
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven CO2 reduction on dye-sensitized NiO photocathodes decorated with palladium nanoparticles†
Ewelina Szaniawskaa,
Anna Wadasa,
Hasina H. Ramanitra b,
Emmanuel A. Fodekeb,
Kamila Brzozowskaa,
Alexandre Chevillot-Biraudb,
Marie-Pierre Santoni*b,
Iwona A. Rutkowska*a,
Mohamed Jouinib and
Pawel J. Kulesza*a
b,
Emmanuel A. Fodekeb,
Kamila Brzozowskaa,
Alexandre Chevillot-Biraudb,
Marie-Pierre Santoni*b,
Iwona A. Rutkowska*a,
Mohamed Jouinib and
Pawel J. Kulesza*a
aFaculty of Chemistry, University of Warsaw, Pasteura 1, PL-02-093 Warsaw, Poland
bUniversité de Paris, ITODYS, CNRS, F-75006 Paris, France. E-mail: marie-pierre.santoni@univ-paris-diderot.fr
First published on 27th August 2020
Abstract
The thin-layer-stacked dye-sensitized NiO photocathodes decorated with palladium nanoparticles (nPd) can be used for the visible-light-driven selective reduction of CO2, mostly to CO, at potentials starting as low as 0 V vs. RHE (compared to −0.6 V in the dark for electrocatalysis). The photosensitization of NiO by the organic dye P1, with a surface coverage of 1.5 × 10−8 mol cm−2, allows the hybrid material to absorb light in the 400–650 nm range. In addition, it improves the stability and the catalytic activity of the final material decorated with palladium nanoparticles (nPd). The resulting multi-layered-type photocathode operates according to the electron-transfer-cascade mechanism. On the one hand, the photosensitizer P1 plays a central role as it generates excited-state electrons and transfers them to nPd, thus producing the catalytically active hydride material PdHx. On the other hand, the dispersed nPd, absorb/adsorb hydrogen and accumulate electrons, thus easing the reductive electrocatalysis process by further driving the separation of charges at the photoelectrochemical interface. Surface analysis, morphology, and roughness have been assessed using SEM, EDS, and AFM imaging. Both conventional electrochemical and photoelectrochemical experiments have been performed to confirm the catalytic activity of hybrid photocathodes toward the CO2 reduction. The recorded cathodic photocurrents have been found to be dependent on the loading of Pd nanoparticles. A sufficient amount of loaded catalyst facilitates the electron transfer cascade, making the amount of dye grafted at the surface of the electrode the limiting parameter in catalysis. The formation of CO as the main reaction product is postulated, though the formation of traces of other small organic molecules (e.g. methanol) cannot be excluded.
1. Introduction
The photophysical and photocatalytic properties of nanocrystalline semiconductor films in hybrid photoelectrode materials are particularly attracting attention in light of photoelectrochemical energy conversion and storage, water splitting (hydrogen evolution), as well as the generation of useful chemicals through the reduction of CO2. Heterogeneous solar-driven thin-film-based technologies, such as Dye-sensitized Photo-Electrosynthetic Cells (DPEC), are particularly promising because they can combine the functions of light harvesting, catalysis, and energy storage (through fuels) in a cost-effective manner.1,2The CO2-reduction reaction (CO2RR) is a representative example of the kinetically-sluggish redox process of environmental importance that proceeds with large overpotentials. Indeed, CO2RR requires breaking strong double bonds in the stable CO2 molecule. Furthermore, the electrochemical and photoelectrochemical conversions of CO2 to useful chemical feedstocks (which include syngas (CO + H2), a known intermediate for further Fischer–Tropsch processes, carbon-based simple organic fuels, or for utility chemicals) are multi-electron and multi-proton processes. Consequently, the design of multifunctional electrode materials (e.g., oxides or metallic nanoparticles) is needed in order to overcome kinetic barriers.3–7
Although CO2RR can be addressed by conventional electrochemical means, the photoelectrochemical conversion of CO2 and water is tempting, presenting different selectivity criteria and possibly lower overpotentials.8–18 However, requirements for photocathodes are pretty exacting: efficient visible light absorption, stability, and fast (interfacial) electron transfers. The last criterion requires a suitable match of the energy levels and long-lived photogenerated charge carriers at the photoelectrochemical (electrode/electrolyte) interfaces. P-type semiconductors, such as copper and nickel oxides, have band edges that are suitably located12 for carrying out reductions such as Hydrogen Evolution Reaction (HER) and CO2RR.17 Due to the existing kinetic limitations and corresponding overpotentials, the conduction band gap edge ought to be positioned at even more negative potentials so as to drive the reduction process successfully.12 The interfacial electron transfer dynamics is facilitated by the appearance of distinct surface states that include terminated crystal lattices and grain boundaries, with dangling bonds favoring the formation of free radicals.5 The activation of CO2 involves the adsorption and weakening of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond, leading to the interfacial reduction of the neutral hydrated CO2 molecule into CO2˙− (or its protonated form HCO2−).19 The CO2RR mechanisms and selectivity depend strongly on the controlled modification of the semiconductor's surface, which is achieved using films (organic, inorganic), nanoparticles, or molecular nanostructures.5,13 In particular, catalytic and co-catalytic species are often needed to drive the high-energy-barrier initial activation step involving the reduction of CO2 to CO2˙− as well as to facilitate the interfacial charge separation by providing trapping sites for electrons, thus decreasing undesirable surface charge recombination.
O double bond, leading to the interfacial reduction of the neutral hydrated CO2 molecule into CO2˙− (or its protonated form HCO2−).19 The CO2RR mechanisms and selectivity depend strongly on the controlled modification of the semiconductor's surface, which is achieved using films (organic, inorganic), nanoparticles, or molecular nanostructures.5,13 In particular, catalytic and co-catalytic species are often needed to drive the high-energy-barrier initial activation step involving the reduction of CO2 to CO2˙− as well as to facilitate the interfacial charge separation by providing trapping sites for electrons, thus decreasing undesirable surface charge recombination.
Copper-based oxides have been investigated as CO2RR photocathodes but their instability caused by photoelectrochemical corrosion tends to limit their practical applications.14–16,18,20–24 On the other hand, NiO is thermally and chemically stable and resists photocorrosion upon illumination.25,26 It has been used as a protective overlayer in photoanodes for solar fuel generation applications.27–29 Interestingly, synthetic approaches allowing the nanoscale engineering of semiconductor photoelectrodes and the preparation of mesoporous NiO films with large surface area are reported.30 Furthermore, based on its conduction band edge position, NiO is capable of driving HER and CO2-reduction under photoelectrochemical conditions.8,27,31,32 However, the photosensitization of NiO is necessary to increase light-harvesting within the visible range due to its wide band gap (ca. 3.6 eV).33 Therefore, the combination of NiO-semiconductor with a dye and a suitable (electro)catalyst is a promising approach to enhance the solar-to-chemical conversion efficiency by decoupling light harvesting, interfacial electron transfers, and bulk charge transport, as well as multi-electronic catalysis.33–35 Indeed, hybrid electrode materials based on NiO have been successfully designed to achieve visible light induced HER or CO2RR (to CO) by photosensitizing the oxide using P1 dye (through carboxylic acid anchoring groups),36 and by adding a catalyst, e.g., the reversible CO2-cycling enzyme carbon-monoxide-dehydrogenase,37 or by combining with a molecular catalyst in solution.38 NiO electrodes have also been sensitized with Ru-dyes and combined with Ni catalyst,39 or with Ru(II)–Re(I)-containing supramolecular cocatalyst.37 In these examples, the catalytic species were chosen for their ability to perform multi-electron catalysis. To achieve this, suitable energy levels are needed for a redox cascade of electron transfers between the components of the photoelectrode. It can be rationalized that upon light absorption, the dye reaches its excited state and injects a hole (h+) into the valence band of NiO; thus, the dye itself undergoes reduction and, in turn, it transfers an electron to the catalyst and, subsequently, to the oxidized species in the electrolyte.36
In the present work, we explore the concept of a hybrid photocathode together with the model of a nanostructured metal electrocatalyst for visible-light-driven CO2RR. While various noble metal heterogeneous catalysts have been examined, palladium nanoparticles could be considered as the state-of-the-art electrocatalysts for CO2RR predominantly to CO and, sometimes, to formate.40–45 The unique interactions of CO2 with Pd, capable of both absorbing and adsorbing hydrogen, are known to favor distinct reaction pathways that affect both the activity and selectivity of CO2RR relative to HER. The design of such photoelectrode materials must be carefully realized in order to favor the intended redox cascade between the components and to minimize competitive charge transfers that reduce the efficiency of the photocathode.46 Consequently, particular attention must be paid to the choice of components and the match between the energy levels and the integration of components within the hybrid 3D-material.38,47,48
2. Experimental section
2.1 Materials
All commercially available chemicals were purchased and used as received: solvents, nickel(II) acetate tetrahydrate, anhydrous nickel(II) chloride, Synperonic® F108 (poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), Sigma Aldrich). Dye P1 was purchased from Dyenamo®. Pd nanoparticles (nPd) (sizes ∼ 6–10 nm) and nanoparticles of platinum (nPt) (sizes ∼ 7–8 nm) were obtained from Sigma-Aldrich and Alfa Aesar, respectively. Sulfuric acid was obtained from POCH (Poland). Nafion perfluorinated resin solution (5 wt%, dissolved in a mixture of lower aliphatic alcohols and water, contains 15–20% water, Sigma-Aldrich) was used in the present work. Sodium sulfate decahydrate (Na2SO4·10H2O, ACS reagent 99%, Sigma-Aldrich), saturated either with argon (pure, Air Products) or carbon dioxide (CO2, 99.99% Air Products), was used to prepare the electrolyte solutions. Fluorine-doped tin oxide (FTO, geometric area, 2.0 cm2) conducting glass (resistivity, ∼15 Ω sq−1; Solaronix®) and glassy carbon (GC, geometric area, 0.071 cm2) were used as the photoelectrode and the working electrode substrates, respectively. Measurements were made at room temperature (20 ± 1 °C).2.2 Physicochemical characterization
X-ray diffraction (XRD) analysis was performed using a PANalytical Empyrean equipped with a multichannel detector (PIXcel 3D) using Cu-Kα radiation in the 20–90° 2θ range. The UV-visible absorption spectra of the thin films (NiO and NiO-P1 on FTO substrate) were recorded on a Cary 4000 spectrophotometer. The morphologies of the electrode surfaces were examined using a Cambridge Supra 40 ZEISS apparatus (acceleration voltage set at 3 kV; top views of surfaces and cross-section for thickness) or a Merlin Zeiss field emission scanning electron microscope (FE-SEM) equipped with two detectors: SE2 and InLens. Elemental analysis was conducted using an energy dispersive spectroscopy (EDS) system on a XFlash Detector 5010 125 eV Quantax Bruker microscope with an acceleration of 15.0 kV and a magnification of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. Transmission electron microscopy (TEM) images of Pd nanoparticles were obtained with a JEM 1400 (JEOL Co., Japan, 2008) transmission electron microscope equipped with a high resolution digital camera (CCD MORADA, SiS-Olympus, Germany). Atomic force microscopy (AFM) was performed using an Easyscan Nanosurface 2.0. Cantilevers with a spring constant of 42 N m−1 and a resonance frequency range of 146–236 kHz, which were tuned prior to further measurement, were employed. The Mott–Schottky plots were obtained from the impedance data generated upon the application of sinusoidal perturbation of frequency ranging from 100 kHz to 1 Hz with 10 mV amplitude. The potential was scanned from 0.895 down to 0.295 V vs. Reversible Hydrogen Electrode (RHE).
000×. Transmission electron microscopy (TEM) images of Pd nanoparticles were obtained with a JEM 1400 (JEOL Co., Japan, 2008) transmission electron microscope equipped with a high resolution digital camera (CCD MORADA, SiS-Olympus, Germany). Atomic force microscopy (AFM) was performed using an Easyscan Nanosurface 2.0. Cantilevers with a spring constant of 42 N m−1 and a resonance frequency range of 146–236 kHz, which were tuned prior to further measurement, were employed. The Mott–Schottky plots were obtained from the impedance data generated upon the application of sinusoidal perturbation of frequency ranging from 100 kHz to 1 Hz with 10 mV amplitude. The potential was scanned from 0.895 down to 0.295 V vs. Reversible Hydrogen Electrode (RHE).
2.3 Fabrication of the NiO film
NiO mesoporous electrodes were prepared on FTO in two steps, following the published procedures: (i) deposition of the compact blocking NiO layer by spin coating (3000 rpm for 30 s) of a Ni(II) precursor solution, followed by sintering at 450 °C (the preparation was done twice, one layer on top of the other);49 (ii) deposition of the mesoporous NiO layer by the ‘Doctor Blade’ method using a Ni(II) precursor solution, followed by calcination at 450 °C.50 Dye-sensitization of the obtained NiO electrodes was performed by soaking in a 0.3 mmol dm−3 P1 dye solution in ethanol for approximately 16 h. The photoelectrodes FTO/NiO-P1 obtained were rinsed with acetonitrile and dried in air.2.4 Preparation of the catalytic system
Suspensions (inks) of metal palladium and platinum nanoparticles were prepared by dispersing 1 mg of the commercial samples through sonication for 120 min in 2.0 cm3 of distilled water to obtain a homogenous mixture. In order to obtain the FTO/NiO-P1/nPd photoelectrodes, further modification of the FTO/NiO-P1 photoelectrodes was done by drop-casting (56 or 112 μdm3) the catalyst inks of nPd onto electrodes (geometric area, ∼2 cm2) and left to dry in air. For the preparation of glassy carbon modified with platinum nanoparticles (used for the anodic stripping voltammetry experiment), 2 μdm3 of the catalyst ink was drop casted over the glassy carbon disk electrode to yield the noble metal loading of 100 μg cm−2.2.5 Electrochemical analysis
Electrochemical measurements (cyclic voltammetry, chronoamperometry experiments) were performed with potentiostats from Bio-Logic SAS (VSP and SP-150) in a 3-electrode setup with an aqueous electrolyte (stated in each case): the counter electrode (Pt grid if not stated otherwise), SCE (3.5 mol dm−3 KCl) as the reference electrode, and the working electrode being the modified (photo)electrode FTO/NiO (the actual active area is measured for each sample in order to determine the measured current density). As a rule, the measured potentials were recalculated at the condition of pH = 0 and expressed against the RHE. The photocatalytic performance of the prepared modified electrodes was determined using a white LED (L.C. RELCO S.p.a., Light Injector, White, 6000 K-6°, 3 W, 4.5 V, 700 mA) calibrated at 0.158 sun (158 W m−2) irradiance using a Si reference solar cell (Fraunhofer ISE): 400–500 nm irradiance is 67.35 (41.8%, D); 500–600 nm irradiance is 64.25 (39.9%, D); 600–700 nm irradiance is 26.14 (16.2%, A); 700–800 nm irradiance is 2.883 (1.8%, D); 800–900 nm irradiance is 0.275 (0.2%, D); 900–1100 nm irradiance is 0.248 (0.2%, D); total irradiance is 161.1 (100%); overall class is D. The experiments were done in 0.1 mol dm−3 sodium sulfate (Na2SO4) electrolyte having pH ≈ 6.4, when the solution was subjected to purging with argon for 20 min (to remove oxygen), and pH ≈ 5.3 when CO2 was bubbled through the cell.3. Results and discussion
3.1 Preparation and characterization of NiO on FTO
The deposition of the NiO film onto the FTO electrode substrate involved, first, the formation of a compact layer, followed by the deposition of a mesoporous layer. It was postulated before that the fabrication of a thin blocking compact-layer on FTO tended to diminish the undesirable loss of charges through recombination.49,50 The actual procedure involved the spin coating of the Ni(II) precursor solution, followed by sintering at 450 °C,49 which was repeated twice to improve the compactness of the layer. The mesoporous NiO over-layer was fabricated by depositing the Ni(II) precursor solution using the ‘Doctor Blade’ method, followed by calcination at 450 °C.50 It is apparent from the SEM cross-section image of the resulting bilayer NiO film (Fig. 1A) that the thicknesses of the inner compact and the outer mesoporous layers are about 200 and 500 nm, respectively. Furthermore, the wide-angle XRD data of the obtained NiO films (Fig. 1B) confirmed the formation of the expected cubic NiO phase (bunsenite, ICSD-53930, see ESI†), with crystallite sizes in the range of 6–8 nm and ∼20 nm for NiO particles, i.e., for the compact and mesoporous layers, respectively. This observation is consistent with that of previous reports.49,50 Nevertheless, the SEM results are consistent with the fairly homogeneous surface morphology, showing very few cracks or defects (see also Fig. S3 and S4†). | ||
| Fig. 1 (A) SEM cross-section image bilayer of the NiO film composed of the blocking (BL) compact inner-layer and the mesoporous outer layer deposited on FTO conducting-glass electrode substrate. (B) Wide-angle XRD pattern for the NiO oxides on FTO: compact layers (first coating, in green, and the second coating in black); the mesoporous over-coating (in purple). | ||
The electrochemical behaviour of the NiO film on the FTO electrode, FTO/NiO, was assessed by performing cyclic voltammetric experiments in 0.1 mol dm−3 Na2SO4 (Fig. S5†). This electrolyte was particularly chosen (instead of carbonate or phosphate buffers) in order to minimize the electrolyte anion adsorption effect. The system exhibited two sets of fairly broad voltammetric peaks, which can be attributed to the approximately reversible redox transitions of NiO.27,51,52 Although the exact nature of the film depends on the preparation method and the experimental conditions, it is reasonable to expect that the oxide and hydroxide species exist at the electrochemical interface,53–55 and the actual electron transfers involve Ni(II) and Ni(III) as well as Ni(III) and Ni(IV) ionic sites. As postulated earlier,27,56 the first oxidation peak at 0.93 V (Fig. S5†) is attributed to the oxidation of Ni(II) to Ni(III), accompanied by the release of proton, whereas the second oxidation peak at 1.26 V is assigned to the oxidation of Ni(III) to Ni(IV). The diagnostic square-wave voltammetric experiment confirmed that both the redox processes involve same amounts of charge and are, herein, monoelectronic (see Fig. S6†). Careful analysis of the cyclic voltammogram in Fig. S5† suggests that the NiO voltammetric peaks described above overlap with the fairly large background currents that can be interpreted in terms of the capacitive or pseudocapacitive properties of the oxide. The feasibility of application of nickel oxides/hydroxides for multiple and reversible charging–discharging in the capacitor-type batteries has been reported before.57 Indeed, our FTO/NiO electrodes exhibited high stability and good reproducibility during prolonged potential cycling (50 cycles in the potential range from 1.54 V to −0.46 V vs. RHE). Regarding the semiconducting properties of nickel oxides, the optical band gap of the NiO film (on FTO) was confirmed by measuring the UV-vis absorption spectra of FTO/NiO and by using the Tauc relationship.58 The band gap energy (Eg) was found to be equal to 3.68 eV, which is consistent with the literature data.49,50 More details are given in the ESI (Fig. S7).†
3.2 Formation and characterization of the P1-dye modified FTO/NiO photoelectrodes
The photosensitization of the NiO semiconducting film with the organic P1-dye was achieved by overnight soaking of the FTO/NiO electrodes in a 0.3 mmol dm−3 ethanolic dye solution. The respective absorption spectra in Fig. S8† shows an increase in the absorbance within the visible region, which is consistent with the presence of the dye on NiO. The anchored dye exhibits a bathochromic shift in its absorbance maximum, from 468 nm observed in solution36 to 497 nm upon chemisorption onto NiO, and is attributed to the coupling of the dye with the semiconductor.36 The estimated surface coverage Γ of the P1-dye is ca. 1.5 × 10−8 mol cm−2, using the data of Fig. S8† and the relationship: A(λairmax) = 1000 × Γ × ε(λsolutionmax), where A(λairmax) is the absorbance of the film measured in air, ε(λsolutionmax) stands for the extinction coefficient of the dye in solution (in mol−1 dm3 cm−1), and Γ stands for the surface coverage (in mol cm−2).59The surface morphology and composition of bare FTO/NiO and the dye-modified (FTO/NiO-P1) electrodes were observed using Field Emission Scanning Electron Microscopy (FE-SEM) (Fig. 2, also S11†). The existence of NiO nanoparticles of ca. 20 nm diameter (Fig. 2A) is consistent with the size determined in the XRD results, as well as with the literature data.60 In case of the FTO/NiO-P1 electrode, the organic matter is visible (by chemical contrast) on the surface (Fig. 2B). To confirm the elemental composition of the organic layer in the FTO/NiO-P1 electrodes, elemental analysis was conducted using Energy Dispersive Spectroscopy (EDS). The elemental mapping showed that Ni and O are uniformly distributed along the NiO bottom layer in the FTO/NiO-P1 arrangement, whereas the S, C and N atoms have only been found in the top layer, thus confirming the presence of the organic dye P1 on the surface (Fig. 2B). The elemental mapping for the stacked arrangement of the FTO/NiO-P1 electrode revealed that Ni and O elements are uniformly distributed along the NiO bottom layer, which is underneath and appears darker in Fig. 2B, identical to the mesoporous structure shown in Fig. 2A (i.e., 20 nm nanoparticles with some pores). On the other hand, the chemical contrast reveals that the S, C, and N atoms are only found in the top layer, which is above the previous layer and appears lighter in Fig. 2B due to chemical contrast (with bigger undefined structures). This confirms the presence of the organic dye P1 on the NiO surface, which appears lighter in Fig. 2B.
 | ||
| Fig. 2 Top-view images of (A) FTO/NiO (from SEM), and (B) FTO/NiO-P1 (from EDS). | ||
The surface morphology of the FTO/NiO-P1 electrodes was probed by atomic force microscopy using the tapping mode (Fig. 3). The surface of the electrodes seems to be rather rough, judging from the differences in the surface levels: 47 nm for FTO/NiO-P1 vs. 13 nm for FTO/NiO. This change in the surface roughness in FTO/NiO-P1 reflects the presence of the adsorbed dye on top of the NiO surface. Most likely, the deposition of the dye yields an irregular surface of the ultra-thin film, which is favored by π-stacking between the dye molecules.
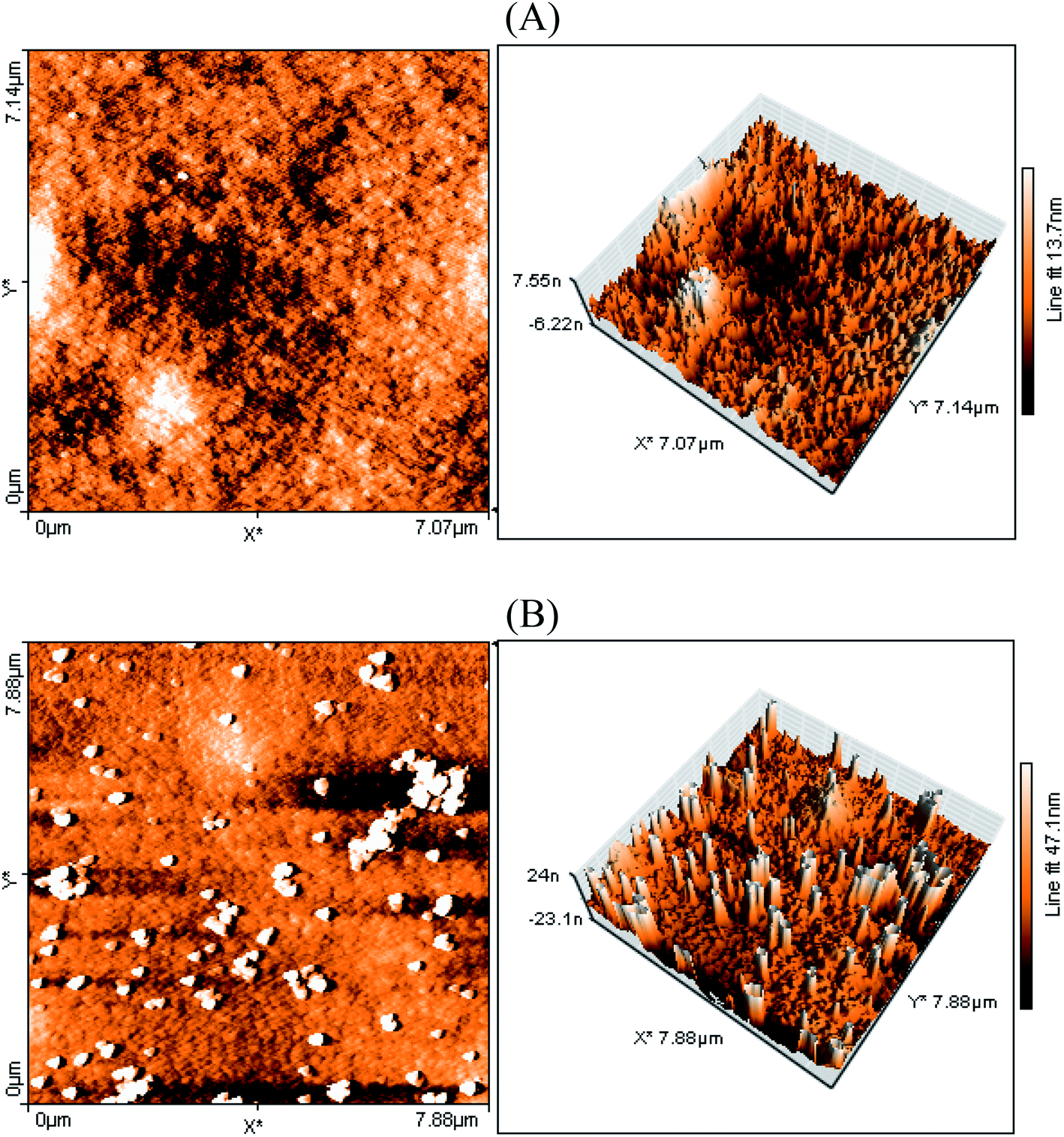 | ||
| Fig. 3 AFM images (in tapping mode) of (A) FTO/NiO, and (B) FTO/NiO-P1. | ||
The flat-band potentials of both the bare and dye-sensitized FTO/NiO electrodes were determined using electrochemical impedance spectroscopy and the Mott–Schottky analysis in aqueous media at various pH.61 In this respect, the feasibility of decoupling of the pH effects and the positions of the band-edge energy levels by formation of the hydrophobic monolayer of an organic dye and, thus, the passivation of the semiconductor surface has been postulated. Here, the passivation is not complete and amounts to 55% of the NiO surface, as shown by the cyclic voltammetry results. The irregular P1-dye layer presents voids that leave some of the NiO surface accessible. We have also determined the flat-band potential, Efb, and the charge carrier (hole) concentration, NA, for the FTO/NiO and FTO/NiO-P1 electrodes considered in the present work. On the basis of the capacitance vs. potential difference measurements, the Efb and NA values were obtained by using the Mott–Schottky equation,58
 | (1) |
For all the samples, the flat-band potentials and the charge carrier concentrations were obtained from the slopes of the extrapolated linear fits of the Mott–Schottky plots (Fig. S11 and S12†). The obtained negative slopes are consistent with a p-type semiconducting behavior. It is noteworthy that the obtained values of both the charge carrier concentrations (densities) and flat-band potentials are comparable to those previously reported for NiO and dye-sensitized NiO photoelectrodes.61 The comparable flat-band potentials determined for FTO/NiO and FTO/NiO-P1, viz., 0.50 and 0.57 V (vs. RHE), respectively, indicate that the anchoring of the P1-dye onto NiO does not significantly modify the energy level of the flat-band potential in the system. Finally, as expected, the charge carrier concentration values have been found to be as high as 4.2 × 1017 and 9.0 × 1015 cm−3 for FTO/NiO and FTO/NiO-P1, respectively.
3.3 Modification of the P1-dye-sensitized FTO/NiO with Pd nanoparticles
The final step in the preparation of the photocathodes for CO2RR is the deposition by drop-casting of the catalyst, palladium nanoparticles (nPd, 6–10 nm). Their TEM image is shown in the inset of Fig. 4. The resulting FTO/NiO-P1/nPd electrodes show the existence of nPd aggregates (lighter colored spots in the system's SEM micrographs, Fig. S13 and 14†). On the whole, the interfacial structure of the photocathode is porous and it is reasonable to expect that palladium exists in contact with the NiO-P1 layer as inter-connected nanostructures (Fig. S13 and 14†). The NiO and Pd nanoparticles have comparable sizes that match the average pore size of the mesoporous layer: 20 vs. 10 nm in diameter, respectively. The partial infiltration of some of the nPd into the NiO-P1 mesoporous layer is possible.8,27 | ||
| Fig. 4 Voltammetric behavior (at 10 mV s−1 scan rate) of palladium nanoparticles (deposited on glassy carbon, loading 100 μg cm−2) recorded in deoxygenated 0.1 mol dm−3 Na2SO4 solutions in the presence (solid line) and absence (dashed line) of CO2. Current densities are provided against the electrode geometric area. | ||
Fig. 4 illustrates the cyclic voltammograms recorded for the nPd deposited (loading, 100 μg cm−2) on the glassy carbon electrode substrate in the argon-saturated 0.1 mol dm−3 Na2SO4 in the presence (solid line) and absence (dashed line) of CO2. It is well established that in a model acid medium such as in 0.5 mol dm−3 H2SO4, the deposits of nPd exhibit characteristic hydrogen sorption/desorption peaks (below 0.25 V)40,62,63 that are also observed here, as well as Pd oxide formation or reduction (above 0.55 V) peaks. In the near-neutral deoxygenated Na2SO4 solution, the sorption of hydrogen (water reduction or proton discharge) is pushed to more negative potentials and starts at about −0.2 V (Fig. 4, dashed line). The coupled phenomenon of hydrogen oxidation and desorption appears in the form of a broad surface-type peak at about 0.45 V. Fairly low anodic currents starting at ca. 0.8 V indicate the formation of a thin oxide layer on the Pd surface (a mix of PdIOH, PdII(OH)2 or PdIIO and, eventually, PdIVO2).64 The appearance of a small peak at about 0.45 V should be correlated with the reduction of previously generated (in anodic scan) Pd oxides. Finally, hydrogen evolution starts at potentials below ca. −0.45 V (Fig. 4, dashed line) and the latter process proceeds, relative to the behavior in the CO2-saturated slightly acidic medium, with ca. 400 mV overpotential.
To evaluate the system's electrocatalytic activity toward CO2RR, the voltammetric electroreduction experiments were carried out in CO2-saturated 0.1 mol dm−3 Na2SO4 (CO2-concentration, ca. 0.033 mol dm−3).65 It is apparent from Fig. 4 (solid line) that CO2RR starts at potentials lower than 0.2 V and yields a reduction peak in the potential range from −0.35 to −0.4 V, i.e., where hydrogen sorption is most operative (compare the solid and dashed lines in Fig. 4). The ability of the Pd nanostructures towards the reversible adsorption/absorption of hydrogen is of importance for the effective hydrogenation (reduction) of CO2. The rapid decrease in the CO2RR peak current, appearing at about −0.4 V (solid line), results from the poisoning of the palladium electrocatalytic interface by CO-type intermediates, which is a process that competes with the sorption of hydrogen. In addition, the formation of formate adsorbates through electrochemically generated Pd-hydrides cannot be excluded here.66
The appearance of relatively smaller peaks (in the reverse oxidation scan) at potentials more positive than 0.7 V (Fig. 4, solid line) should be correlated with overlapping processes such as the oxidation of the CO2RR-products/adsorbates (mostly carbon monoxide, possibly with some formate)40,45 and the oxidation of generated or sorbed hydrogen. It is noteworthy that the oxidation peaks mentioned above (i.e., in the range 0.7 to 1.2 V) are not observed in the absence of CO2 (dashed line). On the whole, the CO2RR voltammetric pattern of Fig. 4 (solid line) is analogous to those reported earlier.40,41,67
3.4 Photoelectrochemical performance of NiO-based systems
First, the (photo)electrochemical behavior of the FTO/NiO system was studied and the respective voltammetric experiments were performed both in dark and upon illumination (with visible light) in the deoxygenated sodium sulfate electrolyte saturated with CO2 (Fig. S15†). As expected from its band gap (∼3.7 eV), light has no effect on the response of FTO/NiO. In Fig. S15,† the voltammetric pattern is in agreement with previous reports describing the behavior of NiO in almost neutral media:38,68 the solution pH drops from 6.4 (Ar) to 5.3 (CO2). Therefore, the appearance of growing reduction currents below −0.3 V (Fig. S15†) could be interpreted in terms of two overlapping processes such as NiO-catalyzed CO2-electroreduction and HER, as postulated earlier.27 Our diagnostic experiments performed with FTO/NiO electrode under electrochemical (dark) conditions, in the presence and absence of CO2 (inset to Fig. S15†), are consistent with the dominating contribution of CO2RR rather than HER, as evidenced by the sharp increase in the cathodic current below −0.3 V. It cannot be excluded that hydrogen-assisted CO2-reduction69 is operative here, with the generation of some Ni0 sites at the NiO surface.The introduction of the visible-light-responsive organic dye P1 onto NiO changes the oxide physicochemical characteristics. Fig. S16† compares the cyclic voltammetric responses of FTO/NiO-P1 recorded in the presence of CO2: the reduction currents (emerging at potentials lower than 0.6 V down to −0.2 V; dashed line) are increased upon irradiation with respect to the dark experiment (solid line). Such significant photocathodic currents (Fig. S16,† inset) indicate that CO2-photoreduction seems to be facilitated by light, which was not observed for FTO/NiO in the absence of the dye (Fig. S15†). Provided that the dye loading is as small as 15 nmol cm−2 and the light intensity is as low as AM 0.1, the net photocurrent density (in the potential range of 0.2–0 V) would be of ca. 5 μA cm−2, which is low but noticeable. It should be noted that due to the incomplete coverage of NiO by the P1-dye (Fig. 2 and 3), some NiO sites are surfacing and can also induce CO2RR and/or HER. However, their contribution is limited and similar in all the samples.
We performed experiments to verify that the conventional (dark) electrocatalytic properties of Pd remain the same, when deposited on glassy carbon (Fig. 4) and on FTO/NiO (for simplicity, not shown here). Fig. 5A and B illustrate the cyclic voltammograms recorded in the presence of CO2 for nPd deposited onto the P1-dye over-layer at two different loadings, 2 and 4 μg cm−2. This information translates to the following relative mass ratios of Pd to P1-dye: 30:1 and 60:1, respectively. The amounts of deposited nPd have been kept low to avoid the excessive darkening of the photocathode. On doubling the loading, a better evaluation of the Pd-induced electrocatalytic effect is feasible. The resulting hybrid photocathodes with smaller (Fig. 5A) and larger Pd loadings (Fig. 5B), abbreviated as FTO/NiO-P1/nPd(x1) and FTO/NiO-P1/nPd(x2), respectively, have been investigated in dark (solid lines) and upon illumination (dashed lines). The fact, that the irradiation with visible light has a significant effect on the electroreduction currents (appearing below 0.7 V), particularly at larger nPd loading (Fig. 5B), clearly suggests that introduction of the nPd catalyst results in further photocurrent enhancement, which should be correlated with light-driven CO2RR. Careful examination of the dark-current corrected (background-subtracted) responses shown in the insets of Fig. 5A and B clearly suggests that the net CO2RR current doubled from −7.5 to −15 μA (at −0.2 V) on increasing the nPd loading twice. This observation supports our view about the catalytic role of Pd deposited onto dye-sensitized p-type NiO cathode and the proposed mechanism: under irradiation, P1 generates excited-state electrons that are transferred to nPd, thus producing the hydride PdHx with catalytically active sites. In this regard, the proposed mechanism resembles the performances of previously reported P1-dye-sensitized NiO photocathodes.38,68 Herein, dispersed palladium nanostructures exhibit charge storage properties, which likely contribute to the distribution of charges and, possibly, to the charge cascade process at the photoelectrochemical interface.18 Importantly, upon illumination, the charge carriers are photogenerated at the NiO-P1 interface and the photoelectroreduction processes are shifted toward less negative potentials (four times more current relative to the performance electrocatalytic experiment, comparison of Fig. S16† and 5A).
 | ||
| Fig. 5 Cyclic voltammetric responses for (A) FTO/NiO-P1/nPd(x1) and (B) FTO/NiO-P1/nPd(x2) photocathodes differing in the Pd loadings (2 and 4 μg cm−2) recorded in 0.1 mol dm−3 Na2SO4 saturated with CO2: in dark (solid line) and upon illumination with AM 0.1 G filter (dash line). Scan rate 20 mV s−1. The insets illustrate the net current densities recorded upon illumination and corrected for dark current. | ||
3.5 Photoelectrochemical behavior of the hybrid decorated photocathode
The photoelectrochemical performance of the FTO/NiO-P1/nPd(x1) photocathode was studied by recording pulsed voltammetric-type current–potential responses upon illumination with visible chopped light (frequency, 0.1 Hz) in the Na2SO4 electrolyte. Fig. 6 illustrates the transient photocurrent generated during the on–off cycles of illumination in the Ar-saturated (black curve) and CO2-saturated (red curve) solutions, respectively. The photocurrent obtained under CO2 is significantly higher than the photocurrent under Ar. Under Ar, the P1-sensitized NiO shows some photocurrent (black curve) due to the photogeneration of charges following light absorption by the dye and subsequent charge separation at the NiO-dye interface. Small current increases observed at potentials lower than 0.1 V (black curve) are due to H sorption and/or HER, which may happen on nPd at potentials below −0.1 V. Judging from the electrocatalytic results of Fig. 5A, the HER process is likely to be suppressed and shifted to even more negative potentials in the presence of CO2, as a result of surface poisoning by CO-type products. Under CO2 (red curve), a higher transient photocurrent is generated, relative to the response in the absence of CO2. This enhancement could be ascribed to the completion of eased electron transfer cascade with the light-driven reduction of CO2, mediated by the nPd catalyst. | ||
| Fig. 6 Voltammetric-type plot of the photocurrent vs. the applied potential recorded for the hybrid FTO/NiO-P1/nPd(x1) photocathode in 0.1 mol dm−3 Na2SO4 electrolyte: saturated with argon (black curve) and saturated with CO2 (red curve). The pulsed responses were recorded upon illumination with chopped light (illumination frequency, 0.1 Hz). Scan rate, 2 mV s−1. | ||
3.6 Verification of the reaction products
When CO2RR is carried out on a Pd catalyst in near-neutral deoxygenated aqueous solutions, CO together with H2 and small amounts of formate are usually produced.40,70,71 To confirm the formation of CO as the main photoelectrochemical reduction product at the hybrid FTO/NiO-P1/nPd(x1) photocathode, anodic stripping-type voltammetric experiments were performed, referring to previous studies.18,72 Here, an additional glassy carbon working electrode with deposited nPt (loading 100 μg cm−2) was placed in the vicinity of the photocathode. After long-term photoelectrochemical reduction of CO2 at the FTO/NiO-P1/nPd(x1) electrode, the photogenerated reaction products were adsorbed and pre-concentrated on the surface of the GC/nPt electrode (see experimental details in the ESI† and the choice of potentials). The pre-concentration potential step prevents the interference originating from the reduction of CO2 on Pt and the adsorption of additional reduction products. Later, the electrode was transferred to a separate deoxygenated 0.5 mol dm−3 solution to execute the actual stripping-type voltammetric oxidation experiment (Fig. 7). Although the latter stripping step can also be performed in near-neutral Na2SO4 electrolyte,72 voltammetric patterns characteristic of electrocatalytic oxidation of the adsorbates of small organic molecules are more unequivocal in acid medium. | ||
| Fig. 7 Voltammetric response of platinum nanoparticles (deposited on glassy carbon; loading, 100 μg cm−2) in deoxygenated 0.5 mol dm−3 H2SO4 (dashed line). Scan rate, 10 mV s−1. Stripping voltammogram for the oxidation of the CO2RR-product adsorbed and pre-concentrated after 15 min of chronoamperometric reduction of CO2 at −0.6 V under illumination (experimental conditions are as that for Fig. 5A). For comparison, the inset illustrates the stripping-type net voltammetric currents for the oxidation of formic acid adsorbate on nPt. | ||
The dashed line in Fig. 7 illustrates the typical response of bare Pt nanoparticles. In the potential range from 0 to 0.25 V, characteristic hydrogen sorption peaks are developed, and at potentials higher than 0.8 V, the oxidation of platinum to platinum oxides takes place (followed by their reduction at 0.7 V). For the in-between hydrogen peaks and the formation of Pt oxides, platinum exists mostly as Pt0 (so called double-layer region).73 The stripping-type voltammetric response (Fig. 7, solid line) of the CO2RR product (generated on the hybrid FTO/NiO-P1/nPd(x1) photocathode), adsorbed/pre-concentrated on the Pt nanoparticles at the neighboring electrode and, immediately, medium-transferred to H2SO4 electrolyte, yields a peak (at ca. 0.6 V; Fig. 7, solid line), which shall be attributed to the oxidation of the CO-adsorbates. The appearance of a single peak (stripping experiment of Pt) implies that CO is the main reaction product, in agreement with other reports of CO2RR on Pd.40,70 The broad peak suggests that in addition to CO, small amounts of other products such as adsorbates or bulk species with aldehyde or alcohol groups can also be generated during CO2 reduction.74 The formation of formate is not likely here because the oxidation of the formic acid adsorbates (deposits) on nPt yields two peaks in the stripping experiment (inset of Fig. 7). The first peak is characteristic of the oxidation of CO (formed due to surface interactions of HCOOH with Pt) and the second peak (∼0.85–0.9 V) stands for the oxidation of HCOOH induced by generated Pt oxides. Here, as no peak is observed in the range of 0.8–0.9 V (Fig. 7), very little or no formate is formed.
3.7 (Photo)electrochemical response of decorated photoelectrodes within an enlarged potential range [+1.2 V; −0.7 V] vs. RHE
The (photo)electrocatalytic response of the decorated photoelectrodes was investigated over a wider range of potentials [+1.2 V; −0.7 V] vs. RHE in order to study the electron transfer cascade at play here, particularly the electron transfers that occur after the formation of D-. The efficiency of these processes depend on their quantum yields and their kinetics. The electrochemical response of the photoelectrodes under CO2 (Fig. 8) displays the reduction of the adsorbed P1 at about −0.51 V vs. RHE (consistent with the literature – Table S1,† and verified with P1 drop-casted on glassy carbon electrode – Fig. S1†). In Fig. 8, the current response is an important feature that depends on the loading of the catalyst on the photoelectrodes. For the FTO/NiO-P1/nPd(x1) electrode (red curve), the cathodic current is higher and starts at a more positive potential than in the FTO/NiO-P1 electrode (black curve). However, the amount of the available catalyst is low (2 μg cm−2), which may limit the catalytic process. In that case, the catalyst becomes the bottleneck in the electron cascade and the formed D- cannot be entirely consumed. This has been confirmed by doubling the amount of catalyst loaded; for the FTO/NiO-P1/nPd(x2) electrode (blue), the quantity of available catalyst is higher (4 vs. 2 μg cm−2) and is not the limiting parameter anymore within the electron transfer cascade. Indeed, this situation is thermodynamically favorable and the reduction of the dye to D- becomes easier, as evidenced by the observed anodic shift of 100 mV (blue curve). The limitation comes, in that case, from the fixed amount of the dye being reduced at the surface of the electrode. This has been confirmed by increasing the light intensity (0.5 sun), which gave the same photocurrents. Besides, the observed increase in the anodic current measured around +1.0 V from FTO/NiO-P1 to FTO/NiO-P1/nPd(x1) and FTO/NiO-P1/nPd(x2) is consistent with a substantial increase in the amount of reduction products formed beforehand. The anodic current increase is less than twice from FTO/NiO-P1/nPd(x1) to FTO/NiO-P1/nPd(x2), which further supports that the amount of active Pd surface available is not the limiting parameter anymore to run the catalysis in the case of nPd(x2). In conclusion, the electron transfer cascade between the components of the decorated photoelectrode performs satisfactorily for CO2RR with sufficient loading of the catalyst. | ||
| Fig. 8 Cyclic voltammograms (second cycle) recorded in 0.1 mol dm−3 Na2SO4 saturated with CO2 and upon illumination, for the photoelectrodes: (black) FTO/NiO-P1; (red) FTO/NiO-P1/nPd(x1); (blue) FTO/NiO-P1/nPd(x2). Scan rate: 10 mV s−1. | ||
4. Conclusions
We report herein on the preparation and characterization of the dye-sensitized-NiO-based photocathodes decorated with nPd for the successful visible-light-induced electrocatalytic reduction of CO2 predominantly to CO, at potentials starting from as low as 0 V vs. RHE. The concept bridges the gap between two classes of photoelectrodes, based on heterogenized-molecular components as well as on electrocatalytic nanoparticles. Among important parameters is the loading of Pd nanoparticles on the dye-sensitized-NiO. A sufficient quantity of loaded catalyst (4 μg cm−2) facilitates the electron transfer cascade, making the fixed amount of dye reduced at the surface of the electrode the limiting parameter for catalysis. Although the proposed system behaved reproducibly and exhibited perfect stability during the time scales of the performed experiments, some loss (ca. 20–30%) of nPd was observed for prolonged photoelectrochemical reduction (12 hours). Practical systems would require stabilization, e.g., with Nafion overcoating. Nevertheless, the present results serve as a proof of concept that show the photoelectrochemical utility of the multi-layered-type photocathode (TOC, Fig. S17†) based on the electron-transfer-cascade. Among important features of the described photoelectrodes is the ability of palladium, upon sorption of hydrogen (interfacial PdHx hydride formation that is correlated with capacitive-type charge storage properties), to selectively catalyze the proton-coupled two-electron reduction of CO2 to CO. The fact that the latter reaction is not subject to the common energy barrier restriction related to the formation of the CO2˙− radical anion (E = −1.9 V vs. RHE)68 is advantageous here.In principle, the proposed hybrid photocathode (CO2RR) can be coupled with a suitable photoanode (for water oxidation) in order to produce a true photoelectrochemical tandem electrolysis cell. It is reasonable to expect that the present concept of the photoelectrode (Fig. S17†) can be extended to many other electrocatalytic systems (e.g., hydrogen evolution and NOx-reduction), provided that they meet the energy level requirements imposed by the electron-transfer-cascade.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Sophie Nowak (Université de Paris) is gratefully acknowledged for the X-ray diffraction analysis of FTO/NiO electrodes. We acknowledge the ITODYS SEM and XRD facilities (Université de Paris, CNRS UMR 7086, Paris, France). This work has been realized under the auspices of Polish-French Collaboration Program “Polonium” (PHC Polonium 2018–2019) and Campus France and the French Embassy in Warsaw are gratefully acknowledged for mobility support. Université Paris Diderot–Université de Paris (Laboratoire ITODYS) and CNRS are gratefully acknowledged for financial support. This work has also been supported by the National Science Center (NCN, Poland) under Opus Project 2018/29/B/ST5/02627.References
- A. Rothschild and H. Dotan, ACS Energy Lett., 2017, 2, 45–51 CrossRef CAS.
- B. D. Sherman, M. V. Sheridan, K.-R. Wee, S. L. Marquard, D. Wang, L. Alibabaei, D. L. Ashford and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 16745–16753 CrossRef CAS PubMed.
- L. Zhang, Z. L. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 36, 11326–11353 CrossRef PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 19, 7610–7672 CrossRef PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS.
- P. J. Kulesza, I. A. Rutkowska and A. Wadas, Encyclopedia of Interfacial Chemistry: Surface Science and Electrochemistry, ed. K. Wandelt, Elsevier, 1st edn, 2018, 5, pp. 521–530 Search PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. S. DuChene, G. Tagliabue, A. J. Welch, X. Li, W.-H. Cheng and H. A. Atwater, Nano Lett., 2020, 20(4), 2348–2358 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, ACS Energy Lett., 2017, 2, 2058–2070 CrossRef CAS.
- E. B. Creel, E. R. Corson, J. Eichhorn, R. Kostecki, J. J. Urban and B. D. McCloskey, ACS Energy Lett., 2019, 4, 1098–1105 CrossRef CAS.
- J. S. DuChene, G. Tagliabue, A. J. Welch, W.-H. Cheng and H. A. Atwater, Nano Lett., 2018, 18, 2545–2550 CrossRef CAS.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- I. A. Rutkowska, E. Szaniawska, J. Taniewicz, A. Wadas, E. Seta, D. Kowalski and P. J. Kulesza, J. Electrochem. Soc., 2019, 166, H3271–H3278 CrossRef CAS.
- C. Janaky, D. Hursán, B. Endrodi, W. Chanmanee, D. Roy, D. Liu, N. R. Tacconi, B. H. Dennis and K. Rajeshwar, ACS Energy Lett., 2016, 1, 332–338 CrossRef CAS.
- J. Ferreira de Brito, A. R. Araujo, K. Rajeshwar and M. V. Boldrin Zanoni, Chem. Eng. J., 2015, 264, 302–309 CrossRef CAS.
- K. Rajeshwar, M. Kabir Hossain, R. T. Macaluso, C. Janáky, A. Varga and P. J. Kulesza, J. Electrochem. Soc., 2018, 165, H3192–H3206 CrossRef CAS.
- E. Szaniawska, I. A. Rutkowska, E. Seta, I. Tallo, E. Lust and P. J. Kulesza, Electrochim. Acta, 2020, 341, 136054 CrossRef CAS.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 36, 11326–11353 CrossRef PubMed.
- G. Ghadimkhani, N. R. Tacconi, W. Chanmanee, C. Janaky and K. Rajeshwar, Chem. Commun., 2013, 49, 1297–1299 RSC.
- J. Ferreira de Brito, A. Alves da Silva, A. J. Cavalheiro and M. V. Boldrin Zanoni, Int. J. Electrochem. Sci., 2014, 9, 5961–5973 Search PubMed.
- W. Siripala, A. Ivanovskaya, T. F. Jaramillo, S. H. Baeck and E. W. McFarland, Sol. Energy Mater. Sol. Cells, 2003, 77, 229–237 CrossRef CAS.
- E. Szaniawsk, K. Bienkowski, I. A. Rutkowska, P. J. Kulesza and R. Solarska, Catal. Today, 2018, 300, 145–151 CrossRef.
- E. Szaniawska, I. A. Rutkowska, M. Frik, A. Wadas, E. Seta, A. Krogul-Sobczak, K. Rajeshwar and P. J. Kulesza, Electrochim. Acta, 2018, 265, 400–410 CrossRef CAS.
- J. He, H. Lindstrom, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- F. Odobel, L. Le Pleux, Y. Pellegrin and E. Blart, Acc. Chem. Res., 2010, 43, 1063–1071 CrossRef CAS PubMed.
- A. Sapi, A. Varga, G. F. Samu, D. Dobo, K. L. Juhasz, B. Takacs, E. Varga, A. Kukovecz, Z. Konya and C. Janaky, J. Phys. Chem. C, 2017, 121, 12148–12158 CrossRef CAS PubMed.
- K. Sun, M. T. McDowell, A. C. Nielander, S. Hu, M. R. Shaner, F. Yang, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. Lett., 2015, 6, 592–598 CrossRef CAS.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- M. P. Dare-Edwards, J. Goodenough, A. Hamnett and N. D. Nicholson, J. Chem. Soc., Faraday Trans., 1981, 77, 643–661 RSC.
- C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- F. Odobel and Y. Pellegrin, J. Phys. Chem. Lett., 2013, 4(15), 2551–2564 CrossRef CAS.
- L. Alibabaei, H. Luo, R. L. House, P. G. Hoertz, R. Lopez and T. J. Meyer, J. Mater. Chem. A, 2013, 1, 4133–4145 RSC.
- W. J. Youngblood, S. H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS.
- P. Qin, H. Zhu, T. Edvinsson, G. Boschloo, A. Hagfeldt and L. Sun, J. Am. Chem. Soc., 2008, 130, 8570–8571 CrossRef CAS PubMed.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- A. Wadas, I. A. Rutkowska, A. Gorczyński, M. Kubicki, V. Patroniak and P. J. Kulesza, Electrocatalysis, 2014, 5, 229–234 CrossRef CAS.
- R.-H. Guo, C.-F. Liu, T.-C. Wei and C.-C. Hu, Electrochem. Commun., 2017, 80, 24–28 CrossRef CAS.
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, ACS Catal., 2018, 8(2), 1510–1519 CrossRef CAS.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137(13), 4288–4291 CrossRef CAS PubMed.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si, F. Yang, S. Miao, J. Wang, G. Wang and X. Bao, Nano Res., 2017, 10, 2181–2191 CrossRef CAS.
- R. Kortlever, C. Balemans, Y. Kwon and M. T. M. Koper, Catal. Today, 2015, 244, 58–62 CrossRef CAS.
- P. J. Cameron and L. M. Peter, J. Phys. Chem. B, 2005, 109(15), 7392–7398 CrossRef CAS PubMed.
- H. Tian, ChemSusChem, 2015, 8, 3746–3759 CrossRef CAS PubMed.
- M. Bonomo, D. Dini and F. Decker, Front. Chem., 2018, 6, 601 CrossRef CAS.
- P. Ho, L. Q. Bao, R. Cheruku and J. H. Kim, Electron. Mater. Lett., 2016, 12, 638–644 CrossRef CAS.
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199(1), 1–7 CrossRef CAS.
- A. G. Marrani, V. Novelli, S. Sheehan, D. P. Dowling and D. Dini, ACS Appl. Mater. Interfaces, 2014, 6, 143–152 CrossRef CAS PubMed.
- B. T. Sone, X. G. Fuku and M. Maaza, Int. J. Electrochem. Sci., 2016, 11, 8204–8220 CrossRef CAS.
- E. A. Gibson, M. Awais, D. Dini, D. P. Dowling, M. T. Pryce, J. G. Vos, G. Boschloo and A. Hagfeldt, Phys. Chem. Chem. Phys., 2013, 15, 2411–2420 RSC.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef CAS.
- D. M. Tench and E. Yeager, J. Electrochem. Soc., 1973, 120, 164–171 CrossRef CAS.
- H. Bode, K. Dehmelt and J. J. Witte, Electrochim. Acta, 1966, 11, 1079–1087 CrossRef CAS.
- N. Bagheri, A. Aghaei, N. Vlachopoulos, M. Skunik-Nuckowska, P. J. Kulesza, L. Häggman, G. Boschloo and A. Hagfeldt, Electrochim. Acta, 2016, 194, 480–488 CrossRef CAS.
- S. R. Morrison, Electrochemistry at semiconductor and oxidized metal electrodes, Plenum Press, New-York, 1980 Search PubMed.
- L. A. Gallagher, S. A. Serron, X. Wen, B. J. Hornstein, D. M. Dattelbaum, J. R. Schoonover and T. Meyer, J. Inorg. Chem., 2005, 44, 2089–2097 CrossRef CAS PubMed.
- Y. Kou, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- Y. Yu, K. A. Click, S.-C. Chien, J. Sun, A. Curtze, L.-C. Lin and Y. Wu, J. Phys. Chem. C, 2019, 123, 8681–8687 CrossRef CAS.
- I. A. Rutkowska, D. Marks, C. Perruchot, M. Jouini and P. J. Kulesza, Colloids Surf., A, 2013, 439, 200–206 CrossRef CAS.
- I. A. Rutkowska, Aust. J. Chem., 2016, 69, 394–402 CrossRef CAS.
- M. Grden, M. Lukaszewski, G. Jerkiewicz and A. Czerwinski, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef CAS.
- M. Jitaru, J. Univ. Chem. Technol. Metall., 2007, 42(4), 333–344 CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- D. Kolbe and W. Vielstich, Electrochim. Acta, 1996, 41, 2457–2460 CrossRef CAS.
- A. Bachmeier, S. Hall, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136(39), 13518–13521 CrossRef CAS PubMed.
- W. Lin, K. M. Stocker and G. C. Schatz, J. Am. Chem. Soc., 2017, 139(13), 4663–4666 CrossRef CAS PubMed.
- R. P. S. Chaplin and A. A. Wragg, J. Appl. Electrochem., 2003, 33, 1107–1123 CrossRef CAS.
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electrochem. Soc., 1990, 137, 1773–1777 Search PubMed.
- A. Wadas, I. A. Rutkowska, M. Bartel, S. Zoladek, K. Rajeshwar and P. J. Kulesza, Russ. J. Electrochem., 2017, 53, 1194–1203 CrossRef CAS.
- C. L. Green and A. Kucernak, J. Phys. Chem. B, 2002, 106, 1036–1047 CrossRef CAS.
- H. Siwek, M. Lukaszewski and A. Czerwinski, Phys. Chem. Chem. Phys., 2008, 10, 3752–3765 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04673f |
| This journal is © The Royal Society of Chemistry 2020 |