
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinear β-amino alcohol catalyst anchored on functionalized magnetite nanoparticles for enantioselective addition of dialkylzinc to aromatic aldehydes†
Carla Sappino‡
 a,
Ludovica Primitivo‡ae,
Martina De Angelisae,
Francesco Righia,
Federica Di Pietroa,
Marika Iannonia,
Luciano Pillonib,
Stefano Vecchio Cipriotic,
Lorenza Suberd,
Alessandra Ricellie and
Giuliana Righi‡*e
a,
Ludovica Primitivo‡ae,
Martina De Angelisae,
Francesco Righia,
Federica Di Pietroa,
Marika Iannonia,
Luciano Pillonib,
Stefano Vecchio Cipriotic,
Lorenza Suberd,
Alessandra Ricellie and
Giuliana Righi‡*e
aDipartimento di Chimica, Sapienza Università di Roma, p.le A. Moro 5, 00185 Roma, Italy. E-mail: carla.sappino@uniroma1.it; ludovica.primitivo@uniroma1.it
bSSPT-PROMAS-MATPRO, ENEA CR Casaccia, Via Anguillarese, 301-00123 Roma, Italy
cDipartimento di Scienze di Base e Applicate per l'Ingegneria, Sapienza Università di Roma, Via del Castro Laurenziano 7, 00161 Roma, Italy
dCNR-ISM, Via Salaria km 29,300-00015 Monterotondo Scalo, Roma, Italy
eCNR-IBPM, c/o Dipartimento Chimica, Sapienza Università di Roma, p.le A. Moro 5, 00185 Roma, Italy. E-mail: giuliana.righi@cnr.it
First published on 12th August 2020
Abstract
A linear β-amino alcohol ligand, previously found to be a very efficient catalyst for enantioselective addition of dialkylzinc to aromatic aldehydes, has been anchored on differently functionalized superparamagnetic core–shell magnetite–silica nanoparticles (1a and 1b). Its catalytic activity in the addition of dialkylzinc to aldehydes has been evaluated, leading to promising results, especially in the case of 1b for which the recovery by simple magnetic decantation and reuse was successfully verified.
Introduction
Although in the last few decades several highly efficient chiral catalysts, able to catalyze a variety of asymmetric transformations in homogeneous catalysis, have been developed, the large-scale use of this technique has not taken hold. It has been calculated that homogeneous catalysis is used in less than 20% of industrially relevant processes.1 The stringent ecological and economical demands for sustainability, embodied in the “green chemistry dictates”, have limited their practical use. The main obstacles result from the difficulty of separating the catalyst from the reaction mixture, leading to expensive and polluting purification steps and to limited possibilities of recovering and reusing the catalyst, often composed of precious chiral moieties. The ongoing development of new asymmetric strategies has exploited techniques belonging to materials chemistry. Indeed, that is how asymmetric catalysis researchers started exploring nanochemistry, a relatively young research field that has undergone an exponential growth in several application areas. For example, nanoparticles composed of a magnetite (Fe3O4) core and a silica (SiO2) shell functionalized to anchor the molecular catalyst can overcome the problem of catalyst recovery through simple magnetic decantation, i.e. attracting the nanoparticles, with the attached catalyst molecules, by a magnet applied to the external flask wall.2 Magnetite nanoparticles, up to around 20 nm in size in fact, exhibit a special form of magnetism called superparamagnetism,3 namely they become magnetic only in the presence of a magnetic field. This behavior has the great advantage that in the absence of a magnetic field the nanoparticles can be well dispersed. For these reasons, there has been enormous interest in the use of magnetic nanoparticles in asymmetric catalysis and several chiral catalysts were immobilized onto such supports.4 In light of the many opportunities and great potential of these combined systems, the number of chiral catalysts immobilized onto magnetic nanoparticles is continuously growing.β-Amino alcohols represent one of the most studied classes of chiral ligands/auxiliaries. They have been used, both in acyclic- and cyclic-derivative form, since the beginning of asymmetric synthesis.5 Despite their wide use in asymmetric catalysis, only a small number of β-amino alcohols have been successfully immobilized on magnetic nanoparticles for catalytic applications.6 We have recently reported the employment of functionalized magnetic nanoparticles as catalysts for the enantioselective Henry reaction. In this context, the nanocatalyst 1b exhibited a promising catalytic activity that remained unchanged in the three catalytic cycles performed.7
Herein we report the results of the employment in the asymmetric dialkylzinc addition to aldehydes both of the homogeneous β-amino alcohol ligand 9 and of its immobilized form, the magnetically recoverable β-amino alcohol nanocatalysts 1a and 1b (Fig. 1).
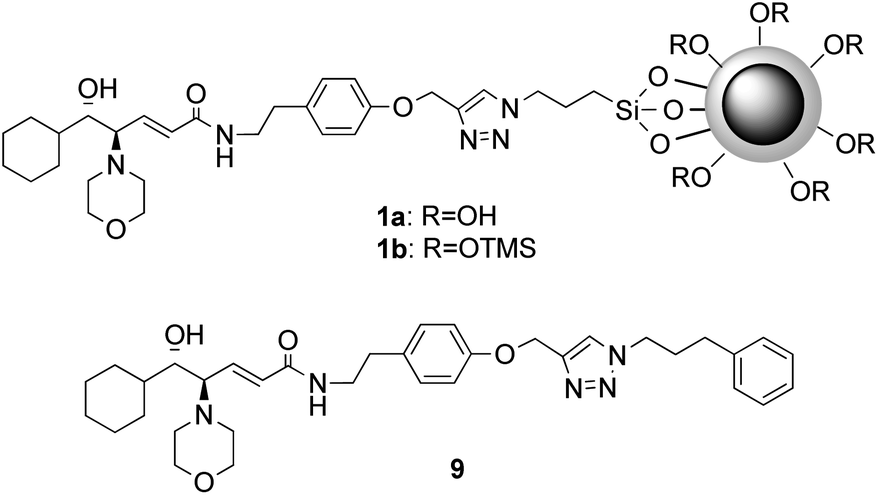 | ||
| Fig. 1 Structure of β-amino alcohol nanocatalysts 1a and 1b and homogeneous β-amino alcohol ligand 9. | ||
Results and discussions
We have recently reported the employment of the β-amino alcohol catalyst 2 (Scheme 1) in the enantioselective addition of diethylzinc to aromatic aldehydes,8 and in light of its broad applicability as chiral catalyst we decided to investigate the catalytic efficiency of its immobilized form.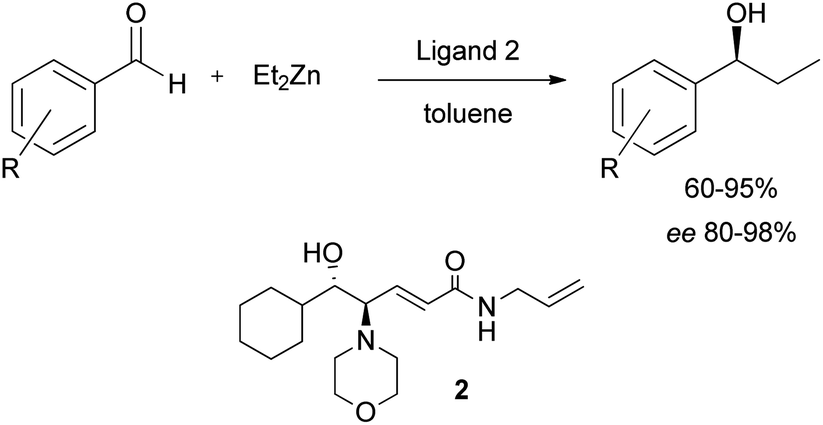 | ||
| Scheme 1 Asymmetric dialkylzinc addition to aldehydes catalysed by ligand 2. | ||
Having extensively explored different anchoring strategies, different functionalities necessary for the bond with the nanoparticle surface, and different suitable spacers, the nanostructured catalyst 1 was designed (Fig. 2).§9
 | ||
| Fig. 2 Immobilization strategy for catalyst 2. | ||
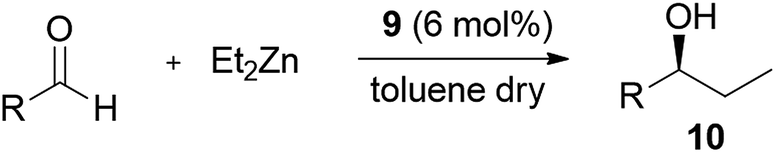
In the amide residue is thus introduced an aromatic and a triazole ring through a CuI-catalyzed azide/alkyne cycloaddition (CuAAC)10 and a nanoparticle having a magnetite (Fe3O4) core. The nanoparticle is coated with a thin silica (SiO2) layer to protect the inner Fe3O4 from oxidation by air and to facilitate the functionalization thanks to the many silanol groups exposed on the surface (Fig. 2).11 Before testing the catalytic efficiency of the nanostructured ligand 1, it was necessary to verify that the introduced differences with respect to the ligand 2 would not affect the catalytic properties. The analogue 9 was designed and obtained through a click reaction between the alkyne 3 and the (3-azidopropyl)benzene 8.¶ The alkyne 3 was synthesized in three steps starting from the acid precursor 5 (Scheme 2).
 | ||
| Scheme 2 (a) EDC/HOAt, DMF, rt, 12 h, 80%; (b) propargyl bromide, K2CO3, CH3CN dry, reflux, 12 h, 83%; (c) morpholine, LiClO4, CH3CN dry, reflux, 12 h, 75%; (d) 8, CuI, DIPEA, THF, rt, 12 h, 89%. | ||
Ligand 9 was then evaluated in the homogeneous phase catalysis test. As shown in Table 1, the addition of Et2Zn to different aldehydes catalyzed by ligand 9 gave results comparable with those obtained with ligand 2.8
|
||||
|---|---|---|---|---|
| Entry | Aldehyde | Yieldb (%) | ee (%) | Product |
| a All the experiments were performed under identical conditions (6 h).b Chemical yields are referred to isolated compounds.c The same results were obtained using CH2Cl2 as solvent. | ||||
| 1 | PhCHO | >95 | 96 | 10a |
| 2 | 4-CNPhCHO | >95 | 76 | 10b |
| 3 | 4-BrPhCHO | >95 | 94 | 10c |
| 4 | 2-ClPhCHO | >95 | 89 | 10d |
| 5 | 2-MePhCHO | >95 | 97 | 10e |
| 6 | 4-MePhCHO | >95 | 96 | 10f |
| 7 | 2-MeOPhCHOc | >95 | 97 | 10g |
| 8 | 3-MeOPhCHO | >95 | 98 | 10h |
| 9 | Cinnamaldehyde | >95 | 88 | 10i |
| 10 | PhCH2CH2CHO | >95 | 87 | 10j |
| 11 | cHexCHO | 70 | 80 | 10k |
Considering the excellent results obtained in the asymmetric catalysis of addition of diethylzinc to aldehydes, we decided also to test the reaction with bulkier organozinc reagents. First, catalyst 9 was tested in the addition of iPr2Zn to benzaldehyde. Benzaldehyde was treated with iPr2Zn and 6 mol% catalyst 9, in dry toluene for 12 h at room temperature. Differently from the Et2Zn addition, the reduction side product benzyl alcohol 12a was collected together with the desired alcohol 11a. However, the use of 9 considerably enhanced the addition, as the mixture ratio moved from substantially only the reduced product collected in absence of the ligand, to 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 for the desired product in the presence of 9; moreover, in the presence of the catalyst, very high enantioselectivity was detected (95.5%) (Table 2).
:12 for the desired product in the presence of 9; moreover, in the presence of the catalyst, very high enantioselectivity was detected (95.5%) (Table 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Aldehyde | Yield 11 (%) | Yield 12 (%) | ee (%) | Product |
| a All the experiments were performed under identical conditions (12 h). | |||||
| 1 | PhCHO (no ligand) | — | 80 | — | |
| 2 | PhCHO | 83 | 11 | 95.5 | 11a |
| 3 | 2-MePhCHO | 53 | 41 | 95 | 11b |
| 4 | 3-MePhCHO | 79 | 16 | 93 | 11c |
| 5 | 4-MePhCHO | 75 | 20 | 86 | 11d |
| 6 | 3-CNPhCHO | 78 | 17 | 87 | 11e |
| 7 | 4-CNPhCHO | 48 | 17 | 98 | 11f |
| 8 | 2-ClPhCHO | 42 | 53 | 81 | 11g |
| 9 | 4-BrPhCHO | 69 | 25 | 88 | 11h |
| 10 | 2-MeOPhCHO | 75 | 20 | 88 | 11i |
| 11 | 3-MeOPhCHO | 83 | 11 | 91 | 11j |

Given the promising results, we evaluated the catalyst 9 in the addition of iPr2Zn to a small selection of aromatic aldehydes. As reported in Table 2, catalyst 9 still exhibits significant catalytic activity, allowing the formation of the addition product, completely absent without catalyst (entry 1), with good yields and satisfactory enantioselectivities. The increased steric hindrance appears to make the addition more difficult, as reflected in the significant formation of the side reduction product. This seems to be confirmed by the lower 11/12 ratio in the case of 2-methylbenzaldehyde and 2-chlorobenzaldehyde whose substituents on the ring are very close to the reaction site (entries 3 and 8).
Once we confirmed the retained catalytic activity, through a CuI-catalyzed azide/alkyne cycloaddition (CuAAC), we immobilized 3 onto azido-modified magnetic nanoparticles 4a and 4b, the latter obtained after treating 4a with hexamethyldisilazane. The superparamagnetic catalysts 1a and 1b were thus obtained with 0.28 mmol g−1 and 0.41 mmol g−1 loading respectively (Scheme 3), as determined by C, H, N elemental analysis (see Experimental section). In fact, as N originates exclusively from the functionalized catalyst, it is possible to determine the mmol of the functionalized catalyst (1a or 1b)/g by dividing the N weight% by its atomic weight, 14, then dividing by 5 as the functionalized catalyst molecule contains five N atoms, and by multiplyng by 10 in order to obtain the mmol in 1 g.
 | ||
| Scheme 3 (a) CuI, DIPEA, THF, r.t., 48 h, 0.28 mmol g−1 loading (1a); 0.41 mmol g−1 loading (1b). | ||
TEM images showed the formation of nanoparticles with diameters in the range 10–15 nm (Fig. 3). HR-TEM images showed lattice fringes attributable both to magnetite (Fe3O4) and maghemite (γ-Fe2O3) phases (Fig. 4), the latter possibly resulting from slight oxidation of magnetite, that, being magnetic as well, does not appreciably influence the superparamagnetic character of the nanoparticles.
 | ||
| Fig. 3 TEM image of functionalized silica–coated magnetite nanoparticles 1b. | ||
 | ||
| Fig. 4 HR-TEM image of functionalized silica–coated magnetite nanoparticles 1b. | ||
The superparamagnetic β-amino alcohol catalyst 1a was initially evaluated in the usual reaction test. Benzaldehyde and diethylzinc were added to a suspension, in toluene, of the magnetic catalyst, and the reaction was mechanically stirred at room temperature for 24 hours. Then the catalyst was easily recovered by magnetic decantation and the supernatant treated as usual. Unfortunately, the analysis of the supernatant revealed that almost no catalysis occurred: the product was collected in 45% yield and with 15% ee (Table 3-entry 1). Considering the poor result observed, we wondered if the vicinal hydroxyl groups exposed on the oxide surface could have interfered with the organometallic reagent, ruining the reaction. Following a reported procedure, we performed a second test adding BuLi to the reaction mixture which was supposed both to react with the vicinal free silanols forming lithium silyloxides and to activate the Et2Zn forming a lithium amino alkoxide species, more reactive than the analogous zinc derivative.12 Once more, these conditions led, in both the concentrations tested, to the desired product in very low yield and mediocre enantioselectivities, even if those values were slightly higher. In addition, a fair amount of 1-phenylpentan-1-ol, derived from the BuLi addition to the aldehyde (entries 2 and 3), was observed. For the same reason, we decided to covalently block the free silanol on the surface by treatment with hexamethyldisilazane, which is able to transform the hydroxyl groups on the nanoparticle surface into trimethylsilyloxide.13 After the treatment with HMDS, we obtained the silylated azido functionalized nanoparticles 4b, which immediately appeared to be much more dispersible in organic solvents. The immobilization of 3 on 4b furnished the nanostructured catalyst 1b that, tested in the usual reaction condition, gave consistently improved results, especially regarding enantioselectivity, although not yet comparable with the homogeneous phase ones. However, noteworthy that the catalytic activity remained about unchanged in the tree catalytic cycles performed.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Aldehyde | Catalyst | Additive | R | Yieldb% | ee% | Product |
| a All the experiments were performed under identical conditions (24 h, r.t.) except for entry 2 and 3 performed at −15 °C.b Chemical yields are referred to isolated compounds. | |||||||
| 1 | PhCHO | 1a | — | Et | 45 | 15 | 10a |
| 2 | PhCHO | 1a | BuLi 0.72 mmol g−1 | Et | 25 | 25 | 10a |
| 3 | PhCHO | 1a | BuLi 0.35 mmol g−1 | Et | 27 | 28 | 10a |
| 4 | PhCHO | 1b | — | Et | 50 | 50 | 10a |
| 5 | PhCHO | 1b I cycle | — | Et | 48 | 50 | 10a |
| 6 | PhCHO | 1b II cycle | — | Et | 47 | 48 | 10a |
| 7 | PhCHO | 1b III cycle | — | Et | 48 | 48 | 10a |
| 8 | 4-Me-PhCHO | 1b | — | Et | 59 | 53 | 10f |
| 9 | 3-MeO-PhCHO | 1b | — | Et | 48 | 58 | 10h |
| 10 | 4-Br-PhCHO | 1b | — | Et | 77 | 47 | 10c |
| 11 | PhCHO | 1b | — | iPr | 46 | 50 | 11a |
| 12 | 4-Me-PhCHO | 1b | — | iPr | 49 | 47 | 11d |
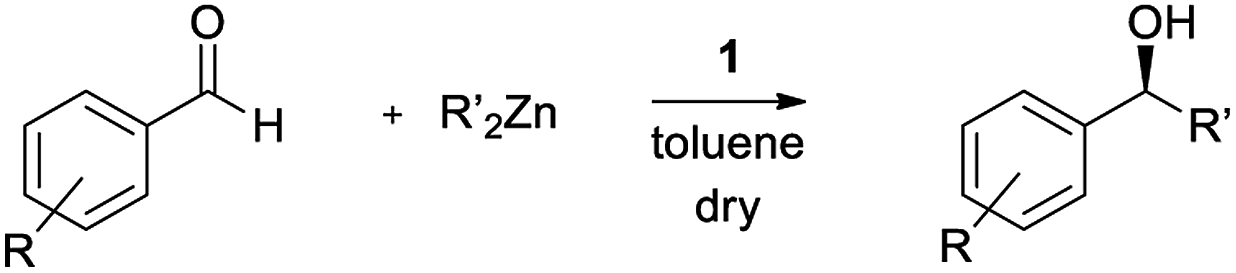
We hypothesized that such a difference in activity between the homogeneous catalyst 9 and its anchored form 1 was due to small amounts of water on the nanoparticle surface incompatible with Et2Zn. To verify it, a sample of well-dried nanostructured catalyst 1b was subjected to thermogravimetric analysis (Fig. 5) to eventually determine the fraction of water (by monitoring the mass loss that occurs heating the sample at a constant rate). Actually, heating up to 100 °C it was possible to observe a mass loss of about 0.3% due to adsorbed water, whereas a mass loss of about 1.5% observed in the range 100–200 °C can be attributed to chemically bonded water. Unfortunately, the nanoparticles can't be subjected to a thermal treatment up to 175 °C to eliminate water, because of the resulting poor dispersibility in solvents caused by aggregation and agglomeration phenomena, probably due to incipient organic decomposition process on the nanoparticle surface.
 | ||
| Fig. 5 TG curve of the nanostructured catalyst 1b. | ||
Conclusions
The structure of the anchorable amino alcohol 3 was designed starting from catalyst 2, whose efficiency had already been reported in the well-established enantioselective amino alcohol-promoted addition of diethylzinc to a variety of aldehydes in the homogeneous phase. We confirmed the retained catalytic activity by employing its analogue 9, adding in particular a triazole ring formed by anchoring the catalyst to the nanoparticles surface. Moreover, its broad applicability as a chiral catalyst was evaluated by carrying out the same reaction with the bulkier diisopropylzinc, still leading to satisfactory results. Given the interesting results obtained, we anchored this type of catalyst on superparamagnetic magnetite nanoparticles. After having chosen a suitable spacer and different functionalized nanoparticle surfaces, the nanostructured catalysts 1a and 1b were selected. Their catalytic activity was evaluated in the addition of dialkylzinc to aldehydes leading to promising results, especially in the case of 1b for which the recovery by simple magnetic decantation and reuse was also successfully verified.A study on the employment of 1b catalyst in different organic reactions is in progress.
Experimental
Unless otherwise stated, commercial reagents purchased from Alfa Aesar, Acros and Aldrich chemical companies were used without further purification. Purification of reaction products was carried out by flash chromatography using Kieselgel F Merck silica gel (230–400 mesh). Thin layer chromatography (TLC) was performed on Kieselgel F 254 pre-coated silica gel plates and visualization was achieved by inspection under UV light (Mineralight UVG 11 254 nm) followed by staining with phosphomolybdic acid dip [polyphosphomolybdic acid (12 g), ethanol (250 mL)] or 2,4-dinitrophenylhydrazine dip [2,4-dinitrophenylhydrazine (4 g) ethanol (100 mL), water (20 mL), sulfuric acid]. 1H NMR spectra were recorded using a Varian Mercury 300 (300 MHz) or a Bruker Avance 400 (400 MHz). Residual solvent peaks were used as internal references for 1H NMR spectra: chloroform (δ 7.26 ppm). Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are reported in Hz. Splitting patterns are designated as s, singlet; bs, broad singlet; d, doublet; bd, broad doublet dd, doublet of doublets; ddd, doublet of doublet of doublets; t, triplet; dt, doublet of triplets; q, quartet; m, multiplet. 13C spectra were recorded using a Varian Mercury 300 (75 MHz) or a Bruker Avance 400 (100 MHz). Chemical shifts (δ) are reported in parts per million (ppm) relative to the internal standard of the residue solvent peak: chloroform (77.00 ppm). Optical rotations were measured on a digital polarimeter Jasco DIP-370 with a cell path length of 1 cm; solution concentrations are reported in grams per 100 mL. Enantiomeric excesses were determined by way of a Agilent 1260 Infinity HPLC system equipped with a diode array detector (DAD) and chiral stationary phase; chiral columns used were Chiralpak IA, Chiralpak IB, Chiralpak IC.Attenuated Total Reflectance-Fourier Transform Infra Red (ATR-FTIR) spectra were recorded on a Shimadzu IR Prestige-21. Elemental analyses for C, H and N were performed on an EA 1110 CHNS–O Element Analyzer. Morphologic and structural investigations were performed by way of a JEOL JEM 2010 Transmission Electron Microscopy (TEM).
The thermal behavior of a powder sample of 1b was studied from room temperature to 180 °C by a simultaneous Stanton Redcroft Thermogravimetry/Heat Flux Differential Thermal Analysis apparatus (STA625 model). The instrument is equipped with two Al crucibles (one for the sample and one for the reference, which was empty). After calibration with sapphire this instrument is able to provide heat flow data as a DSC unit (with lower sensitivity). So, the thermogravimetry (TG) experiments were carried out under purging inert Ar atmosphere of 50 mL min−1 and heating rate of 10 °C min−1, providing the mass loss as a function of temperature. About 8 mg of the sample, precisely weighed with an accuracy of ±0.001 mg, was considered. Calibration of temperature was performed by measuring the onset melting temperature of high purity indium.
The synthesis and characterization of compounds 3, 6, 7, 8, 9, the preparation of magnetite/silica core–shell nanoparticles (4a and 4b) and the nanostructured catalysts (1a and 1b) have been reported in our previous publication.7
General procedure for the addition of dialkylzinc to aldehydes catalyzed by free ligand in homogeneous phase
To a solution of the chiral amino alcohol (0.06 mmol, 6 mol%) in toluene dry (2 mL) the aldehyde (1 mmol) was added under argon atmosphere at room temperature. The mixture was then cooled to 0 °C. 2.2 mmol of dialkylzinc (2.2 mL of a 1.0 M hexane solution of Et2Zn or 2.2 mL of a 1.0 M toluene solution of iPr2Zn) were added dropwise. Then the reaction mixture was allowed to reach the room temperature and stirred for 6 hours. The reaction was quenched by the addition of a saturated NH4Cl solution (10 mL). The mixture was then extracted with CH2Cl2. The combined organic extracts were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (Hex/EtOAc 80:20).
General procedure for the addition of dialkylzinc to aldehydes catalyzed by functionalized nanoparticles
0.25 mmol of aldehyde was added, under argon atmosphere and at room temperature, to a dispersion of the catalyst-loaded nanoparticles (6 mol%) in 3 mL of dry toluene. The mixture was mechanically agitated for 20 min and then cooled to 0 °C. 0.8 mmol of dialkylzinc (0.8 mL of a 1.0 M hexane solution of Et2Zn or 0.8 mL of a 1.0 M toluene solution of iPr2Zn) was added dropwise. Then the reaction mixture was allowed to reach the room temperature and mechanically agitated for 24 hours. The reaction vessel was placed over an external magnet until the reaction mixture became transparent, and the solution was separated from the nanoparticles. The reaction was quenched by the addition of a saturated NH4Cl solution (3 mL) and then extracted with CH2Cl2. The combined organic extracts were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (Hex/EtOAc 80:20) as eluent. The recovered nanoparticles were repeatedly washed with CH2Cl2 and toluene and then they were stored in 3 mL of anhydrous toluene.
:2, 0.9 mL min−1, 258 nm, minor 12.1 min and major 12.4 min). 1H NMR (400 MHz, CDCl3) δ 7.40–7.23 (m, 5H, Ph), 4.60 (t, J = 6.6 Hz, 1H, CHOH), 1.90–1.67 (m, 3H, CH2, OH), 0.92 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 150.2, 132.3, 126.7, 119.0, 111.0, 75.1, 32.1, 9.9.:5, 0.8 mL min−1, 220 nm, minor 7.6 min and major 8.3 min). 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.4 Hz, 1H, Ph), 7.27–7.12 (m, 3H, Ph), 4.87 (t, J = 6.4 Hz, 1H, CHOH), 2.34 (s, 3H), 1.82–1.70 (m, 3H, CH2, OH), 0.99 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 142.9, 142.3, 134.7, 130.5, 127.3, 126.4, 125.3, 72.2, 31.0, 19.2, 10.5.:3, 0.8 mL min−1, 280 nm, major 10.9 min and minor 11.7 min). 1H NMR (400 MHz, CDCl3) δ 7.33–7.21 (m, 2H), 6.99–6.85 (m, 2H), 4.80 (t, J = 6.6 Hz, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) OH), 3.84 (s, 3H, OCH3), 2.61 (bs, 1H, OH), 1.87–1.76 (m, 2H, C2CH3), 0.96 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 156.7, 132.5, 128.2, 127.1, 120.8, 110.6, 72.4, 55.3, 30.2, 10.5.:5, 1 mL min−1, 220 nm, minor 9.9 min and major 10.5 min). 1H NMR (400 MHz, CDCl3) δ 7.25 (t, J = 8.1 Hz, 1H, Ph), 6.92–6.89 (m, 2H, Ph), 6.81 (ddd, J = 8.3, 2.5, 1.0 Hz, 1H, Ph), 4.55 (t, J = 6.6 Hz, 1H, CHOH), 3.80 (s, 3H, OCH3), 2.03 (bs, 1H, OH), 1.85–1.68 (m, 2H, CH2), 0.92 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 159.8, 146.5, 129.5, 118.4, 113.0, 111.5, 76.0, 55.3, 31.9, 10.2.:2, 1 mL min−1, 254 nm, minor 13.0 min and major 14.9 min). 1H NMR (400 MHz, CDCl3) δ 7.41–7.37 (m, 2H, Ph), 7.35–7.29 (m, 2H, Ph), 7.27–7.22 (m, 1H, Ph), 6.58 (d, J = 15.9 Hz, 1H, PhCH), 6.20 (dd, 1H, J = 15.9, 6.7 Hz, CCHOH), 4.22 (dd, 1H, J = 12.3, 6.2 Hz, COH), 1.72–1.61 (m, 3H, C2CH3 + OH), 0.98 (t, J = 7.5 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 136.9, 132.4, 130.6, 128.7, 127.8, 126.6, 74.6, 30.4, 9.9.:1, 0.8 mL min−1, 220 nm, minor 5.5 min and major 5.9 min). 1H NMR (400 MHz, CDCl3) δ 3.27 (ddd, J = 8.9, 5.4, 3.9 Hz, 1H, CHOH), 1.86–0.91 (m, 17H, c-Hex, CH2CH3, OH), 0.94 (t, 3H, J = 7.4 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 77.8, 43.3, 29.5, 27.9, 27.0, 26.7, 26.5, 26.4, 10.4.:1, 1 mL min−1, 220 nm, T = 30 °C, major 8.0 min and minor 8.6 min).1H NMR (300 MHz, CDCl3) δ 7.40–7.20 (m, 5H, Phe), 4.36 (d, 1H J = 6.9 Hz, CHOH), 2.10 (bs, 1H, OH), 2.03–1.87 (m, 1H, C(CH3)2), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.80 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (75 MHz, CDCl3) δ 143.8, 128.3, 127.5, 126.7, 80.2, 35.4, 19.1, 18.3.:2, 1 mL min−1, 220 nm, T = 30 °C, major 6.8 min and minor 7.1 min).1H-NMR (300 MHz, CDCl3) δ: 7.49–7.35 (m, 1H, Phe); 7.31–7.06 (m, 4H, Phe); 4.63 (d, 1H, J = 6.7 Hz, COH); 2.34 (s, 3H, CH3–Phe); 2.02–1.91 (m, 1H, C(CH3)2); 1.74 (bs, 1H, OH); 1.04 (d, 3H, J = 6.67 Hz, CH3); 0.85 (d, 3H, J = 6.67 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 142.3, 135.1, 130.4, 127.2, 126.2, 126.2, 75.9, 34.7, 19.6, 19.5, 18.0.:2, 1 mL min−1, 220 nm, T = 30 °C, major 6.9 min and minor 7.2 min) 1H NMR (300 MHz, CDCl3) δ 7.40–6.96 (m, 4H, Phe), 4.31 (d, 1H, J = 6.9 Hz, COH), 2.36 (s, 3H, CH3–Phe), 2.04–1.86 (m, 1H, C(CH3)2), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.80 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 143.8, 137.9, 128.3, 128.2, 127.4, 123.8, 80.2, 35.3, 21.6, 19.2, 18.4.:2, 1 mL min−1, 220 nm, T = 30 °C, major 6.8 min and minor 7.3 min).1H NMR (300 MHz, CDCl3) δ 7.32–7.09 (m, 4H, Phe), 4.32 (d, 1H, J = 7.0 Hz, COH), 2.35 (s, 3H, CH3–Phe), 2.02–1.88 (m, 1H, C(CH3)2), 1.85 (bs, 1H, OH), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.79 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 140.8, 137.1, 129.0, 126.6, 80.1, 35.3, 21.2, 19.1, 18.5.:2, 1 mL min−1, 220 nm, T = 30 °C, major 19.8 min and minor 22.4 min). 1H-NMR (300 MHz, CDCl3) δ: 7.63 (t, 1H, J = 1.7 Hz, Phe), 7.58–7.53 (m, 2H, Phe), 7.47–7.41 (m, 1H, Phe) 4.46 (d, 1H, J = 6.2 Hz, COH) 2.01–1.83 (m, 1H, C(CH3)2); 1.60 (bs, 1H, OH); 0.95 (d, 3H, J = 6.7 Hz, CH3); 0.84 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 145.1, 131.2, 131.1, 130.3, 129.1, 119.1, 112.4, 78.8, 35.5, 18.9, 17.7.:2, 1 mL min−1, 220 nm, T = 30 °C, major 20.8 min and minor 22.2 min). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, 2H, J = 8.2 Hz, Phe), 7.19 (d, 2H, J = 8.2 Hz, Phe), 4.33 (d, 1H, J = 7.0 Hz, COH), 2.96–2.84 (m, 1H, C(CH3)2), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.79 (d, J = 6.8 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 149.4, 132.3, 127.6, 119.2, 111.2, 79.1, 35.6, 19.2, 17.8.:2, 1 mL min−1, 220 nm, r.t., minor 7.3 min and major 8.5 min). 1H NMR (400 MHz, CDCl3) δ 7.51 (dd, 1H, J = 7.7, 1.7 Hz, Ph), 7.35–7.26 (m, 2H, Ph), 7.22–7.16 (m, 1H, Ph), 4.91 (d, 1H, J = 6.0 Hz, COH), 2.12–1.99 (m, 1H, C(CH3)2), 1.84 (bs, 1H, OH), 0.97 (d, 3H, J = 6.7 Hz, CH3), 0.93 (d, 3H, J = 6.9 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 141.4, 130.8, 129.5, 128.4, 128.2, 126.9, 75.6, 34.2, 19.5, 17.2.:1, 1 mL min−1, 220 nm, T = 30 °C, major 8.8 min and minor 9.2 min). 1H NMR (300 MHz, CDCl3) δ 7.44 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 4.31 (d, J = 6.6 Hz, 1H), 2.11 (bs, 1H), 1.95–1.83 (m, 1H), 0.95 (d, J = 6.7 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 142.6, 131.3, 128.4, 121.2, 79.3, 35.3, 18.9, 18.1.:2, 1 mL min−1, 220 nm, T = 30 °C, major 7.8 min and minor 8.4 min). 1H NMR (400 MHz, CDCl3) δ 7.32–7.18 (m, 2H, Ph), 6.95 (td, 1H, J = 7.4, 1.1 Hz, Ph), 6.88 (d, 1H, J = 8.2 Hz, Ph), 4.51 (d, 1H, J = 7.4 Hz, COH), 3.84 (s, 3H, OCH3), 2.23 (bs, 1H, OH), 2.13–1.97 (m, 1H, C(CH3)2), 1.03 (d, 3H, J = 6.7 Hz, CH3), 0.80 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 156.7, 131.7, 128.2, 128.1, 120.6, 110.6, 76.8, 55.3, 34.2, 19.6, 18.5.:2, 1 mL min−1, 220 nm, T = 30 °C, major 13.4 min and minor 17 min). 1H NMR (400 MHz, CDCl3) δ 7.28–7.22 (m, 1H, Ph), 6.91–6.87 (m, 2H, Ph), 6.84–6.79 (m, 1H, Ph), 4.34 (d, 1H, J = 6.9 Hz, COH), 3.81 (s, 3H, OCH3), 2.00–1.89 (m, 1H, C(CH3)2), 1.77 (bs, 1H, J = 9.7 Hz, OH), 1.00 (d, CH3, J = 6.7 Hz, 3H), 0.81 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 159.7, 145.6, 129.3, 119.1, 112.9, 112.2, 80.1, 55.3, 35.4, 19.2, 18.3.
OH), 3.84 (s, 3H, OCH3), 2.61 (bs, 1H, OH), 1.87–1.76 (m, 2H, C2CH3), 0.96 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 156.7, 132.5, 128.2, 127.1, 120.8, 110.6, 72.4, 55.3, 30.2, 10.5.:5, 1 mL min−1, 220 nm, minor 9.9 min and major 10.5 min). 1H NMR (400 MHz, CDCl3) δ 7.25 (t, J = 8.1 Hz, 1H, Ph), 6.92–6.89 (m, 2H, Ph), 6.81 (ddd, J = 8.3, 2.5, 1.0 Hz, 1H, Ph), 4.55 (t, J = 6.6 Hz, 1H, CHOH), 3.80 (s, 3H, OCH3), 2.03 (bs, 1H, OH), 1.85–1.68 (m, 2H, CH2), 0.92 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 159.8, 146.5, 129.5, 118.4, 113.0, 111.5, 76.0, 55.3, 31.9, 10.2.:2, 1 mL min−1, 254 nm, minor 13.0 min and major 14.9 min). 1H NMR (400 MHz, CDCl3) δ 7.41–7.37 (m, 2H, Ph), 7.35–7.29 (m, 2H, Ph), 7.27–7.22 (m, 1H, Ph), 6.58 (d, J = 15.9 Hz, 1H, PhCH), 6.20 (dd, 1H, J = 15.9, 6.7 Hz, CCHOH), 4.22 (dd, 1H, J = 12.3, 6.2 Hz, COH), 1.72–1.61 (m, 3H, C2CH3 + OH), 0.98 (t, J = 7.5 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 136.9, 132.4, 130.6, 128.7, 127.8, 126.6, 74.6, 30.4, 9.9.:1, 0.8 mL min−1, 220 nm, minor 5.5 min and major 5.9 min). 1H NMR (400 MHz, CDCl3) δ 3.27 (ddd, J = 8.9, 5.4, 3.9 Hz, 1H, CHOH), 1.86–0.91 (m, 17H, c-Hex, CH2CH3, OH), 0.94 (t, 3H, J = 7.4 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 77.8, 43.3, 29.5, 27.9, 27.0, 26.7, 26.5, 26.4, 10.4.:1, 1 mL min−1, 220 nm, T = 30 °C, major 8.0 min and minor 8.6 min).1H NMR (300 MHz, CDCl3) δ 7.40–7.20 (m, 5H, Phe), 4.36 (d, 1H J = 6.9 Hz, CHOH), 2.10 (bs, 1H, OH), 2.03–1.87 (m, 1H, C(CH3)2), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.80 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (75 MHz, CDCl3) δ 143.8, 128.3, 127.5, 126.7, 80.2, 35.4, 19.1, 18.3.:2, 1 mL min−1, 220 nm, T = 30 °C, major 6.8 min and minor 7.1 min).1H-NMR (300 MHz, CDCl3) δ: 7.49–7.35 (m, 1H, Phe); 7.31–7.06 (m, 4H, Phe); 4.63 (d, 1H, J = 6.7 Hz, COH); 2.34 (s, 3H, CH3–Phe); 2.02–1.91 (m, 1H, C(CH3)2); 1.74 (bs, 1H, OH); 1.04 (d, 3H, J = 6.67 Hz, CH3); 0.85 (d, 3H, J = 6.67 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 142.3, 135.1, 130.4, 127.2, 126.2, 126.2, 75.9, 34.7, 19.6, 19.5, 18.0.:2, 1 mL min−1, 220 nm, T = 30 °C, major 6.9 min and minor 7.2 min) 1H NMR (300 MHz, CDCl3) δ 7.40–6.96 (m, 4H, Phe), 4.31 (d, 1H, J = 6.9 Hz, COH), 2.36 (s, 3H, CH3–Phe), 2.04–1.86 (m, 1H, C(CH3)2), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.80 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 143.8, 137.9, 128.3, 128.2, 127.4, 123.8, 80.2, 35.3, 21.6, 19.2, 18.4.:2, 1 mL min−1, 220 nm, T = 30 °C, major 6.8 min and minor 7.3 min).1H NMR (300 MHz, CDCl3) δ 7.32–7.09 (m, 4H, Phe), 4.32 (d, 1H, J = 7.0 Hz, COH), 2.35 (s, 3H, CH3–Phe), 2.02–1.88 (m, 1H, C(CH3)2), 1.85 (bs, 1H, OH), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.79 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 140.8, 137.1, 129.0, 126.6, 80.1, 35.3, 21.2, 19.1, 18.5.:2, 1 mL min−1, 220 nm, T = 30 °C, major 19.8 min and minor 22.4 min). 1H-NMR (300 MHz, CDCl3) δ: 7.63 (t, 1H, J = 1.7 Hz, Phe), 7.58–7.53 (m, 2H, Phe), 7.47–7.41 (m, 1H, Phe) 4.46 (d, 1H, J = 6.2 Hz, COH) 2.01–1.83 (m, 1H, C(CH3)2); 1.60 (bs, 1H, OH); 0.95 (d, 3H, J = 6.7 Hz, CH3); 0.84 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 145.1, 131.2, 131.1, 130.3, 129.1, 119.1, 112.4, 78.8, 35.5, 18.9, 17.7.:2, 1 mL min−1, 220 nm, T = 30 °C, major 20.8 min and minor 22.2 min). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, 2H, J = 8.2 Hz, Phe), 7.19 (d, 2H, J = 8.2 Hz, Phe), 4.33 (d, 1H, J = 7.0 Hz, COH), 2.96–2.84 (m, 1H, C(CH3)2), 1.01 (d, 3H, J = 6.7 Hz, CH3), 0.79 (d, J = 6.8 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 149.4, 132.3, 127.6, 119.2, 111.2, 79.1, 35.6, 19.2, 17.8.:2, 1 mL min−1, 220 nm, r.t., minor 7.3 min and major 8.5 min). 1H NMR (400 MHz, CDCl3) δ 7.51 (dd, 1H, J = 7.7, 1.7 Hz, Ph), 7.35–7.26 (m, 2H, Ph), 7.22–7.16 (m, 1H, Ph), 4.91 (d, 1H, J = 6.0 Hz, COH), 2.12–1.99 (m, 1H, C(CH3)2), 1.84 (bs, 1H, OH), 0.97 (d, 3H, J = 6.7 Hz, CH3), 0.93 (d, 3H, J = 6.9 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 141.4, 130.8, 129.5, 128.4, 128.2, 126.9, 75.6, 34.2, 19.5, 17.2.:1, 1 mL min−1, 220 nm, T = 30 °C, major 8.8 min and minor 9.2 min). 1H NMR (300 MHz, CDCl3) δ 7.44 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 4.31 (d, J = 6.6 Hz, 1H), 2.11 (bs, 1H), 1.95–1.83 (m, 1H), 0.95 (d, J = 6.7 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 142.6, 131.3, 128.4, 121.2, 79.3, 35.3, 18.9, 18.1.:2, 1 mL min−1, 220 nm, T = 30 °C, major 7.8 min and minor 8.4 min). 1H NMR (400 MHz, CDCl3) δ 7.32–7.18 (m, 2H, Ph), 6.95 (td, 1H, J = 7.4, 1.1 Hz, Ph), 6.88 (d, 1H, J = 8.2 Hz, Ph), 4.51 (d, 1H, J = 7.4 Hz, COH), 3.84 (s, 3H, OCH3), 2.23 (bs, 1H, OH), 2.13–1.97 (m, 1H, C(CH3)2), 1.03 (d, 3H, J = 6.7 Hz, CH3), 0.80 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 156.7, 131.7, 128.2, 128.1, 120.6, 110.6, 76.8, 55.3, 34.2, 19.6, 18.5.:2, 1 mL min−1, 220 nm, T = 30 °C, major 13.4 min and minor 17 min). 1H NMR (400 MHz, CDCl3) δ 7.28–7.22 (m, 1H, Ph), 6.91–6.87 (m, 2H, Ph), 6.84–6.79 (m, 1H, Ph), 4.34 (d, 1H, J = 6.9 Hz, COH), 3.81 (s, 3H, OCH3), 2.00–1.89 (m, 1H, C(CH3)2), 1.77 (bs, 1H, J = 9.7 Hz, OH), 1.00 (d, CH3, J = 6.7 Hz, 3H), 0.81 (d, 3H, J = 6.8 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 159.7, 145.6, 129.3, 119.1, 112.9, 112.2, 80.1, 55.3, 35.4, 19.2, 18.3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dott. Daniele Mirabile Gattia (ENEA) for XRD measurement.Notes and references
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- L. M. Rossi, N. J. S. Costa, F. P. Silvia and R. Wojcieszak, Green Chem., 2014, 16, 2906–2933 RSC.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- (a) R. Dalpozzo, Green Chem., 2015, 17, 3671–3686 RSC; (b) J. Safaei-Ghomi and S. Zahedi, Appl. Organomet. Chem., 2015, 29, 566–571 CrossRef CAS.
- D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS PubMed.
- F. Michalek, A. Lagunas, C. Jimeno and M. A. Pericas, J. Mater. Chem., 2008, 18, 4692 RSC.
- C. Sappino, L. Primitivo, M. De Angelis, M. Oneto Domenici, A. Mastrodonato, I. Ben Romdan, C. Tatangelo, L. Suber, L. Pilloni, A. Ricelli and G. Righi, ACS Omega, 2019, 4, 21809–21817 CrossRef CAS PubMed.
- C. Sappino, A. Mari, A. Mantineo, M. Moliterno, M. Palagri, C. Tatangelo, L. Suber, P. Bovicelli, A. Ricelli and G. Righi, Org. Biomol. Chem., 2018, 16, 1860–1870 RSC.
- C. Sappino, Design and synthesis of new nanostructured chiral catalyst, PhD thesis, Sapienza University of Rome, 2017.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- J. Fraile, J. A. Mayoral, J. Serrano, M. A. Pericàs, L. Solà and D. Castellnou, Org. Lett., 2003, 5, 4333–4335 CrossRef CAS PubMed.
- V. Slavov, A. R. Sanger and K. T. Chuang, J. Phys. Chem. B, 2000, 104, 983–989 CrossRef.
- M. Asami and S. Inoue, Bull. Chem. Soc. Jpn., 1997, 70, 1687–1690 CrossRef CAS.
- Y. S. Shih, R. Boobalan, C. Chen and G.-H. Lee, Tetrahedron: Asymmetry, 2014, 25, 327–333 CrossRef CAS.
- Z.-L. Wu, H.-L. Wu, P.-Y. Wu and B.-J. Uang, Tetrahedron: Asymmetry, 2009, 20, 1556–1560 CrossRef CAS.
- W. Wu, S. Liu, M. Duan, X. Tan, C. Chen, Y. Xien, Y. Lan, X. Dong and X. Zhang, Org. Lett., 2016, 18, 2938–2941 CrossRef CAS PubMed.
- W. K. Yang and B. T. Cho, Tetrahedron: Asymmetry, 2000, 11, 2947–2952 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04554c |
| ‡ These authors contributed equally to this work. |
| § A ligand derived from the immobilization on nanoparticles of a simpler structure [(4R,5S,E)-5-cyclohexyl-5-hydroxy-4-morpholino-N-(prop-2-yn-1-yl)pent-2-enamide] instead of 3 had been previously tested leading to poorer results in the catalytic reaction. |
| ¶ Not commercially available, synthesized from 3-phenyl-1-propanol in 2 steps: (a) PBr3, CH2Cl2 dry, 0 °C, 12 h; (b) NaN3, DMSO dry, 60 °C, 12 h, 31% total etc. |
| This journal is © The Royal Society of Chemistry 2020 |