
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO-free, aqueous mediated, instant and selective reduction of nitrobenzene via robustly stable chalcogen stabilised iron carbonyl clusters (Fe3E2(CO)9, E = S, Se, Te)†
Charu Sharma,
Avinash Kumar Srivastava ,
Aditi Soni,
Sangeeta Kumari and
Raj Kumar Joshi*
,
Aditi Soni,
Sangeeta Kumari and
Raj Kumar Joshi*
Department of Chemistry, Malaviya National Institute of Technology, Jaipur 302017, Rajasthan, India. E-mail: rkjoshi.chy@mnit.ac.in
First published on 1st September 2020
Abstract
Highly stable and thermally robust iron chalcogenide carbonyl clusters Fe3E2(CO)9 (E = S, Se or Te) have been explored for the reduction of nitrobenzene. A 15 min thermal heating of an aqueous solution of nitrobenzene and hydrazine hydrate in the catalytic presence of Fe3E2(CO)9 (E = S, Se or Te) clusters yield average to excellent aniline transformations. Among the S, Se and Te based iron chalcogenised carbonyl clusters, the diselenide cluster was found to be most efficient and produce almost 90% yield of the desired amino product, the disulfide cluster was also found to be significantly active, produce the 85% yield of amino product, while the ditelluride cluster was not found to be active and produced only 49% yield of the desired product. The catalyst can be reused up to three catalytic cycles and it needs to be dried in an oven for one hour prior to reuse for the best results. The developed method is inexpensive, environmentally benign, does not require any precious metal or a high pressure of toxic CO gas and exclusively brings the selective reduction of the nitro group under feasible and inert free conditions.
Introduction
Since the initial application of an iron catalyst by Reppe and Vetter1 in 1953, iron-based catalysis has influenced the establishment of various catalysts based on 4d and 5d transition elements. However, before Reppe and Vetter's pioneering work, Fe in the form of iron carbonyl had been known since 1891.2,3 Being the second most abundant metal in the earth's crust, Fe is always under the limelight to explore as a catalyst for a variety of organic reactions. Since, an efficient, cost-effective, fast, clean and selective catalyst is needed in modern research, iron is the most attractive option available in the surroundings.4 However, catalysts derived from 4d and 5d transition metals and other rare earth metals are more efficient than 3d transition metals, but also expensive in meeting present and upcoming societal demands.4 Zero-valent iron in the form of Fe(CO)5 is one of the least expensive forms and of ubiquitous availability. In situ activation and catalysis by Fe(CO)5 require a strong base, moreover, the di-sodium tetracarbonyl ferrate Na2Fe(CO)4 and iron tetra carbonyl hydride H2Fe(CO)4 species were formed during the reaction, these species are highly temperature and light sensitive and drastically affect the productivity of the reaction.5 Nevertheless, due to the high catalytic potential of Fe(CO)5, it has been extensively used for various carbonylation reactions as well as as for high-nuclearity clustere formations.6–11 Electron-transfer,12 hydrogenation,13 hydride transfer14 and electrochemical methods15 are some of the important available methodologies to furnish distinct amines by utilizing a nitro precursor. The reduction of nitrobenzene has gained tremendous interest due to its hazardous effect on the environment. The process of reduction is quite complicated as the nitro group reduction advances in stages and stops due to the formation of hydroxylamine and azoarene as side products at an intermediate stage.16 Traditionally, catalytic transfer hydrogenation has been employed in the presence of Ru,17 Rh,18 Pd,19 or Ni (ref. 20) metal catalysts. Very few studies have utilized inexpensive Co,21 Fe,22 or Zn (ref. 23) metals for transfer hydrogenation; however, economic consequences strongly favour the iron-based system due to its greater environmental sustainability.Traditionally, a plentiful inexpensive iron powder was used for nitro reduction, but it has low efficiency and produces a large amount of wastage. The nano-form of zero-valent iron (nZVI) was used for the reduction of nitro derivatives, but it was unsuccessful, as the catalytic activity of NPs is significantly related to their specific morphology, which is difficult to produce in bulk. Moreover, the formation of nZVI is a dynamic, time-consuming and laborious process. Apart from that, the accumulation of nZVI reduces its mobility, dispersibility and simultaneously reduces its catalytic reactivity.24 Zero-valent metal carbonyl clusters have some unique inherent properties of catalysis and thermal stability. Moreover, the absence of large bulk phases results in a high surface to volume ratio, which is beneficial for catalysis, since it minimizes the reaction rate per unit amount of catalyst, which likewise limits the cost.25 Metal carbonyls and clusters have been used for a wide range of catalysis: in particular, for carbonylation,26 C–C coupling,27 hydroformylation,28 hydroesterification,29 reduction of nitroaranes30–32 and many more value-added organic transformations.6–11 Initially (Scheme 1), Pittman and co-workers in 1979 utilised Rh6(CO)12 for nitro reduction.33 Later, in 2014, Kaneda and co-workers used a composite dendrimer of Rh5 carbonyl cluster and polyamine composite for nitro reduction.34 Recently in 2017, Lipshutz and co-workers used carbonyl iron powder under aqueous micellar catalysis condition.35 Fe(CO)5 and Fe3(CO)12 have also been used for nitro reduction, but all the methods require rather drastic reaction condition, including long duration, high pressure of CO gas, handling of highly toxic Fe(CO)5, reducing agent and various supporting reagents for to promote the reaction.36
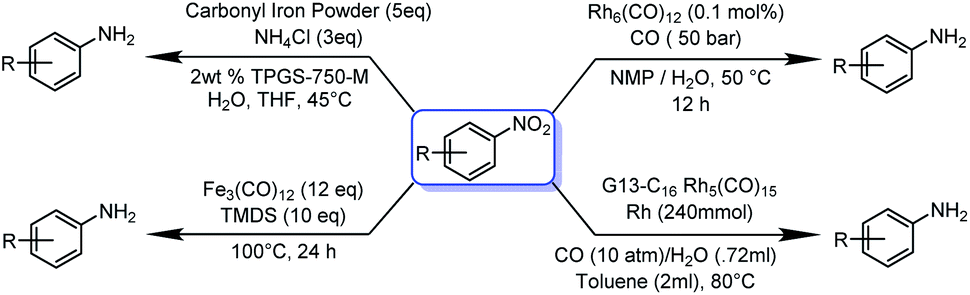 | ||
| Scheme 1 Previous reports on metal carbonyl catalysed nitro reduction reactions. Most metal carbonyl catalysed nitro reduction reactions require a high pressure of CO and H2(g). | ||
Recently, chalcogen and metal chalcogenide complexes have come under the spotlight, these complexes are continuously breaking the stereotype of air and moisture sensitive metal complexes. Due to good electron donor properties of chalcogen atoms, chalcogenide complexes have emerged as a potential alternative to phosphine complexes. Their ability to withstand air/moisture and their permanence during catalysis makes them suitable catalysts for long-duration reactions, which include C–C couplings37,38 C–N couplings39 and other organic transformations.40
In addition to gaseous hydrogen, several other reducing agents have been used that allow the efficient reduction of nitrobenzene when used in combination with metals, including boranes,41 NaBH4,42 silanes43 and hydrazine.44–46 Hydrazine hydrate is used as a very suitable reducing reagent as it generates only N2 as a by-product. It is fairly safe and easy to handle compared with its unstable anhydrous form. Many nitro reduction protocols using a combination of iron complexes and hydrazine hydrate have been published in the last few years.47–49
This paper is designed to report a selective reduction of nitroarenes with hydrazine, catalysed by zero-valent Fe3Se2(CO)9 and Fe3S2(CO)9 carbonyl clusters. These clusters are robustly stable, insensitive towards air, moisture, water and sunlight, can be stored for a long time and can be easily synthesized on a large scale. The present method produces significant transformations and does not generate any waste (Scheme 2). The catalyst is found to be efficient and brings the desired transformation in just 15 min. Moreover, it works smoothly in aqueous media at 110 °C and is applicable for a large variety of functionalised nitrobenzenes.
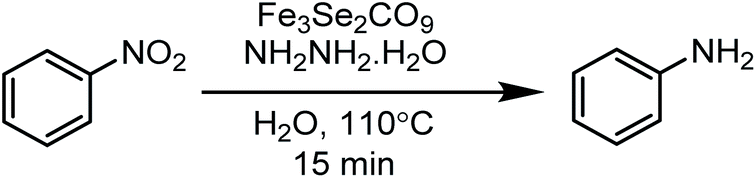 | ||
| Scheme 2 Nitro reductions catalysed by an Fe3Se2(CO)9 cluster. | ||
Results and discussion
Iron complexes have been established as excellent catalysts for various organic transformations, our group has also exploited quite a few iron based organic transformations.6,47 The literature encouraged us to investigate the catalytic activity of chalcogen-stabilized iron carbonyl complexes. This is the first report which utilises Fe3E2(CO)9 (E = S, Se or Te) clusters for the reduction of nitrobenzene and its derivatives to their corresponding arylamines. In the very first reaction, a dioxane solution of nitrobenzene and hydrazine (a source of hydrogen) with an Fe3Se2(CO)9 (3 mol%) cluster was refluxed with continuous monitoring of the reaction on a TLC. After 6 h, complete consumption of reactant along with the significant formation of the desired reduction product was experienced. The reaction was reinvestigated in the absence of Fe-catalyst, but no chemical transformation was recorded. Then, the reaction was optimized in search of the best suitable parameters for maximum transformation (Table 1). Initially, the amount of iron catalyst was optimised: 1 mol% of the catalyst failed to initiate the reaction, while 1.5 mol% of the catalyst started to produce the desire product in below to average amount (29%), however, the yield was improved from 29 to 47, 68 and 89% with 2, 2.5 and 3 mol% of the Fe-catalyst, respectively. No substantial improvement in the yield of aniline was experienced with 3.5 or 4 mol% of catalyst loading. Hence, 3 mol% of catalyst was the ideal amount for a creditable transformation (Table 1, entries 2–8). The other chalcogen-stabilised iron carbonyl clusters Fe3S2(CO)9 and Fe3Te2(CO)9 were also explored for the present reaction (Table 1, entries 9 and 10). It was observed that Fe3S2(CO)9 shows almost comparable catalytic efficiency to the selenium analogue, (82% yield of aniline), while the Fe3Te2(CO)9 was not efficient as it yielded average (49%) transformation of the desire product. Hence, the order of catalytic efficiency for these catalysts is Fe3Se2(CO)9 ≈ Fe3S2(CO)9 > Fe3Te2(CO)9. The difference in catalytic efficiency may be due to the electron-donor properties of S/Se/Te.37
|
|||||||
|---|---|---|---|---|---|---|---|
| S. no. | Solvent | Fe cat | Cat, mol% | N2H4, mmol | Temp., °C | Time (min) | Yielda, % |
| a Isolated yields, (a) Fe3S2(CO)9, (b) Fe3Se2(CO)9, (c) Fe3Te2(CO)9, optimised conditions: nitrobenzene (1 mmol), N2H4·H2O (2 mmol), catalyst Fe3Se2(CO)9 (3 mol%), temperature 110 °C, time 15 min, solvent water. | |||||||
| 1 | H2O | — | — | 2 | 110 | 15 | nd |
| 2 | H2O | b | 1 | 2 | 110 | 15 | nd |
| 3 | H2O | b | 1.5 | 2 | 110 | 15 | 29 |
| 4 | H2O | b | 2 | 2 | 110 | 15 | 47 |
| 5 | H2O | b | 2.5 | 2 | 110 | 15 | 68 |
| 6 | H2O | b | 3 | 2 | 110 | 15 | 89 |
| 7 | H2O | b | 3.5 | 2 | 110 | 15 | 90 |
| 8 | H2O | b | 4 | 2 | 110 | 15 | 90 |
| 9 | H2O | a | 3 | 2 | 110 | 15 | 85 |
| 10 | H2O | c | 3 | 2 | 110 | 15 | 49 |
| 11 | H2O | b | 3 | 0.5 | 110 | 15 | 19 |
| 12 | H2O | b | 3 | 1 | 110 | 15 | 37 |
| 13 | H2O | b | 3 | 1.5 | 110 | 15 | 69 |
| 14 | H2O | b | 3 | 2 | 110 | 15 | 87 |
| 15 | H2O | b | 3 | 2.5 | 110 | 15 | 88 |
| 16 | H2O | b | 3 | 3.0 | 110 | 15 | 90 |
| 17 | H2O | b | 3 | 2 | 80 | 360 | 85 |
| 18 | H2O | b | 3 | 2 | 120 | 10 | 88 |
| 19 | H2O | b | 3 | 2 | 130 | 7 | 87 |
| 20 | H2O | b | 3 | 2 | 150 | 2 | 89 |
| 21 | 2-Propanol | b | 3 | 2 | 110 | 15 | 27 |
| 22 | Butanol | b | 3 | 2 | 110 | 15 | 75 |
| 23 | sec-Butanol | b | 3 | 2 | 110 | 15 | 69 |
| 24 | EtOH | b | 3 | 2 | 110 | 15 | 79 |
| MeOH | b | 3 | 2 | 110 | 15 | 82 | |
| 25 | Toluene | b | 3 | 2 | 110 | 15 | 45 |
| 26 | Dimethyl formamide | b | 3 | 2 | 110 | 15 | 5 |
| 27 | Dioxane | b | 3 | 2 | 110 | 15 | 19 |
In a further attempt to optimize the amount of hydrazine hydrate which was used as a hydrogen source in the reaction, a 0.5 mmol amount of hydrazine produced 19% yield of aniline, while increasing the amount of hydrazine significantly increased the yield of aniline; however, no considerable improvement was observed beyond the 2 mmol amount of hydrazine (Table 1, entries 11–16). Here, 2 mmol of hydrazine hydrate serves as the optimal amount to achieve the desired quantity of hydrogen for the reaction. During the temperature optimization, the reaction was found a bit sluggish at below 60 °C; moreover, it took 6 h to produce 85% transformation at 80 °C (Table 1, entry 17). But, increasing the temperature, the duration of the reaction is drastically reduced, and at 110 °C, the reaction took just 15 min to brings the 90% transformation, while at 120, 130 and 150 °C, the reaction was completed in just 10, 7 and 2 min, respectively (Table 1, entries 18–20). In the present method, water was used as the solvent, and the reaction was refluxed between 100 and 110 °C. The solvent was also optimized, and the highest yield was achieved in water, which is a green and economical solvent.
Reaction also shows the significant transformations in methanol, ethanol, butanol, and sec-butyl alcohols however, isopropyl alcohol was not found suitable as it produces only 27% product (Table 1, entries 21–25). Other organic solvents, including toluene, 1,4-dioxane and dimethylformamide produce 45%, 19% and 5% yields of aniline, respectively (Table 1, entries 26–27). Besides hydrazine, other sources of hydrogen (Table 2), including isopropyl alcohol with KOH, and acetic acid or formic acid with triethylamine, were considered, but none of them mimicked the reaction. Moreover, not even a trace of product was observed when the reaction mixture was refluxed for up to 12 h. An insignificant transformation was obtained in an isopropyl alcohol and KOH solution, and it was not worth considering.
| S. no. | Reagents | % yield |
|---|---|---|
| a Nitrobenzene (1 mmol), catalyst Fe3Se2(CO)9 (3 mol%), temperature 110 °C, time 15 min, solvent water. | ||
| 1 | Isopropyl alcohol and KOH | <20 |
| 2 | Acetic acid with triethylamine | — |
| 3 | Formic acid with triethylamine | — |
| 4 | Hydrazine | 89 |
Substrate scope
In order to check the scope of the reaction, various functionally different nitroarenes were screened at the optimized reaction parameters (Table 3). In general, most of the reactions produced excellent yield of the desired amine products. However, some variation was also observed with the change in the position of the functional group at the benzene ring. In comparison to para- and meta-substitution, the ortho-functionalised nitrobenzene derivatives show a bit less transformation. The electronic nature of the functional groups also influences the desired transformation, and a slightly better yield was obtained with electron-withdrawing group attached to nitrobenzene. The initial reaction of nitrobenzene produces an excellent 89% yield of reductive product, i.e., aniline (1). Furthermore, the substrate scope was extended to halide derivatives of nitrobenzene: 4-chloro-, 4-bromo- and 4-iodonitrobenzene react smoothly and produce 86%, 84% and 83% yield of the respective aniline products. The position of the substituent also influences the productivity: a slightly reduced yield was obtained with 2-bromonitro benzene (5, 79%). 2,5-di-bromonitrobenzene also reacted efficiently and produced an excellent yield (6, 82%) of the respective product. Various electron-withdrawing groups were also tested for the present reaction, para-hydroxy and para-cyanonitrobenzene both yield highly significant transformations (7, 87%) and (8, 85%) of the respective aniline products. The 1,4-dinitrobenzene reacted under the optimised conditions to give an outstanding yield with a selective reduction of one nitro group, and para-nitroaniline (9) was isolated with 88% transformation. Moreover, a 75% transformation of 1,4-diaminobenzene (10) was observed when the same reaction was repeated with an increased quantity of hydrazine. A slightly reduced reduction of 2-aminonitrobenzene to 1,2-diaminobenzene (11, 76%) was observed due to the ortho positions. However, a significantly enhanced yield (12, 86%) of the meta-substituted aminonitrobenzene was recorded. A similar reduced trend was also observed with other ortho- and para-functional derivatives. The ortho- and para-derivatives of methyl nitrobenzene produced 71% (13) and 73% (14) transformation of their respective products. A marginally increased yield was recorded with para-methoxy nitrobenzene (15, 80%). Functional group interference was expected, but para-nitro acetophenone produced an excellent 83% (16) yield of desired product. Furthermore, the reaction of para-nitro benzyl alcohol conveniently react to produce 79% (17) of the corresponding product. Similarly, 1-nitronaphthalene also participated well in the reaction to produce 87% (18).| a Reaction conditions: Fe3Se2(CO)9 (3 mol%), nitrobenzene derivatives (1 mmol), N2H4·H2O (2 mmol), temperature 110 °C, time 15 min, solvent water, isolated yields. |
|---|
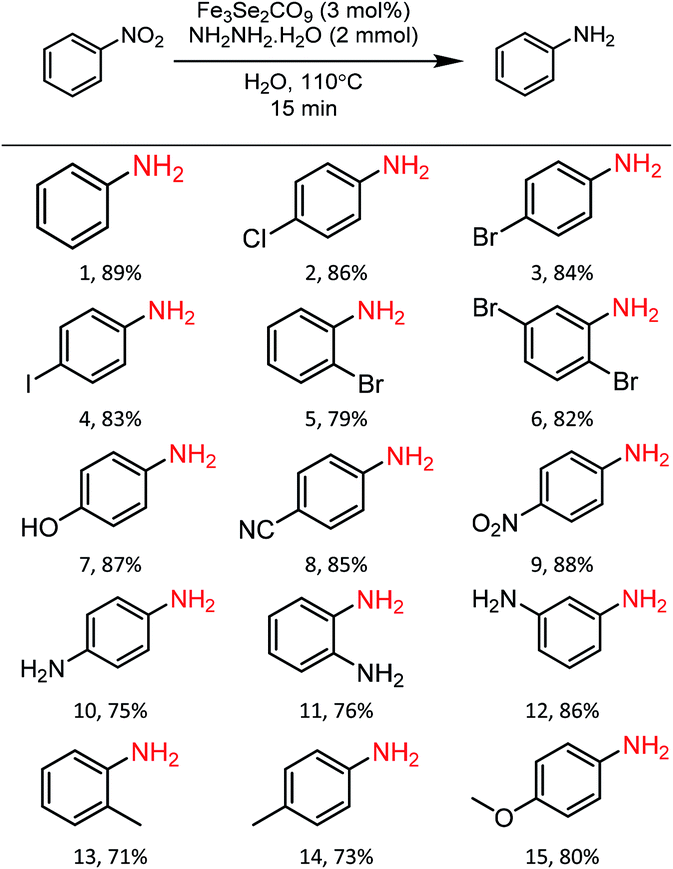 |
Catalyst recycling
The catalyst was isolated from the organic layer and used again with an aqueous layer for another catalytic cycle. In the second and third catalytic cycles, 63 and 46% transformations of the desired product were obtained, respectively. This indicates the decreasing catalytic efficiency of the catalyst. Besides this, it was also noted that there was 20% loss of catalyst in each catalytic run. This may be due to the presence of moisture on the surface of the catalyst, as a broad signal for moisture (at 3409 cm−1) was recorded in the FTIR spectrum of the used catalyst (Fig. S1, ESI†). Therefore, the catalyst has to be dried in an oven for 1 h before using it for the next catalytic cycle.Plausible mechanism
While performing control experiments it was observed that the reaction has good selectivity towards the electron-withdrawing functionalities. A reaction mixture of 4-methylnitrobenzene and 4-cyanonitrobenzene (1 equivalent of each) under optimised reaction condition produces para-cyano aniline (73%) as a major product and para-methyl aniline (27%) as minor product (Scheme 3).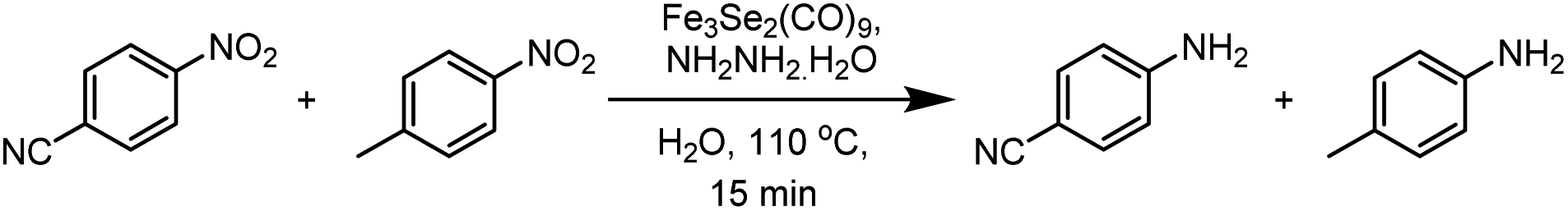 | ||
| Scheme 3 Control experiment showing affinity towards the withdrawing group. | ||
In another control experiment maintaining similar reaction conditions and the same reagents, after 15 minutes of the reaction, furthermore 2 equiv. of hydrazine was added and the reaction was again run for next 15 minutes. After two consecutive cycles, 91 and 56% yields of p-cyano aniline and p-methyl aniline were obtained respectively. In the second run, the required hydrazine was added, but the activity of the catalyst was considerably compromised. Therefore only a marginal increase in the yield of p-cyano aniline was observed, while due to the significant quantity of unreacted 4-methyl nitrobenzene it showed a significant increase in transformations.
However, neither a detailed study conducted nor any intermediate species were isolated during the present transformation, but a tentative mechanism has been proposed on the basis of evidence available in the literature (Scheme 4). There are strong evidences in the literature suggests that Fe(0) and Se both are individually excellent catalysts for nitro reduction.48–55
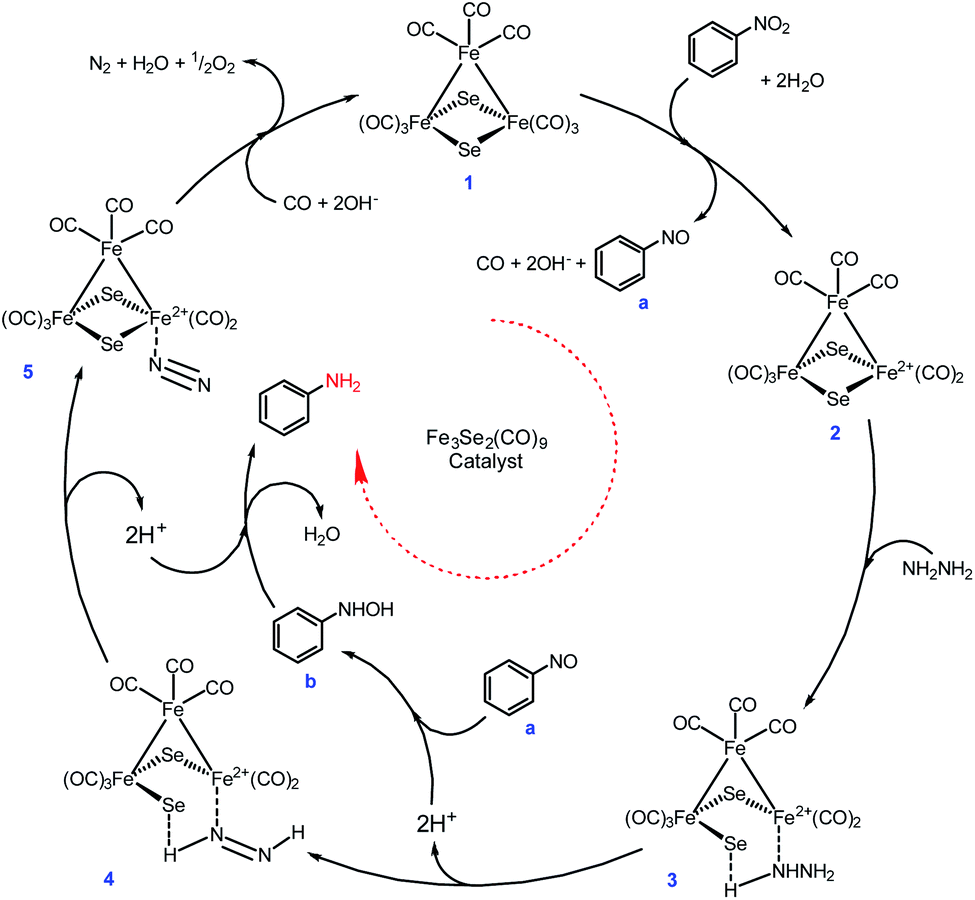 | ||
| Scheme 4 A plausible mechanism for the nitro reduction. | ||
Initially, the Fe-cluster (1) interacts with nitrobenzene in the presence of water and is oxidised and releases the required electrons with the simultaneous loss of CO to form intermediate (2), which facilitates the formation of nitrosobenzene (a).53 The hydrazine coordinates with intermediate (2) to produce (3). The interaction of the proton present in the close vicinity of the bridged Se in intermediate (3) leads to the loss of the Fe–Se bond. The deprotonation of intermediate (3) results in intermediate (4). Moreover, a simultaneous protonation of nitrosobenzene forms intermediate arylhydroxylamine (b). Further consecutive deprotonation of intermediate 4 and protonation of arylhydroxylamine form intermediate 5 and arylamine, respectively. Loss of N2 and reunion of CO with intermediate 5 regenerate the catalyst (1). The nitro reduction is possible via either four or six electron transfer. The six electron transfer strongly needs an alkaline medium47 and during the six electron transfer, nitrosobenzenes may undergo condensation. This is quite rare, but the formation of azoxybenzene followed by azobenzene and hydrazobenzene to aniline is frequently observed.50
Experimental section
General procedure for the catalytic synthesis
In a clean reaction tube were placed the Fe3Se2(CO)9 catalyst (13.5 mg, 3 mol%) and the derivative of nitroarenes (1 mmol). To this were added hydrazine hydrate (64 μL, 2 mmol) and water as a solvent, the reaction tube was heated with continuous stirring at 110 °C for 15 min. After completion of the reaction, the reaction mixture was cooled to room temperature, and the desired organic product was extracted in an organic layer of ethyl acetate. The organic layer was then dried over anhydrous Na2SO4, and ethyl acetate was evaporated on a rotavapor at reduced pressure to obtain the crude product. Finally, the crude product was subjected to a chromatographic work-up and further purification and spectral characterization.Conclusion
In conclusion, a robustly stable, reusable, highly economical, easy to synthesise and zero-valent iron chalcogenide carbonyl cluster Fe3Se2(CO)9 has been explored for the reduction of nitroarenes. The chalcogen-stabilised Fe-cluster brings about an excellent transformation to a range of functionalised nitroarenes under the inert free conditions and in an aqueous medium. The methodology was found to be strongly feasible and worked in the green solvent water. Moreover, it also avoids the use of a precious metal catalyst or supporting reagents. The catalyst can be reused for three catalytic cycles. The present methodology also provides a selective but temperature-dependent timeframe (2–15 min) to accomplish the reaction. This is the first report where zero-valent chalcogen-stabilised iron carbonyl clusters were explored for nitro reduction in the absence of high CO pressure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Raj K. Joshi thanks the CSIR (01(2996)/19/EMR-II) for financial assistance. Charu Sharma, Sangeeta Kumari and Avinash K. Srivastava thank MNIT for the research fellowships. Aditi Soni thanks UGC for a research fellowship. The authors also acknowledge the MRC, MNIT for providing characterisation facilities.Notes and references
- W. Reppe, H. Vetter and J. Liebigs, Ann. Chem., 1953, 582, 133 CAS.
- L. Mond and F. Quinke, J. Am. Chem. Soc., 1891, 59, 604 RSC.
- M. Berthelot, C. R. Hebd. Seances Acad. Sci., 1891, 112, 1343 Search PubMed.
- C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- L. Vancea and W. A. G. Graham, J. Organomet. Chem., 1977, 134, 219–227 CrossRef CAS.
- R. K. Joshi and N. Satrawala, Tetrahedron Lett., 2017, 58, 2931–2935 CrossRef CAS.
- P. Mathur, R. K. Joshi, B. Jha, A. K. Singh and S. M. Mobin, J. Organomet. Chem., 2010, 695, 2687–2694 CrossRef CAS.
- P. Mathur, R. K. Joshi, D. K. Rai, B. Jha and S. M. Mobin, Dalton Trans., 2012, 41, 5045–5054 RSC.
- P. Mathur, D. K. Rai, R. K. Joshi, B. Jha and S. M. Mobin, Organometallics, 2014, 33, 3857–3866 CrossRef CAS.
- P. Mathur, B. Jha, A. Raghuvanshi, R. K. Joshi and S. M. Mobin, J. Organomet. Chem., 2012, 712, 7–14 CrossRef CAS.
- B. N. Jha, A. Raghuvanshi, R. K. Joshi, S. M. Mobin and P. Mathur, Appl. Organomet. Chem., 2017, 31, e3805 CrossRef.
- S. C. Jensen, S. B. Homan and E. A. Weiss, J. Am. Chem. Soc., 2016, 138, 1591–1600 CrossRef CAS PubMed.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed.
- O. Beswick, I. Yuranov, D. T. L. Alexander and L. Kiwi-Minsker, Catal. Today, 2015, 249, 45–51 CrossRef CAS.
- H. Li, C. Huang, Y. Li, W. Yang and F. Liu, RSC Adv., 2019, 9, 22523–22530 RSC.
- R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982–7984 RSC.
- R. G. D. Noronha, C. C. Romão and A. C. Fernandes, J. Org. Chem., 2009, 74, 6960–6964 CrossRef PubMed.
- H. Berthold, T. Schotten and H. Hönig, Synthesis, 2002, 1607–1610 CAS.
- R. J. Rahaim and R. E. Maleczka, Org. Lett., 2005, 7, 5087–5090 CrossRef CAS PubMed.
- S. K. Mohapatra, S. U. Sonavane, R. V. Jayaram and P. Selvam, Org. Lett., 2002, 4, 4297–4300 CrossRef CAS PubMed.
- U. Sharma, P. Kumar, N. Kumar, V. Kumar and B. Singh, Adv. Synth. Catal., 2010, 352, 1834–1840 CrossRef CAS.
- A. Agrawal and P. G. Tratnyek, Environ. Sci. Technol., 1995, 30, 153–160 CrossRef.
- F. A. Khan and C. Sudheer, Tetrahedron Lett., 2009, 50, 3394–3396 CrossRef CAS.
- T. Phenrat, N. Saleh, K. Sirk, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2007, 41, 284–290 CrossRef CAS PubMed.
- L. Jos de Jongh, Physical properties of metal cluster compounds, Model systems for nanosized metal particles, Wiley-VCH, New York, 1999, pp. 1434–1453 Search PubMed.
- G. Gonzalez-Moraga, Cluster Chemistry: Introduction to the Chemistry of Transition Metal and Main Group Element Molecular Clusters, 1993 Search PubMed.
- M. B. Thathagar, J. E. Eleshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886–2890 CrossRef CAS PubMed.
- S. Pandey, K. Vipin Raj, D. R. Shinde, K. Vanka, V. Kashyap, S. Kurungot, C. P. Vinod and S. H. Chikkali, J. Am. Chem. Soc., 2018, 140(12), 4430–4439 CrossRef CAS PubMed.
- A. M. Lennertz, J. Laege, M. J. Mirbach and A. Saus, J. Organomet. Chem., 1979, 171, 203–207 CrossRef CAS.
- I. Sorribes, G. Wienhofer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794 CrossRef CAS PubMed.
- E. Pedrajas, I. Sorribes, A. L. Guschin, Y. A. Laricheva, K. Junge and M. Beller, ChemCatChem, 2017, 9, 1128–1134 CrossRef CAS.
- E. Pedrajas, I. Sorribes, K. Junge, M. Beller and R. Llusar, ChemCatChem, 2015, 7, 2675–2681 CrossRef CAS.
- R. Ryan, G. M. Wilemon, M. P. Dalsanto and C. U. Pittman, J. Mol. Catal., 1979, 5, 319–330 CrossRef CAS.
- Z. Maeno, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2014, 50, 6526 RSC.
- N. R. Lee, A. A. Bikovtseva, M. C. Clerget, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 6518–6521 CrossRef CAS PubMed.
- Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511–9514 CrossRef CAS PubMed.
- K. N. Sharma, N. Satrawala, A. K. Srivastava, M. Ali and R. K. Joshi, Org. Biomol. Chem., 2019, 17, 8969–8976 RSC.
- K. N. Sharma, N. Satrawala and R. K. Joshi, Eur. J. Inorg. Chem., 2018, 16, 1743–1751 CrossRef.
- C. Sharma, A. K. Srivastava, K. N. Sharma and R. K. Joshi, Org. Biomol. Chem., 2020, 18, 3599–3606 RSC.
- K. N. Sharma, M. Ali, A. K. Srivastava and R. K. Joshi, J. Organomet. Chem., 2019, 879, 69–77 CrossRef CAS.
- J. W. Bae, Y. J. Cho, S. H. Lee, C. O. M. Yoon and C. M. Yoon, Chem. Commun., 2000, 1857–1858 RSC.
- X. Lin, M. Wu, D. Wu, S. Kuga, T. Endoe and Y. Huang, Green Chem., 2011, 13, 283–287 RSC.
- K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769–1771 RSC.
- R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A. E. Surkus, M. M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972–10974 RSC.
- S. Kim, E. Kim and B. M. Kim, Chem.–Asian J., 2011, 6, 1921–1925 CrossRef CAS PubMed.
- D. Cantillo, M. M. Moghaddam and C. O. Keppe, J. Org. Chem., 2013, 78, 4530–4542 CrossRef CAS PubMed.
- A. K. Srivastava, M. Ali, S. Siangwata, N. Satrawala, G. S. Smith and R. K. Joshi, Asian J. Chem., 2020, 8, 377–384 Search PubMed.
- H. Alper and J. T. Edward, Can. J. Chem., 1970, 48, 1543–1549 CrossRef CAS.
- T. Miyata, K. Kondo, S. Murai, T. Hirashima and N. Sonoda, Angew. Chem., Int. Ed., 1980, 19, 1008 CrossRef.
- A. Agarwal and P. G. Tratyek, Environ. Sci. Technol., 1996, 30, 153–160 CrossRef.
- X. Liu and S. Lu, J. Mol. Catal. A: Chem., 2004, 212, 127–130 CrossRef CAS.
- Y. Keum and Q. Li, Chemosphere, 2004, 54, 255–263 CrossRef CAS PubMed.
- V. Popat and N. Padhiyar, IJCEA, 2013, 4, 401–405 CrossRef CAS.
- H. Lee, B. Kim, Y. Park, S. Kim and S. Jung, J. Nanomater., 2015, 392537 Search PubMed.
- L. Shao, Y. Li, J. Lu and X. Jiang, Org. Chem. Front., 2019, 6, 2999–3041 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04491a |
| This journal is © The Royal Society of Chemistry 2020 |