
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne pot synthesis of trifluoromethyl aryl sulfoxides by trifluoromethylthiolation of arenes and subsequent oxidation with hydrogen peroxide†
Monika Horvat,
Gregor Kodrič,
Marjan Jereb and
Jernej Iskra*
and
Jernej Iskra*
Faculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia. E-mail: jernej.iskra@fkkt.uni-lj.si
First published on 18th September 2020
Abstract
Hydrogen peroxide was used for oxidation of various aryl trifluoromethyl sulfides. Trifluoroacetic acid was used as an activating solvent that enables non-catalyzed oxidation and increases selectivity for sulfoxide formation. As shown by oxidation of thianthrene TFA enhances electrophilic character of the oxidant and further oxidation of sulfoxide group is blocked. We have joined trifluoromethylthiolation of arenes using a modified Billard reagent (p-ClPhNHSCF3) with oxidation of aryl trifluoromethyl sulfides using 1.2 equiv. of 30% aqueous hydrogen peroxide and this one-pot process has superior yields than would have been obtained in a two step process.
Introduction
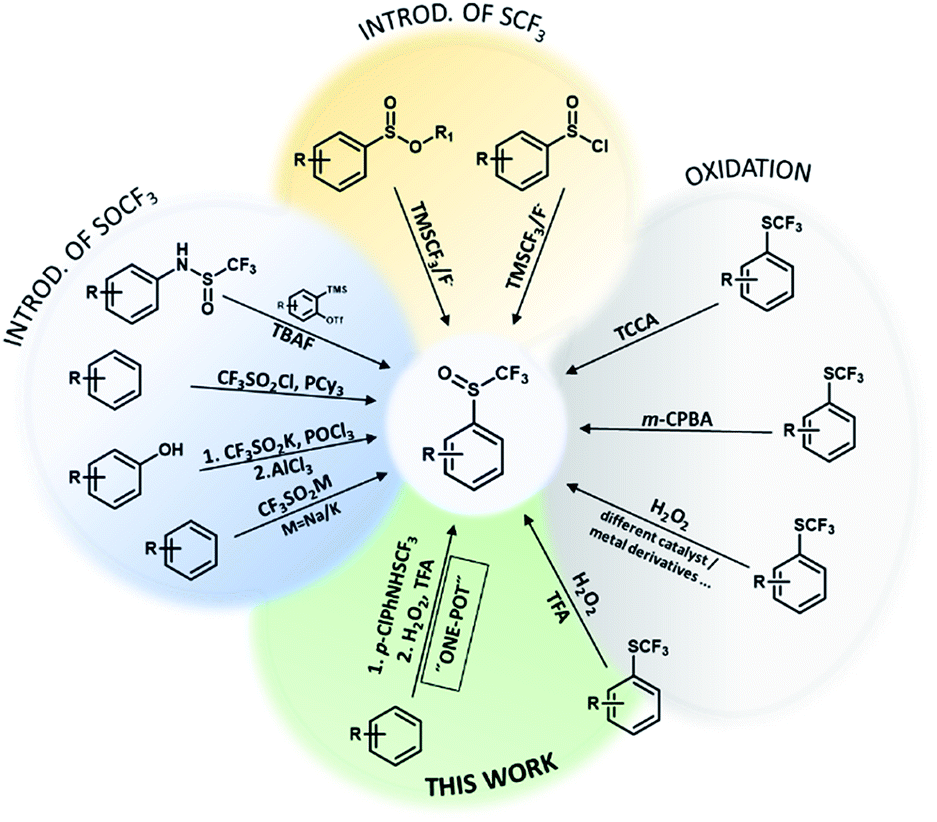
Organofluorine compounds have great potential in the development of new materials, bioactive compounds, agrochemicals, pharmaceutical compounds and in many other areas.1,2 They are interesting because of their chemical, physical and physiological properties.3 The trifluoromethylsulfanyl group (SCF3) has a high lipophilicity parameter (1.44) and a strong electron-withdrawing effect.4,5 It has a slightly weaker electron-withdrawing character (Hammett constant: σm = 0.40, σp = 0.50) than the trifluoromethyl group (CF3) (Hammett constant: σm = 0.43, σp = 0.54). Functional groups SOCF3 and SO2CF3 have even stronger electron-withdrawing character (SOCF3: σm = 0.63, σp = 0.69; SO2CF3: σm = 0.79, σp = 0.93) than CF3.6 Organic compounds with SOCF3 and SO2CF3 groups are not as common as SCF3 compounds due to their problematic synthesis. However, they show pharmacological and biological activities (antibacterial, antimalarial, anti-pneumonia and nervous anorexia treatment) and they are already on the market e.g. fipronil, ponazuril.7,8Trifluoromethyl aryl sulfoxides can be prepared directly from different aromatic molecules by sulfinylation of aromatics by triflinate salts in acidic medium,9 by CF3SO2Cl/PCy3 reagent10 and by thia-Fries rearrangement process in the presence of AlCl3 (Scheme 1).11 In 1999 Langlois reported on the synthetic method for trifluoromethanesulfinates and trifluoromethanesulfinamides using the CF3SO2Na/POCl3 system.12 They can also be prepared with 1-(trifluoromethylsulfinyl)pyrrolidine-2,5-dione in good yields.13 Allylic trifluoromethanesulfenates can be prepared from allylic alcohols using [N–SCF3]+ reagent via a [2,3]-sigmatropic rearrangement.14 ortho-Trifluoromethanesulfinyl anilines can be synthesized by intermolecular C–N addition of amides and S–N addition of sulfinamides to arynes (Scheme 1).15 Opposite strategy is introduction of CF3 group into sulfinic esters or sulfinyl halides using TMSCF3 reagent and fluoride activator.16,17
 | ||
| Scheme 1 Different ways for preparation of aryl trifluoromethyl sulfoxides. | ||
Researchers have discovered new processes for the selective oxidation of sulfides to corresponding sulfoxides, which are easy to handle, green and cost-effective. Trifluoromethyl sulfides are less reactive for oxidation and various oxidizing agents such as m-CPBA, CF3CO3H, NaIO4, TCCA, cyclic diacyl peroxide, Oxone, MoO2Cl2(OPPH3)2/Cu(NO3)2 have been used and only two examples with H2O2 (F20TPPFe and TFA) were reported (Scheme 1).3,18–22 As trifluoromethyl sulfides are less reactive, selectivity of oxidation is problematic and overoxidation to sulfones occurs readily. The problem with most oxidants is the formation of toxic waste and environmentally harmful by-products (metal salts, reagent residues).23 Most of them are expensive, oxidation is very temperature sensitive and overoxidation can lead to the formation of unfavorable by-product (trifluoromethyl sulfone).18 In recent years, research has focused on the development of an effective, simple and selective route for the synthesis of trifluoromethyl sulfoxides under mild reaction conditions using safe and clean oxidation processes. H2O2 is an attractive oxidant, inexpensive, soluble in water and many organic solvents, and environmentally friendly, since water is the only theoretical by-product.23,24 Oxidation with H2O2 is useful for the synthesis of pharmaceuticals and agrochemicals, which requires high chemical purity.24,25 The problem with hydrogen peroxide is that it reacts slowly with organic compounds and must be activated. There are many reports of the hydrogen peroxide based oxidation of sulfides to corresponding sulfoxides under appropriate activation conditions (nucleophilic, electrophilic and radical activation) using transition metal or organic catalyst,23,26–28 while H2O2 was also activated with fluorinated alcohols29 (Scheme 1).
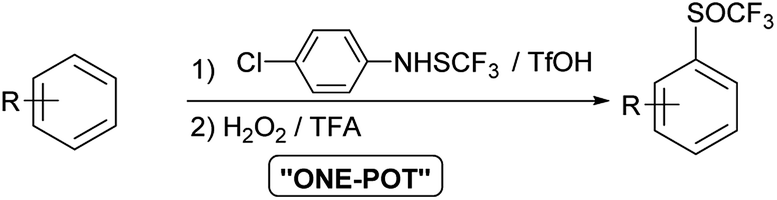
Studies on oxidation of aromatic trifluoromethyl sulfides with H2O2 are limited. In 2019 Yagupolskii reported on the oxidation protocol for the oxidation of CF3S to the CF3S(O) group using 15% hydrogen peroxide in trifluoroacetic acid. During the oxidation no further unwanted oxidation to trifluoromethyl sulfone occurred. This method has some disadvantages, such as the incomplete conversion of the trifluoromethyl sulfides to sulfoxides. In a few cases the conversion was complete, in others 87–98%.30 The method for direct synthesis of trifluoromethyl sulfoxides from aromatic molecules by introduction of SCF3 group followed by oxidation has not yet been investigated. Due to the limited set of available aryl trifluoromethyl sulfides, the one-pot procedure is very desirable. The trifluoromethylsulfanyl group (SCF3) can be introduced into the aromatic molecules with various electrophilic reagents.6,31,32 One of the most commonly and widely used reagent is the Billard reagent PhNHSCF3,33,34 while reagent p-ClPhNHSCF3 is suited for the use on wider range of arenes.35 We present in this report effective, simple and highly selective method for the one-pot synthesis of various aryl trifluoromethyl sulfoxides from different activated and deactivated aromatic molecules by trifluoromethylthiolation with the stable and easy to use reagent p-ClPhNHSCF3,35 followed by oxidation with H2O2 in TFA (Scheme 2).
 | ||
| Scheme 2 One-pot synthesis of trifluoromethyl sulfoxides from different aromatic molecules. | ||
Results and discussion
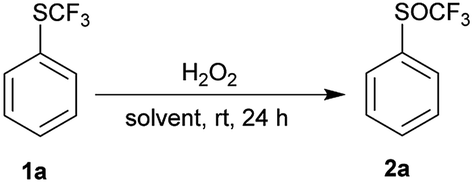
Phenyl trifluoromethyl sulfide 1a was chosen as model substrate for an initial study on the effect of reaction conditions on the selective oxidation of the trifluoromethylsulfanyl group (SCF3) with hydrogen peroxide as an oxidant. The sulfur atom is deactivated for oxidation because of the electron-withdrawing CF3 group and must be activated. We examined the effect of different solvents, the concentration of hydrogen peroxide, transition metals, Brønsted and Lewis acids on the oxidation of the sulfur atom.First, we investigated the oxidation of trifluoromethyl sulfide 1a to the corresponding trifluoromethyl sulfoxides 2a or sulfones 3a with 30% H2O2 in different solvents such as non-polar solvent (PhCH3), polar aprotic solvents (DCM, EtOAc, MeCN), polar protic solvents (EtOH, i-PrOH) and fluorinated alcohols (TFE, HFIP) (Table 1). A solution of trifluoromethyl sulfide 1a in selected solvent and 2 equiv. of 30% aqueous hydrogen peroxide was stirred at room temperature for 24 h. Afterwards the conversion was determined by 1H NMR spectroscopy of the crude reaction mixture. The experimental results in Table 1 show that a small amount of product 2a was formed only in the HFIP. In other solvents oxidation did not occur.
|
|||
|---|---|---|---|
| Entry | Solvent | Conv.b [%] | |
| a Reaction conditions: phenyl trifluoromethyl sulfide 1a (0.5 mmol), H2O2 (1.0 mmol, 30–100%), solvent (2 mL), rt, 24 h.b Conversion to product was determined by 1H NMR.c TFE – 2,2,2-trifluoroethanol, HFIP – 1,1,1,3,3,3-hexafluoropropan-2-ol.d Reflux temperature. | |||
| 1 | Non-polar | PhCH3 | 0 |
| 2 | Polar aprotic | DCM, EtOAc, MeCN | 0 |
| 3 | Polar protic | EtOH, i-PrOH | 0 |
| 4 | Fluorinated alcoholsc | TFE | 0 |
| 5 | HFIP (30%) | 12 | |
| 6 | HFIP (50%) | 15 | |
| 7 | HFIP (100%) | 16 | |
| 8 | HFIPd (30%) | 50 | |
Fluorinated alcohols strongly activate hydrogen peroxide by their high ionizing power, strong hydrogen bond donor ability and weak hydrogen bond acceptor strength. HFIP has a stronger effect than TFE. It is known that the activation of 30% hydrogen peroxide by HFIP is strong enough to selectively oxidize sulfides to sulfoxides.36 Furthermore, HFIP deactivates sulfoxide for further oxidation to sulfone by its interaction with sulfoxide group. In the case of sulfide 1a, the deactivating effect of the trifluoromethyl group outweights activation by HFIP. We have tried to increase the reaction with more concentrated hydrogen peroxide (50% and 100%). Surprisingly, the concentration of H2O2 had only a minor influence on the conversion (Table 1, entries 5–7). Only at the reflux temperature 50% of 2a were formed.
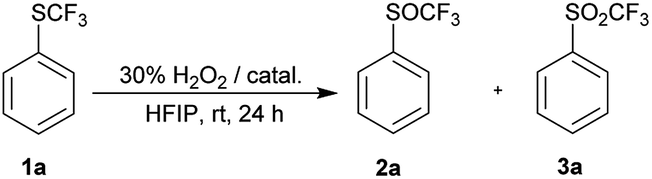
Since the activation by HFIP itself was not strong enough to achieve complete oxidation of 1a, we decided to investigate the effect of stronger activators, such as metal catalysts, Brønsted and Lewis acids. We used only 1.2 equiv. of H2O2 and 10 mol% catalyst in these reactions to reduce over-oxidation. Strong Brønsted acids catalyzed the oxidation of 1a and complete conversion was observed in 24 hours at room temperature, but trace amounts of sulfone 3a were also formed (Table 2, entries 1–3). TFA – a weaker acid, was not so effective, while no reaction occurred in acetic acid (entries 4 and 5). Activation with Lewis acid BF3·OEt2 led to a similar result as with strong acids. Selected metal-catalyzed oxidation depended on the metal H2WO4 was the least selective catalyst, MTO did not give complete conversion, while FeCl3 gave similar results to TFA (Table 2, entries 7–9).
|
||||
|---|---|---|---|---|
| Entry | Activator | Conv.b [%] | Product selectivity [%] | |
| 2a | 3a | |||
| a Reaction conditions: phenyl trifluoromethyl sulfide 1a (0.5 mmol), H2O2 (0.6 mmol, 30%), catalyst (10 mol%), HFIP (2 mL), rt, 24 h.b Conversion to product was determined by 1H NMR.c 1 mol% of activator. | ||||
| 1 | HCl | 100 | 99 | 1 |
| 2 | H2SO4 | 100 | 99 | 1 |
| 3 | TfOH | 100 | 99 | 1 |
| 4 | TFA | 11 | 100 | 0 |
| 5 | AcOH | 0 | 0 | 0 |
| 6 | BF3·Et2O | 99 | 98 | 2 |
| 7 | H2WO4 | 100 | 95 | 5 |
| 8 | CH3ReOc, pyridinec | 97 | 100 | 0 |
| 9 | FeCl3, pyridine | 10 | 100 | 0 |
Since TFA had some catalytic activity, we decided to use it as a solvent for oxidation of 1a to 2a. To a solution of trifluoromethyl sulfide 1a in TFA 1.2 equiv. of 30% aqueous hydrogen peroxide was added. The reaction mixture was stirred at room temperature and 1a was completely oxidized within three hours with formation of trifluoromethyl sulfoxide 2a and sulfone 3a in ratio 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Table 3, entry 1). Analogous oxidation of 1a in AcOH was slower and conversion after 48 hours reaction was 80%. Promising results in TFA lead to further optimization. The use of an equimolar amount of H2O2 led to a selective formation of sulfoxide 2a with 96% conversion in 3 h at room temperature (Table 3, entry 2), however reaction did not proceed to completion even after prolonged reaction time (Table 3, entry 3). The reaction at 0 °C was slower and a higher conversion was achieved with a higher excess of H2O2 (Table 3, entries 4–7). The quantitative conversion was achieved with two equivalents of the oxidant, but a small amount of over-oxidized product 3a was present in the reaction mixture. The quantitative and selective conversion of 1a to 2a was achieved with 1.2 equiv. of H2O2 at 0 °C in 6 hours (Table 3, entry 8). Sulfone 3a was difficult to form in TFA and harsher conditions were required – 2.4 equiv. of hydrogen peroxide at 60 °C led to formation of sulfone 3a with a yield of 96% (Table 3, entry 9).
:3 (Table 3, entry 1). Analogous oxidation of 1a in AcOH was slower and conversion after 48 hours reaction was 80%. Promising results in TFA lead to further optimization. The use of an equimolar amount of H2O2 led to a selective formation of sulfoxide 2a with 96% conversion in 3 h at room temperature (Table 3, entry 2), however reaction did not proceed to completion even after prolonged reaction time (Table 3, entry 3). The reaction at 0 °C was slower and a higher conversion was achieved with a higher excess of H2O2 (Table 3, entries 4–7). The quantitative conversion was achieved with two equivalents of the oxidant, but a small amount of over-oxidized product 3a was present in the reaction mixture. The quantitative and selective conversion of 1a to 2a was achieved with 1.2 equiv. of H2O2 at 0 °C in 6 hours (Table 3, entry 8). Sulfone 3a was difficult to form in TFA and harsher conditions were required – 2.4 equiv. of hydrogen peroxide at 60 °C led to formation of sulfone 3a with a yield of 96% (Table 3, entry 9).
|
|||||
|---|---|---|---|---|---|
| Entry | Temp. | Equiv. H2O2 | Conv.b [%] | Product selectivity [%] | |
| 2 | 3 | ||||
| a Reaction conditions: phenyl trifluoromethyl sulfide 1 (0.5 mmol), H2O2 (0.5–1.2 mmol, 30%), TFA (2 mL), 3 h.b Conversion to product was determined by 1H NMR.c Reaction time: 6 h.d Isolated yield. | |||||
| 1 | Rt | 1.2 | 100 | 97 | 3 |
| 2 | Rt | 1.0 | 96 | 100 | 0 |
| 3 | Rt | 1.0c | 94 | 100 | 0 |
| 4 | 0 °C | 1.0 | 65 | 100 | 0 |
| 5 | 0 °C | 1.2 | 67 | 100 | 0 |
| 6 | 0 °C | 1.5 | 84 | 100 | 0 |
| 7 | 0 °C | 2.0 | 100 | 99 | 1 |
| 8 | 0 °C | 1.2c | 100 | 100 | 0 |
| 9 | 60 °C | 2.4 | 100 | 4 | 96 (95%)d |
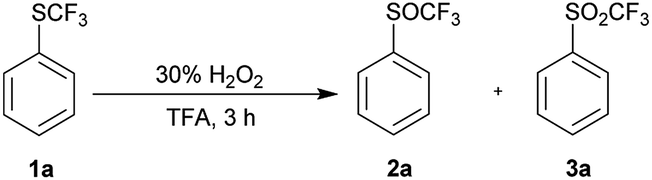
We applied the knowledge gained in initial studies to oxidize various aromatic trifluoromethyl sulfides 1. Due to limited commercial availability, a library of trifluoromethyl sulfides 1 was prepared from various activated and deactivated aromatic substrates using our method with p-ClPhNHSCF3.35 The best conditions for the preparation of 2a (1.2 equiv. of 30% H2O2, 0 °C, TFA) were applied. To a solution of trifluoromethyl sulfide 1 in TFA, 1.2 equiv. of 30% H2O2 was added and the reaction mixture was stirred at 0 °C until the sulfide was consumed, as determined by GC-MS. In all cases a selective and quantitative transformation to trifluoromethyl sulfoxide 2 was observed and we prepared thirteen trifluoromethyl sulfoxides 2 (Scheme 3) with electron-donating and electron-withdrawing groups on the aromatic ring in 77–95% yield. The oxidation tolerated the presence of hydroxyl, acetyl and hydroxymethyl groups on the aryl ring. On the other hand, the formyl group is not tolerated and p-formylphenyl trifluoromethyl sulfide was converted into a complex mixture of products. The phenanthryl derivative 1m was also oxidized in 85% yield to the corresponding sulfoxide 2m.
 | ||
| Scheme 3 Selective oxidation of aryl trifluoromethyl sulfides 1. | ||
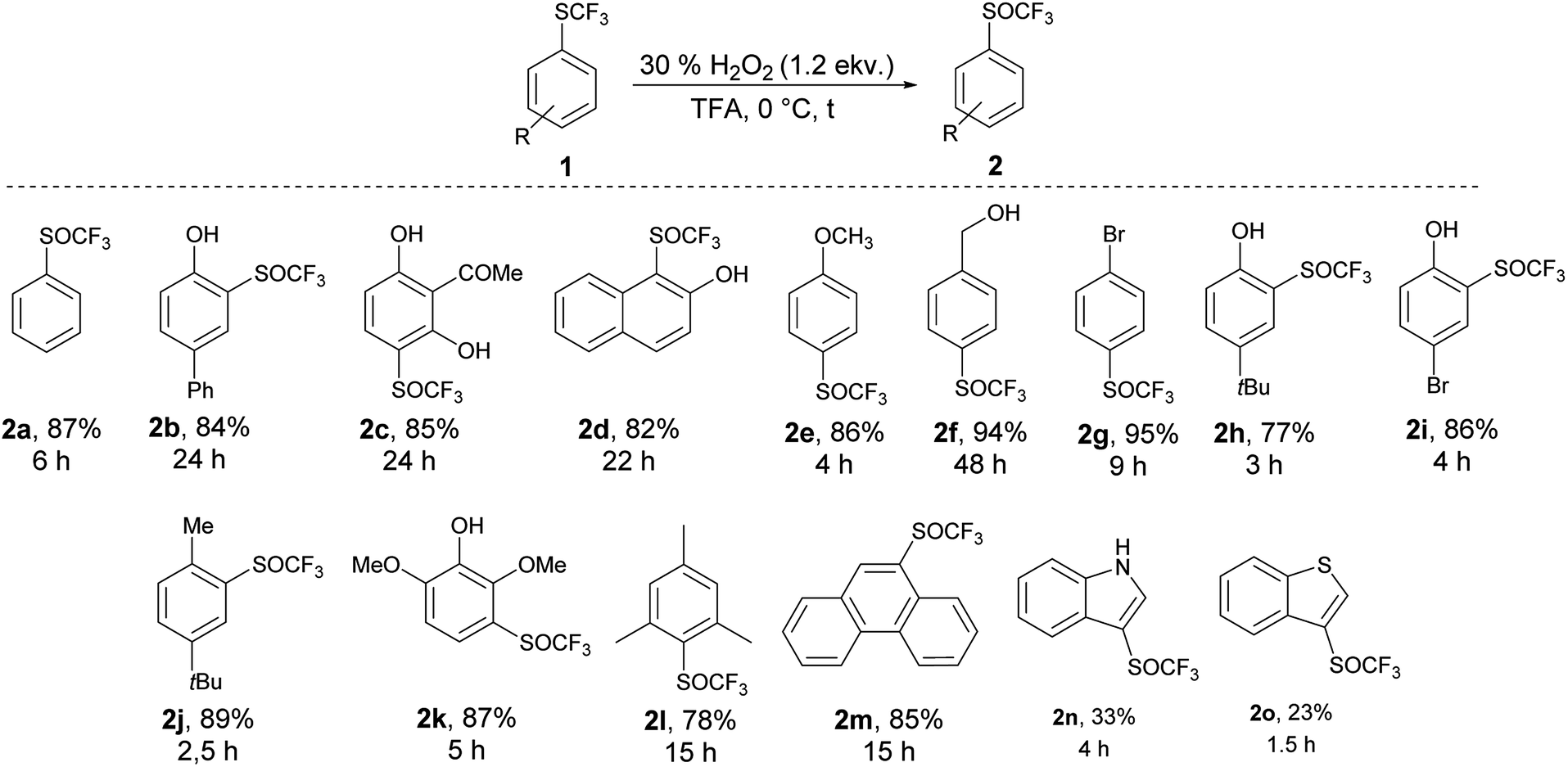
An introduction of the SCF3 group into heteroaromatic ring is difficult and pyridine, imidazole and benzothiazole failed to react under these reaction conditions. On the other hand, pyrrole was converted into a complex mixture of products. Furthermore, sulfur atom in SCF3 group is very deactivated for oxidation leading to problems with selectivity of oxidation of substrates with various functional groups. We made a further study on the series – benzofuran, benzothiophene and indole to see how heteroatoms affects the oxidation. Trifluoromethylthiolation of benzofurane did not occur under the classical reaction conditions and only starting compound was recovered. Indole and benzothiophene were selectively transformed to the corresponding trifluoromethylthiolated derivatives 1n and 1o, respectively.35 Indole was selectively and quantitatively trifluoromethylthiolated to give 3-((trifluoromethyl)thio)-1H-indole 1n, which was oxidized under the standard reaction conditions to the desired trifluoromethyl sulfoxide 2n and isolated in 33% yield (Scheme 3). A lower yield is attributed to the formation of a complex mixture of products. 3-((Trifluoromethyl)thio)benzo[b]thiophene 1o has been completely oxidated, but oxidation occurred on both sulfur atoms and a mixture of 2o and 3-((trifluoromethyl)thio)benzo[b]thiophene 1-oxide 2o′ in the ratio 34:66 was formed. We isolated both products, however the side product 2o′ is not stable and starts to decompose during isolation procedure into the product 2o′′ (Scheme 4). This surprising reaction could be a result of a hetero Diels–Alder additions followed be a hydride shift. The product 2o′′ also decomposes slowly into a complex mixture of products.
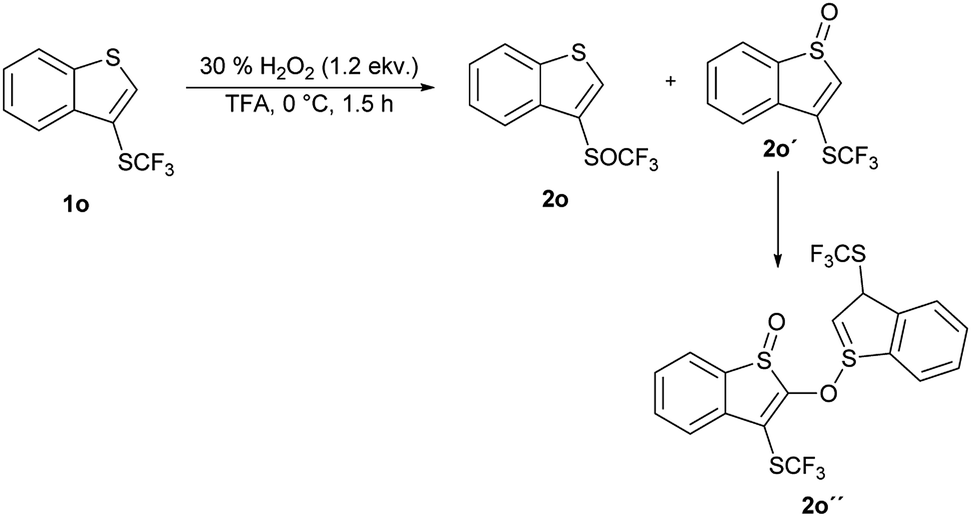 | ||
| Scheme 4 Oxidation of 3-((trifluoromethyl)thio)benzo[b]thiophene 1o. | ||
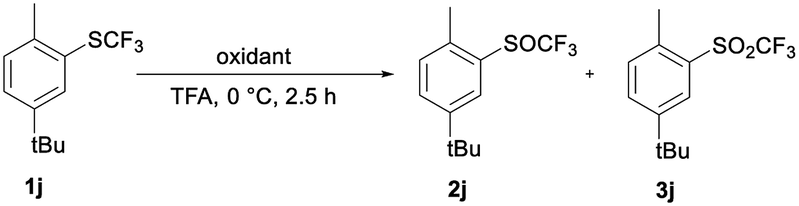
TFA as solvent is a very effective activator of H2O2. As in template catalysis in fluorinated alcohols,29 formation of hydrogen bonds between ArSCF3 1, TFA and H2O2 could be the reason for the activation. Furthermore, the interaction between sulfoxide 2 and TFA could reduce the nucleophilicity of the sulfur atom and deactivate the SOCF3 group for further oxidation to the sulfones.29 To test the specific interaction of TFA and H2O2 in selective oxidation, we compared the results of the oxidation of sulfide 1j with H2O2 and with meta-chloroperoxybenzoic acid (m-CPBA).37
Table 4 shows data for the oxidation of sulfide 1j to sulfoxide 2j or sulfone 3j with m-CPBA and with 30% H2O2. 1j is oxidized selectively and quantitatively with 1.2 equiv. of 30% H2O2 in TFA in 2.5 hours at 0 °C. An analogous reaction with m-CPBA is less effective and less selective in TFA. Under the same reaction conditions the conversion is only 22%, while a significant amount of sulfone 3j is already formed.
To further elucidate the role of trifluoroacetic acid in this reaction we have turned to thianthrene 4 as a model substrate. Adam and his coworkers have studied the oxidation of thianthrene 5-oxide (SO) as a mechanistic probe to determine the electronic character of various oxidising agents. Electrophilic oxidants oxidize SO at the sulfide S-atom to give sulfoxide SOSO, while nucleophilic oxidants react preferentially at the sulfoxide S-atom to afford sulfone SSO2.38,39
We compared selectivity of oxidation of thianthrene 4 with m-CPBA and with H2O2 in three solvents – DCM, MeCN and TFA. As presented in Fig. 1, oxidation with 2 equiv. of m-CPBA in DCM and MeCN proceeded at room temperature with 100% conversion. The main product was sulfoxide SO 5, while some sulfone SSO2 7 already formed. Reaction in TFA was performed at 0 °C and the main reaction channel was oxidation of both sulfur atoms with formation of SOSO 6 with the major stereoisomer being cis-SOSO 6a. The result is in accordance with the effect of TFA.40 No formation of SSO2 7 was observed. Oxidation of thianthrene S 4 with H2O2 proceeds only in TFA. Contrary to oxidation in MeCN and DCM, cis-SOSO 6a was the major product in reaction in TFA and the selectivity for 6a/6b was higher at 0 °C. Even after prolonged reaction time at room temperature no sulfone 7 is formed. Results show that TFA promotes oxidation through electrophilic process, which is evident by higher selectivity towards formation of sulfoxide products.
 | ||
| Fig. 1 Selectivity of oxidation of thianthrene 4 (reaction conditions: 4 (0.5 mmol), oxidant (1.0 mmol), solvent (5 mL), time. Product distribution was determined by 1H NMR spectroscopy). | ||
TFA therefore plays an important role in the activation of hydrogen peroxide for the oxidation of ArSCF3 and in the deactivation of further oxidation to sulfone. TFA could activate the electrophilic character of H2O2 for oxidation by hydrogen bonding, while the same interaction with the oxygen atom of the sulfoxide group would make the S-atom in SOCF3 more electrophilic and thus less reactive.
The same activation by TFA could also be effective in the reaction of trifluoromethylthiolation of aromatic molecules with the ArNHSCF3 reagent. Our goal was to combine both reactions – the introduction of SCF3 group and its oxidation – in a one-pot process to incorporate SOCF3 group into the aromatic ring.
With these optimized reaction conditions in hand, we have investigated the possibility of selective and quantitative synthesis of trifluoromethyl sulfoxides 2 from different aromatic and heteroaromatic molecules 1′ in one step. The reaction conditions for the preparation of trifluoromethyl sulfides 1 were taken from the reported reaction conditions.35 A solution of substrate 1′, reagent p-ClPhNHSCF3, activator TfOH and solvent was stirred for 20 h at room temperature. The choice of the solvent depended on the reactivity of the arene (DCM, hexane or TFA). After completion of the reaction, TFA was added to the reaction mixture and cooled to 0 °C. 1.2 equiv. of 30% aqueous hydrogen peroxide were added and the reaction time was followed by GC-MS.
Ten different products were prepared by one-pot process and the reactions were quantitative and selective as in both separate processes (Scheme 5). In general, this one-pot process generated the corresponding products in high yields (78–93%), with the exception of heteroaromates 2n (37%) and 2o (25%). Trifluoromethyl sulfoxides 2 were obtained in quantitative yields, without any trace of trifluoromethyl sulfone 3. Compared to the classical two-step process, the one-pot method is faster, cheaper, easier to carry out and gives better yields (yields were 2 to 13% higher than in the classical two-step process).
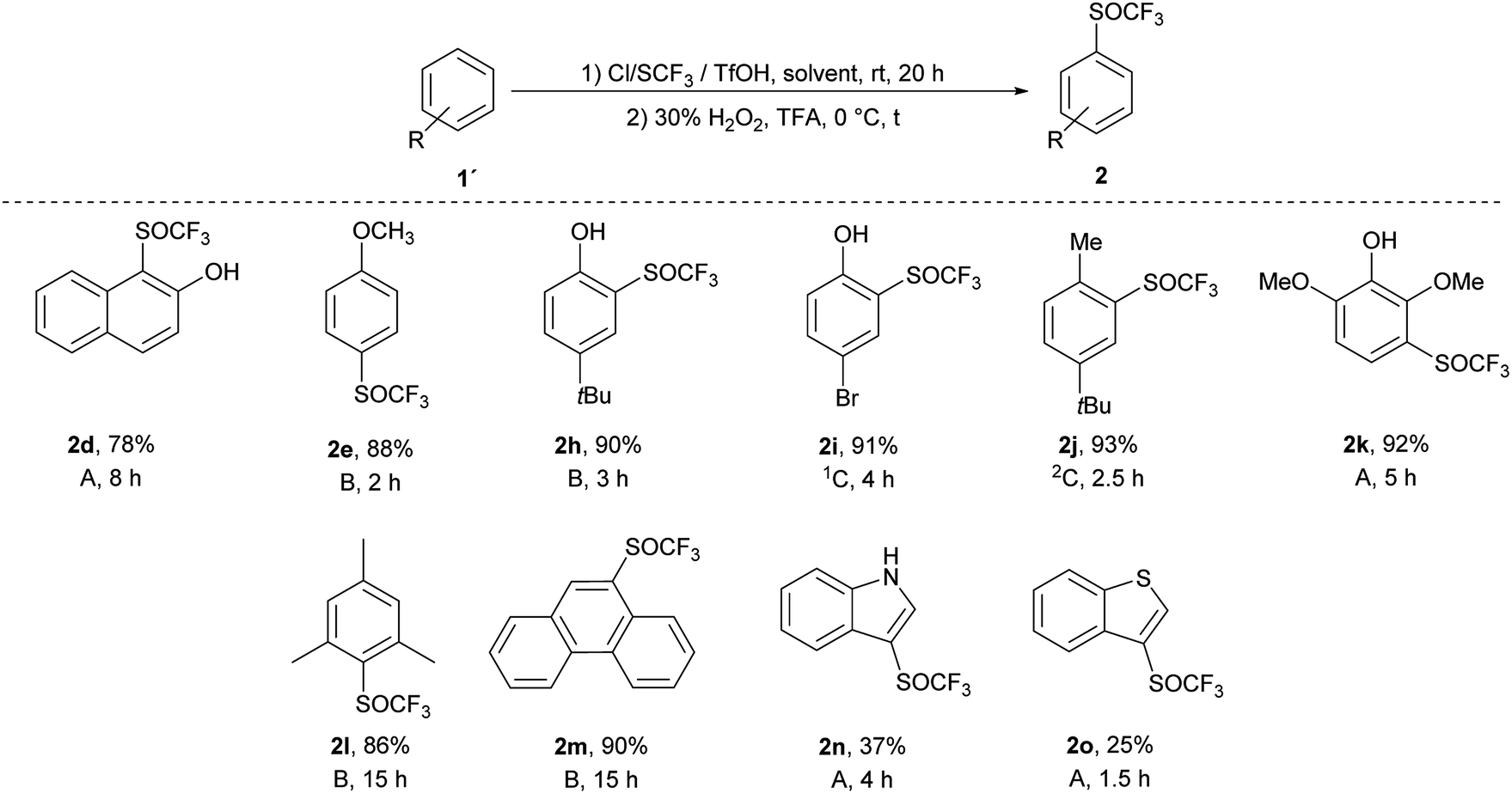 | ||
| Scheme 5 Isolated yields of the trifluoromethyl sulfoxides 2. (1) (Method A: 1a:Cl/SCF3:TfOH = 1:1.3:1.3, solvent: DCM; method B: 1a:Cl/SCF3:TfOH = 1:1.3:1.3, stepwise addition of Cl/SCF3, solvent: DCM; method C: 1a:Cl/SCF3:TfOH = 1:2:3; stepwise addition of Cl/SCF3 and TfOH, solvent: 1hexane/2TFA), rt, 20 h. (2) H2O2 (30%), TFA (5 mL), t, 0 °C. | ||
Conclusions
In summary, we have developed the highly selective oxidation of aryl trifluoromethyl sulfides to the corresponding sulfoxides under metal-free conditions with 30% H2O2. The oxidation was performed in the presence of 30% aqueous hydrogen peroxide as oxidant and TFA as solvent and activator. TFA proved to be effective in activating hydrogen peroxide and increasing selectivity by deactivating further oxidation to sulfone. Furthermore, we have investigated a new method for the direct introduction of SOCF3 functional group into various aromatic molecules. This method allows a simple and efficient synthesis of different trifluoromethyl sulfoxides from aromatic molecules by one-pot trifluoromethylthiolation with p-ClPhNHSCF3 reagent followed by oxidation with 30% H2O2. Mild reaction conditions and generally excellent yields were observed (78–93%).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of the Slovenian Research Agency (P1-0134, P1-0230) is greatly appreciated. The authors are grateful to the stuff of The Centre for Research Infrastructure at the Faculty of Chemistry and Chemical Technology (IC UL FCCT).Notes and references
- G. K. S. Prakash and Z. Zhang, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds, ed. F. R. Leroux and A. Tressaud, Elsevier, 2017, pp. 289–337, DOI:10.1016/B978-0-12-803740-9.00011-1.
- X. Pannecoucke and T. Besset, Org. Biomol. Chem., 2019, 17, 1683–1693 RSC.
- X. H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS.
- S. Barata-Vallejo, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 7150–7182 RSC.
- F. Toulgoat and T. Billard, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds, ed. H. Groult, F. R. Leroux and A. Tressaud, Elsevier, 2017, pp. 141–179, DOI:10.1016/B978-0-12-803740-9.00006-8.
- H. Chachignon and D. Cahard, Chin. J. Chem., 2016, 34, 445–454 CrossRef CAS.
- V. N. Boiko, Beilstein J. Org. Chem., 2010, 6, 880–921 CrossRef.
- D. Bonnet-Delpon and J.-P. Bégué, Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons, 2008 Search PubMed.
- C. Wakselman, M. Tordeux, C. Freslon and L. Saint-Jalmes, Synlett, 2001, 550–552 CrossRef CAS.
- H. Chachignon and D. Cahard, J. Fluorine Chem., 2017, 198, 82–88 CrossRef CAS.
- X. Chen, M. Tordeux, J.-R. Desmurs and C. Wakselman, J. Fluorine Chem., 2003, 123, 51–56 CrossRef CAS.
- T. Billard, A. Greiner and B. R. Langlois, Tetrahedron, 1999, 55, 7243–7250 CrossRef CAS.
- V. D. Romanenko, C. Thoumazet, V. Lavallo, F. S. Tham and G. Bertrand, Chem. Commun., 2003, 1680–1681 RSC.
- M. Maeno, N. Shibata and D. Cahard, Org. Lett., 2015, 17, 1990–1993 CrossRef CAS.
- Z. Liu and R. C. Larock, J. Am. Chem. Soc., 2005, 127, 13112–13113 CrossRef CAS.
- R. P. Singh, G. Cao, R. L. Kirchmeier and J. n. M. Shreeve, J. Org. Chem., 1999, 64, 2873–2876 CrossRef CAS.
- V. N. Movchun, A. A. Kolomeitsev and Y. L. Yagupolskii, J. Fluorine Chem., 1995, 70, 255–257 CrossRef CAS.
- R.-Y. Tang, P. Zhong and Q.-L. Lin, J. Fluorine Chem., 2007, 128, 636–640 CrossRef CAS.
- S. Gan, J. Yin, Y. Yao, Y. Liu, D. Chang, D. Zhu and L. Shi, Org. Biomol. Chem., 2017, 15, 2647–2654 RSC.
- H. Marom, S. Antonov, Y. Popowski and M. Gozin, J. Org. Chem., 2011, 76, 5240–5246 CrossRef CAS.
- L. V. Sokolenko, I. I. Maletina, L. M. Yagupolskii and Y. L. Yagupolskii, Synlett, 2010, 2075–2078 CrossRef CAS.
- E. Baciocchi, M. F. Gerini and A. Lapi, J. Org. Chem., 2004, 69, 3586–3589 CrossRef CAS.
- A. Goti and F. Cardona, in Green Chemical Reactions. NATO Science for Peace and Security Series (Series C: Environmental Security), ed. P. Tundo and V. Esposito, Springer Netherlands, Dordrecht, 2008, pp. 191–212 Search PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- A. S. Rao, H. R. Mohan and J. Iskra, in Encyclopedia of Reagents for Organic Synthesis, ed. P. Fuchs, Wiley, 2013, DOI:10.1002/047084289X.rh040.pub2.
- K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS.
- S. Matavos-Aramyan, S. Soukhakian and M. H. Jazebizadeh, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 195, 181–193 CrossRef CAS.
- F. Shi, M. K. Tse, H. M. Kaiser and M. Beller, Adv. Synth. Catal., 2007, 349, 2425–2430 CrossRef CAS.
- J. P. Begue, D. Bonnet-Delpon and B. Crousse, Synlett, 2004, 18–29 CAS.
- V. L. Sokolenko, K. R. Orlova, A. A. Filatov, L. Y. Yagupolskii, E. Magnier, B. Pégot and P. Diter, Molecules, 2019, 24, 1249–1261 CrossRef.
- X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS.
- Z. Huang and N. Shibata, in New Horizons of Process Chemistry: Scalable Reactions and Technologies, 2017, pp. 163–178, DOI:10.1007/978-981-10-3421-3_12.
- M. Jereb and K. Gosak, Org. Biomol. Chem., 2015, 13, 3103–3115 RSC.
- Q. Glenadel, S. Alazet and T. Billard, J. Fluorine Chem., 2015, 179, 89–95 CrossRef CAS.
- M. Horvat, M. Jereb and J. Iskra, Eur. J. Org. Chem., 2018, 3837–3843 CrossRef CAS.
- K. S. Ravikumar, Y. M. Zhang, J. P. Begue and D. Bonnet-Delpon, Eur. J. Org. Chem., 1998, 2937–2940 CrossRef CAS.
- A. S. Rao, H. R. Mohan and A. Charette, in Encyclopedia of Reagents for Organic Synthesis, 2005, DOI:10.1002/047084289X.rc140.pub2.
- E. Eržen, J. Koller and B. Plesničar, J. Org. Chem., 2001, 66, 5155–5162 CrossRef.
- W. Adam, W. Haas and G. Sieker, J. Am. Chem. Soc., 1984, 106, 5020–5022 CrossRef CAS.
- W. Adam and D. Golsch, J. Org. Chem., 1997, 62, 115–119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04621c |
| This journal is © The Royal Society of Chemistry 2020 |