
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSide-by-side comparison of Notch- and C83 binding to γ-secretase in a complete membrane model at physiological temperature†
Budheswar Dehuryab,
Ning Tanga,
Rukmankesh Mehra a,
Tom L. Blundellb and
Kasper P. Kepp*a
a,
Tom L. Blundellb and
Kasper P. Kepp*a
aDepartment of Chemistry, Technical University of Denmark, DK-2800 Kongens Lyngby, Denmark. E-mail: kpj@kemi.dtu.dk; Tel: +45 45252409
bDepartment of Biochemistry, University of Cambridge, Tennis Court Road, CB2 1GA, UK
First published on 24th August 2020
Abstract
γ-Secretase cleaves the C99 fragment of the amyloid precursor protein, leading to formation of aggregated β-amyloid peptide central to Alzheimer's disease, and Notch, essential for cell regulation. Recent cryogenic electron microscopy (cryo-EM) structures indicate major changes upon substrate binding, a β-sheet recognition motif, and a possible helix unwinding to expose peptide bonds towards nucleophilic attack. Here we report side-by-side comparison of the 303 K dynamics of the two proteins in realistic membranes using molecular dynamics simulations. Our ensembles agree with the cryo-EM data (full-protein Cα-RMSD = 1.62–2.19 Å) but reveal distinct presenilin helix conformation states and thermal β-strand to coil transitions of C83 and Notch100. We identify distinct 303 K hydrogen bond dynamics and water accessibility of the catalytic sites. The RKRR motif (1758–1761) contributes significantly to Notch binding and serves as a “membrane anchor” that prevents Notch displacement. Water that transiently hydrogen bonds to G1753 and V1754 probably represents the catalytic nucleophile. At 303 K, Notch and C83 binding induce different conformation states, with Notch mostly present in a closed state with shorter Asp–Asp distance. This may explain the different outcome of Notch and C99 cleavage, as the latter is more imprecise with many products. Our identified conformation states may aid efforts to develop conformation-selective drugs that target C99 and Notch cleavage differently, e.g. Notch-sparing γ-secretase modulators.
Introduction
Alzheimer's disease (AD), the major neurodegenerative disease that leads to gradual loss of episodic memory, cognition, life quality and autonomy, affects tens of millions of people worldwide, yet its biochemical complexity has so far prevented causal treatment, with only modest delays in disease progression offered by current drugs on the market.1–3 The consistent failures of much heralded drug candidates has led to a surge in fundamental research rethinking the causal mechanisms of this devastating disease.4–9Senile plaques in patient brains consisting of aggregated β-amyloid (Aβ) are a central pathological hallmark of AD.10–16 The Aβ peptides are formed by sequential cleavage of β-amyloid precursor protein (APP), first by β-secretase which removes the ectodomain and produces the C-terminal fragment C99, and then by the catalytic presenilin (PS) subunit of γ-secretase, which trims C99 to Aβ within the membrane.15,17–19 Because of the slow, consecutive trimming of C99, the released Aβ peptides differ in size.15,20–23 The shorter Aβ peptides, notably Aβ38 and Aβ40, are probably benign, whereas longer Aβ42 and Aβ43 peptides are more hydrophobic, aggregation-prone, membrane-interacting, and toxic to cells.12,24,25 Mutations in the genes coding for APP, PS1, and PS2 cause early-onset familial AD, which supports the involvement of Aβ.26–29 The most persistent feature of these mutations is an increase in the Aβ42/Aβ40 ratio resulting from C99 cleavage,15 which correlates with the clinical severity of mutation, implying a clear relationship to disease.30–33
The γ-secretase complex also cleaves the Notch receptor and thereby initiates one of the most important signalling pathways in eukaryote cells, central to multiple cell differentiation processes, embryogenesis, and neuronal function and neuron death.34–37 Cleavage of Notch by γ-secretase occurs in a manner similar to C99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 and releases the Notch intracellular domain (NICD), which plays an important role in learning and memory.37,39 As for C99, Notch-cleavage can occur at multiple positions near the intracellular membrane border, but most of the resulting fragments are rapidly degraded according to the N-end rule.39,40 A number of studies have reported that atypical Notch signalling results in neurodevelopment defects, cancer, and acne inversa.41,42
38 and releases the Notch intracellular domain (NICD), which plays an important role in learning and memory.37,39 As for C99, Notch-cleavage can occur at multiple positions near the intracellular membrane border, but most of the resulting fragments are rapidly degraded according to the N-end rule.39,40 A number of studies have reported that atypical Notch signalling results in neurodevelopment defects, cancer, and acne inversa.41,42
γ-Secretase consists of four subunits: PS1 or PS2, presenilin enhancer 2 (PEN-2), anterior pharynx defective 1A or 1B (APH-1A, APH-1B), and nicastrin (NCT).43,44 The catalytic subunit PS1/PS2 contains nine transmembrane helices (TM) and harbours the two catalytic aspartate residues (Asp257 in TM6 and Asp385 in TM7), which constitute the active site of γ-secretase.18,45,46
γ-Secretase is a primary therapeutic target for AD as it controls the Aβ production: assuming that inhibition of γ-secretase activity would lower Aβ production and thus be beneficial for AD, a large number of γ-secretase inhibitors have been developed.47–50 Unfortunately, none of them have shown significant endpoints, and side effects were observed in some trials.4 This failure can be due to several reasons: one is that the amyloid hypothesis is incorrect or should be supplemented, for example by accounting for metabolic and inflammatory pathways.6,7,51,52 Another is that Aβ is involved in AD but cannot be reduced to this role indiscriminately as it serves important functions in the brain.53,54 A third is that the amyloid hypothesis is correct but that inhibition of γ-secretase prevents the cleavage of substrates such as Notch.55–58 Non-selective γ-secretase inhibition may thus in fact contribute to cognitive decline.59
Realizing these problems and staying with the amyloid hypothesis, Notch-sparing γ-secretase modulators have become a major research focus.5,60 However, the molecular basis of selectivity of such γ-secretase modulators is not understood, and this limits their further development.61 Since the dynamic conformational changes in both enzymes and substrates are critical to the intramembrane proteolysis and thus, the molecular basis for selective Notch-sparing drugs, a strict comparison of the normal-temperature dynamics of the wild-type protein–substrate complexes in a complete membrane is of interest. The present paper reports such comparative molecular dynamics simulations (MD) as an effort to aid Notch-sparing drug-design.
This aim is of particular current interest and feasibility since new structures of γ-secretase bound to the shorter C83 analogue of C99 and Notch-100 have been solved by cryo-electron microscopy (cryo-EM) in 2019, revealing major changes upon substrate binding of the two substrates.62,63 The corresponding dynamics at physiologically relevant temperature on this structural background are of major interest, since some potentially important conformational states freeze out in the cryo-EM samples.64 The cryo-EM structures also necessarily feature cysteine mutations for cross-linking the substrate and mutation of a catalytic aspartate to alanine (D385A) to avoid substrate turnover.
The present study reports the computer-simulated dynamics of wild-type (after restoring the engineered mutations) C83- and Notch-100-bound γ-secretase in lipid bilayers, evaluated by triplicate MD simulations at the microsecond time scale. The topologies and backbone coordinates agree excellently with the cryo-EM data, but additionally reveal substantial and distinct thermal conformational dynamics of the regulatory loops and PS1 helices crucial for substrate binding, including distinct lipid binding sites and effects of pathogenic mutations on the stability of both the C83 and Notch-100 γ-secretase complexes. Most notably, the Notch-bound protein remains consistently in the closed state with shorter Asp–Asp distance and inferred more complete cleavage in the wild type. We identify these differences as those that should be central to any drug design of Notch-sparring γ-secretase modulators for treatment of AD.
Computational methods
Model preparation
This work presents new all-atom simulations of the γ-secretase–Notch complex in a realistic membrane model. Comparison is made throughout to simulations of the C83 complex adopted from a previous study of 6IYC at 303 K.64 The present work followed the exact same protocol in order to enable a strict comparison of the normal temperature dynamics for the two substrates. The cryo structure features C83, which binds more tightly than the natural substrate C99. When the structure came out, we set out to isolate the possible composition effects of (1) missing membrane (this membrane protease is only active in the membrane, the cryo data do not include a full bilayer); (2) temperature dynamics (the cryo data represent the global minima, with other modes frozen out); (3) mutation (the protein is mutated as discussed above, the real protein is wild type); (4) substrate model (the natural substrate is C99, the cryo substrate is C83). All effects must be separated out one at the time. We have systematically studied all these effects to understand the effect of the various composition approximations.64,65 The present data represent the missing comparison directly relevant to the experimental data (but changed to wild type). We have emphasized direct comparison of experimental 6IYC and 6IDF for the wild type proteins, avoiding the potential impact of the C99/C83 modification, which remains theoretical.Specifically, the cryo-EM structure of γ-secretase with Notch100 (6IDF)63 bound was processed using BIOVIA Discovery Studio Visualizer version 4.5 (BIOVIA DSV). First, the heteroatoms including ligands and lipids were removed as they represent fragmented coordinates of the complex chemical composition on the sample film. Both in our previous64 and present work, the mutations Q112C, D385A (in PS1 of 6IYC and 6IDF), and P1728C (in Notch-100 of 6IDF) were modelled back to the wild type (WT) residues using the mutagenesis protocol of the PyMOL program.66 The missing side chains of the visible backbone residue atoms in the two structures were modelled using the WHAT-IF web server.67,68 Finally, the structure was checked for bad contacts and the refined structures were used to prepare lipid bilayer systems. The catalytic residues in the cryo-EM-structures 6IYC and 6IDF represent the mutants with one Asp changed to Ala to avoid substrate turnover during data collection, and are deprotonated states without a charge relay, as shown previously.64 All pKa values computed so far both for the experimental structures and for different simulated conformation states maintain that both aspartates should be deprotonated, although this may change e.g. upon ion binding.64 We also emphasize that the prediction method may not account accurately for the role of proton charge relays in hydrogen-bonded systems, which are also inaccessible to experiment at this point due to the mutation requirement discussed above.
The complete membrane-protein–water systems used for MD simulation were prepared using the CHARMM-GUI server (http://www.charmm-gui.org/).69 The two enzyme–substrate complexes were orientated using the PPM server (http://opm.phar.umich.edu).70 The POPC lipid membrane dimensions were built based on an XY length of 115 Å for proper embedding of the complete enzyme–substrate complexes. Both systems were arranged in rectangular boxes because of the large NCT subunit, which makes simulation cells with explicit all-atom lipid and water atoms very large. The length of the Z axis was defined based on the following parameters: water thickness 22.5 nm; hydration number (number of water molecules per one lipid molecule) 50; and percent ratio of water/lipid ratio 50% w/w. The systems were built using the replacement method with lipid ring penetration checking. Salt strength can substantially affect the electrostatics and conformational properties of proteins71–74 and may thus change the conformation-sensitive of γ-secretase dynamics.75 Accordingly, the systems were converted to a physiological (0.15 M) NaCl concentration using the Monte Carlo placement method.
Molecular dynamics simulations
The files produced as described above were used as input to the GROMACS molecular dynamics program version 2018.5.76 Parameters and topology were assigned according to the CHARMM36m force field77 and default CHARMM TIP3P water model,78,79 which was used for simulation as in previous work, to enable strict comparison.64,65,80–82 The systems were energy minimized for 5000 steps. Subsequently, we pre-equilibrated the system with two instances of temperature coupling for 25000 steps each (with a time interval of 1 fs), then two steps of temperature and pressure coupling with 25000 steps each (1 fs), and finally, two 50000-step equilibrations with a time interval of 2 fs at constant temperature and pressure. In all systems Nose–Hoover temperature thermostat83 was applied to maintain temperature with a time constant of 1 ps and the semi-isotropic Parrinello–Rahman barostat applied to maintain the pressure at 1 bar with a coupling time constant of 5 ps. All bonds to hydrogen atoms were constrained by the LINCS algorithm.84
In all cases, we used the Verlet cut-off scheme when calculating the long-range non-bonded interaction energies and the Particle Mesh Ewald (PME) to calculate the electrostatic potential energies with periodic boundary conditions. The parameters used for long-range interactions were a non-bonded cut-off of 12 Å and a force-based switch parameter value of 10 Å. The final MD production simulations were performed in triplicate for both systems, with each triplicate simulation running for 500 ns with a time-step of 2 fs. The six simulations were initiated using random velocity seeds to make them statistically independent (time-uncorrelated). All simulations were performed at a near-physiological temperature of 303.15 K, because the dynamics of conformation-sensitive membrane proteins are quite sensitive to temperature.64
Dynamic trajectory analysis
For all six simulated systems, the intrinsic stabilities were monitored by plotting the backbone root-mean-squared deviations (RMSD) of the structures from their respective starting configurations, the radius of gyration (Rg), and the solvent accessible surface area (SASA). In addition, the root-mean-squared fluctuations (RMSF) of the protein Cα atoms, the secondary structure of the C83, Notch100 and PS1 subunits, and the conformation states obtained from principal component analysis (PCA) were analyzed using the final equilibrated trajectories from 200–500 ns, since the first part of MD trajectories of large systems carries artefacts from the start configuration setup.The collective motions of the protein–membrane–water systems were analysed by extracting the top principal components (PCs) using the GROMACS utility toolkits applied to the main-chain atoms, and MODE-TASK.85 We performed singular value decomposition PCA on alpha carbons (1355 atoms for C83 and 1363 for Notch100) for both systems, and the trajectories were prepared by removing periodicity and water molecules. The lipid binding occupancy was determined using VMD.86 The water molecules accessing the catalytic residues within 5 Å were identified. To understand the water hydrogen-bond dynamics in the catalytic site, we computed the number of hydrogen bond forming amino acids. RMSD-based clustering was performed using the cluster analysis method implemented in Gromacs,87 with a cut-off of 0.2 nm.
The membrane parameters, including deuterium order parameters and the densities of the membrane components, were analysed over the full trajectories using gmx order and gmx density modules of GROMACS. We were specifically interested in assuring the realism of the protein–membrane system and the lipid membrane and thus computed the density profile of the POPC head, tail, and phosphate groups as well as the TIP3P water. The membrane area, area per lipid and membrane thickness were calculated using the FATSLiM program.88
Binding free energies
The binding free energies of the substrate-bound γ-secretase–complexes were calculated using the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method.89 We employed the g_mmpbsa program90 to compute the ΔGbind of each complex using 500 snapshots obtained from the last 300 ns trajectories at equal interval of time. The free energy of binding involves the components described in eqn (1):| G = Gbond + Gele + GvdW + Gpol + Gnpol − TS | (1) |
Computing stability effects (ΔΔGMUT) of pathogenic PS1 mutations
In order to evaluate whether the structural differences between the bound substrates have any probable energy effects of significance, we computed the pathogenic non-synonymous mutations of the TM region of PS1, following the procedure previously described.91 Specifically, we computed the change in folding free energy (ΔΔGMUT, measured in kcal mol−1) upon mutation using the four well-known structure-based methods I-Mutant 3.0,92 FoldX 5.0,93 POPMUSIC 2.1,94 and mCSM.95 We computed ΔΔGMUT both for the two experimental structures (6IYC and 6IDF converted into wild type, both otherwise unchanged) and for the representative structures obtained from clustering analysis of our three independent simulations for each protein–substrate complex. In case of I-Mutant, the ΔΔGMUT were computed using the default parameters. FoldX calculations were carried out using the repair function with and the average ΔΔGMUT were based on five runs. Since the methods use different sign conventions, for comparison in the main paper, we used a common sign convention of ΔΔGMUT > 0 being destabilizing; all results reported follow this convention.Results and discussion
Normal-temperature dynamics in a lipid bilayer
The all-atom MD simulations reported in the present work supplement the cryo-EM data: while maintaining closely the experimental structural coordinates, the simulated structures represent the wild-type systems, not the cysteine mutant and in particular the catalytic D385A mutation, which is likely to affect the substrate binding dynamics. The cryo data represent a rapidly frozen sample mainly in the global conformation state, with minor conformations with fast time scales frozen out. Even if partly accounted for during image processing and model building, the dynamics are more meaningfully discussed at physiological temperature.64,96,97 The membrane also plays an important role in the protein–substrate dynamics98 that increases at physiological temperature,64 and this needs to be assessed beyond the mixed chemical composition of the experimental cryo sample. Since the substrate dynamics are the key to understand C99 and Notch processing and thus, the molecular basis for selective Notch-sparing drugs, a strict comparison of the normal-temperature dynamics of the wild-type protein–substrate complexes in a complete membrane is of interest.The studied γ-secretase–C83 and γ-secretase–Notch complexes, shown in Fig. 1A and B respectively, were simulated each in triplicate for ½ μs, for a total of 3 μs. A detailed comparison of the 303 K-temperature dynamics of the TM substrate domains (sequences in Fig. 1 below the structures) is of particular interest. Whereas large loop motions and major conformational changes (e.g. partial unfolding events) occur on longer time scales, the helix–helix interactions that control the catalytic pocket and substrate processing occur on the 100 ns time scale,99 as confirmed by repeated transitions and broad helix tilt angle distributions and Asp–Asp distances on these time scales.65,81 According to the FIST (Fit–Stay–Trim) model, these helix motions convert between the open substrate-binding site (weak “grabbing” by the “fist”) giving imprecise and reduced substrate cleavage, releasing longer toxic Aβ, and the semi-open state (with tighter “grabbing”) that fits substrates better and gives more precise and extensive cleavage, and thus shorter Aβ.60,65,80,91,100
 | ||
| Fig. 1 Simulated γ-secretase models bound to C83 and Notch100 within a lipid bilayer. (A) Wild-type γ-secretase–C83 complex. (B) Wild-type γ-secretase–Notch100. The subunits nicastrin (green), PS1 (blue), APH1-A (magenta), PEN2 (yellow), and the substrates C83/Notch100 (orange) are coloured differently. The small spheres represent the head groups of the POPC molecules. The trans-membrane sequences of the two substrates are shown below the structures (acc: accessibility and hyd: hydrophobicity). The arrows show the position of the cleavage sites in C83 and Notch100. The images were generated using ENDScript2.0. | ||
To capture these effects as accurately as possible, we simulate the systems with all atoms including hydrogens, since the helix–helix side chain interactions are dominated by hydrogen encounters in the hydrophobic, salt bridge, and hydrogen bond interactions. Coarse-grained models at longer time scales importantly assess overall topological dynamics but do not capture these interaction energies and thus cannot describe the substrate binding dynamics accurately without hydrogens present. Thus, for our specific purpose, explicit all-atom dynamics at 500 ns are the relevant trade-off between system compositional precision and sampling extent.
Below, we discuss in detail the similarities and differences of C83 and Notch100 dynamics, with emphasis on substrate secondary structure, the conformations of the catalytic aspartates and the size of the active site, the dynamics of regulatory hydrophilic loop 1 (HL1) and the substrate recruiting PAL motif. This analysis may serve as a normal-temperature membrane-specific picture of the dynamics of substrate binding as a supplement to the cryo-EM data, which may hopefully aid the understanding of the relative binding affinity of the two substrates to γ-secretase, which we consider central to any development of effective Notch-sparing γ-secretase-based therapies.
In order to test the realism of the overall simulated membrane-embedded γ-secretase–substrate systems, we computed the membrane order parameters, the density profile of the POPC bilayer, and the membrane thickness and area per lipid (Fig. S1†). The calculated order parameters are in agreement with the experimental101 and MD-simulated102,103 data for POPC membrane systems. Specifically, the computed Sn1 parameters displayed a minimum at the third carbon position and an extreme at carbons 5–6, and then a downward shift along the palmitoyl chain. We also observed a double-bump structure with a dip at the carbon 10 position in Sn2 parameters, which is also seen experimentally.101 As expected we observed a slight deviation in the Scd for unsaturated double bonds in the sn-2 oleoyl tail, probably due to the incorrect prediction by the gmx tool. In addition, we also observed slightly larger area per lipid as compared to pure bilayers,103,104 which may be due to the number of lipids used in our study and the very large γ-secretase complex interacting with the lipids via many discrete TM residues. Except for these minor variations, the data in Fig. S1† show that the lipid bilayer model provides a realistic basis for estimating the impact of the membrane on the substrate binding dynamics on the background of the experimental cryo-EM structures.
The conformational stability of the membrane-embedded proteins was evaluated by computing the backbone RMSD (Fig. 2), Rg, and SASA relative to the starting structure of each simulation (see ESI text and Fig. S2–S4†). These data indicate that each system attained equilibrium, with excellent agreement between the computed and experimental backbone atom coordinates of the transmembrane domains.
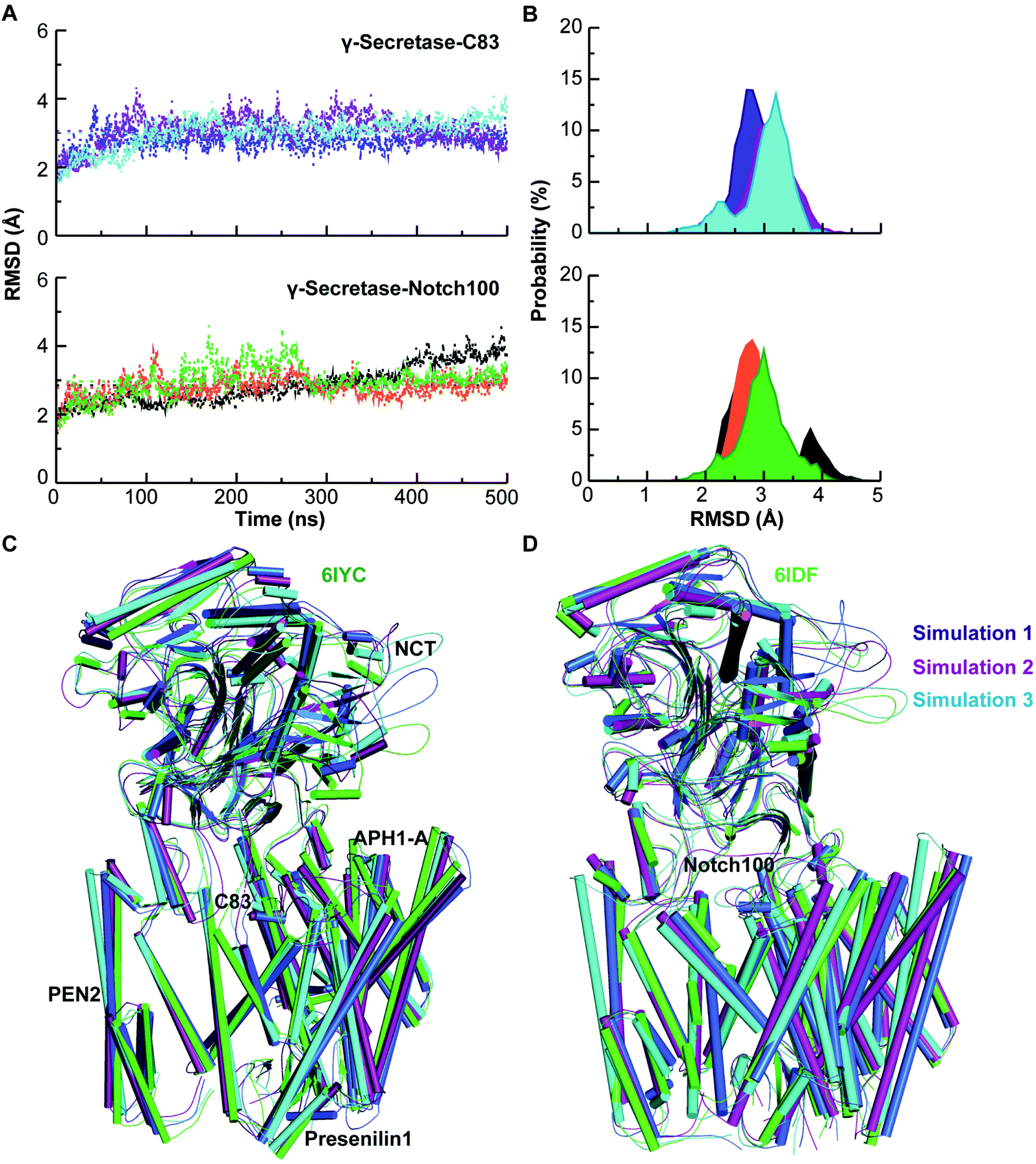 | ||
| Fig. 2 Dynamical stability and comparison of experimental and simulated structures. (A) Backbone Cα-RMSD of triplicate γ-secretase–C83 and Notch100 simulations relative to the initial structure. Blue, purple and cyan lines represent simulation 1, 2, and 3 for γ-secretase–C83. Black, orange, and green lines represent simulation 1, 2, and 3 for γ-secretase–Notch100. (B) RMSD of γ-secretase–C83 and γ-secretase–Notch100. (C) Experimental γ-secretase–C83 (6IYC, green) compared to structures for simulation 1 (blue), 2 (purple), and 3 (cyan). (D) Experimental γ-secretase–Notch100 (6IDF, green) compared to simulation 1 (blue), 2 (purple), and 3 (cyan). | ||
Dynamics of substrate-bound γ-secretase subunits
To identify differences in the structural dynamics, we computed the residue-wise Cα RMSF of each subunit of the γ-secretase–substrate complexes averaged over each trajectory of the three triplicate simulations (Fig. S5†). The RMSF of NCT-ECD in γ-secretase–C83 displayed several high peaks as expected and discussed above, whereas the region Phe240–Glu245, which interacts with the extracellular part of C83, displayed small fluctuations (1.3–1.6 Å). NCT-ECD motions are important because they likely affect the dynamic stability of γ-secretase–Aβ assemblies and thereby modulate Aβ length,105 and some γ-secretase modulators tend to bind at the extracellular interface between NCT and PS1.106 The dynamics of Ser241 and Ile242 in the proximity of NCT interacting with Lys12 of C83 (corresponding to Lys699 in APP) were explored by measuring the minimum distance between these residues (Fig. S6†). The substantial thermal fluctuations should be viewed in the light that the C83 structure has some missing residues and cross-linking of PS1 (Q112C) to the C83 (V8C) might affect this region. Some γ-secretase modulators are reported to bind in TM1 and HL1 of PS1 close to NCT (Fig. S6†), which interacts with the TM of APP.105,107 The binding induces a conformational change in PS1.108 Unlike PS1 fAD mutations, γ-secretase modulators probably increase the stability of the γ-secretase–Aβ complex and decrease the premature dissociation of Aβ, which results in additional trimming of the substrate.109In γ-secretase–C83, the hydrophilic loop 2 (HL2) and the extended PS1–TM6a fluctuations were larger than in γ-secretase–Notch100, i.e. these dynamics are markedly different. In contrast, the dynamics of the hydrophilic extracellular loop HL1 involved in substrate gating110 was relatively similar for both enzyme–substrate complexes, although it represents the binding sites of some γ-secretase modulators.111 Nevertheless, we expect a selective drug to engage the two enzyme–substrate complexes differently, as discussed below.
In PS1, we generally observed large movements of TM2, TM6a and TM9, consistent with previous findings.65,80,112 Tominaga and co-workers found that the distance between TM6 and TM7 relate to Aβ42 production.113 In both systems, the CTF of PS1 fluctuated less than the NTF. This is probably because a large part of C83 TM is surrounded by the cavity formed by TM2, TM3, and TM5 of the NTF. The TMs of both substrates are relatively inflexible because the membrane dampens their motions. However, the extracellular N-terminal of Notch100 (positioned outside the membrane), which harbours a small helix, is more flexible than C83, in agreement with NMR data for the free substrates.114,115 In contrast, the Cα-RMSF values for the catalytic Asp257 and Asp385 and the PAL (Pro433–Leu435) motif indicate that the C83-complex is distinctly more flexible than the Notch100-complex, which could be relevant for distinguishing the two modes of substrate recruitment and binding. We note that the C83 N-terminal region has five missing residues (D694–698N), but since they are located in the extracellular space and highly flexible,65 this is unlikely to impact the local substrate TM dynamics. As compared to the apo form (5FN2), the four sub-units of γ-secretase-substrate bound conformations displayed similar trends in RMSF with a few exceptions in the loop regions bridging the TM helices.116
Multi-state ensembles of C83 and Notch100–γ-secretase complexes
To characterize the overall conformational states and capture the coupled motions, we employed PCA (for details see Materials and methods section†). As shown in Fig. S7,† PC1, PC2 and PC3 represent most of essential motions of the enzyme–substrate complexes. The trace values of the co-variance matrix of C83 systems were 28.55, 28.29 and 36.11 nm2 whereas for the Notch100 it was 38.27, 25.18 and 38.19 nm2 indicating multi-state behaviour of both systems. As shown from the projected PCs (Fig. S8 and S9†), both protein complexes are clearly multi-state systems with at least three major conformational states. One state is caused by the NCT breathing mode and another by the absence of the maturation loop HL2 that releases the PS1-TM movements required for substrate processing, which is not possible in the immature state with an intact HL2, before autoproteolysis (all proteins studied here are in the mature form with N-terminal and C-terminal PS1).112Porcupine plots based on PC1 (Fig. 3) and PC2 (Fig. S10†) quantify these differences, most notably the NCT-ECD inward-outward breathing mode, which probably is responsible for the now well-established role of NCT in substrate recognition.65,117 The dynamic conformational behaviour of NCT-ECD observed in our study is in agreement with the MD studies of apo-form γ-secretases.116,118 We observed differential flexibility in PEN2 (Fig. 3C and F) as compared to the other subunits. Bai et al.75 reported that γ-secretase can exist in different conformational states with respect to the distance between catalytic aspartates and the relative position of PS1 and PEN-2. We also observed tilting of APH-1A, in agreement with the cryo-EM structures (Fig. 3A, D, E and F). In contrast, the TM segments of PS1, which are positioned close to PEN-2 and APH-1A, displayed stable interactions during most of the simulation time. PEN-2 and APH-1A are likely to substantially aid PS1 stabilization and activation.119 Kaiser–Meyer–Olkein (KMO) indices and cosine contents of the first PCs ranged from 0.72 to 1 indicating acceptable local sampling, while the squared cosine content (0.58, 0.58, 0.86 for PC1 in C83 and 0.71, 0.64 and 0.70 for PC1 in Notch100 for the three independent simulations) indicates good local convergence within the subspace defined by the original experimental structures; we note that this does not imply global ergodic sampling but only good local sampling around the experimental states.
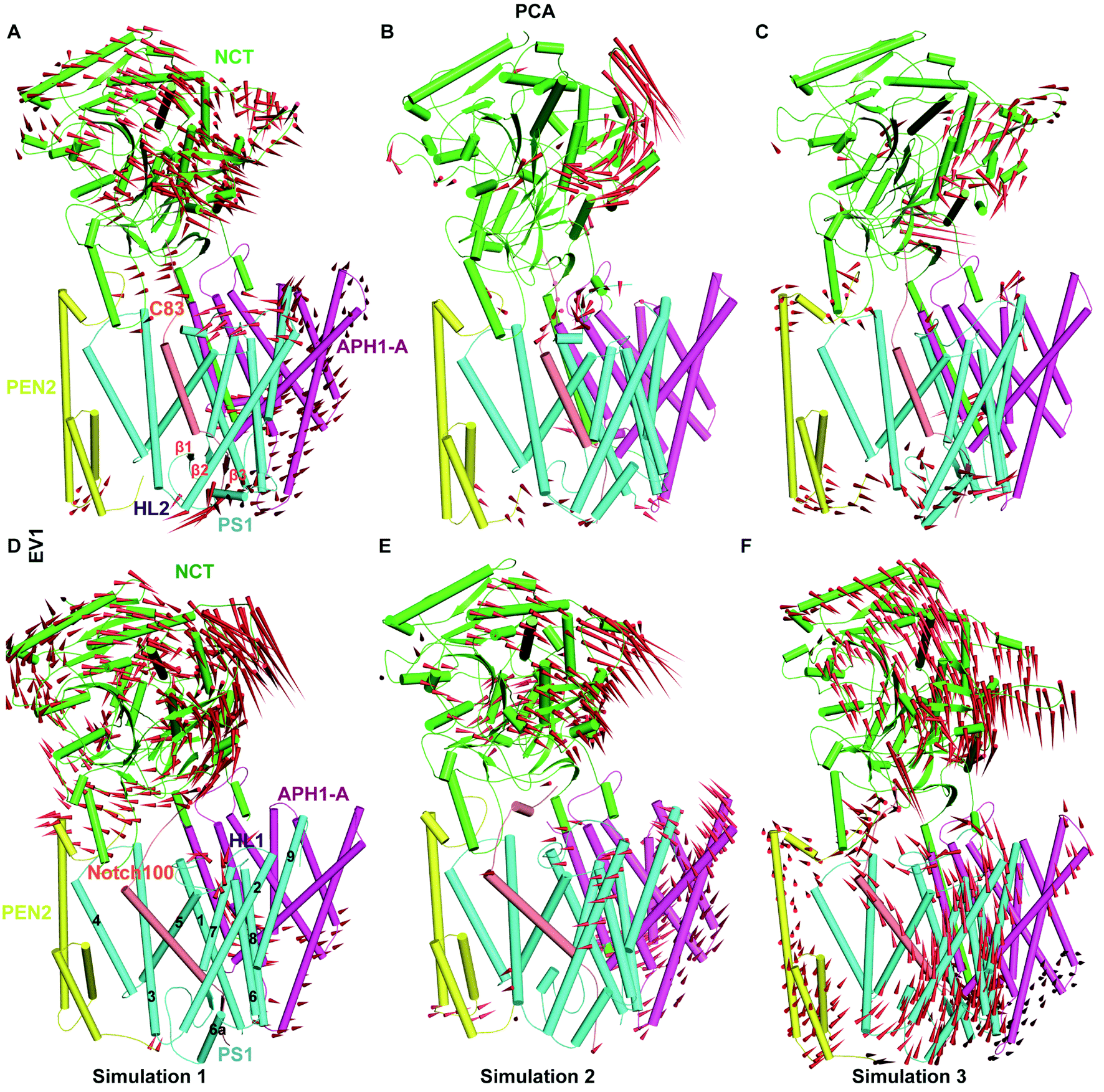 | ||
| Fig. 3 Main atomic dynamics of γ-secretase–C83 and Notch100 complexes. (A–C) The movement of NCT (green), PS1 (cyan), APH1-A (purple), PEN2 (yellow), and C83 (tint grey) of γ-secretase–C83 for simulation 1 (A), 2, (B), and 3 (C). (D–F) The movement of the NCT, PS1, APH1-A, PEN2, and Notch100 (tint grey) of γ-secretase–Notch100 for simulation 1 (D), 2, (E), and 3 (F). The red arrows represent the direction and amplitude of motion. The TM domains in PS1 are marked 1, 2, 3, 4, 5, 6, 6a, 7, 8 and 9. | ||
Fig. 4 shows the main representative structures obtained from clustering analysis of each simulation, compared to the experimental cryo structures as in Fig. 2C and D, but zooming in on the catalytic PS1 subunits. Fig. S11† shows the corresponding analysis for the complete trans-membrane region of the full protein–substrate complexes, documenting the very good agreement with the experimental topologies (shown in green). Careful comparison of the 303 K structures of the two complexes indicate differences in the β1 (along with TM6a) and β2 strand (connected with TM7) (most notable in simulation 1, Fig. 4B), which may affect differential binding and subsequent cleavage of the substrates. In one simulation of both γ-secretase–C83 (simulation 2, Fig. 4C) and γ-secretase–Notch100 (simulation 1, Fig. 4G) we observed complete loss of the three β-strands in PS1 and the substrates. Bound Notch100 (Fig. 4J) displayed more structural variation than C83 (Fig. 4E) but we saw some loss of β3-strand in both C83 (Fig. 4C) and Notch100 (Fig. 4G).
 | ||
| Fig. 4 Comparison of 303 K-simulated PS1 and experimental cryo-EM structures. (A) Superimposed view of simulated and experimental (6IYC) structures of γ-secretase–C83. (B–D) Ensemble-representative simulated PS1–C83 structures. (E) Zoomed-in view of the catalytic residues in the three simulated structures of 6IYC (catalytic and PAL residues are shown as sticks). (F) Superimposed view of simulated and experimental (6IDF) γ-secretase–Notch100. (G–I) Ensemble-representative simulated PS1–Notch100 structures. (J) Zoomed-in view of the catalytic residues in the three simulated structures of 6IDF. Blue, purple and cyan represent simulation 1, 2, and 3 (deep salmon color represents the substrates); the experimental structures are shown in green. | ||
Different dynamical sizes of Notch- and C83-bound catalytic pockets
In order to understand the differential membrane dynamics of the two enzyme–substrate complexes, we monitored the distance between the Cα of Asp257 and Asp385, which defines the open (large Cα–Cα distance) and compact (least Cα–Cα distance) conformation states of the catalytic pocket that control substrate cleavage pathways according to the FIST model (Fig. 5A–D). The Asp–Asp distance (∼11.5 Å averaged over the three simulation systems) for the C83-complex (experimental 6IYC: 10.6 Å) is slightly higher than for the Notch100 conformation (∼10.1 Å averaged over the three simulations; experimental 6IDF: 10.4 Å). Both the temperature and the membrane expand the protein in the membrane, explaining the difference between the cryo-EM and simulated data (simulated cooled structures look similar to the experimental cryo structures).64 We note that the distances agree well with our previous MD studies of full-length C99 inside γ-secretase with a complete, matured HL2 present,65 i.e. the C83–C99 difference and presence of HL2 do not affect this important distance directly, but probably affect overall substrate affinity. | ||
| Fig. 5 Dynamics of the catalytic Asp257–Asp385, large hydrophilic loop 1 (HL1) and PAL motif. Left panel: γ-secretase–C83 complex and right panel: γ-secretase–Notch100 complex. (A) Orientation and Cα–Cα distance between the catalytic aspartates in γ-secretase–C83 compared to the experimental 6IYC. (B) Orientation and Cα–Cα distance between the aspartates in γ-secretase–Notch100 compared to 6IDF (blue: simulation 1, purple: simulation 2 and cyan: simulation 3). (C) Dynamics of the Cα–Cα distance between Asp257 and Asp385 in γ-Secretase–C83. (D) Dynamics of the Cα–Cα distance between Asp257 and Asp385 in γ-secretase–Notch100. The magenta line displays the experimental distance. (E) Minimum distance between the terminal residues of HL1 in γ-secretase–C83. (F) Minimum distance between the terminal residues of HL1 in γ-secretase–Notch100. (G) Minimum distance between the terminal PAL residues (Pro433 and Leu435) in γ-secretase–C83. (H) Minimum distance between Pro433 and Leu435 in γ-secretase–Notch100. | ||
We also monitored minimum-distance (using gmx mindist) between the terminal residues of HL1 (Ser104–Glu123 in γ-secretase–C83 and Notch100 complexes) at the membrane interface of TM1 and TM2 of PS1 (Fig. 5E and F). The separation was much larger in the Notch100 complex than in C83–γ-secretase consistently for all three independent simulations. Similarly, the end-to-end separation of the PAL motif (Pro433–Ala434–Leu435) was measured as the minimum distance between Pro433 and Leu435 in PS1. We observed a clear difference in its extension in γ-secretase–C83 and γ-secretase–Notch100 (Fig. 5G and H), mainly caused by a changed conformation of Leu432. Mutation of this residue may affect PAL motif and abolishes the activity of the enzyme.62,63 The residue forms a strong backbone hydrogen bond with the Notch and C83 β-strand, which probably contributes to the role of the PAL motif in substrate recognition.120
The dynamic variation in the distance between the catalytic aspartates and the cleavage sites of the substrates are relevant to the cleavage mechanism of C83 and Notch100. These variations are portrayed in Fig. 6. ε-Cleavage occurs either at Thr719–Leu720 (associated with the Aβ48 pathway) or at Leu720–Val721 (Aβ49 pathway). We observed major differences in the distance between the catalytic Asp257 and these cleavage sites of C83, whereas Asp385 was generally relatively fixated with respect to the cleavage sites. The imprecise cleavage resulting in two pathways i.e., Aβ49 or Aβ48 requires a relatively open and dynamic substrate pocket, according to the FIST model.60,80,121 Asp385 is closer to the cleavage site at V1754 in the Notch100-complex (Fig. 6; average distance below ∼5 Å).
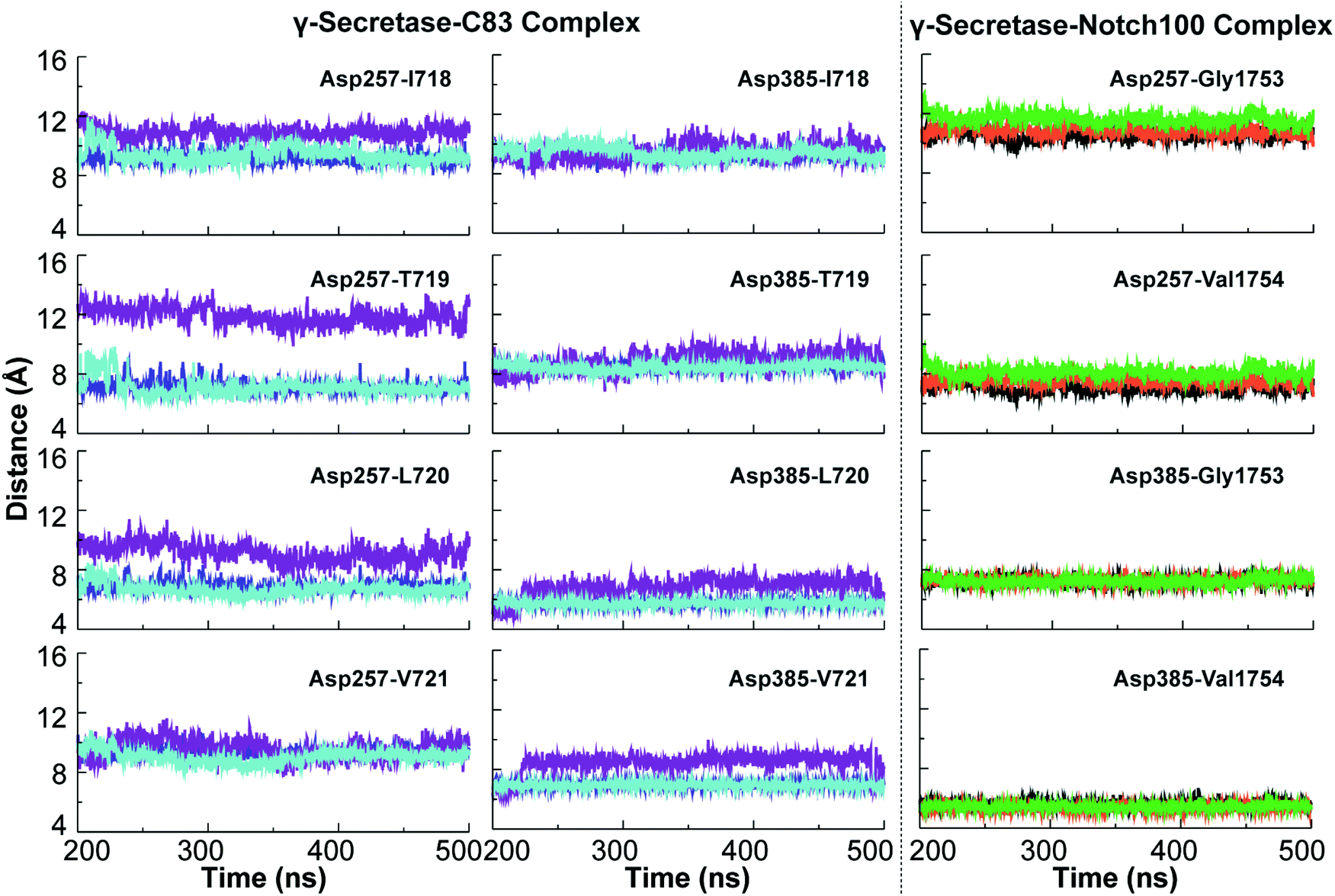 | ||
| Fig. 6 Simulated minimum distances between catalytic Asp257/Asp385 and cleavage sites of C83 and Notch100. Left panels: average distances between the catalytic residues of PS1 and cleavage sites in C83 (Leu720–Val721 giving the Aβ49 pathway and Thr719–Leu720 giving the Aβ48 pathway). Right panel: average distances between the catalytic residues and cleavage sites of Notch100 (Gly1753 and Val1754). | ||
Hydrogen bond dynamics, water accessibility, and lipid binding
How γ-secretase enables access of nucleophilic water within the lipid bilayer remains an open question that largely depends on the room-temperature hydrogen-bond dynamics. The close encounter of Asp385 to the substrate discussed above suggests that this residue can possibly hydrogen bond and engage in proton transfer with the substrate directly or via a catalytic water molecule, since a short distance is required for proton transfer.116 The mechanism of γ-secretase probably involves a critical formation of an initial hydrogen-bond or full proton transfer between the catalytic water molecule and the aspartate to enhance water's nucleophilicity,122 and after establishment of the tetrahedral intermediate, we can expect the proton to be relayed back to the N-terminus of the newly formed cleavage product.123,124In order to understand the thermal dynamics of these hydrogen bonds in more detail, we measured the dynamics of hydrogen bonds and compared them for the pairs NCT–C83/Notch100, PS1–C83/PS1–Notch100, POPC–PS1, C83/Notch100–POPC, catalytic Aspartates (Asp257/Asp385)–water (TIP3) as highlighted in Fig. 7 and S12.† The hydrogen bonds between POPC lipids and the substrates (C83 and Notch100) displayed substantial dynamics, which average only to the most stable conformation in the cryo-EM data (Fig. S12†).
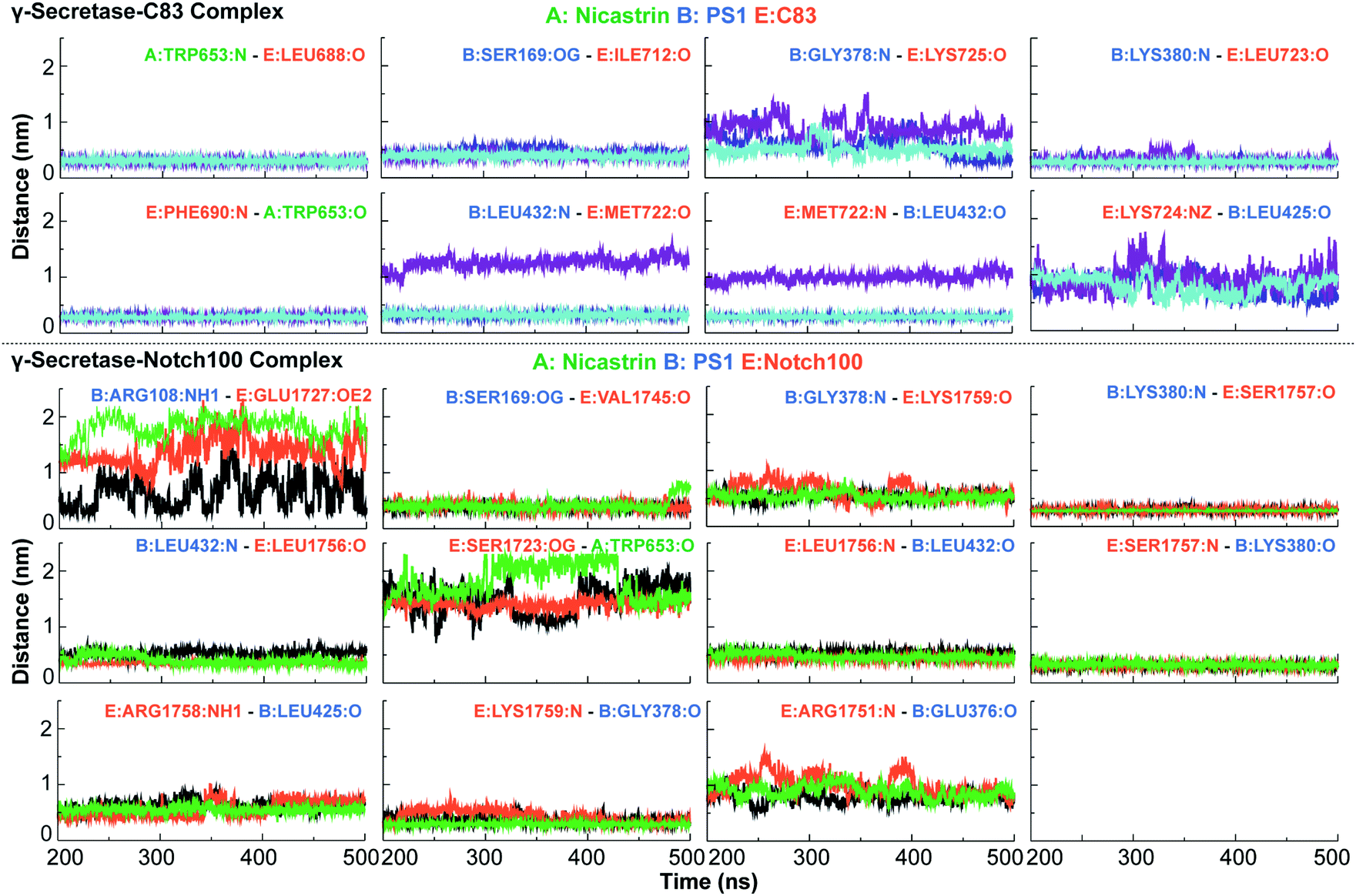 | ||
| Fig. 7 Important hydrogen bond dynamics of NCT ECD and PS1 interacting with C83 and Notch100. A: NCT, B: presenilin 1 and E: C83 or Notch100. The residues of C83 and Notch100 are numbered according to their position in corresponding substrates. | ||
We computed the water-mediated hydrogen-bond dynamics in both systems: the C83 complex displayed more water interaction than Notch100 in the same regions, which may affect substrate selectivity. In particular, by computing the water molecules within 5 Å of the Asp257/Asp385 residues (average numbers/frame in Fig. S13†), we found that more water molecules access the two catalytic residues in the C83 complex than in the Notch100 complex. The surrounding residues within 5 Å (Fig. S14†) indicated substantial differences in hydrogen bonding to water molecules. We propose that the different water accessibility could be an important determinant of the relative affinity (selectivity) of the two substrates, of relevance to developing modulators. In both complexes, we also observed that Asp257 forms more hydrogen bonds with water as compared to Asp385. In addition, the solvent exposed residues Leu721 (of C83), Gly1753 and Val1754 (of Notch100) interacted considerably with water, with the space available for these hydrogen bonds being more restricted in the Notch complex. Supporting this analysis is also the SASA of each of these residues in both systems during the last 300 ns (Fig. S15 and S16†).
Hydrogen bonds mediated by the POPC lipid head-groups were analyzed by measuring the retention time to specific head groups (Fig. S17 and S18†). Many stable POPC lipid binding regions around hydrophobic TM helices were clearly visible (Fig. S17†). Some residues of the NCT ECD and the substrate interface displayed lipid binding. Some lipids bound between the TM domains of NCT and near APH-1A (mostly aromatic residues) were very far from the active site. However, in PS1, the substrate-binding cavity formed by TM2, TM3, and TM5 of γ-secretase–C83 possesses a lipid binding site (Fig. S18†), interestingly close to many residues known to be mutated in early-onset familial AD.32 The main lipid binding sites near the substrate-binding pocket reside at HL1, TM2, TM6, TM6a and TM9,21,125 which supports the hypothesis that membrane dynamics plays a major role in substrate recognition and cleavage. The residues binding to POPC lipids (Table S2†) is in congruence with several previous MD studies by other research groups.116,118,126
Specific interactions of C83 and Notch 100 with γ-secretase
With the similarities and differences in molecular recognition of C83 and Notch100 by γ-secretase in mind, we computed the intermolecular contacts of the ensembles and measured the hydrogen bond pairs for each trajectory (Fig. 7–9). The important hydrogen bonds and electrostatic contacts are summarized in Tables S3 and S4†. Glu280, which is located in the loop connecting TM6a and the strand β1, interacted consistently with the positively charged triplet lysine anchor motif (Lys724–Lys726) of the C83 complex (Table S3†). The mutation of this residue (E280A) has been observed in hundreds of early-onset AD patients; we suggest its mechanism of action is to disturb the anchor motif and thereby increase the mobility of the substrate, creating looser “grabbing” that enhances the imprecise cleavage leading to less activity and relatively longer Aβ according to the FIST model. Strong electrostatic charge interactions may aid the positioning of the substrates.127 Apart from the NCT interface contacts, we also observed hydrogen-bonds between TM6a or the HL2 loop and C-terminal end of C83. The tri-lysine anchor of C83 resides just C-terminal to the initial (ε) cleavage site after Thr719, and the catalytic pocket of γ-secretase is enriched with negatively charged residues. Polybasic regions C-terminal to the TM helix are common to most γ-secretase substrates and are conserved in both C83 and the Notch family.128 | ||
| Fig. 8 Interactions between NCT, PS1 and C83 obtained from ensemble-representative cluster structures. (A) Simulation 1. (B) Simulation 2. (C) Simulation 3 (NCT: green; PS: blue; and C83: salmon). The hydrogen-bonds (shown in magenta dotted lines) are displayed for clarity. The NCT residues interacting with substrate are green, while PS1 is blue and C83 residues are orange. The interacting residues are shown in ball and stick format. | ||
 | ||
| Fig. 9 Intermolecular contacts between NCT, PS1 and Notch100 obtained from the top-ranked cluster. (A) Simulation 1. (B) Simulation 2. (C) Simulation 3. The hydrogen bonds (shown in magenta dotted lines) are displayed for clarity. The NCT residues interacting with substrate are labelled green, while PS1 is blue and Notch100 residues are orange. The interacting residues are shown in ball and stick format. | ||
In the Notch100 complex, the TM helix (residues 1734–1750) interacted with TMs of PS1 via several van der Waals interactions (Table S4†). Gly1753 at the C-terminal of the Notch100 TM helix formed a hydrogen bond with Gly384 in the GXGD motif of PS1 (Fig. 9); the absence of side chains in Gly384 probably is an important feature of proteolytic activity of γ-secretase, possibly due to water co-localization: the AD-associated mutation G384A exhibits a 90% loss of activity and a 170-fold higher Aβ42/Aβ40 ratio compared to wild-type γ-secretase in an in vitro assay,32 which we suggest could easily be due to loss of a co-localized water involved in the water hydrogen bond network discussed above.
Leu432 of PS1, which precedes the PAL motif (Pro433–Ala434–Leu435), formed backbone hydrogen bonds with Notch100 (Table S4†). This observation is consistent with the reported role of the PAL motif in substrate recognition.120 The primary cleavage site for Notch100 is located between Gly1753 and Val1754, which also formed hydrogen bonds with the GXGD motif of PS1 (notably with Gly382 and Gly384), and thus perhaps important for substrate binding and for orienting the scissile peptide bonds towards the active site. Finally, we note that we consistently observed a strong hydrogen bond between the catalytic Asp385 and Leu1755 close to cleavage site (Val1754). Altogether, several amino acids from PS1 interact differently with the TM helices of C83 and Notch100. A direct interaction between NCT ECD and C83 (Fig. 8) provide a possibility that this part of NCT affects the stability of the enzyme–substrate complex which in turn would modulate the Aβ length, according to the FIST model80,121 and experimental evidence.105
To understand the energetic difference in substrate recognition, we computed the binding free energy for both complexes, using the equilibrated snapshots from the last 300 ns, with the MM/PBSA approach (Table S5†). We found that the energetic contributions from different components of Notch and C83 were similar in nature. The free-energy decomposition analysis (Fig. S19†) indicated that the contributing residues are generally identical in all the replicates with only minor deviations. The positively charged motif (RKRR 1758–1761) of Notch100 contributed significantly to overall binding (average of −75.2, −74.9, and −72.3 kJ mol−1 for the three simulations) supporting its role as a “membrane anchor” of Notch. The positive charged residues at the C-terminus of the TM region of the Notch family are fully conserved indicating the important function of this anchoring.
TM-helix conformations of γ-secretase-substrate complexes
As the last part of our analysis, to understand the differences in recognition of the substrates, we measured the deviation in tilt angles of TMs (Fig. 10 and Table S6†) and secondary structure changes (Fig. S20–S22†). Substantial structural rearrangements were observed in the substrate-binding regions of PS1 that are likely to be induced by differential substrate binding (Fig. 4 and S11†). Our computed tilt angle distributions of TM helices in PS1 (Fig. 10 and Table S6†) showed marked differences, most importantly in TM2, TM5, TM6, and TM6a. Not only the variation in tilt angles but also the prevalent tilt angle of each TM helix varied between the ensembles; we expect the cryo-EM data to reflect an average of these motions. These differences along with Cα–Cα distance of catalytic pairs define the open (large Cα–Cα distance) and compact states (small Cα–Cα distance) that control the pathways and extent of cleavage according to the FIST model.65,80 TM2, TM6, and TM9 are involved in initial substrate binding.129 | ||
| Fig. 10 Distribution of tilt angles of transmembrane domains (TMs) of PS1 and C83 and Notch100. The tilt angles were computed using the Cα coordinates of the TMs. (A) Tilt angles in γ-secretase–C83. (B) Tilt angles in γ-secretase–Notch100. | ||
As seen from Fig. 4 and S11,† we observed a complete loss of three-stranded β-sheet (formed by PS1 and C83) in one simulation (simulation 2) and a partial loss of one β-strand (extended towards TM7) in another simulation (simulation 3). For β1 extended from TM6a of PS1, we observed the same residues forming β-sheet in only two conformations, while in the second replica, it was converted to a turn. We also observed loss of β-strand in the Notch100 complex simulations. The structural ensembles analyzed using VMD (Fig. S21–S22†) indicated that the β-strands disappear at different time points and often converted to coil. The β-sheet is probably favoured at cryogenic temperature, both from thermodynamic considerations and from actual cold vs. hot simulations.64
It has been suggested that the transmembrane substrate helix of intramembrane proteases unwinds during binding to enable the catalytic aspartates to access the peptide bonds,130 and solution NMR chemical shifts indicate that this is also the case for cleavage of C99.131 Such a mechanism requires helix-destabilizing residues to partially unzip during substrate binding. The recent cryo-EM structure has been claimed to show such a partial unwinding within the Notch-TMD.63 β-strands in TM6 and TM7 of PS1, which were observed in our previous MD simulations of apo-γ-secretase,80,112 indicate a possible role of β-strands in γ-secretase PS1 physiological temperature dynamics. The evolution of secondary structure for each trajectory (Fig. S21 and S22†) clearly indicates that C83 and Notch100 display consistent partial helix–coil transitions, which could be consistent with such partial unwinding events, and the cryo-EM structures probably feature an extended helix because it is a cooled average of these dynamic motions that we observe here.
Effect of pathogenic PS1 mutations on the stability of the enzyme–substrate complexes
Several studies have shown that PS1 mutations that cause familial AD tend to increase the Aβ42/Aβ40 ratio, which correlates with clinical severity of the mutation.32,100 According to the amyloid hypothesis, an increased Aβ42/Aβ40 ratio may be pathogenic, whereas the presenilin hypothesis implies that the mutations cause disease by loss of other functions of presenilin, which could include Notch processing.132,133 To put the structural differences discussed above into energy context, we estimated the effect on the folding stability ΔΔGMUT of the enzyme–substrate complexes caused by 149 PS1 mutations both using the experimental 6IYC and 6IDF structures (after converting to wild type with no other changes made) and our simulated structures in the membrane for each substrate. In order to account for method variations, we compared four methods: I-Mutant, FoldX, POPMUSIC and mCSM.The results, summarized in Fig. 11, suggest that pathogenic mutations consistently destabilize the enzyme–substrate complexes both with C83 and Notch bound. These results are largely similar for the experimental and computed structures, with some local differences (Fig. S23–S25†) and are consistent with kinetic data that indicate that pathogenic mutations destabilize γ-secretase–APP/Aβn interactions.106 Thus, we conclude that any destabilizing effect of PS1 mutation on the C83/C99 complex is likely to have a similar effect on the stability of the Notch complex. Interestingly, a few exceptions, shown as outliers in Fig. 11, indicate that not all mutations affect the two substrate-bound proteins similarly, indicating again that local variations may be of interest to selective targeting of the two enzyme–substrate complexes. Most AD-causing mutations in PS1 and APP cluster at the interface between the enzyme and substrate, supporting the idea that altered substrate binding and processing contribute to AD.
 | ||
| Fig. 11 Computed stability effects (ΔΔGMUT, kcal mol−1) for 149 pathogenic PS1 mutations of substrate-bound γ-secretase (6IYC and 6IDF). The median is shown as a white dot inside the violin plot. (A and D) Computed ΔΔGMUT for 6IYC and 6IDF using (A) I-Mutant, (B) FoldX, (C) POPMUSIC and (D) mCSM. (E–H) Linear correlation between ΔΔGMUT of the 149 mutations for 6IYC and 6IDF computed using (E) I-Mutant, (F) FoldX, (G) POPMUSIC and (H) mCSM. | ||
Conclusions
Previous studies have revealed that substrate binding changes the γ-secretase structure both in the cryo data75,134 and during normal temperature dynamics.65,80 These changes are markedly influenced by the membrane,65,116,126 and more at normal temperature than at cryo temperature.64 AD-associated PS1 mutations are likely to destabilize γ-secretase–substrate complexes, thereby favoring a more open bound state with lower activity and cleavage precision, leading to production of longer Aβs, which could underly pathogenesis.80,106,121 Notch-sparing modulators of γ-secretase should contribute their binding affinity to stabilize the ternary complexes, favoring the compact innocent conformation state leading to high processing of C99 without impairing the Notch cleavage notably.60 For these reasons, a detailed comparative study of the dynamics that distinguish these substrates in a membrane model at physiological temperature is of major interest. Some main features of this comparison are summarized in Table 1. Our MD simulations are in agreement with and complement the cryo-EM data, considering the average experimental resolution of ∼2.6 Å,62,63 and more importantly provide the dynamics in the context of the 303 K interactions with the lipid molecules, which contribute to the substrate binding dynamics.| Complexes | MD systems | RMSD (nm) | Rg (nm) | SASA (nm2) | RMSF (nm) | Distance (nm) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nicastrin | PS1 | APH1-A | PEN2 | C83/Notch100 | Asp–Asp | |||||
| a Values within parenthesis represent the standard deviations. | ||||||||||
| γ-Secretase–C83 | 1 | 0.28 (0.02) | 4.03 (0.02) | 604.6 (6.40) | 0.14 (0.05) | 0.12 (0.04) | 0.13 (0.03) | 0.13 (0.02) | 0.11 (0.03) | 1.15 (0.02) |
| 2 | 0.31 (0.03) | 4.01 (0.02) | 609.8 (5.46) | 0.14 (0.05) | 0.11 (0.04) | 0.12 (0.03) | 0.12 (0.03) | 0.14 (0.04) | 1.15 (0.04) | |
| 3 | 0.30 (0.03) | 3.99 (0.02) | 613.2 (7.24) | 0.15 (0.06) | 0.13 (0.03) | 0.13 (0.04) | 0.18 (0.05) | 0.12 (0.02) | 1.14 (0.03) | |
| γ-Secretase–Notch100 | 1 | 0.30 (0.05) | 3.97 (0.02) | 613.5 (7.48) | 0.17 (0.06) | 0.10 (0.03) | 0.13 (0.04) | 0.16 (0.05) | 0.16 (0.09) | 1.00 (0.02) |
| 2 | 0.28 (0.02) | 3.95 (0.02) | 611.7 (4.29) | 0.13 (0.04) | 0.10 (0.03) | 0.12 (0.03) | 0.13 (0.04) | 0.14 (0.05) | 1.01 (0.02) | |
| 3 | 0.30 (0.03) | 3.94 (0.04) | 621.0 (7.05) | 0.15 (0.05) | 0.15 (0.04) | 0.13 (0.04) | 0.22 (0.05) | 0.21 (0.09) | 1.02 (0.02) | |
We consistently observed β-strand formation in several solvent-exposed parts of PS1, in agreement with previous studies.80,112 Cryo-EM data have also recently supported the presence of β-strand formation in PS1.63 However, at physiological temperature, the recognition sheet formation seems to weaker than at cryo temperature, as we find that temperature destabilizes it, consistent with our findings for the C83-complex.64
We find that the RKRR motif (1758–1761) contributes significantly to Notch binding and serves as a “membrane anchor”, which restricts the displacement of Notch toward the extracellular side of the membrane. We find that some water molecules can transiently hydrogen bond to G1753 and V1754 at physiological temperature (but not at cryo temperature); these water molecules probably represent the catalytic nucleophiles. We also found that the span of HL1 at the membrane interface of TM1 and TM2 of PS1 is larger in the Notch100 complex than in C83–γ-secretase in all three independent simulations, indicating a likely hotspot for binding selective modulators. Similarly, the extension of the PAL motif (Pro433–Ala434–Leu435) differed in the two enzyme–substrate complexes because of a change in the conformation of Leu432.
At physiologically relevant temperature, Notch and C83 binding induces different conformation states not seen in the cryo-EM data: one state is more open and the other is more compact. Notch is mostly represented by the more closed state, as best defined by the catalytically important Asp–Asp distances (Table 1). These findings can explain the different outcome of cleaving Notch and C99 by γ-secretase, specifically, the more imprecise cleavage of C9922 in a more open state leading to more diverse cleavage products.135 To put these structural differences into a more relevant context, we computed the stability effect of 149 PS1 mutations using four different computational methods, to account for method variation. The methods consistently indicate that the mutations destabilize both the Notch and C83 complexes. However, a few mutations affect the complexes differently (Fig. 11), illustrating local variations in the substrate-bound structures.
We suspect that any selective targeting of C99 over Notch will require an emphasis on these different conformation states rather than the average densities seen at cryo temperature, and thus our structural dynamic insights could aid the development of selective drugs targeting C99 cleavage over Notch cleavage, i.e. “Notch-sparing” γ-secretase modulators that specifically modulate C99 cleavage by γ-secretase without interfering with normal healthy processing of Notch.136
Conflicts of interest
All authors hereby declare that they have no competing interests, neither financial nor non-financial, related to this work.Acknowledgements
The authors acknowledge computer time from DTU high-performance computing facility, Lyngby, DTU. The Danish Council for Independent Research|Natural Sciences (DFF), grant case 7016-00079B, and the Novo Nordisk Foundation, grant NNF17OC0028860, are gratefully acknowledged for supporting this work.References
- K. Blennow, M. J. de Leon and H. Zetterberg, Lancet, 2015, 368, 387–403 CrossRef.
- C. Ballard, S. Gauthier, A. Corbett, C. Brayne, D. Aarsland and E. Jones, Lancet, 2011, 377, 1019–1031 CrossRef.
- E. Nichols, C. E. I. Szoeke, S. E. Vollset, N. Abbasi, F. Abd-Allah, J. Abdela, M. T. E. Aichour, R. O. Akinyemi, F. Alahdab, S. W. Asgedom, A. Awasthi, S. L. Barker-Collo, B. T. Baune, Y. Béjot, A. B. Belachew, D. A. Bennett, B. Biadgo, A. Bijani, M. S. Bin Sayeed, C. Brayne, D. O. Carpenter, F. Carvalho, F. Catalá-López, E. Cerin, J. Y. J. Choi, A. K. Dang, M. G. Degefa, S. Djalalinia, M. Dubey, E. E. Duken, D. Edvardsson, M. Endres, S. Eskandarieh, A. Faro, F. Farzadfar, S. M. Fereshtehnejad, E. Fernandes, I. Filip, F. Fischer, A. K. Gebre, D. Geremew, M. Ghasemi-Kasman, E. V. Gnedovskaya, R. Gupta, V. Hachinski, T. B. Hagos, S. Hamidi, G. J. Hankey, J. M. Haro, S. I. Hay, S. S. N. Irvani, R. P. Jha, J. B. Jonas, R. Kalani, A. Karch, A. Kasaeian, Y. S. Khader, I. A. Khalil, E. A. Khan, T. Khanna, T. A. M. Khoja, J. Khubchandani, A. Kisa, K. Kissimova-Skarbek, M. Kivimäki, A. Koyanagi, K. J. Krohn, G. Logroscino, S. Lorkowski, M. Majdan, R. Malekzadeh, W. März, J. Massano, G. Mengistu, A. Meretoja, M. Mohammadi, M. Mohammadi-Khanaposhtani, A. H. Mokdad, S. Mondello, G. Moradi, G. Nagel, M. Naghavi, G. Naik, L. H. Nguyen, T. H. Nguyen, Y. L. Nirayo, M. R. Nixon, R. Ofori-Asenso, F. A. Ogbo, A. T. Olagunju, M. O. Owolabi, S. Panda-Jonas, V. M. d. A. Passos, D. M. Pereira, G. D. Pinilla-Monsalve, M. A. Piradov, C. D. Pond, H. Poustchi, M. Qorbani, A. Radfar, R. C. Reiner, S. R. Robinson, G. Roshandel, A. Rostami, T. C. Russ, P. S. Sachdev, H. Safari, S. Safiri, R. Sahathevan, Y. Salimi, M. Satpathy, M. Sawhney, M. Saylan, S. G. Sepanlou, A. Shafieesabet, M. A. Shaikh, M. A. Sahraian, M. Shigematsu, R. Shiri, I. Shiue, J. P. Silva, M. Smith, S. Sobhani, D. J. Stein, R. Tabarés-Seisdedos, M. R. Tovani-Palone, B. X. Tran, T. T. Tran, A. T. Tsegay, I. Ullah, N. Venketasubramanian, V. Vlassov, Y. P. Wang, J. Weiss, R. Westerman, T. Wijeratne, G. M. A. Wyper, Y. Yano, E. M. Yimer, N. Yonemoto, M. Yousefifard, Z. Zaidi, Z. Zare, T. Vos, V. L. Feigin and C. J. L. Murray, Lancet Neurol., 2019, 18, 88–106 CrossRef.
- E. Karran and J. Hardy, Ann. Neurol., 2014, 76, 185–205 CrossRef CAS PubMed.
- B. De Strooper and L. Chávez Gutiérrez, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 419–437 CrossRef CAS PubMed.
- K. P. Kepp, J. Alzheimer's Dis., 2017, 55, 447–457 Search PubMed.
- K. Herrup, Nat. Neurosci., 2015, 18, 794–799 CrossRef CAS PubMed.
- S. L. Sensi, A. Granzotto, M. Siotto and R. Squitti, Trends Pharmacol. Sci., 2018, 39, 1049–1063 CrossRef CAS PubMed.
- J. R. Harrison and M. J. Owen, Br. J. Psychiatry, 2016, 208, 1–3 CrossRef PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- C. L. Masters, D. C. Gajdusek and C. J. J. Gibbs, Brain, 1981, 104, 535–558 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- Y. Zhang, R. Thompson, H. Zhang and H. Xu, Mol. Brain, 2011, 4, 3 CrossRef CAS PubMed.
- D. J. Selkoe, Trends Cell Biol., 1998, 8, 447–453 CrossRef CAS PubMed.
- D. M. Bolduc, D. R. Montagna, M. C. Seghers, M. S. Wolfe and D. J. Selkoe, eLife, 2016, 5, e17578 CrossRef PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- Y. M. Li, M. Xu, M. T. Lai, Q. Huang, J. L. Castro, J. DiMuzlo-Mower, T. Harrison, C. Lellis, A. Nadin, J. G. Neduvelli, R. B. Register, M. K. Sardana, M. S. Shearman, A. L. Smith, X. P. Shi, K. C. Yin, J. A. Shafer and S. J. Gardell, Nature, 2000, 405, 689–694 CrossRef CAS PubMed.
- M. S. Wolfe, W. Xia, B. L. Ostaszewski, T. S. Diehl, W. T. Kimberly and D. J. Selkoe, Nature, 1999, 398, 513–517 CrossRef CAS PubMed.
- A. Bhattarai, S. Devkota, S. Bhattarai, M. S. Wolfe and Y. Miao, ACS Cent. Sci., 2020, 6, 969–983 CrossRef CAS PubMed.
- A. Fukumori, R. Fluhrer, H. Steiner and C. Haass, J. Neurosci., 2010, 30, 7853–7862 CrossRef CAS PubMed.
- H. Steiner, A. Fukumori, S. Tagami and M. Okochi, Cell Stress, 2018, 2, 292–310 CrossRef PubMed.
- M. Takami, Y. Nagashima, Y. Sano, S. Ishihara, M. Morishima-Kawashima, S. Funamoto and Y. Ihara, J. Neurosci., 2009, 29, 13042–13052 CrossRef CAS PubMed.
- T. E. Golde, S. Estus, L. H. Younkin, D. J. Selkoe and S. G. Younkin, Science, 1992, 255, 728–730 CrossRef CAS PubMed.
- K. Pauwels, T. L. Williams, K. L. Morris, W. Jonckheere, A. Vandersteen, G. Kelly, J. Schymkowitz, F. Rousseau, A. Pastore, L. C. Serpell and K. Broersen, J. Biol. Chem., 2012, 287, 5650–5660 CrossRef CAS PubMed.
- S. Chemuru, R. Kodali and R. Wetzel, J. Mol. Biol., 2016, 428, 274–291 CrossRef CAS PubMed.
- N. Brouwers, K. Sleegers and C. Van Broeckhoven, Ann. Med., 2008, 40, 562–583 CrossRef CAS PubMed.
- E. Levy-Lahad, W. Wasco, P. Poorkaj, D. M. Romano, J. Oshima, W. H. Pettingell, C. E. Yu, P. D. Jondro, S. D. Schmidt and K. Wang, et al., Science, 1995, 269, 973–977 CrossRef CAS PubMed.
- E. I. Rogaev, R. Sherrington, E. A. Rogaeva, G. Levesque, M. Ikeda, Y. Liang, H. Chi, C. Lin, K. Holman and T. Tsuda, Nature, 1995, 376, 775–778 CrossRef CAS PubMed.
- D. C. Ryman, N. Acosta-Baena, P. S. Aisen, T. Bird, A. Danek, N. C. Fox, A. Goate, P. Frommelt, B. Ghetti, J. B. S. Langbaum, F. Lopera, R. Martins, C. L. Masters, R. P. Mayeux, E. McDade, S. Moreno, E. M. Reiman, J. M. Ringman, S. Salloway, P. R. Schofield, R. Sperling, P. N. Tariot, C. Xiong, J. C. Morris and R. J. Bateman, Neurology, 2014, 83, 253–260 CrossRef CAS PubMed.
- M. Dimitrov, J.-R. Alattia, T. Lemmin, R. Lehal, A. Fligier, J. Houacine, I. Hussain, F. Radtke, M. Dal Peraro, D. Beher and P. C. Fraering, Nat. Commun., 2013, 4, 2246 CrossRef PubMed.
- T.-H. H. Xu, Y. Yan, Y. Kang, Y. Jiang, K. Melcher, H. E. Xu, T.-H. H. Xu, H. E. Xu, K. Melcher, Y. Jiang and Y. Yan, Cell Discovery, 2016, 2, 16026 CrossRef CAS PubMed.
- L. Sun, R. Zhou, G. Yang and Y. Shi, Proc. Natl. Acad. Sci., 2016, 114, E476–E485 CrossRef PubMed.
- N. Tang and K. P. Kepp, J. Alzheimer's Dis., 2018, 66, 939–945 CAS.
- S. Artavanis-Tsakonas, M. D. Rand and R. J. Lake, Science, 1999, 284, 770–776 CrossRef CAS PubMed.
- E. R. Andersson, R. Sandberg and U. Lendahl, Development, 2011, 138, 3593–3612 CrossRef CAS PubMed.
- C. Siebel and U. Lendahl, Physiol. Rev., 2017, 97, 1235–1294 CrossRef CAS PubMed.
- J. D. Lathia, M. P. Mattson and A. Cheng, J. Neurochem., 2008, 107, 1471–1481 CrossRef CAS PubMed.
- M. Okochi, H. Steiner, A. Fukumori, H. Tanii, T. Tomita, T. Tanaka, T. Iwatsubo, T. Kudo, M. Takeda and C. Haass, EMBO J., 2002, 21, 5408–5416 CrossRef CAS PubMed.
- R. Kopan and M. X. G. Ilagan, Cell, 2009, 137, 216–233 CrossRef CAS PubMed.
- D. J. Gibbs, J. Bacardit, A. Bachmair and M. J. Holdsworth, Trends Cell Biol., 2014, 24, 603–611 CrossRef CAS PubMed.
- A. P. Weng, A. A. Ferrando, W. Lee, J. P. Morris, L. B. Silverman, C. Sanchez-Irizarry, S. C. Blacklow, A. T. Look and J. C. Aster, Science, 2004, 306, 269–271 CrossRef CAS PubMed.
- B. Wang, W. Yang, W. Wen, J. Sun, B. Su, B. Liu, D. Ma, D. Lv, Y. Wen, T. Qu, M. Chen, M. Sun, Y. Shen and X. Zhang, Science, 2010, 330, 1065 CrossRef CAS PubMed.
- T. Sato, T. S. Diehl, S. Narayanan, S. Funamoto, Y. Ihara, B. De Strooper, H. Steiner, C. Haass and M. S. Wolfe, J. Biol. Chem., 2007, 282, 33985–33993 CrossRef CAS PubMed.
- W. T. Kimberly, M. J. LaVoie, B. L. Ostaszewski, W. Ye, M. S. Wolfe and D. J. Selkoe, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6382–6387 CrossRef CAS PubMed.
- H. Steiner, K. Duff, A. Capell, H. Romig, M. G. Grim, S. Lincoln, J. Hardy, X. Yu, M. Picciano, K. Fechteler, M. Citron, R. Kopan, B. Pesold, S. Keck, M. Baader, T. Tomita, T. Iwatsubo, R. Baumeister and C. Haass, J. Biol. Chem., 1999, 274, 28669–28673 CrossRef CAS PubMed.
- W. T. Kimberly, W. Xia, T. Rahmati, M. S. Wolfe and D. J. Selkoe, J. Biol. Chem., 2000, 275, 3173–3178 CrossRef CAS PubMed.
- J. Cui, X. Wang, X. Li, X. Wang, C. Zhang, W. Li, Y. Zhang, H. Gu, X. Xie, F. Nan, J. Zhao and G. Pei, Cell Discovery, 2015, 1, 15021 CrossRef CAS PubMed.
- A. Sogorb-Esteve, M. S. García-Ayllón, M. Llansola, V. Felipo, K. Blennow and J. Sáez-Valero, Mol. Neurobiol., 2018, 55, 5047–5058 CrossRef CAS PubMed.
- T. E. Golde, E. H. Koo, K. M. Felsenstein, B. A. Osborne and L. Miele, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2898–2907 CrossRef CAS PubMed.
- B. De Strooper, Cell, 2014, 159, 721–726 CrossRef CAS PubMed.
- H. Lee, X. Zhu, R. J. Castellani, A. Nunomura, G. Perry and M. A. Smith, J. Pharmacol. Exp. Ther., 2007, 321, 823–829 CrossRef CAS PubMed.
- K. P. Kepp, Neurobiol. Aging, 2019, 80, 46–55 CrossRef CAS PubMed.
- M. A. Smith, G. Casadesus, J. A. Joseph and G. Perry, Free Radicals Biol. Med., 2002, 33, 1194–1199 CrossRef CAS PubMed.
- K. P. Kepp, Prog. Neurobiol., 2016, 143, 36–60 CrossRef CAS PubMed.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van Der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS PubMed.
- T. E. Ballard, K. R. Bales, F. Wang, D. S. Johnson, N. Pozdnyakov, Y.-M. Li, C. J. Crump, S. S. Sisodia and S. V. Castro, Biochemistry, 2012, 51, 7209–7211 CrossRef PubMed.
- V. Coric, S. Salloway, C. H. Van Dyck, B. Dubois, N. Andreasen, M. Brody, C. Curtis, H. Soininen, S. Thein, T. Shiovitz, G. Pilcher, S. Ferris, S. Colby, W. Kerselaers, R. Dockens, H. Soares, S. Kaplita, F. Luo, C. Pachai, L. Bracoud, M. Mintun, J. D. Grill, K. Marek, J. Seibyl, J. M. Cedarbaum, C. Albright, H. H. Feldman and R. M. Berman, JAMA Neurol., 2015, 72, 1324–1333 CrossRef PubMed.
- R. S. Doody, R. Raman, M. Farlow, T. Iwatsubo, B. Vellas, S. Joffe, K. Kieburtz, F. He, X. Sun, R. G. Thomas, P. S. Aisen, E. Siemers, G. Sethuraman and R. Mohs, N. Engl. J. Med., 2013, 369, 341–350 CrossRef CAS PubMed.
- C. J. Crump, D. S. Johnson and Y. M. Li, Biochemistry, 2013, 52, 3197–3216 CrossRef CAS PubMed.
- N. Tang, A. K. Somavarapu and K. P. Kepp, ACS Omega, 2018, 3, 18078–18088 CrossRef CAS.
- A. Hall and T. R. Patel, Prog. Med. Chem., 2014, 53, 101–145 CrossRef CAS PubMed.
- R. Zhou, G. Yang, X. Guo, Q. Zhou, J. Lei and Y. Shi, Science, 2019, 363, eaaw0930 CrossRef CAS PubMed.
- G. Yang, R. Zhou, Q. Zhou, X. Guo, C. Yan, M. Ke, J. Lei and Y. Shi, Nature, 2019, 565, 192–197 CrossRef CAS PubMed.
- R. Mehra, B. Dehury and K. P. Kepp, Phys. Chem. Chem. Phys., 2020, 22, 5427–5438 RSC.
- B. Dehury, N. Tang and K. P. Kepp, Biochem. J., 2019, 476, 1173–1189 CrossRef CAS PubMed.
- W. L. DeLano, CCP4 Newsletter on protein crystallography, 2002, 40, 82–92 Search PubMed.
- G. Vriend, J. Mol. Graphics, 1990, 8, 52–56 CrossRef CAS PubMed.
- R. Rodriguez, G. Chinea, N. Lopez, T. Pons and G. Vriend, Bioinformatics, 1998, 14, 523–528 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- M. A. Lomize, I. D. Pogozheva, H. Joo, H. I. Mosberg and A. L. Lomize, Nucleic Acids Res., 2012, 40, D370–D376 CrossRef CAS PubMed.
- K. P. Jensen, J. Phys. Chem. B, 2008, 112, 1820–1827 CrossRef CAS PubMed.
- K. P. Jensen and W. L. Jorgensen, J. Chem. Theory Comput., 2006, 2, 1499–1509 CrossRef CAS PubMed.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS PubMed.
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed.
- X. Bai, E. Rajendra, G. Yang, Y. Shi and S. H. Scheres, Elife, 2015, 4, 551–560 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2016, 14, 71–73 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- A. D. J. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- A. K. Somavarapu and K. P. Kepp, ACS Chem. Neurosci., 2017, 8, 2424–2436 CrossRef CAS PubMed.
- B. Dehury, N. Tang, T. L. Blundell and K. P. Kepp, RSC Adv., 2019, 9, 20901–20916 RSC.
- B. Dehury, N. Tang and K. P. Kepp, J. Biomol. Struct. Dyn., 2019, 1–15 Search PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- C. Ross, B. Nizami, M. Glenister, O. Sheik Amamuddy, A. R. Atilgan, C. Atilgan and Ö. T. Bishop, Bioinformatics, 2018, 34, 3759–3763 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. Van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- S. Buchoux, Bioinformatics, 2017, 33, 133–134 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- R. Mehra and K. P. Kepp, Arch. Biochem. Biophys., 2019, 678, 108168 CrossRef CAS PubMed.
- E. Capriotti, P. Fariselli and R. Casadio, Nucleic Acids Res., 2005, 33, W306–W310 CrossRef CAS PubMed.
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33, W382–W388 CrossRef CAS PubMed.
- Y. Dehouck, J. M. Kwasigroch, D. Gilis and M. Rooman, BMC Bioinf., 2011, 12, 151 CrossRef PubMed.
- D. E. V. Pires, D. B. Ascher and T. L. Blundell, Bioinformatics, 2014, 30, 335–342 CrossRef CAS PubMed.
- J. Dong, A. Hubel, J. C. Bischof and A. Aksan, J. Phys. Chem. B, 2009, 113, 10081–10087 CrossRef CAS PubMed.
- K. V. Dunlop, R. T. Irvin and B. Hazes, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 80–87 CrossRef PubMed.
- L. Dominguez, L. Foster, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci., 2016, 113, E5281–E5287 CrossRef CAS PubMed.
- M. Karplus and J. A. McCammon, Nat. Struct. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- N. Tang, B. Dehury and K. P. Kepp, J. Chem. Inf. Model., 2019, 59, 858–870 CrossRef CAS PubMed.
- J. Seelig and J. L. Browning, FEBS Lett., 1978, 92, 41–44 CrossRef CAS.
- X. Zhuang, E. M. Dávila-Contreras, A. H. Beaven, W. Im and J. B. Klauda, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 3093–3104 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- J. Seelig and N. Waespe-Sarcevic, Biochemistry, 1978, 17, 3310–3315 CrossRef CAS PubMed.
- D. Petit, M. Hitzenberger, S. Lismont, K. M. Zoltowska, N. S. Ryan, M. Mercken, F. Bischoff, M. Zacharias and L. Chávez-Gutiérrez, EMBO J., 2019, 38, e101494 CrossRef PubMed.
- M. Szaruga, B. Munteanu, S. Lismont, S. Veugelen, K. Horré, M. Mercken, T. C. Saido, N. S. Ryan, T. De Vos, S. N. Savvides, R. Gallardo, J. Schymkowitz, F. Rousseau, N. C. Fox, C. Hopf and B. De Strooper, Cell, 2017, 170, 443–456 CrossRef CAS PubMed.
- K. Takeo, S. Tanimura, T. Shinoda, S. Osawa, I. K. Zahariev, N. Takegami, Y. Ishizuka-Katsura, N. Shinya, S. Takagi-Niidome, A. Tominaga, N. Ohsawa, T. Kimura-Someya, M. Shirouzu, S. Yokoshima, S. Yokoyama, T. Fukuyama, T. Tomita and T. Iwatsubo, Proc. Natl. Acad. Sci., 2014, 111, 10544–10549 CrossRef CAS PubMed.
- X. Wang, J. Cui, W. Li, X. Zeng, J. Zhao and G. Pei, J. Alzheimer's Dis., 2015, 47, 927–937 CAS.
- M. Okochi, S. Tagami, K. Yanagida, M. Takami, T. S. Kodama, K. Mori, T. Nakayama, Y. Ihara and M. Takeda, Cell. Reprogram., 2013, 3, 42–51 CrossRef CAS PubMed.
- S. Takagi-Niidome, T. Sasaki, S. Osawa, T. Sato, K. Morishima, T. Cai, T. Iwatsubo and T. Tomita, J. Neurosci., 2015, 35, 2646–2656 CrossRef CAS PubMed.
- T. Cai, M. Yonaga and T. Tomita, J. Neurosci., 2017, 37, 12272–12280 CrossRef CAS PubMed.
- A. K. Somavarapu and K. P. Kepp, Neurobiol. Dis., 2016, 89, 147–156 CrossRef CAS PubMed.
- A. Tominaga, T. Cai, S. Takagi-Niidome, T. Iwatsubo and T. Tomita, J. Neurosci., 2016, 36, 1362–1372 CrossRef CAS PubMed.
- C. L. Deatherage, Z. Lu, J. H. Kim and C. R. Sanders, Biochemistry, 2015, 54, 3565–3568 CrossRef CAS PubMed.
- C. R. Sanders, C. L. Deatherage, S. Ma, Z. Lu, M. W. Voehler, J. A. Smith, B. M. Kroncke and R. L. McFeeters, Sci. Adv., 2017, 3, e1602794 CrossRef PubMed.
- R. Aguayo-Ortiz, C. Chávez-García, J. E. Straub and L. Dominguez, Chem. Sci., 2017, 8, 5576–5584 RSC.
- D. M. Bolduc, D. R. Montagna, Y. Gu, D. J. Selkoe and M. S. Wolfe, Proc. Natl. Acad. Sci., 2016, 113, E509–E518 CrossRef CAS PubMed.
- M. Hitzenberger and M. Zacharias, Front. Chem., 2019, 6, 640 CrossRef PubMed.
- N. Watanabe, S. Takagi, A. Tominaga, T. Tomita and T. Iwatsubo, J. Biol. Chem., 2010, 285, 19738–19746 CrossRef CAS PubMed.
- C. Sato, S. Takagi, T. Tomita and T. Iwatsubo, J. Neurosci., 2008, 28, 6264–6271 CrossRef CAS PubMed.
- A. K. Somavarapu and K. P. Kepp, J. Neurochem., 2016, 137, 101–111 CrossRef CAS PubMed.
- E. Erez, D. Fass and E. Bibi, Nature, 2009, 459, 371–378 CrossRef CAS PubMed.
- X. Li, S. Dang, C. Yan, X. Gong, J. Wang and Y. Shi, Nature, 2013, 493, 56–61 CrossRef PubMed.
- M. Hitzenberger and M. Zacharias, ACS Chem. Neurosci., 2019, 10, 1826–1840 CrossRef CAS PubMed.
- A. Fukumori and H. Steiner, EMBO J., 2016, e201694151 Search PubMed.
- R. Aguayo-Ortiz, J. E. Straub and L. Dominguez, Phys. Chem. Chem. Phys., 2018, 20, 27294–27304 RSC.
- Y. Yan, T. H. Xu, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1412–1424 CrossRef CAS PubMed.
- T. H. Xu, Y. Yan, Y. Kang, Y. Jiang, K. Melcher and H. E. Xu, Cell Discovery, 2016, 2, 16026 CrossRef CAS PubMed.
- S. Li, W. Zhang and W. Han, ACS Chem. Neurosci., 2017, 8, 1279–1290 CrossRef CAS PubMed.
- J. Ye, U. P. Davé, N. V Grishin, J. L. Goldstein and M. S. Brown, Proc. Natl. Acad. Sci., 2000, 97, 5123–5128 CrossRef CAS PubMed.
- N. Clemente, A. Abdine, I. Ubarretxena-Belandia and C. Wang, Sci. Rep., 2018, 8, 12411 CrossRef PubMed.
- J. Shen and R. J. Kelleher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 403–409 CrossRef CAS PubMed.
- R. J. Kelleher and J. Shen, Proc. Natl. Acad. Sci., 2017, 114, 629–631 CrossRef CAS PubMed.
- N. Elad, B. De Strooper, S. Lismont, W. Hagen, S. Veugelen, M. Arimon, K. Horré, O. Berezovska, C. Sachse and L. Chávez-Gutiérrez, J. Cell Sci., 2015, 128, 589–598 CrossRef CAS PubMed.
- S. Tagami, M. Okochi, K. Yanagida, A. Ikuta, A. Fukumori, N. Matsumoto, Y. Ishizuka-Katsura, T. Nakayama, N. Itoh and J. Jiang, Mol. Cell. Biol., 2008, 28, 165–176 CrossRef CAS PubMed.
- J. Trambauer, R. M. Rodríguez Sarmiento, A. Fukumori, R. Feederle, K. Baumann and H. Steiner, EMBO Rep., 2020, 21, e47996 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The data required to reproduce the present computational work and scrutinize details of the analysis are included in the file named “suppinfo.pdf”. Furthermore, the full representative structures from cluster analysis are available as three separate PDB files for all three independent simulations. See DOI: 10.1039/d0ra04683c |
| This journal is © The Royal Society of Chemistry 2020 |