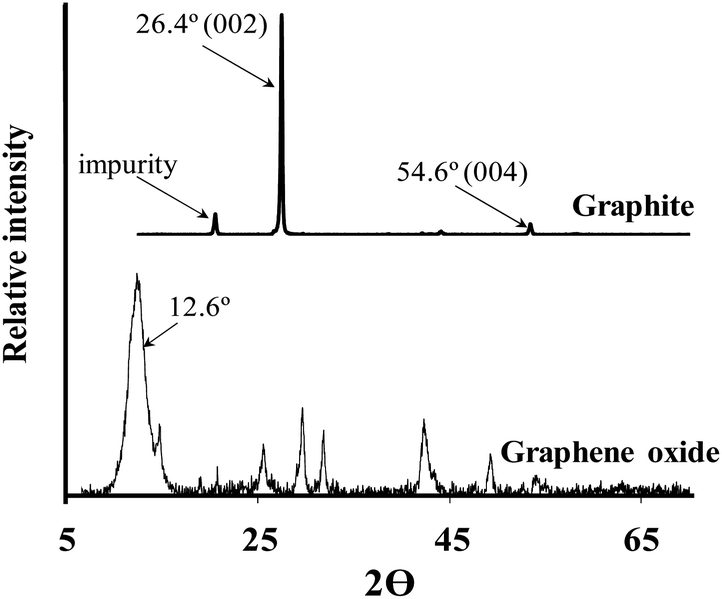 | ||
| Fig. 1 PXRD spectra of graphite starting material (above) and graphene oxide (below). | ||
To visually inspect the nanocomposites on a nanoscale, Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) was employed. Fig. 2 shows the Scanning Electron Micrographs (SEM) of graphite (A and B) and graphene oxide (C and D) respectively. Like the pages of a book these nanostructures organise in layers. By oxidising graphite the goal was the intercalation of oxygen functionalities between layers such that more individualised sheets could form when dispersed in water.
 | ||
| Fig. 2 SEM of graphite (top: (A) and (B)) and GO (bottom: (C) and (D)) showing separation of layers at the nanoscale. | ||
The deposition of Pd NPs on graphene oxide can be seen as quasi-spherical bright dots on SEM (A) and dark dots on TEM (B) respectively, see Fig. 3. The Pd NPs were detected on both the external surface, and some of the particles are intercalated between the GO lamellae. All particles fall within the nano range and were monodispersed on the G with minimal agglomeration. The histogram accompanied in the figure shows the particle size distribution as obtained by TEM. From the images, the lamellar structure of GO can be clearly distinguished.
 | ||
| Fig. 3 SEM (A) and TEM (B) of Pd/GO and a histogram showing the particle diameter distribution of Pd NPs on GO. | ||
Fig. 4 shows the respective TEM images of Fe3O4/GO and Pd/Fe3O4/GO, the SEM is presented in Fig. S4 and S5 in the ESI.† Fe3O4 NPs are shown to be deposited on GO in clusters, and not as evenly dispersed as the Pd NPs on GO (Fig. 3). Some Fe3O4 NPs also appear not to be anchored onto the GO. The histogram of the Fe3O4 NPs' particle diameters shows slightly large diameter size distribution than the Pd NPs. However, no histogram was constructed for the NPs of Pd/Fe3O4/GO, since it includes both Fe3O4 NPs and Pd NPs and there is no way to discern between them for measurements.
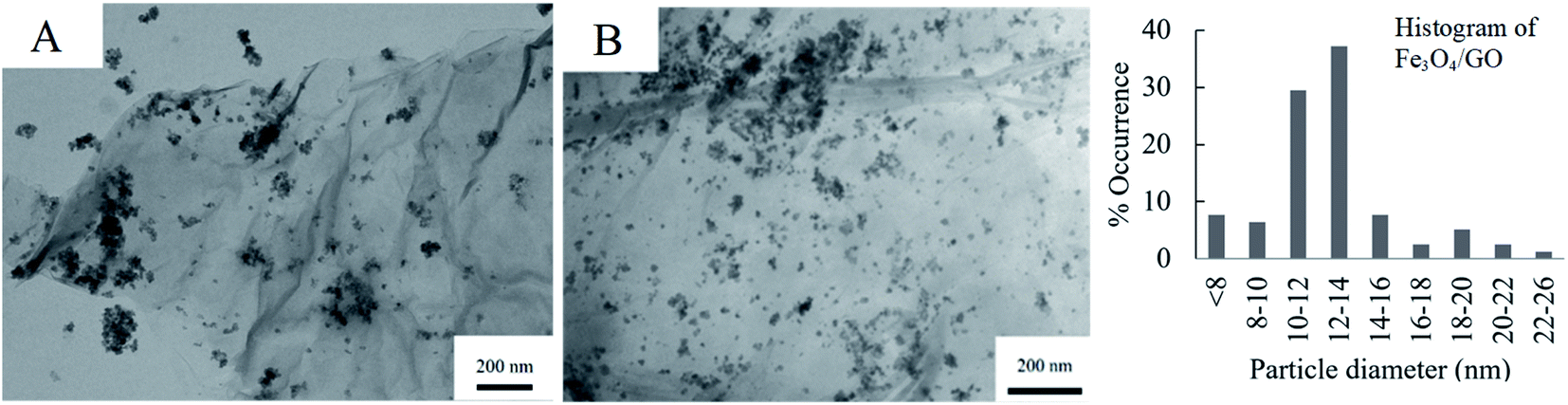 | ||
| Fig. 4 TEM images of (A) Fe3O4/GO (and its histogram) and (B) Pd/Fe3O4/GO. | ||
Fig. 5 shows the SEM and TEM micrographs of Pd/CNT/GO. The long, thin strands of the CNT and are uniformly distributed between the sheets of GO. The strands of the CNT appear entangled and has a diameter of ca. 17 nm. In some instances, it seems like Pd NPs are not only deposited on the basal planes of GO sheets, but also on the CNTs. The particle size of the Pd NPs formed on the Pd/CNT/GO is in average much larger and has a larger size distribution compared to both Pd/GO and Pd/Fe3O4/GO.
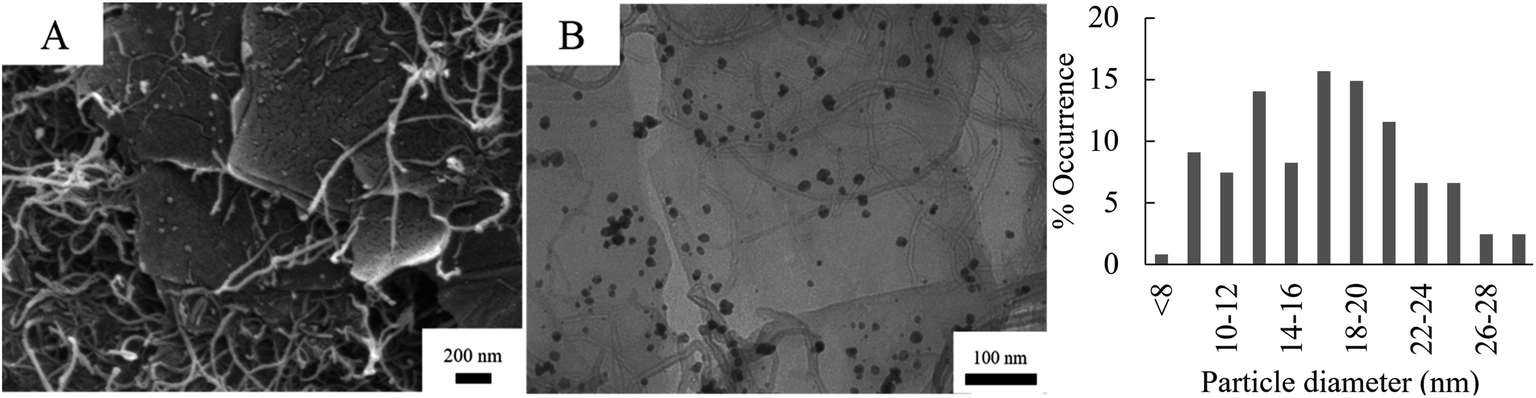 | ||
| Fig. 5 SEM (A) and TEM (B) of Pd/CNT/GO with accompanying particle diameter distribution histogram of the Pd NPs. | ||
Table 6 is a summary of mean particle diameters, standard deviations as well as the minimum and maximum diameter of the Pd and Fe3O4 NPs as measured from the TEM images. All the nano-particles fall within the nano range, with mean diameters spanning from ca. 11 nm to 18 nm. Fe3O4 NP were prepared neat (row 1 in Table 6), before being deposited on GO. In each of the three nanocomposites where the Fe3O4 NPs' sizes were measured, the particles were therefore quite similar. Pd NPs, on the other hand, were synthesized separately for each respective GO-precursor, creating more size variability among nanocomposites. The Pd NPs deposited on the CNT/GO nanocomposite revealed the biggest mean particle diameter of 17.24 nm.
| Particle measured | Mean particle diameter (nm) | Standard deviation | Min. particle diameter (nm) | Max. particle diameter (nm) | |
|---|---|---|---|---|---|
| Fe3O4 NPs | Fe3O4 | 11.39 | 3.250 | 6.05 | 20.29 |
| Fe3O4/GO | Fe3O4 | 12.58 | 3.409 | 6.01 | 25.92 |
| Pd/GO | Pd | 11.27 | 3.809 | 3.27 | 26.45 |
| Pd/CNT/GO | Pd | 17.24 | 5.251 | 7.81 | 31.57 |
The thermal stability of the key intermediates and the Pd-containing nanocomposites were analysed by thermogravimetric analysis (TGA) from room temperature to 800 °C at a rate of 10 °C min−1 under N2 atmosphere (see Fig. S6–S9 in the ESI†). Graphite is thermally stable, remaining unchanged across the entire temperature range, with no significant change in mass. GO shows three mass loss steps upon heating. The first small step occurring below 150 °C (with an onset temperature of ca. 41 °C), can be attributed to solvent evaporation, including that of water molecules physisorbed onto the compound surface.52 This step reoccurs consistently for all of the nanocomposites, with the exception of Pd/Fe3O4/GO. Following this, graphene oxide continues to decompose in two additional steps. The second decomposition is in accordance with literature and is explained by the expulsion of oxygen functional groups, likely in the forms of CO and CO2, resulting in the disintegration of the graphene matrix.53,54 In last step, GO is finally burned out. This massive mass loss stands in contrast to the thermogram of graphite, showing that graphite was oxidised to produce GO.
Other than the small mass loss due to solvent evaporation at onset temperature of 40.71 °C, Pd/GO only has a single significant decomposition step and retains up to 70% of its mass up to 800 °C. This mass loss is likely still due to the removal of remaining labile oxygen-containing functional groups after the reduction step necessary to deposit Pd NPs. Interestingly, Pd/GO is thermally more stable than GO with no sharp drop in mass. There is, however, a slow and steady mass loss due to the decomposition of more stable oxygen functionalities, however no burn out of Pd/GO was detected within this temperature range.
The thermogram of CNT is stable over the temperature range, similar to graphite, since it also only consists of thermostable “rolled-up” carbon sheets. The thermogram of Pd/CNT/GO has the same form as Pd/GO, discussed above. Similar to Pd/GO, there is substantial mass lost due to the removal of labile oxygen-containing functional groups on GO at ca. 200 °C, as well as a slow and steady mass loss at higher temperatures. Pd/CNT/GO shows a slightly larger fraction of solid residue of ca. 75% as opposed to the ca. 70% of Pd/GO. This is attributed to the additional CNT present in the sample.
Like graphite, Fe3O4 is impressively stable at high temperatures, remaining relatively unchanged in mass. The nanocomposite Fe3O4/GO, Pd/Fe3O4/GO and Pd/CNT/Fe3O4/GO, shows an initial mass loss due to solvent evaporation, followed by a ca. 13.8% mass loss attributed to the release of CO and CO2,55 followed by a slow and steady mass loss as the temperature was ramped. For the Pd-containing nanocomposites, there is a sharp decline in mass at 614–701 °C, representing about 9% of the sample's mass. According to literature PdO is thermally stable at low temperatures, while at elevated temperatures it decomposes to Pd0 and O2.45 In our study, however, this trend is only observable when the Pd is combined with Fe3O4 in a nanocomposite (Pd/Fe3O4/GO and Pd/CNT/Fe3O4/GO).
All thermal decomposition steps of compounds as analysed by TGA are shown in Table 7. The table shows the mass lost (%) as well as the onset and offset temperatures (°C) of the respective steps.
| Compound | Step 1 | Step 2 | Step 3 | Step 4 | Total mass loss | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mass lost (%) | Onset and offset (°C) | Mass lost (%) | Onset and offset (°C) | Mass lost (%) | Onset and offset (°C) | Mass lost (%) | Onset and offset (°C) | ||
| GO | 5.62 | 45.90 | 20.95 | 150.85 | 56.81 | 406.77 | 83.38 | ||
| 142.13 | 275.70 | 636.16 | |||||||
| Pd/GO | 2.20 | 40.71 | 18.35 | 127.88 | 20.55 | ||||
| 118.38 | 282.81 | ||||||||
| Pd/CNT/GO | 1.27 | 40.59 | 13.87 | 131.06 | 1.60 | 317.40 | 16.74 | ||
| 118.61 | 277.15 | 386.29 | |||||||
| Fe3O4/GO | 1.48 | 40.66 | 14.18 | 135.54 | 15.56 | ||||
| 116.07 | 256.93 | ||||||||
| Pd/Fe3O4/GO | 7.96 | 124.61 | 8.76 | 613.98 | 16.72 | ||||
| 246.95 | 701.19 | ||||||||
| Pd/CNT/Fe3O4/GO | 0.42 | 40.28 | 8.29 | 127.26 | 2.22 | 296.63 | 7.82 | 613.61 | 18.75 |
| 113.11 | 265.42 | 387.35 | 712.35 | ||||||
X-ray Photoelectron Spectroscopy analysis (XPS) was conducted on the four nanocomposites and their precursors. In each case, the expected composite elements were detected and curves fittings was simulated accordingly. The overarching purpose of XPS analysis was quantification of elemental composition, done in conjunction with ICP results, as well as to illuminate the oxidation states of respective elements.
The C 1s, O 1s and Pd 3d areas of Pd/GO as well as the Fe 2p spectra of Fe3O4 NPs are shown in Fig. 6, as examples of the simulated curve fittings. Carbon (C 1s) spectra were typically simulated with two distinct photoelectron lines, the main peak of the simulated adventitious carbon was shifted to ca. 284.8 eV to compensate for charging. Adventitious carbon was abundant, as expected, which overlaps with sp2-hybridised carbon. The C–O bonds (that includes carboxyl, epoxide and other oxygen functionalities) of the graphene oxide structure are located at ca. 286.8 eV. This is in correlation with reported XPS data of the C–O binding energy (286.7 eV).56 An envelope curve covering all the oxygen species is simulated at ca. 532.7 eV.
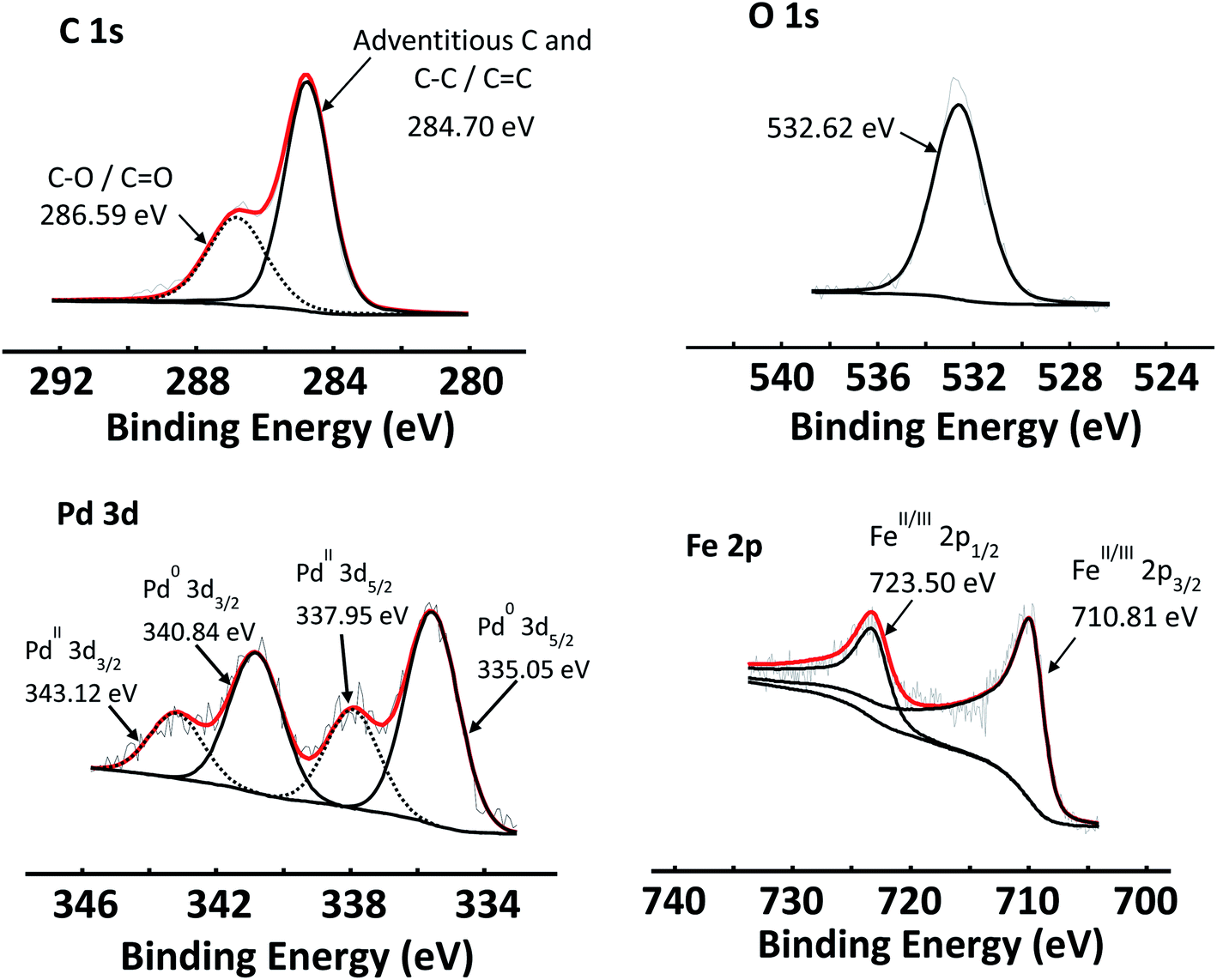 | ||
| Fig. 6 XPS spectra of C 1s, O 1s and Pd 3d photoelectron areas of Pd/GO, as well as the Fe 2p of Fe3O4 NPs. | ||
The Pd 3d photoelectron lines of all the catalysts revealed that Pd was present in both the 0 and II oxidation states in varying ratios (see Table 8). Fig. 6 shows the Pd 3d area of Pd/GO as an example. The Pd0 3d5/2 photoelectron line is located at ca. 335.6 eV, while PdII 3d5/2 photoelectron line is located at ca. 338.0 eV, correlating well with literature.57–59 See the ESI† for the Pd binding energies of each of the Pd-containing nanocomposites.
:Pd(II) for each catalyst as determined by XPS analysis. As well as Pd-content in each catalyst as determined by ICP analysis and apparent optical band energy, 
| Compound | Ratio Pd0:PdII |
% Pd0 | Ratio Fetot:Pdtot |
% Pd/collective mass |
|
|---|---|---|---|---|---|
| GO–Pd | 2.63:1 |
72.0 | — | 8.7 | 1.38 |
| GO–Fe3O4–Pd | 2.49:1 |
71.3 | 1:0.3 |
9.1 | 2.89 |
| GO–Fe3O4–Pd (spent) | 7.12:1 |
87.7 | 1:0.2 |
— | — |
| GO–CNT–Pd | 2.76:1 |
73.0 | — | 8.1 | 1.63 |
| GO–Fe3O4–CNT–Pd | 3.36:1 |
77.1 | — | 7.9 | 2.79 |
One of the primary uses of XPS is to elucidate the oxidation states of elements. This is especially helpful concerning metals like Pd, since in this study Pd NPs constitute the catalyst's active site, and certain oxidation states can be selectively better suited for catalysis. The oxidation state ratios of Pd(0) to Pd(II) among the catalysts were found to vary from 2.49:1 to 3.36:1 (see Table 8). The two CNT-containing nanocomposites showed marginally higher Pd0 content, this phenomenon is also reported in literature.60
From Table 8's second and third entries it can be deduced that not only did the Pd to Fe ratio of Pd/Fe3O4/GO decrease after catalytic use, but it also underwent significant reduction from Pd(II) to Pd(0), with the initial oxidation state ratio of 2.49:1 of Pd(0) to Pd(II) changing to 7.12:1. It is reasonable to expect that the amount of oxygen-moieties on the graphene sheets also decreased, although this was not measured.
In reference to Fig. 6, the Fe 2p photoelectron lines of all the Fe3O4-containing samples were simulated with two main photoelectron envelopes at ca. 710.8 eV and 723.0 eV for the Fe 2p3/2 and Fe 2p1/2 photoelectron lines with a spin orbit splitting of 12.2 eV. In literature the Fe 2p3/2 photoelectron lines of Fe3O4 is normally reported as one peak and not deconvoluted. The binding energies determined here, correlates well with reported binding energy positions.61,62 It has been reported in literature that Fe 2p3/2 photoelectron lines of Fe3O4 (alternatively written as FeO·Fe2O3) does not present with a shake-up feature.61 The absence of a shake feature for the Fe 2p photoelectron lines was also established in this study.
However, the Fe 2p photoelectron lines of the Fe3O4, could be deconvoluted for the FeII and FeIII species using the fitting parameters described by Biesinger et al.63 see Fig. S10 in the ESI.†
Table 8 also contains the Pd content by mass% for each respective catalyst as determined by ICP-OES. The average Pd content is 8.6% with Pd/Fe3O4/GO having the highest Pd content (9.1%) by a small margin. Overall there is not significant variance in Pd loading.
The apparent optical band energy,  of the four nanocomposites was determined from their absorption spectra, see Table 8 and the ESI† on how it was determined and apparent optical band energy,
of the four nanocomposites was determined from their absorption spectra, see Table 8 and the ESI† on how it was determined and apparent optical band energy, 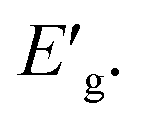
The Pd-containing nanocomposites were analysed for their catalytic activity for the reduction of nitroarene compounds in the presence of an excess of NaBH4. Three different nitrophenols used is: 4-nitrophenol (4-NP), 3-nitrophenol (3-NP) and 2-nitrophenol (2-NP), catalysis resulted in their respective aminophenols (AP). [The three starting materials and their resulting products' chemical structures are shown in Fig. S15 in the ESI†]. In each case, the reaction was followed by UV/Vis spectroscopy and a corresponding calibration curve was set up to assist with calculations (see Fig. S16 in the ESI†). The equation obtained from the calibration curve (y = 0.0797x) directly relates the observed absorbance of the 4-NP (y) to its concentration (ppm) in the reaction mixture (x). From the concentration, the amount of moles of starting material (left at a certain time) can be elucidated and used to calculate catalyst efficiency. The apparent turnover frequency (TOF′) was determined from the eqn (1):
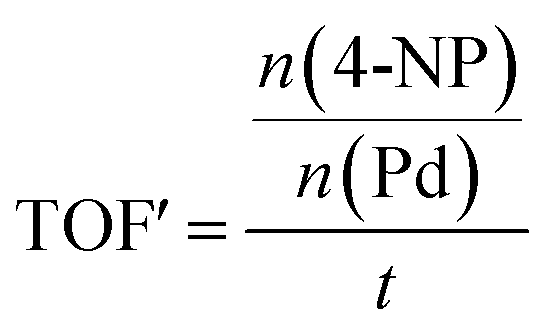 | (1) |
The term apparent is used, since the total amount of Pd present in the sample (as determined by ICP-OES) is used and not just the exposed Pd.
The amount of moles of Pd (active site) was obtained from ICP-OES results, by multiplying the mg Pd/mg catalyst with the mass of the catalyst used. For example, for Pd/GO the 0.087 mg Pd/mg catalyst was found by ICP, and 45 mg catalyst was used, thus 0.087 × 45 = 0.03915 mg Pd is present. Using the molecular mass of Pd, which is 106.42 g mol−1, the amount of moles Pd was calculated to be 3.68 × 10−7 mol. The TOF′ was calculated at time t = 100 s (100 s after the reaction was initiated) for all the nanocomposites, for ease of comparison.
Fig. 7 displays the UV/Vis absorption spectra of the first cycle of the catalytic reduction of 4-NP using Pd/Fe3O4/GO, followed over time. It shows the decline in absorbance at ca. 400 nm, representing the depletion of 4-NP. This decline is accompanied by an increase of absorbance at ca. 300 and 230 nm, representing the formation of the product, 4-AP.
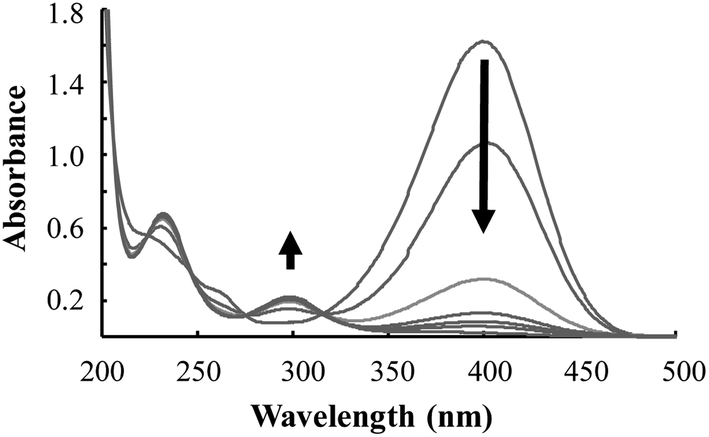 | ||
| Fig. 7 The UV/Vis absorption spectra of the reduction of 4-NP by Pd/Fe3O4/GO. | ||
Table 9 summarises the TOF′ of all the catalysts and the three different; 2-NP, 3-NP and 4-NP (bar graphs are show in Fig. S17–S20 in the ESI†). For the reduction of 4-NP, after the first cycle, the catalyst was separated from the reaction mixture (by centrifugation or the application of an external magnet), washed with distilled water and dried in vacuum to be used again. From Table 9, it is clear that for the first cycle of the reduction of 4-NP, Pd/CNT/Fe3O4/GO showed the highest catalytic activity (at the 100 s mark), while Pd/Fe3O4/GO resulted in the lowest TOF′. The addition of the Fe3O4 (which was added to render the catalyst magnetic) has a negative impact on the catalysis when comparing Pd/GO and Pd/Fe3O4/GO. The addition of CNT to Pd/GO (resulting in Pd/CNT/GO), on the other hand, has resulted in a TOF′ more than double that of Pd/GO. This implies that the accessibility to the active catalytic moiety (the Pd NPs) was greatly improved by adding the CNT or that the CNT possibly assisted in the electron transfer resulting in a higher TOF′. Interestingly, when Fe3O4 was added to the composite containing CNT, contrary to expectations, the TOF′ increased even more. However, when relating the TOF′ (of the first cycle) with the % Pd0 present, a directly proportional relationship is obtained, this implies that Pd0 is a better catalyst for the reduction of 4-NP. This is also true for 2-NP, see Fig. S21 and S22 in the ESI† for the correlations. The results obtained in this study (for the first catalytic cycle) compares well and even performs better than some reported TOF by other catalysts, Pd on Fe3O4 cellulose composite (8.3 × 10−5 s−1),64 Pd NPs supported within the cryogels (2.18 × 10−5 to 14.7 × 10−5 s−1),65 and PdCu bimetallic nanocrystals (1.31 × 10−6 and 4.25 × 10−6).66
| pKa67 | |||
|---|---|---|---|
| Nitrophenol | 2-NP | 3-NP | 4-NP |
| pKa-values | 7.23 | 8.36 | 7.15 |
| TOF′ (h−1) | ||||
|---|---|---|---|---|
| Nitrophenol | 2-NP | 3-NP | 4-NP cycle 1 | 4-NP cycle 2 |
| Pd/GO | 5.30 × 10−5 | 2.74 × 10−4 | 2.31 × 10−5 | 2.76 × 10−5 |
| Pd/Fe3O4/GO | 8.45 × 10−5 | 2.17 × 10−4 | 0.98 × 10−5 | 1.88 × 10−5 |
| Pd/CNT/GO | 2.56 × 10−4 | 8.09 × 10−6 | 5.36 × 10−5 | 1.42 × 10−5 |
| Pd/CNT/Fe3O4/GO | 8.19 × 10−4 | 3.73 × 10−5 | 8.22 × 10−5 | 2.31 × 10−5 |
All four nanocomposites were tested for a second catalytic cycle for the reduction of 4-NP after being recovered and washed. The CNT-containing nanocomposites showed a ca. 3.6 times decrease in activity (as indicated by the TOF′). Whereas, the nanocomposites, Pd/GO and Pd/Fe3O4/GO showed a slight increase in activity. Having established in the first catalytic cycle, that the Pd0 is catalytically more active than the unreduced Pd, could possibly explain the improvement of the TOF′. Since it was shown from the XPS of the used Pd/Fe3O4/GO, that the ratio of Pd0:PdII increased after the first catalytic cycle.
Even though the TOF′ for 2-NP is considerably higher (ranging from 1.9 to 35 times) than that for 4-NP, they give a similar pattern in terms of the catalytic performance of the respective nanocomposites, which is opposite to that of 3-NP. In the case of the latter it is Pd/GO, rather than Pd/CNT/Fe3O4/GO, that shows superior activity as measured by TOF′ at 100 s.
In organic chemistry it is known that the positions of substituents on a benzene ring can affect reaction reactivity. According to some sources in literature it would be expected for the meta-substituted substrate (3-NP) to be more reactive compared to para- and ortho-substituted species (4-NP and 2-NP), since its resonance structure is less stable.68–70 In this study TOF's were highest for 2-NP instead, although followed closely by those for 3-NP.
For composites Pd/GO and Pd/Fe3O4/GO, a directly proportional relationship was established between the pKa of the nitrophenols and the TOF′ values (see Table 9 and Fig. 8). The higher the pKa, the stronger the base and the greater its ability to accept a proton. Since reduction involved the reaction with H+ (proton), a higher nitrophenol pKa is expected to result in a higher TOF′.
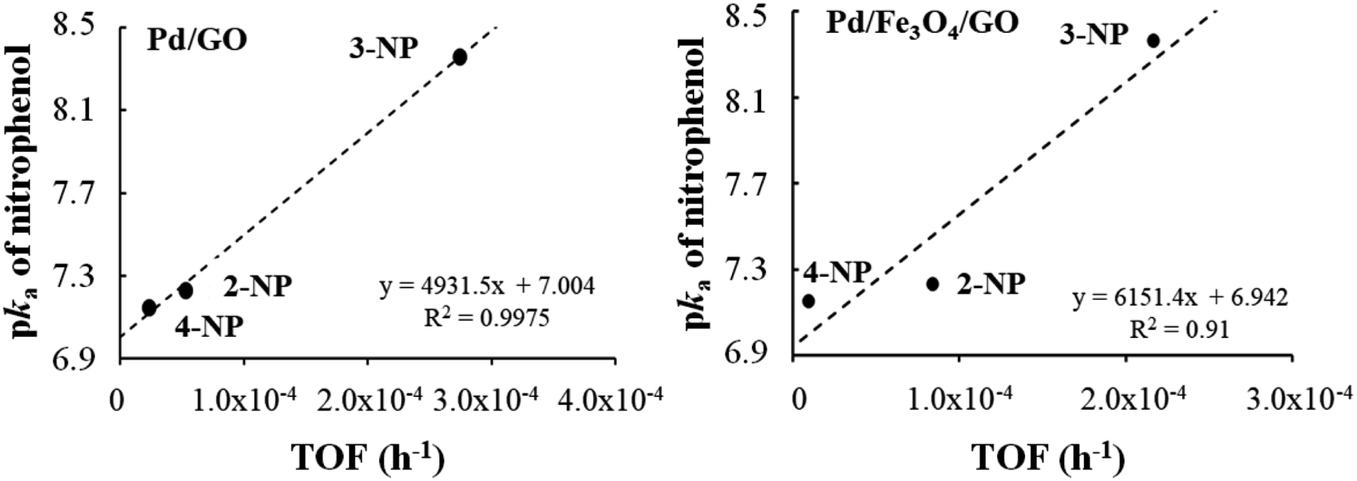 | ||
| Fig. 8 Relationship between the pKa of the nitrophenols and the TOF′ (h−1) found for nanocomposite Pd/GO–Pd (left) and Pd/Fe3O4/GO (right). | ||
Conclusion
SEM images and PXRD analysis confirmed the increase of interlayer spacing between graphene layers during the preparation of GO. While ATR-FTIR and TGA confirmed the presence of the oxygen-containing functional groups that cause this separation. It follows that, although it is clear that the interlayer spaces increased, single-layer graphene oxide was not observed.According to TEM images, the particle sizes of Fe3O4 NPs varied from less than 8 nm up to 22 nm in diameter and showed mild aggregation. More uniform dispersion can be improved by surface modification of Fe3O4 NPs, which would also improve the chemical stability of Fe3O4.19
Depositing of the catalytically active Pd NPs was confirmed by electron microscopy, since the Pd NPs were visible on the nano scale. The nanoparticles were also spherical and well dispersed with mean diameters being 11.3, and 17.2 nm for GO–Pd, and GO–CNT–Pd, respectively.
Graphite, CNT and Fe3O4 were found to be thermally stable, with the TGA showed less than 5% mass lost up to 800 °C. Therefore, when CNT and Fe3O4 were added onto the GO-based nanocomposite they contributed to the nanocomposite's thermal stability. The same was found after the addition of Pd NPs. Since all synthesized composites contained GO, they showed thermal stability up to ca. 120 °C, which is the onset of GO's decomposition.
It was found that for the reduction of 4-NP and 2-NP, Pd/CNT/Fe3O4/GO gave the highest TOF′ values, that is, 8.22 × 10−5 h−1 and 8.19 × 10−4 h−1 respectively. For the reduction of 3-NP Pd/GO showed the highest TOF′ of 2.74 × 10−4 h−1. This is not explainable by the resonance structures of the respective nitrophenols, however, some correlations between the pKa of nitrophenol and catalyst TOF′ were found in the cases of Pd/GO and Pd/CNT/Fe3O4/GO. The results indicated that Pd0 is superior to PdII for the catalysis of this specific reaction and therefore to obtain a high Pd0:PdII ratio is of paramount importance. This leads to the following point concerning recyclability.
As an example, Pd/Fe3O4/GO was analysed by XPS before and after catalysis, which showed lowering of the Pd:Fe ratio. In addition to the reduction of PdII to Pd0 which also took place during catalysis, with the initial Pd0:PdII ratio of 2.49:1 increasing to 7.12:1 post-catalysis. This explains the slight increase in catalyst efficiency for the second cycle of catalysis for Pd/GO and Pd/Fe3O4/GO since Pd0 is catalytically more active than PdII. This has major implications considering GO is the catalyst support, since it contains many oxygen functionalities and necessarily also undergoes reduction during this process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to acknowledge generous financial support from the National Nanoscience Postgraduate Teaching and Training Platform, Sasol, the NRF and UFS during the course of this study.References
- A. Ruíz-Baltazar, R. Esparza, G. Rosas and R. Pérez, J. Nanomater., 2015, 1–8 Search PubMed.
- A. R. Hajipour and Z. Tavangar-Rizi, Appl. Organomet. Chem., 2017, 31, e3701 CrossRef.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583 RSC.
- L.-M. Yang, V. Bačić, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, J. Am. Chem. Soc., 2015, 137, 2757–2762 CrossRef CAS.
- B. Song, Y. Zhou, H.-M. Yang, J.-H. Liao, L.-M. Yang, X.-B. Yang and E. Ganz, J. Am. Chem. Soc., 2019, 141, 3630–3640 CrossRef CAS.
- J.-H. Liu, L.-M. Yang and E. Ganz, ACS Sustainable Chem. Eng., 2018, 6, 15494–15502 CrossRef.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 11944–11952 RSC.
- J. Liu, L. Yang and E. Ganz, Energy Environ. Mater., 2019, 2, 193–200 CrossRef.
- J.-H. Liu, L.-M. Yang and E. Ganz, RSC Adv., 2019, 9, 27710–27719 RSC.
- L. Xu, L.-M. Yang and E. Ganz, Theor. Chem. Acc., 2018, 137, 98 Search PubMed.
- L.-M. Yang, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, Phys. Chem. Chem. Phys., 2015, 17, 17545–17551 RSC.
- L.-M. Yang, I. A. Popov, T. Frauenheim, A. I. Boldyrev, T. Heine, V. Bačić and E. Ganz, Phys. Chem. Chem. Phys., 2015, 17, 26043–26048 RSC.
- J. Yang, X. Wang, Y. Qu, X. Wang, H. Huo, Q. Fan, J. Wang, L. Yang and Y. Wu, Adv. Energy Mater., 2020, 2001709 CrossRef.
- R. Rajesh, E. Sujanthi, S. Senthil Kumar and R. Venkatesan, Phys. Chem. Chem. Phys., 2015, 17, 11329–11340 RSC.
- J. Chen, Y. Zhang, M. Zhang, B. Yao, Y. Li, L. Huang, C. Li and G. Shi, Chem. Sci., 2016, 7, 1874–1881 RSC.
- X. Ma, H. Tao, K. Yang, L. Feng, L. Cheng, X. Shi, Y. Li, L. Guo and Z. Liu, Nano Res., 2012, 5, 199–212 CrossRef CAS.
- N. Ye, Y. Xie, P. Shi, T. Gao and J. Ma, Mater. Sci. Eng., C, 2014, 45, 8–14 CrossRef CAS.
- S. Yamamoto, H. Kinoshita, H. Hashimoto and Y. Nishina, Nanoscale, 2014, 6, 6501–6505 RSC.
- P. Nayak, S. P. Nair and S. Ramaprabhu, Microchim. Acta, 2016, 183, 1055–1062 CrossRef CAS.
- W. Sun, X. Lu, Y. Tong, Z. Zhang, J. Lei, G. Nie and C. Wang, Int. J. Hydrogen Energy, 2014, 39, 9080–9086 CrossRef CAS.
- K. Shaughnessy and R. DeVasher, Curr. Org. Chem., 2005, 9, 585–604 CrossRef CAS.
- P. Zhao, X. Feng, D. Huang, G. Yang and D. Astruc, Coord. Chem. Rev., 2015, 287, 114–136 CrossRef CAS.
- W.-W. Liu, S.-P. Chai, A. R. Mohamed and U. Hashim, J. Ind. Eng. Chem., 2014, 20, 1171–1185 CrossRef CAS.
- M. A. Atieh, O. Y. Bakather, B. Al-Tawbini, A. A. Bukhari, F. A. Abuilaiwi and M. B. Fettouhi, Bioinorg. Chem. Appl., 2010, 2010, 1–9 CrossRef.
- W. W. Liu, S. P. Chai, A. R. Mohamed and U. Hashim, J. Ind. Eng. Chem., 2014, 20, 1171–1185 CrossRef CAS.
- K. Yan, T. Lafleur and J. Liao, J. Nanopart. Res., 2013, 15, 1906 CrossRef.
- S. H. Aboutalebi, S. Aminorroaya-Yamini, I. Nevirkovets, K. Konstantinov and H. K. Liu, Adv. Energy Mater., 2012, 2, 1439–1446 CrossRef CAS.
- S. H. Aboutalebi, S. Aminorroaya-Yamini, I. Nevirkovets, K. Konstantinov and H. K. Liu, Adv. Energy Mater., 2012, 2, 1439–1446 CrossRef CAS.
- Y. Pan, H. Bao and L. Li, ACS Appl. Mater. Interfaces, 2011, 3, 4819–4830 CrossRef CAS.
- F. Yang, A. Feng, C. Wang, S. Dong, C. Chi, X. Jia, L. Zhang and Y. Li, RSC Adv., 2016, 6, 16911–16916 RSC.
- W. Zhang, X. Li, R. Zou, H. Wu, H. Shi, S. Yu and Y. Liu, Sci. Rep., 2015, 5, 11129 CrossRef.
- K.-S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS.
- X. Zhou, J. Qiao, L. Yang and J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 CrossRef.
- V. K. Gupta, N. Atar, M. L. Yola, Z. Üstündağ and L. Uzun, Water Res., 2014, 48, 210–217 CrossRef CAS.
- Y.-C. Chang and D.-H. Chen, J. Hazard. Mater., 2009, 165, 664–669 CrossRef CAS.
- X. Kong, H. Zhu, C. Chen, G. Huang and Q. Chen, Chem. Phys. Lett., 2017, 684, 148–152 CrossRef CAS.
- L. K. Parrott and E. Erasmus, Catal. Lett., 2018, 148, 2008–2018 CrossRef CAS.
- E. Botha, M. Landman, P. H. van Rooyen and E. Erasmus, Inorg. Chim. Acta, 2018, 482, 514–521 CrossRef CAS.
- E. Erasmus, J. Electron Spectrosc. Relat. Phenom., 2018, 223, 84–88 CrossRef CAS.
- S. J. Gerber and E. Erasmus, Mater. Chem. Phys., 2018, 203, 73–81 CrossRef CAS.
- S. J. Gerber and E. Erasmus, Transition Met. Chem., 2018, 43, 409–420 CrossRef CAS.
- R. Rajesh and R. Venkatesan, J. Mol. Catal. A: Chem., 2012, 359, 88–96 CrossRef CAS.
- A. Misra, P. K. Tyagi, M. K. Singh and D. S. Misra, Diamond Relat. Mater., 2006, 15, 385–388 CrossRef CAS.
- A. Misra, P. Tyagi, P. Rai and D. S. Misra, J. Nanosci. Nanotechnol., 2007, 7, 1820–1823 CrossRef CAS.
- T. Yan, H. Zhang, D. Huang, S. Feng, M. Fujita and X.-D. Gao, Nanomaterials, 2017, 7, 59 CrossRef.
- Z. Çiplak, N. Yildiz and A. Çalimli, Fullerenes, Nanotubes, Carbon Nanostruct., 2015, 23, 361–370 CrossRef.
- N. B. A. Nanditha and J. Manokaran, Res. J. Chem. Environ., 2014, 18, 54–61 Search PubMed.
- L. Yang, J. Tian, J. Meng, R. Zhao, C. Li, J. Ma and T. Jin, Molecules, 2018, 23, 562 CrossRef.
- K. Zhang, Y. Zhang and S. Wang, Sci. Rep., 2013, 3, 3448 CrossRef.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- S. N. Alam, N. Sharma and L. Kumar, Graphene, 2017, 06, 1–18 CrossRef CAS.
- L. Li, M. Chen, G. Huang, N. Yang, L. Zhang, H. Wang, Y. Liu, W. Wang and J. Gao, J. Power Sources, 2014, 263, 13–21 CrossRef CAS.
- R. Justin, K. Tao, S. Román, D. Chen, Y. Xu, X. Geng, I. M. Ross, R. T. Grant, A. Pearson, G. Zhou, S. MacNeil, K. Sun and B. Chen, Carbon, 2016, 97, 54–70 CrossRef CAS.
- F. T. Johra, J.-W. Lee and W.-G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- H.-Z. Tan, Z.-Q. Wang, Z.-N. Xu, J. Sun, Z.-N. Chen, Q.-S. Chen, Y. Chen and G.-C. Guo, Catal. Sci. Technol., 2017, 7, 3785–3790 RSC.
- E. Erasmus, J. W. Niemantsverdriet and J. C. Swarts, Langmuir, 2012, 28, 16477–16484 CrossRef CAS.
- E. Erasmus, S. Afr. J. Chem., 2013, 66, 216–220 CAS.
- V. M. Shinde, E. Skupien and M. Makkee, Catal. Sci. Technol., 2015, 5, 4144–4153 RSC.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- E. Erasmus, J. Claassen and W. Van der Westhuizen, Water SA, 2016, 42, 442 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- T. Baran, Catal. Lett., 2019, 149, 1721–1729 CrossRef CAS.
- D. Berillo and A. Cundy, Carbohydr. Polym., 2018, 192, 166–175 CrossRef CAS.
- H. Park, D. A. Reddy, Y. Kim, S. Lee, R. Ma, M. Lim and T. K. Kim, Appl. Surf. Sci., 2017, 401, 314–322 CrossRef CAS.
- K. A. Kacprzak, I. Czekaj and J. Mantzaras, Phys. Chem. Chem. Phys., 2012, 14, 10243 RSC.
- R. Begum, R. Rehan, Z. H. Farooqi, Z. Butt and S. Ashraf, J. Nanopart. Res., 2016, 18, 231 CrossRef.
- H. Kang, M. Kim and K. H. Park, Appl. Catal., A, 2015, 502, 239–245 CrossRef CAS.
- Y. Tuo, G. Liu, B. Dong, J. Zhou, A. Wang, J. Wang, R. Jin, H. Lv, Z. Dou and W. Huang, Sci. Rep., 2015, 5, 13515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04715e |
| This journal is © The Royal Society of Chemistry 2020 |