
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAncillary ligand electro-activity effects towards phenyl acetylene homocoupling reaction by a nickel(II) complex of a non-innocent O-amino phenol ligand: a mechanistic insight†
Mina Nasibipoura,
Elham Safaei *a,
Marziyeh Sadat Masoumpourb and
Andrzej Wojtczakc
*a,
Marziyeh Sadat Masoumpourb and
Andrzej Wojtczakc
aDepartment of Chemistry, College of Sciences, Shiraz University, 71454, Shiraz, Iran. E-mail: e.safaei@shirazu.ac.ir
bDepartment of Chemistry, Estahban Higher Education Center, Estahban 74519-44655, Iran
cNicolaus Copernicus University, Faculty of Chemistry, 87-100 Torun, Poland
First published on 25th June 2020
Abstract
A new Ni(II) complex, was synthesized from the reaction of a non-innocent o-aminophenol ligand, and Ni(OAc)2. The crystal structure of NiIIL2NIS (in which, IS stands for iminosemiquinone radical ligand with cyanide (shown by N in NIS) substituent on phenolate rings) exhibits the square planar environment of Ni(II). The complex has been crystalized in the monoclinic system and Ni(II) was surrounded by two oxygen and two nitrogen atoms of two ligands. Variable-temperature magnetic susceptibility measurement for crystalline samples of complex shows the effective magnetic moment per molecule (μeff) of near zero and the diamagnetic nature of the complex (S = 0) which emphasize that strong antiferromagnetic coupling prevailed between the two unpaired electrons of LNIS ligands and Ni(II) high spin electrons. The complex is EPR silent which confirms the diamagnetic character of the Ni(II) complex. Electrochemical measurement (CV) indicates the redox-active character of ligand and metal. NiIIL2NIS complex proved to be effective for free metal- or base counterpart homocoupling of phenyl acetylene at room temperature. To the best of our knowledge, this is the first example of using Ni(II) complex without using any reducing agent due to the promotion ancillary effect of non-innocent o-aminophenol ligand which acts as an “electron reservoir” and can reversibly accept and donate electrons in the catalytic cycle. The theoretical calculation confirms the magnetostructure, electronic spectrum and confirmed the suggested mechanism of phenyl acetylene homocoupling with emphasis on the role of non-innocent ligand electro-activity and the effect of ligand substituent on the efficiency and stability of the complex.
Introduction
In a metal complex, the coordination of ligand to the metal center affects the properties of a complex. To organize new coordination complexes with suitable applications, designing new multifunctional ligands is the first and most important feature. Redox non-innocent ligands, which are able to be easily oxidized or reduced by one or more electrons in a ping-pong mechanism, can participate in the electron transfer reactions and therefore they have an important effect on the redox states of the central metal being coordinated. These ligands are able to modify the electrochemical potentials of the metal by stabilizing unusual oxidation states of metal ions, or by participating in these processes actively.1–7 Hence, non-innocent ligands are of great interest because of their unique properties which they may offer to their metal complexes, including magnetic exchange, optical and electronic properties, as well as reactivity.1,8–14One of the most archetypal examples of such species is o-aminophenol. Once deprotonated, o-aminophenol ligands can in fact accommodate three stable oxidation states: o-aminophenolate, o-iminobenzosemiquinone and o-iminobenzoquinone (Scheme 1).15 The o-aminophenols are one of the well-known non-innocent ligands that can form a wide range of complexes and in recent years. Some complexes of their one and two electron reduced forms have been reported by research groups.16–23 Redox-active metal complexes can show interesting reactivity towards organic molecules in various electron transfer processes that involve the metal centers and ligands.
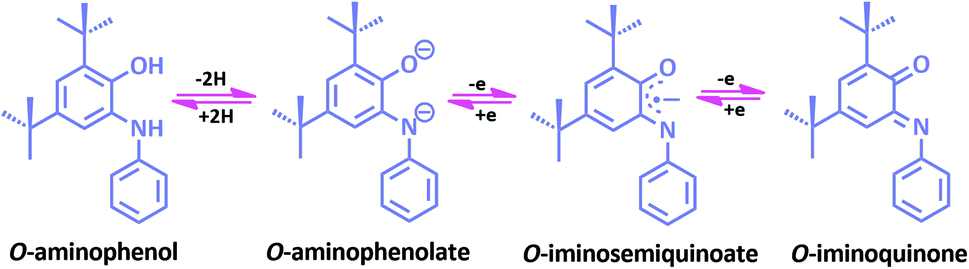 | ||
| Scheme 1 Oxidation states of the bidentate o-aminophenol. | ||
Scientists have focused on 3,5-di-tert-butyl-o-aminophenols, due to their tert-butyl substituents role in lowering the oxidation potential of the ligand and facilitating its oxidation and preventing oxidative decomposition. In other words, the electron-donating property of tert-butyl substituents enabled the deprotonated ligand to efficiently stabilize high oxidation states of the complex.
Many catalytic processes require ligands and metals which have the ability to change their oxidation state to facilitate desirable chemical transformations.1,3 In this regard, in o-aminophenolate complexes the redox transformations are modulated by the o-aminophenol ligand which can be considered as an “electron reservoir” and can reversibly accept and donate electrons.
There is increasing interest in the use of Ni catalysts because of their high efficiency and low costs. Among these catalytic systems, the nickel catalyzed cross- and homocoupling reactions widely used nowadays for the formation of C–C bonds have experienced significant developments.24–50
Homocoupling of terminal alkynes that include the formation of 1,3-diynes, has possibly become one of the most applied tools for the creation of a C–C bond. This reaction was investigated by Glaser and since its discovery in 1869,51 has experienced great improvement. This process was further developed by Eglinton and Hay.52–54 Since then, lots of modified copper-mediated Glaser–Eglinton–Hay coupling reactions have been broadly used in the synthesis of 1,3-diyne derivatives. There have been fewer thorough investigations of the ability of nickel complexes for the terminal alkynes homocoupling reactions in contrast to copper mediated reactions. Nickel-catalyzed homocoupling reaction was first pioneered by Rhee and co-workers.55
Homocoupling reaction generally follows an oxidative addition, transmetallation and reductive elimination catalytic cycle in which the use of electron-donating ligands helps the reaction and can promote first and last steps. For the Ni-catalyzed C–C homocoupling reactions, the Ni(0) is generally considered as catalytically active species and the mechanism follows a Ni(0)–Ni(II) catalytic cycle. The direct use of Ni(0) complexes as pre-catalysts, such as [Ni(cod)2] and [Ni(PPh3)4], is the simplest way. Still, such nickel sources are very difficult to handle and control because of their air and moisture sensitivity and high toxicity. Moreover, they are costly and more expensive than normal Pd sources. So the air and thermal stable Ni(II) complexes are often used as precatalysts activated to generate the active Ni(0) species in situ. The most commonly used Ni sources are the phosphine-coordinated nickel(II) halides, such as NiCl2(PPh3)2, NiCl2(PCy3)2, NiCl2(dppf), and NiCl2(dppe).44–46 In contrast to Pd(II), Ni(II) cannot readily be reduced to Ni(0) by the added base or solvent in the reaction system. A solution to this problem is treating the Ni(II) complexes with additional reducing agents such as Zn or n-BuLi and additional ligands for in situ generating Ni(0).24–44
In the current work, we used the o-aminophenol ligand, H2LNAP, which displays interesting redox properties and prepare its nickel complex (Scheme 2). This ligand coordinates in bidentate fashion and presents the neutral complex with square planar geometry of general formula [NiL2NIS].
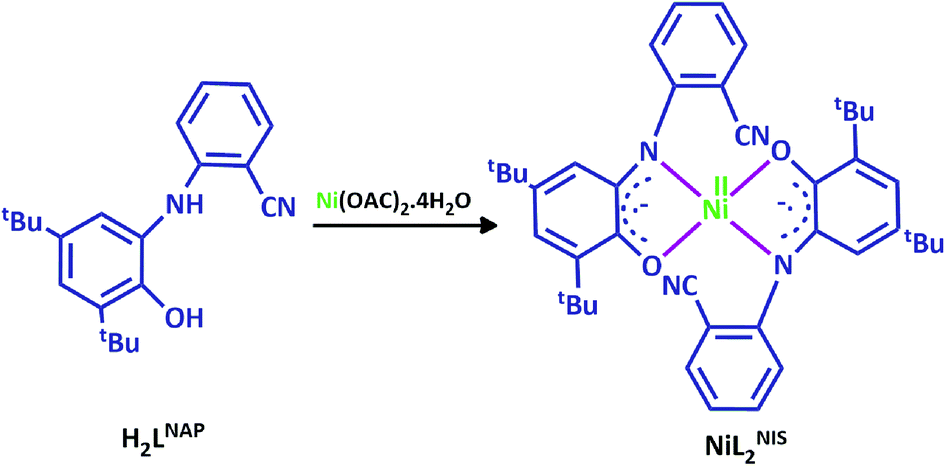 | ||
| Scheme 2 The structure of H2LNAP and the associated NiIIL2NIS complex. | ||
As a part of our ongoing effort we were interested to develop a new facile protocol for nickel catalyzed homocoupling reactions at room temperature in which use of hard-to-handle nickel(II) sources without any additional reducing agents would be obviated. We are interested in replacing any additive with the electroactive ligand coordinated to Ni(II) center.
Results and discussion
Synthesis and characterization of H2LNAP
The H2LNAP ligand possessing a redox-active aminophenolate function was synthesized via concentration of 3,5-di-tert-butylcatechol and 2-aminobenzonitrile as described in literature.56 H2LNAP was characterized by IR and elemental analysis. The free ligand H2LNAP exhibits characteristic IR bands at![[small nu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e1dd.gif) = 3421 cm−1 (νO–H stretch) and = 3354 cm−1 (νN–H vibrational stretches) and = 2222 cm−1 (–CN group).
= 3421 cm−1 (νO–H stretch) and = 3354 cm−1 (νN–H vibrational stretches) and = 2222 cm−1 (–CN group).
Synthesis and characterization of NiIIL2NIS
Dark green crystals of NiIIL2NIS were prepared with 86% yield by treating H2LNAP with Ni(OAc)2·4H2O in the presence of triethylamine. Elemental analytical data of the complex correspond to the calculated values for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal:ligand ratio. The IR spectrum of the complex indicates that strong and sharp νO–H stretch of the ligand disappears. Lack of band for the phenolic proton indicates ligation of the deprotonated phenol moiety and its coordination to Ni(II) center. The observed band in the region of around 1600–1700 cm−1 is attributed to the radical-anion form of o-iminobenzosemiquinone ligands. The phenolic ν(C–O) stretching band appears at = 1254 cm−1. The bands that appear at = 2869, 2916, and 2954 cm−1 belong to tert-butyl groups and confirm the presence of the 2,4-ditert-butylphenolate moiety in the coordinating ligand. The band due to ν(C
:2 metal:ligand ratio. The IR spectrum of the complex indicates that strong and sharp νO–H stretch of the ligand disappears. Lack of band for the phenolic proton indicates ligation of the deprotonated phenol moiety and its coordination to Ni(II) center. The observed band in the region of around 1600–1700 cm−1 is attributed to the radical-anion form of o-iminobenzosemiquinone ligands. The phenolic ν(C–O) stretching band appears at = 1254 cm−1. The bands that appear at = 2869, 2916, and 2954 cm−1 belong to tert-butyl groups and confirm the presence of the 2,4-ditert-butylphenolate moiety in the coordinating ligand. The band due to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching was observed in the range of 1400–1500 cm−1. In addition, the ν(C–H) bands of benzylic CH2 group is observed in around 2500 cm−1. The band appearing at = 2226 indicates the presence of –CN group and no ligation from N atom to Ni.
C) stretching was observed in the range of 1400–1500 cm−1. In addition, the ν(C–H) bands of benzylic CH2 group is observed in around 2500 cm−1. The band appearing at = 2226 indicates the presence of –CN group and no ligation from N atom to Ni.
In the 1H NMR of the complex, we can see the t-Bu and benzylic hydrogen's in around 1 ppm. The peaks placed between 6–8 ppm are related to the phenolic and cyano benzyl hydrogen's and the peak area are correspondence to the number of predicted hydrogen's in the complex structure. 14N NMR shows the two kind of nitrogen of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and –CN functional groups.
N and –CN functional groups.
Crystal structure of NiIIL2NIS
Crystallographic data for the complex are given in Table 1. Selected bond distances and angles are listed in Table 2. Complete crystallographic data, and bond distances, angles and torsion angles [°] are given in supplemental Tables S1, S2 and S3† respectively.| Empirical formula | C42H48N4NiO2 |
| Formula weight | 349.78 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions | a = 16.469(2) b = 8.1558(8) c = 16.897(2), α = 90 β = 118.733(17) γ = 90 |
| Volume | 1990.1(5) |
| Z/Z′ | 2/0.5 |
| Temperature | 293(2) K |
| Density (calculated) | 1.167 Mg m−3 |
| Crystal size | 0.529 × 0.356 × 0.122 mm3 |
| Absorption coefficient | 0.525 mm−1 |
| Reflections collected | 13001 |
| Independent reflections | 4554 [R(int) = 0.0568] |
| Goodness-of-fit on F2 | 0.977 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0462, wR2 = 0.1037 |
| R indices (all data) | R1 = 0.0742, wR2 = 0.1146 |
| O1–Ni1–O1#1 | 180.0 |
|---|---|
| a Symmetry transformations used to generate equivalent atoms: #1 − x, 1 − y, −z. | |
| O1–Ni1–N1 | 85.13(6) |
| O1#1–Ni1–N1 | 94.87(6) |
| C1–O1–Ni1 | 113.91(12) |
| Ni1–O1 | 1.8365(13) |
| Ni1–N1 | 1.8516(15) |
| C1–C2 | 1.425(3) |
| C2–C3 | 1.379(3) |
| C4–C5 | 1.364(3) |
| C5–C6 | 1.419(3) |
| O1–C1 | 1.317(2) |
| C6–N1 | 1.353(2) |
| C12–C13 | 1.441(3) |
| C13–N2 | 1.139(3) |
The asymmetric unit of the structure consists of a half of NiIIL2NIS molecule (Fig. 1), with one LNIS ligand and the central Ni(II) positioned on the center of symmetry. Therefore the other half of the molecule is generated by the center of symmetry [1 − x,−y + 1, −z]. Due to such molecular symmetry, the coordination sphere NiN2O2 has a square-planar geometry with bi-dentate LNIS coordinated via its aminophenolate moiety, and the five-membered chelate ring formed by LNIS is flat. The nitrile substituent is not involved in the coordination of Ni(II). Such coordination mode was also detected for CuL2NIS.57
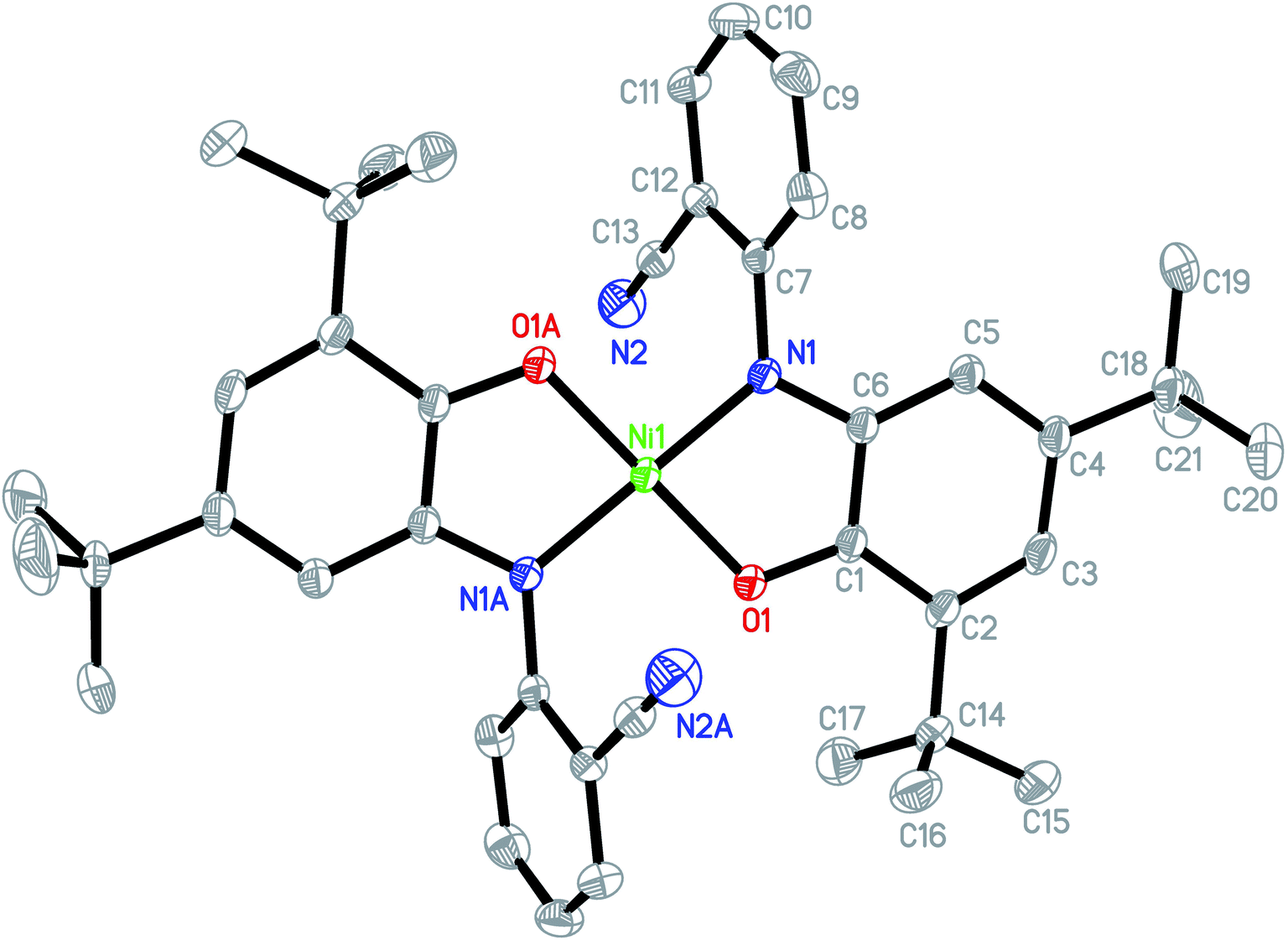 | ||
| Fig. 1 Molecular structure of NiIIL2NIS, H atoms have been omitted for clarity. Thermal ellipsoids are set at 30% probability. | ||
In the coordination sphere, the angles O1–Ni1–N1 and N1–Ni1–O1 [1 − x,−y + 1, −z] are 85.13(6) and 94.87(6)°, respectively (Table 2). The Ni1–O1 coordination bond of 1.8365(13) Å is slightly longer than Ni1–N1 1.8516(15) Å. In the aminophenolic moiety, C2–C3 bonds 1.379(3) and C4–C5 1.364(3) Å are significantly longer that other ring bonds ranging from 1.419(3) to 1.428(3), reflecting the localization of double bonds at C2–C3 and C4–C5. The O1–C1 and C6–N1 distances of 1.317(2) and 1.353(2) Å, respectively, correspond to double bonds. The observed coordination bond lengths and distribution of the double bonds indicate that LNIS is found in the iminosemiquinone radical mono anion form, similar to that found in the Cu complex.57
Conformation of the LNIS ligand is described with the C1–C6–N1–C7 and C6–N1–C7–C8 torsion angles of 179.18(16) and −68.7(2)°. The dihedral angle between two phenyl rings of LNIS is 69.46(12)°. Geometry of the nitrile group is typical. Analysis of the crystal packing reveals the interaction between two almost parallel nitrile–phenyl moieties. That interaction involves the nitrile C13–N2 substituent and the phenyl ring C7–C12 [−x, −1/2 + y, −1/2 − z], the N⋯Cg distance to the ring gravity center being 3.326(3) Å, although the C–N⋯Cg angle is 98.0(2)°. Also, the intermolecular C19–H19C⋯N2 [−x, 1/2 + y, −1/2 − z] interaction is found, with the C⋯N distance of 3.400(4) Å.
Magnetic susceptibility and EPR measurements
Variable-temperature magnetic susceptibility measurement for crystalline samples of NiIIL2NIS complex was carried out with an applied magnetic field of 1000 Oe in the temperature range 1.8–300 K (Fig. 2). The magnetic properties were measured similarly and corrected for temperature independent paramagnetism prior to analysis. The low value of χT, <0.01 emu K mol−1 over nearly the entire temperature range, suggests strong antiferromagnetic coupling of both ligand radical spins with that of the central high-spin Ni(II) ion. This spin distribution results in a singlet (S = 0) ground state as shown in Scheme 3. The non-zero χT value indicates the presence of uncoupled spins in the sample as an impurity (0.7%), as is common for many spin-coupled systems. The slight increase in χT at temperatures above 250 K indicates some thermal population of the higher-spin state.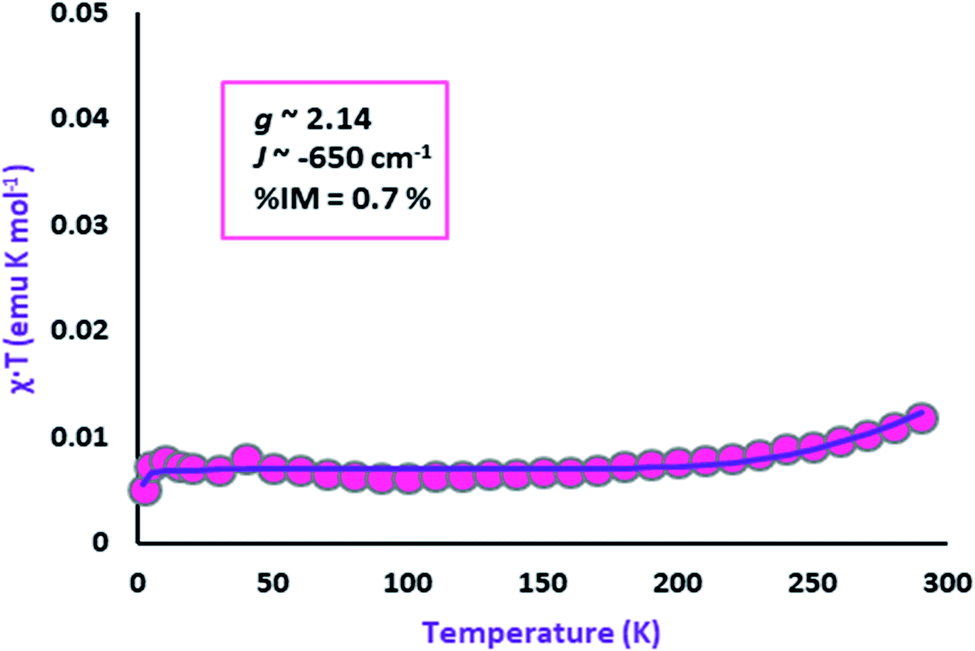 | ||
| Fig. 2 Variation of effective magnetic moment (μeff) with variation in temperature for the complex NiIIL2NIS. | ||
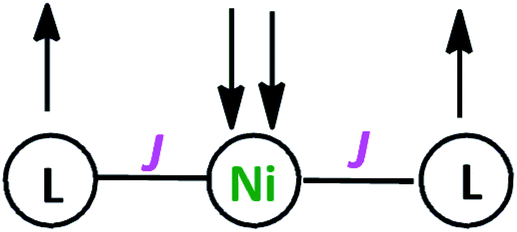 | ||
| Scheme 3 The Schematic representation of antiferromagnetic coupling between ligands and Ni(II) spins. | ||
As in the case of the Cu complex,57 only one spin–spin coupling parameter J (∼−650 cm−1) was included in the model, as well as a single effective g value (∼2.14) for all of the paramagnetic centres, due to the minimal population of the high-spin state at 300 K. The modelled values of these parameters should be considered to be only approximate.
Electrochemistry
Cyclic voltammogram (CV) of complex NiIIL2NIS has been recorded in CH2Cl2 solution containing 0.10 M NBu4ClO4 as supporting electrolyte at a glassy carbon as working electrode and an Ag/AgNO3 reference electrode at room temperature. NiIIL2NIS is composed of a Ni(II) ion and two iminosemiquinone moieties. The complex underwent two quasi-reversible one-electron redox processes in the anodic and cathodic studied range (Fig. 3 and Scheme 4) which can be assigned as the radical-ligand based iminosemiquinone/iminoquinone (NiIIL2NIS/NiIILNISLNIQ) and iminosemiquinone/amidophenoxide (NiIIL2NIS/NiIILNISLNAP) redox couples consistent with reports on similar complexes.58–63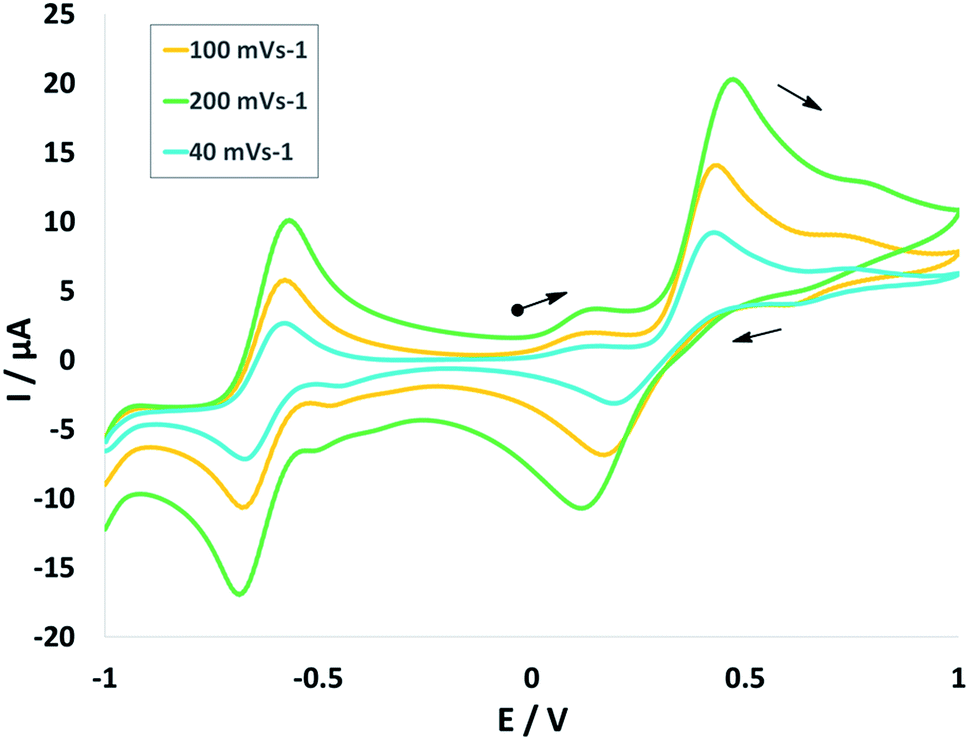 | ||
| Fig. 3 Cyclic voltammograms of NiIIL2NIS. Conditions: 1 mM complex, 0.1 M NBu4ClO4, scan rate 40, 100, 200 mV s−1, CH2Cl2, 298 K. | ||
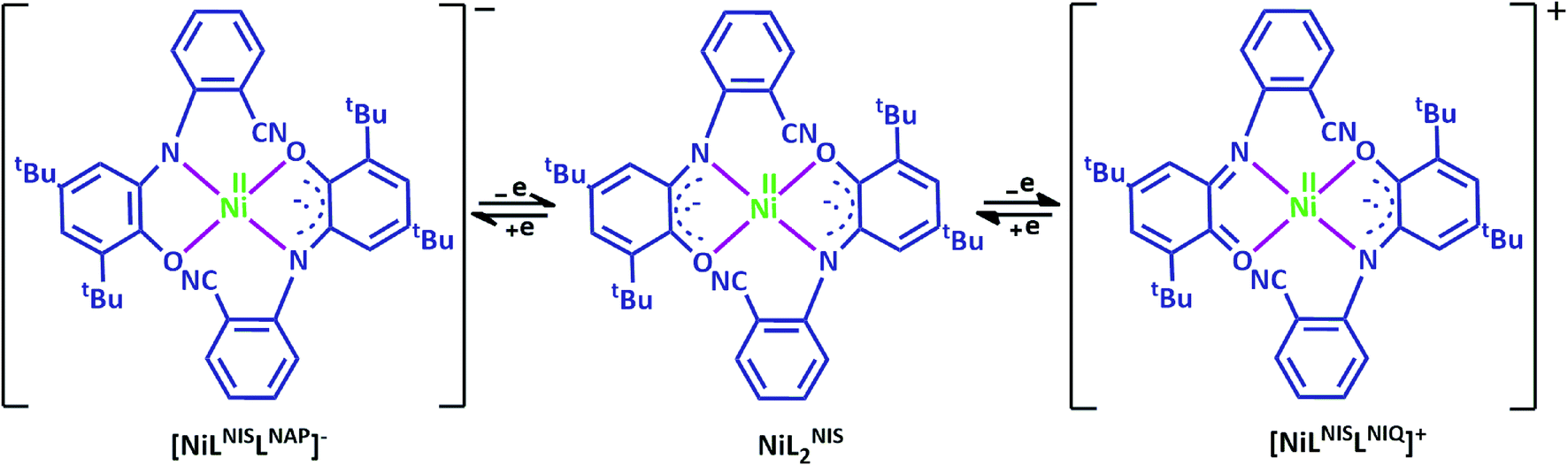 | ||
| Scheme 4 The possible redox states of the NiIIL2NIS complex. | ||
This process correspond to the following equation:
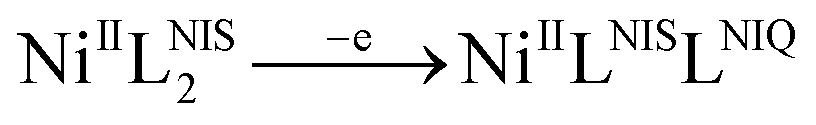
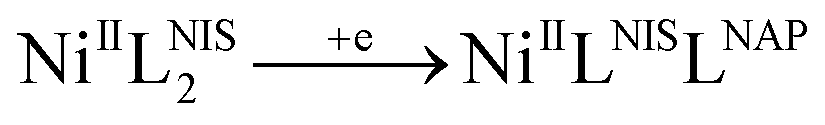
The observed electrochemical behavior is typical for similar square planar complexes of two o-phenylenediamnine ligands.62
Similar complex of Zn(LISQ)2 (without –CN substituent),63 demonstrates only two ligand-centered reduction stages. Since Zn(II) is not redox active metal, the potential peaks of −0.17 and −0.77 are not related to the metal centered reduction.
NiIIL2NIS complex shows one ligand centered oxidation peak potential at +0.47 V and one reduction peak potential at −0.68 V compatible to the observed peak for Zn(LISQ)2 complex, respectively. Comparison the electrochemical behavior of NiIIL2NIS with the similar structure of NiII(LISQ)2 (without –CN substituent synthesized by Wieghardt group)64 shows that NiIIL2NIS oxidation is more difficult than that of NiII(LISQ)2 (+0.04 V) due to the presence of cyanide groups, while the reduction potential in NiII(LISQ)2 has been observed in more negative peak potential of −1.07 and −1.64 V. In the other world, the reduction is easier in NiIIL2NIS comparing to its cyanide free congener. The role of cyanide group in the redox potential tuning can be observe in the catalytic process and also in the stability and reactivity of intermediates in theoretical mechanism cycle. Variation of the peak potentials with increasing the scan rates confirms the quasi reversible electrochemical processes in NiIIL2NIS.
Electronic spectroscopy
The electronic absorption (UV-Vis/NIR) spectrum of the complex NiIIL2NIS was recorded in dichloromethane at room temperature (25 °C) in the range of 200–700 nm (Fig. 4). The spectrum displays three important absorption bands for the complex. An intense band in the higher energy region (313 nm) is attributed to the intra-ligand π → π* transition involving the phenolate group and the absorption band at 395 nm corresponded to the ligand to-metal charge transfer (LMCT) from phenolate orbitals to d orbitals of Ni(II), what is typical for these kinds of complexes. The absorption band at lower energy (600 nm) was due to metal-to-ligand (MLCT) charge transfer.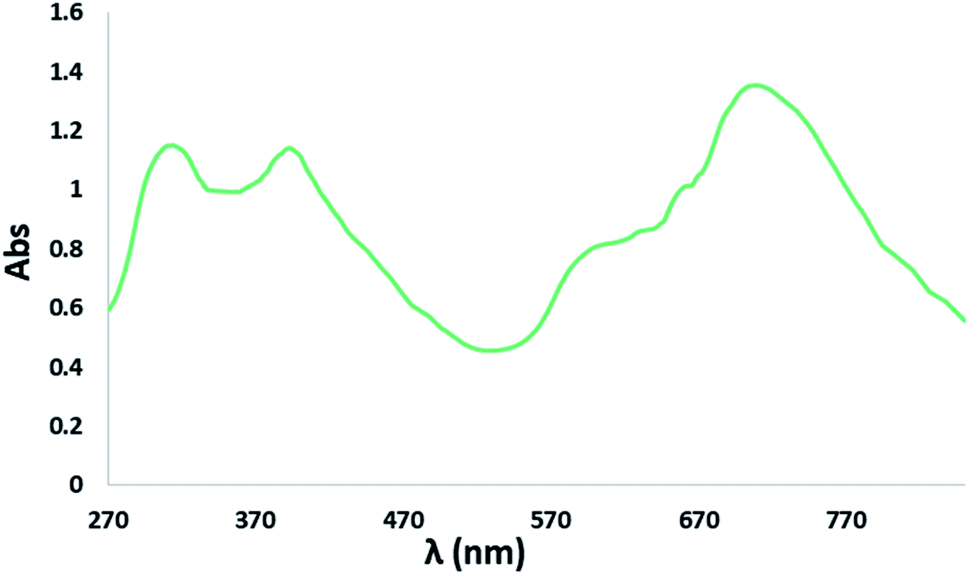 | ||
| Fig. 4 Electronic spectrum of 0.2 mM CH2Cl2 solutions of NiIIL2NIS. | ||
The theoretical analysis of the spectrum has been investigated and describe later.
Catalytic activity of NiIIL2NIS complex for homocoupling of phenyl acetylene derivatives
Continuing our previous research in C–H activation processes,62 we were interested in studying the catalytic activity of NiIIL2NIS in the homo coupling of terminal alkynes by evaluating the efficiency of the catalyst in the homocoupling reaction of various types of phenyl acetylenes (Scheme 5). Before addressing the main goal of this report, the optimized conditions for homocoupling reaction were figured out after optimizing investigations. Various control experiments were carried out to optimize the reaction conditions with respect to different parameters: solvent (THF, EtOH, CH3CN and toluene), base (KOH, K2CO3, Na2CO3, Cs2CO3), time and catalyst amount. Phenyl acetylene was typically used as a standard substrate. Based on results shown in Table 3, THF is better solvent than the other three solvents, in both kinetic and thermodynamic approaches.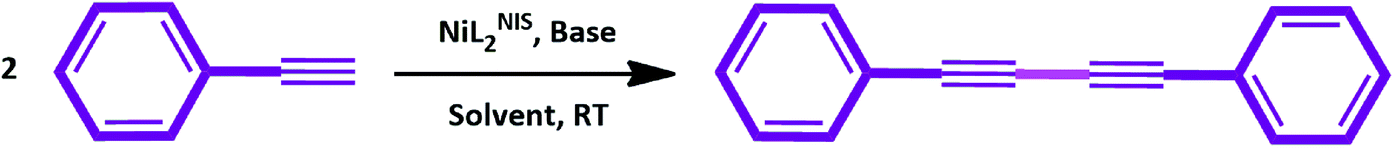 | ||
| Scheme 5 Catalytic activity of NiIIL2NIS in homocoupling of phenyl acetylene. | ||
| Entry | Base | Solvent | Catalyst | Time (h) | Conversion (%) |
|---|---|---|---|---|---|
| 1 | Cs2CO3 | THF | 2 mol% | 4 | 100 |
| 2 | Cs2CO3 | Acetonitrile | 2 mol% | 4 | 80 |
| 3 | Cs2CO3 | THF | 3 mol% | 2 | 100 |
| 4 | KOH | THF | 2 mol% | 4 | 90 |
| 5 | KOH | Acetonitrile | 2 mol% | 6 | 70 |
| 6 | KOH | Toluene | 2 mol% | 6 | 50 |
| 7 | KOH | ETOH | 2 mol% | 5 | 50 |
| 8 | KOH | THF | 3 mol% | 2 | 100 |
| 9 | K2CO3 | THF | 2 mol% | 6 | 75 |
| 10 | Na2CO3 | THF | 2 mol% | 6 | 70 |
It considers that THF affects the stability of activated complex in the rate determining step of the reaction. Our results show that both KOH and Cs2CO3 are effective bases, and KOH was chosen due to its availability and lower price. The amount of catalyst was investigated and 3 mol% of NiIIL2NIS was used as optimized value. The homocoupling reaction time was a short time of 2 h.
Table 3 summarizes the results of the homocoupling of phenyl acetylene under various reaction conditions. These data reveal that (3 mol% NiIIL2NIS/2 mmol KOH/in THF) seemed to be the most suitable for gaining excellent yields of the desired product and gives the most satisfactory yields of biphenyl products. After finding optimized reaction conditions, the scope of this catalytic coupling approach was explored with various substituted phenyl acetylenes (Table 4). All phenyl acetylenes underwent oxidation to desired biphenyl compounds in excellent yields. It considers that the substituent on the phenyl acetylene doesn't considerably affect the coupling reaction yield.
| Substrate | Product | CNV (%) |
|---|---|---|
| a Reaction condition: NiIIL2NIS (3 mol%), phenyl acetylene derivates (1 mmol), KOH (2 mmol), THF (3 mL), time (2 h).b Reaction condition: catalyst without CN group ([NiII(LISQ)2]),64 phenyl acetylene (1 mmol), KOH (2 mmol), THF (3 mL), time (3.5 h). | ||
 |
 |
100a |
 |
 |
100a |
 |
 |
100a |
 |
 |
100a |
 |
 |
100a |
 |
 |
94a |
 |
 |
80b |
It's worth to say that the comparison of the reaction yield and time of NiIIL2NIS with NiII(LISQ)2 (ref. 64) show a better activity of the NiIIL2NIS complex with cyanide group. The effect of cyanide will be discussed in the mechanism and theoretical parts.
The effect of ligand and complex system was investigated by the blank tests with Ni(II) acetate/KOH and H2L/Ni(II) acetate/KOH catalytic systems respectively and low yield of product was achieved in both cases (Table 5). These results demonstrate that the metal–ligand cooperation in the structure of the complex has an important role in the catalytic activity of NiIIL2NIS in the homocoupling reaction (Table 5).
| Entry | Catalytic system | Time (h) | Conversion (%) |
|---|---|---|---|
| 1 | Ni(OAc)2/KOH | 10 | 25 |
| 2 | Ni(OAc)2/H2LKOH | 10 | 35 |
The activity of synthesized catalyst was compared with other catalysts which had been recommended in the literature. Table 6 (entries 1–9) exhibits clearly that the catalyst NiIIL2NIS has much more activity than others. Nickel-catalyzed homocoupling reactions have been developed considerably.
| Entry | Catalyst | Solvent/T (°C) | Time (h) | Yield (%) | Additive | Ref., year |
|---|---|---|---|---|---|---|
| a Ni-microwave induced nanotubes. | ||||||
| 1 | Ni(CO)4 lithium phenylacetylide | THF/−30 | 15 | 48 | I2–MeOH | Ref. 55, 1969 |
| 2 | [Ni(PPh3)2Cl2] PhBr | DMF/50 | 20 | 28–89 | Ph3P Zn | Ref. 44, 1997 |
| 3 | [Ni(PMe3)2Cl2] lithium acetylide | THF/20 | 1 | 90 | PMe3 | Ref. 65, 1989 |
| 4 | [Ni(PPh3)2Cl2] phenylethynyllithium | THF/reflux | 12 | 38 | PMe3 | Ref. 66, 1994 |
| 5 | NiCl2·6H2O phenylacetylene | THF/90 | 20 | 93 | Cu I TMEDA | Ref. 48, 2008 |
| 6 | Acyclic bipyridyl NiCl2 phenylacetylene | THF/78 | 99 | n-BuLi | Ref. 45, 2010 | |
| 7 | Ni-MINTa | MW(140–170) | 15–45 min | 89–95 | DABCO | Ref. 42, 2012 |
| 8 | (H)2Phen(Me)2.NiCl2 2-iodohex-1-ene | DMF/RT | 1 | 91 | Zr(cp)2Cl2 Mn–LiCl | Ref. 43, 2011 |
| 9 | [Ni(dppe)Cl2] | DCE/RT | 4 | 97 | Ag2O | Ref. 47, 2015 |
| 10 | NiIIL2NIS phenyl acetylene | THF/RT | 2 | 95 | — | This work |
Comparison of catalytic properties of all complexes in Table 6 (entries 1–10) led us to the novel property of NiIIL2NIS catalyst. It's worth to say that, classical coupling protocols employ palladium or nickel catalysis concept that typically requires Pd(0) or Ni(0) catalysts or complexes of Ni(II) which can be regenerated, activated or ‘switched-on’ by the addition of stoichiometric metallic reducing agents, particularly zinc(0), manganese(0), Cu(I) and Ag(I) salts, organometallic complexes or any kind of additive such as base, I2 or PPh3. In recent years, scientist's effort led to improve the reaction yield, kinetic of the reaction, loading of catalyst, decreasing the amount of waste material such as metal and organic waste and by-products produced by traditional methods.43–45,47,48,55,65,66
In our contribution in these efforts, we have been tried to develop innovate and greener protocols. We haven't used any reducing agents or additives due to the presence of a redox active non innocent ligand which plays a crucial role in switching the oxidation state of complex and regenerate it in the time of oxidative addition/reductive elimination processes.
To the best of our knowledge, this is the first report of a Ni(II) complex without using it as bimetallic systems of Ni(II)/M(Zn(0)), Mn(0), Cu(I), Ag(I) and an organometal complex or any kind of additive such as base, I2 or PPh3. This phenomenon confirms the fact that non-innocent o-aminophenol ligand acts as an “electron reservoir” and can accept and donate electrons in catalytic cycle reversibly. Therefore, the synthesized catalyst can handle the reaction progress better than other reported nickel catalysts under mild conditions.
Based on our observations, we proposed a plausible mechanistic pathway for this reaction (Scheme 6). In the first step phenyl acetylene has been deprotonated by KOH to phenyl acetylide. Then this species will be coordinated to Ni(II) center and one of the non-innocent iminosemiquinone LNIS ligands undergoes changes in oxidation state and iminosemiquinonate/iminoquinonate complex of [NiIILNISLNIQ]+ is achieved to keep the total oxidation state of the complex unchanged. Then the resulted complex is coordinated with the second phenyl acetylide species and forms four coordinate [NiIIL2NIQ]2+ complex with two acetylide species occupied 5th and 6th positions which return to the first NiIIL2NIS complex via reductive elimination.
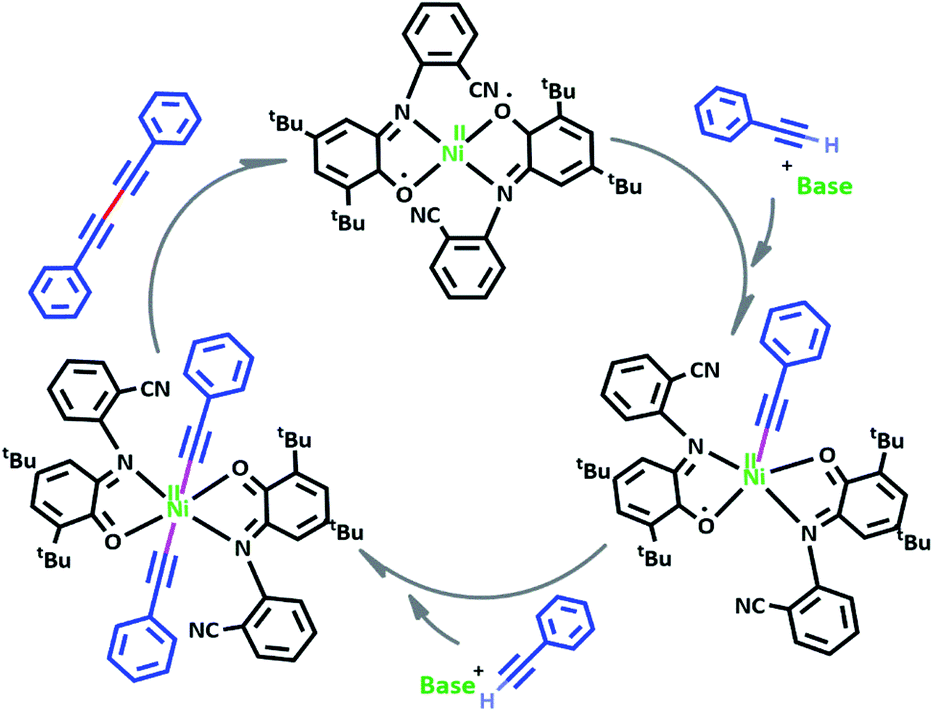 | ||
| Scheme 6 Plausible reaction mechanism. | ||
With considering this possible mechanism and comparing the Table 5 results (entries 1 and 2 for NiIIL2NIS and NiII(LISQ)2 complexes, respectively), we can see the effect of cyanide group on the more favorable addition of acetylide species to the Ni(II) center in NiIIL2NIS complex. In addition based on electrochemical results, the possible faster changing and tuning of ligand oxidation states of [NiIIL2NIQ]2+ intermediate to the primary complex after reductive elimination makes NiIIL2NIS more efficient catalyst. The quantitative description of cyanide effect on this process based on kinetic stability is discussed in the next part.
Computational details
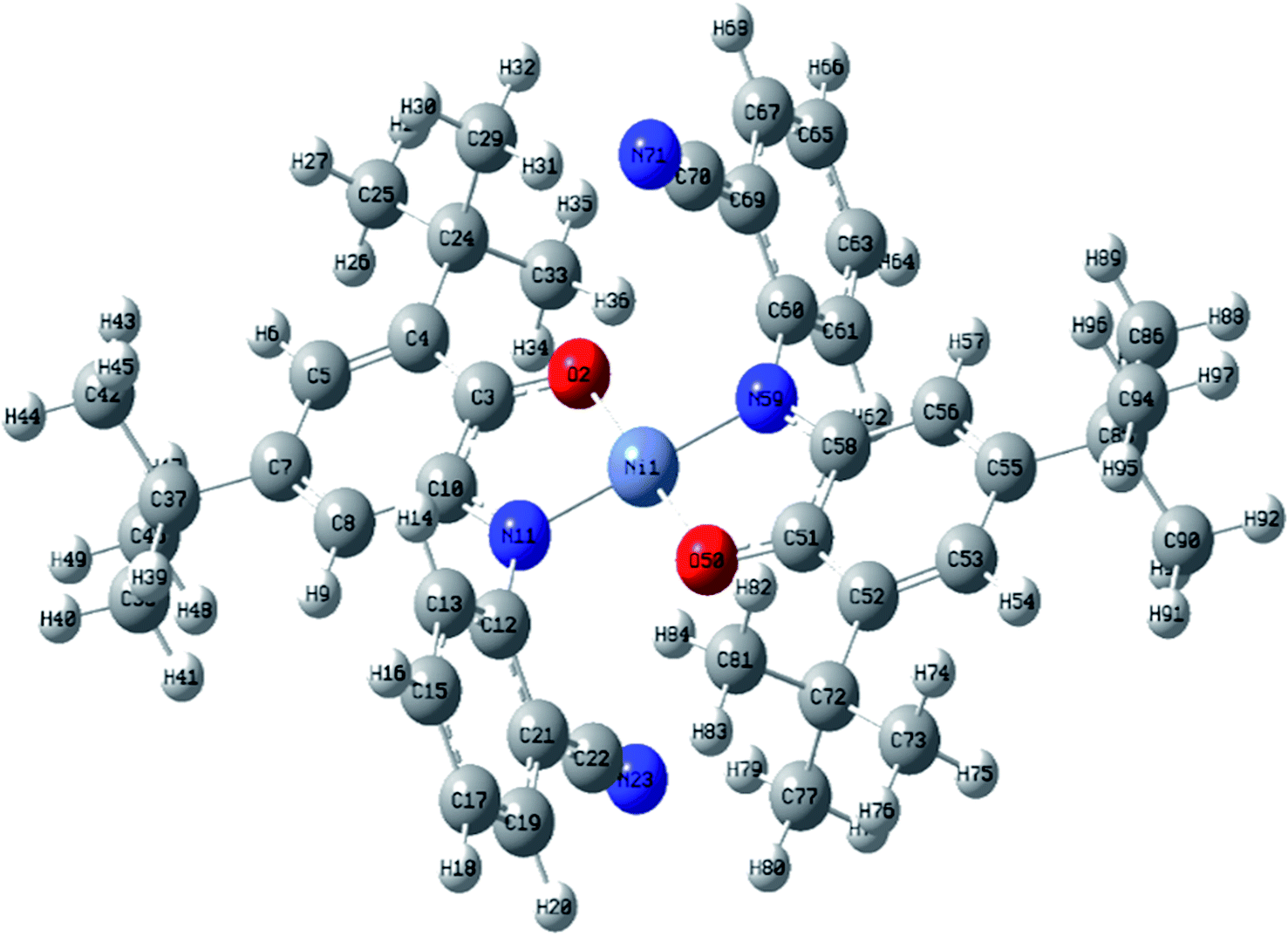 | ||
| Fig. 5 The Optimized structure of catalyst, NiIIL2NIS. Selected bond lengths [Å] and angles [°]: Ni(1)–O(2) 1.83, Ni(1)–N(11) 1.84, C(3)–C(4) 1.43, C(4)–C(5) 1.38, C(7)–C(8) 1.38, C(8)–C(10) 1.42, O(2)–C(3) 1.30, C(10)–N(11) 1.35, C(21)–C(22) 1.44, C(22)–N(23) 1.16, O(2)–Ni(1)–O(50) 180.0, O(2)–Ni(1)–N(11) 85.75, O(50)–Ni(1)–N(11) 94.25, C(3)–O(2)–Ni(1) 113.75. | ||
| Eexc (ev) | TD-DFT λ (nm) | Osc. strength (f) | Key transitions | Character | Exp. λ (nm) |
|---|---|---|---|---|---|
| 1.11 | 1114 | 0.5327 | (72%) HOMO → LUMO | Ni(d)/L(π) → L(π*) | |
| 1.94 | 640 | 0.0039 | (60%) HOMO → LUMO+1 | Ni(d)/L(π) → L(π*) | 600 |
| 2.45 | 507 | 0.0896 | (70%) HOMO−2 → LUMO | L(π) → Ni(d)/L(π*) | |
| 3.08 | 402 | 0.0335 | (68%) HOMO−3 → LUMO | L(π) → L(π*) | 395 |
| 3.72 | 333 | 0.0947 | (62%) HOMO → LUMO+3 | Ni(d)/L(π) → L(π*) | 313 |
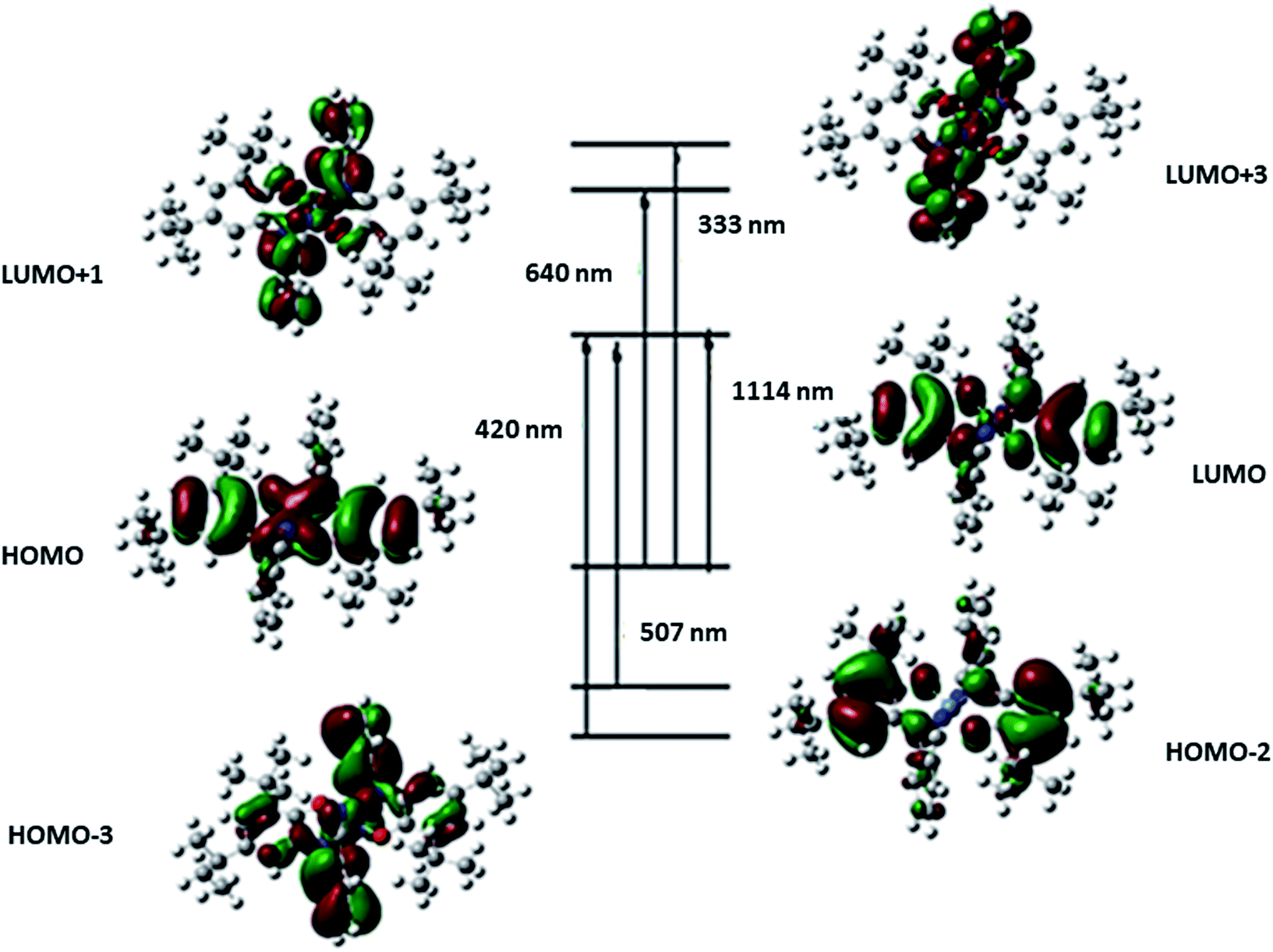 | ||
| Fig. 6 Molecular orbitals corresponding to TD-DFT excitations for catalyst NiIIL2NIS. | ||
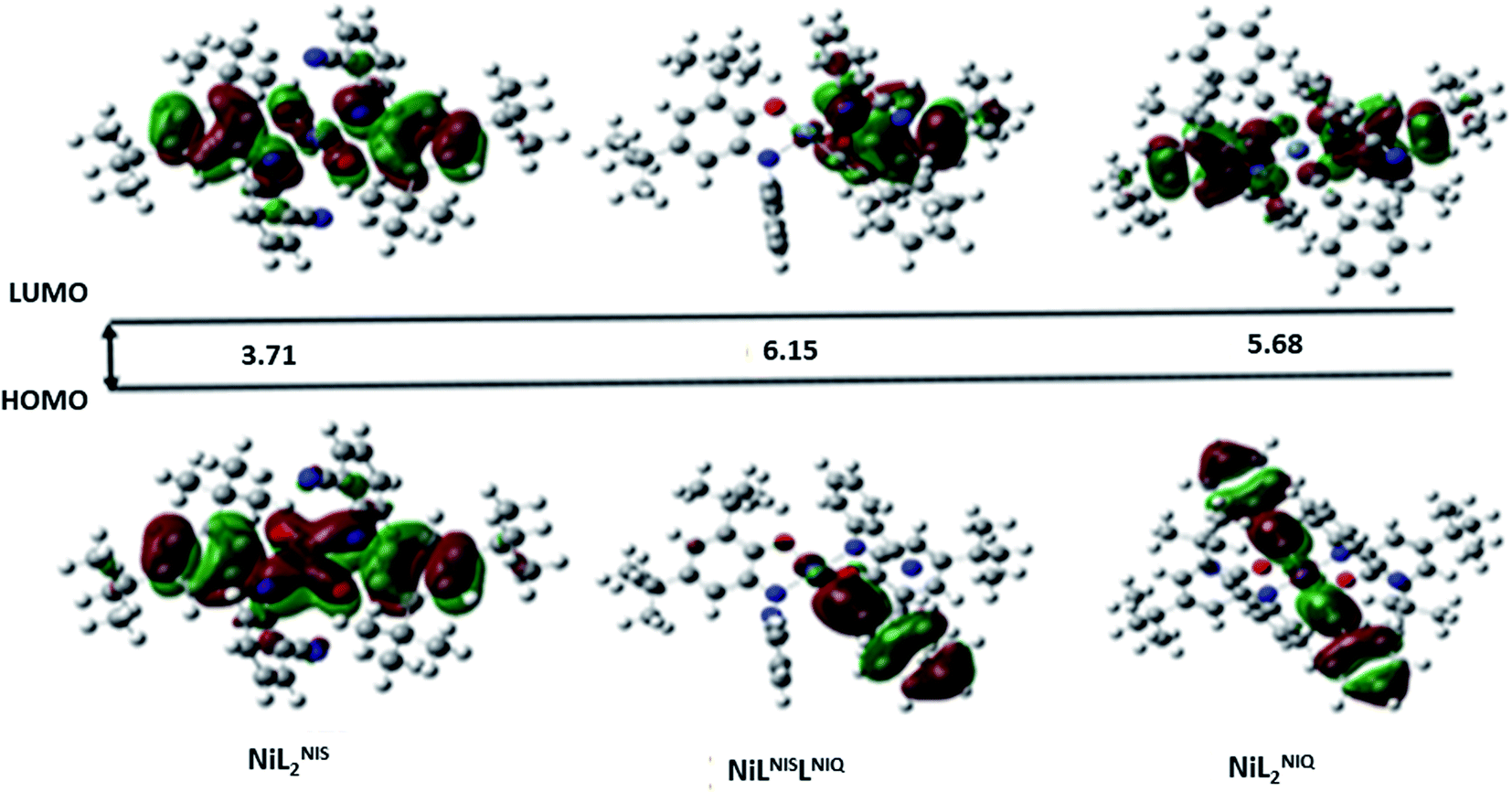 | ||
| Fig. 7 Schematic representation of the calculated frontier molecular orbitals (FMOs) of the species involved in the proposed catalytic cycle with their energy levels (ev) in solution (Tetrahydrofuran (THF)). | ||
As presented in Scheme 7, the suggested mechanism for phenyl acetylene homocoupling reaction includes three steps. The first step is the addition of one phenyl acetylate ligand in alkaline media to Ni(II) center to form NiIILNISLNIQ complex. In other words, the oxidation state of one of the non-innocent o-iminobenzosemiquinonate (ISQ−) ligands in catalyst, NiIIL2NIS, changes and converts to o-iminobenzoquinone (IBQ) ligand in NiIILNISLNIQ complex. In the next step, second phenyl acetylide ligand can be coordinated to NiIILNISLNIQ to form NiIIL2NIQ complex. The optimized structure of NiIIL2NIQ complex in the solution indicated that two phenyl acetylate ligands are coordinated with Ni(II) in trans isomer form and both non-innocent o-aminophenol ligands are in o-iminobenzoquinone (IBQ) oxidation state. Our calculation shows that the cis isomer in THF solution is not stationary point (the cis structure is unstable structure). Finally, NiIIL2NIQ complex undergoes a homocoupling reaction to form biphenyl acetylene through the corresponding saddle point, TS structure, with an imaginary frequency along the reaction coordinate as shown in Scheme 7 and regenerating catalyst, NiIIL2NIS, to restart the cycle. Fig. 7 shows that the HOMO and LUMO of NiIILNISLNIQ and NiIIL2NIQ are primarily located on phenyl acetylide and o-aminophenol ligands, respectively. It is worth mentioning that the binding of phenyl acetylide ligand to Ni(II) center form MOs which have phenyl acetylide character, and they are better electron acceptors in comparison with MOs of uncoordinated phenyl acetylide. The DFT calculations were also used to calculate the distribution of electrostatic potential (EPS) for all complexes presented in the catalytic cycle in THF (Fig. 8).
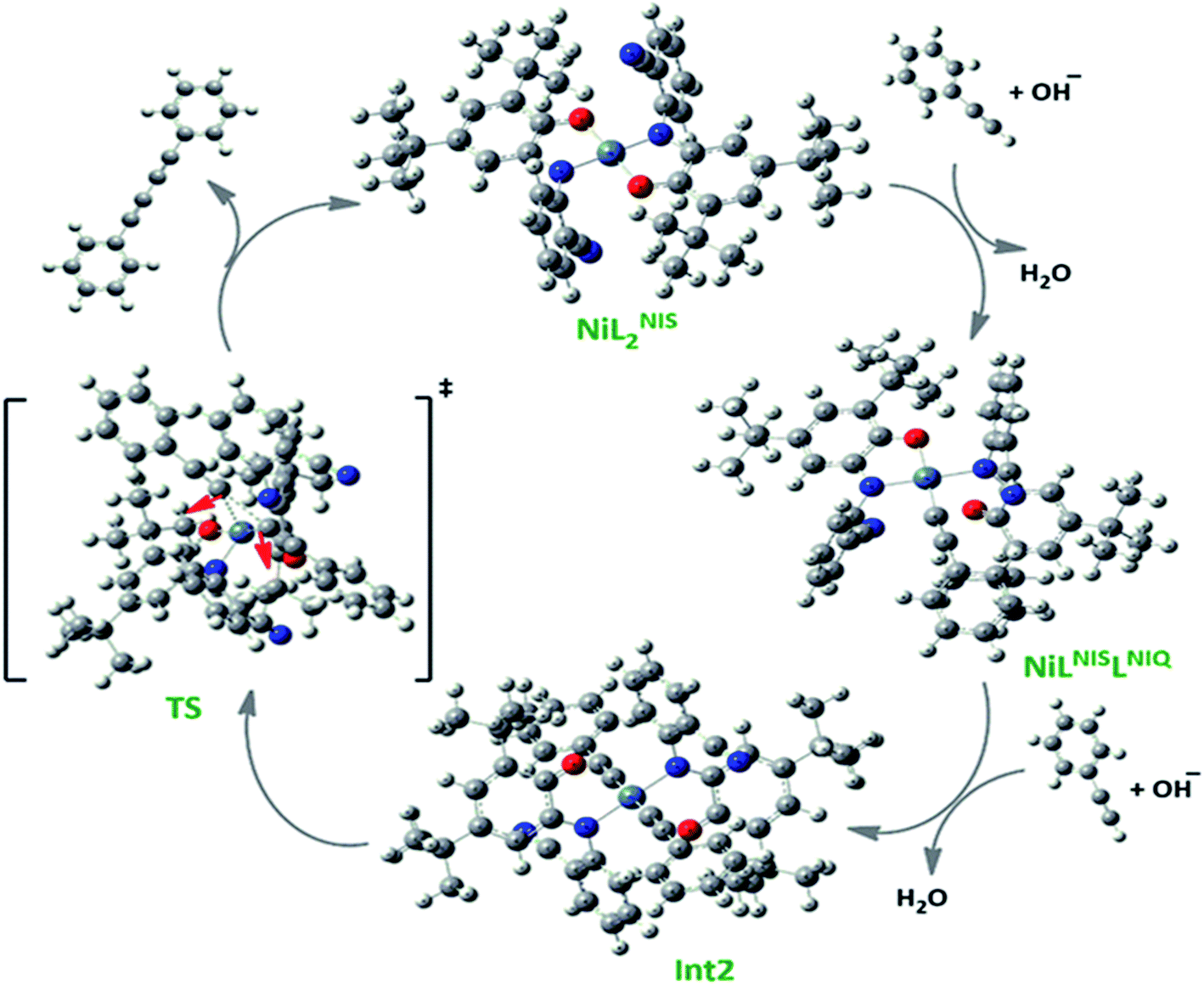 | ||
| Scheme 7 The optimized structures of the species involved in the catalytic cycle in the solution phase (THF). | ||
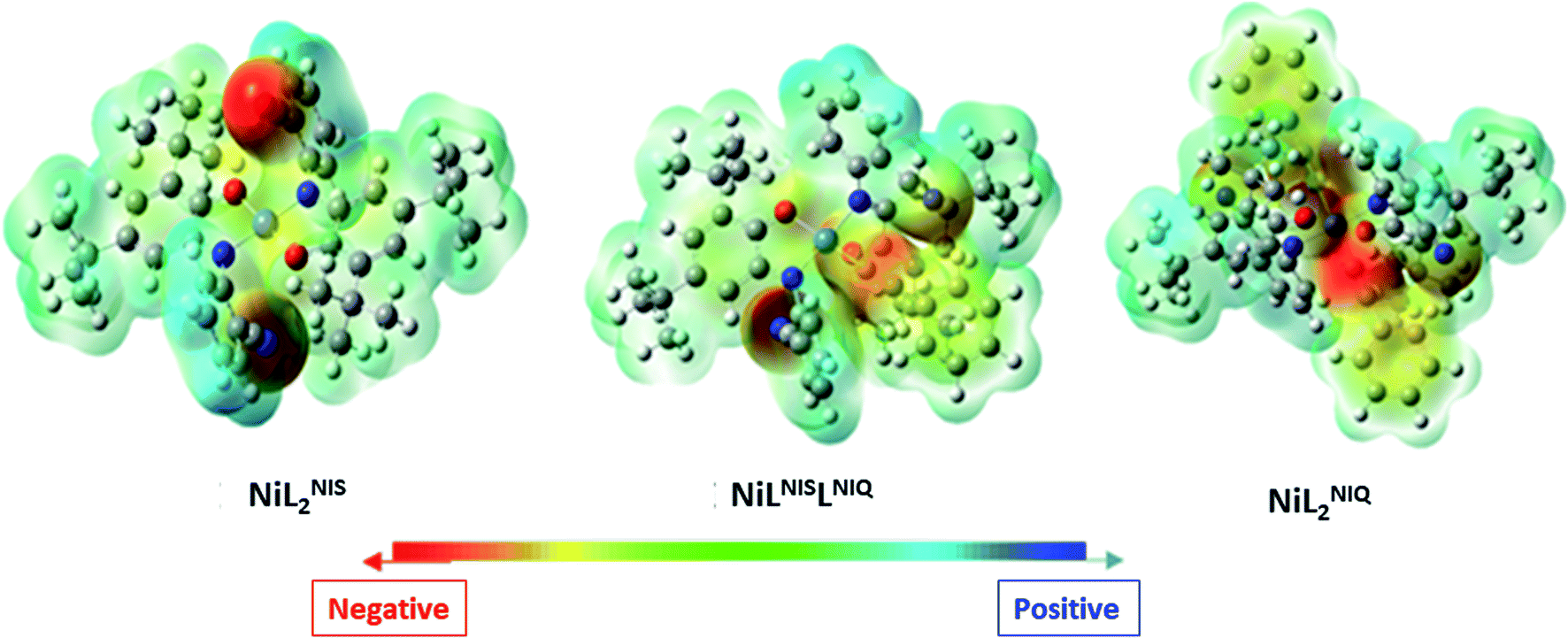 | ||
| Fig. 8 Distributions of electrostatic potential (ESP) for different species (isovalue = 0.0004) in solution (Tetrahydrofuran (THF)). | ||
Distributions of ESP for all species are consistent with the distribution of their FMOs, and confirm again the proposed mechanism for the catalytic homocoupling reaction. As shown in Fig. 8, when the catalyst undergoes two successive phenyl acetylide additions to produce NiIIL2NIQ, electronic density is distributed on the phenyl acetylide ligand and the electrostatic potential is negative on two phenyl acetylide ligands.
Red arrows in TS structure show the imaginary vibrational mode of TS along the reaction coordinate kinetic stability of the intermediates (stability with respect to the activated complex) has a crucial influence on the catalytic cycles. The HOMO–LUMO energy separation has been used as a simple indicator of kinetics stability.67 For a compound with large HOMO–LUMO gap is energetically unfavourable to extract electron from a low-lying HOMO or to add electron to a high-lying LUMO, and then to form transition state. Therefore, this compound has high kinetic stability and low chemical reactivity.68 In our catalytic cycle, NiIIL2NIQ complex is one of the reaction intermediates which undergoes a homocoupling reaction via TS structure to form biphenyl acetylene product. In order to better evaluate the efficiency of the catalyst for the homocoupling reaction, the energy of molecular orbitals for the NiIIL2NIQ complex with and without cyanide group of NiII(LISQ)2 (ref. 64) and with methoxy group of NiIIL2OMe in the non-innocent o-iminobenzosemiquinonate (ISQ−) ligand were calculated. HOMO–LUMO energy separation were 5.68 eV, 6.06 eV and 5.88 eV for intermediate NiIIL2NIQ complex with and without cyanide group and with methoxy group, respectively. According to these results, NiIIL2NIQ complex has lower kinetic stability and therefore, has higher chemical reactivity than the others. The Gibbs free energy changes for the formation of biphenyl acetylene product from the intermediate NiIIL2NIQ complexes with and two other mentioned complexes were also calculated using DFT calculation. The Gibbs free energy changes for the reaction were −70.67 kcal mol−1, −49.70 kcal mol−1 and −49.69 kcal mol−1 for NiIIL2NIS, NiII(LISQ)2 and NiIIL2OMe, respectively (Table 8). These results are in consistence with experimental results (Table 8) and demonstrate that the NiIILNIS is also, thermodynamically more desirable than the others.
| HOMO (Hartree) | LUMO (Hartree) | Bandgap (ev) | |
|---|---|---|---|
| NiIIL2NIS | −0.22849 | −0.09202 | 3.713349 |
| NiII(LISQ)2 | −0.22288 | −0.08449 | 3.765592 |
| NiIIL2OMe | −0.21924 | −0.08081 | 3.76668 |
Experimental
Materials and methods
All reagents and solvents were acquired from commercial companies (Merck, Sigma-Aldrich, Acros, and Fluka) and used as supplied, except those for electrochemical measurements. Elemental analysis (CHN) were carried out on Thermo Finnigan-Flash 1200. IR spectra were recorded on a FT-IR Bruker Vector spectrophotometer. Electronic spectra were recorded on a Cary 5000 spectrophotometer. Cyclic voltammetry (CV) was made with a computer controlled electrochemical system (Metrohm Autolab B.V., Utrecht, the Netherlands) with a NOVA 1.7 software equipped with an Ag wire reference electrode, a glassy carbon working electrode, and a Pt counter electrode with 0.1 M NBu4ClO4 solutions in CH2Cl2. Ferrocene was used as an internal standard. Magnetic measurements were obtained from powder samples of solid material in the temperature range between 2–290 K by using a Quantum Design SQUID magnetometer MPMS-7. 1H and 15N NMR spectra of NiIILNIS were collected with Bruker 700 spectrometer (700 MHz) in CDCl3, reference CH3NO2.Synthesis of H2LNAP
Ligand was synthesized according to the literature.56 A mixture of 3,5-di-tert-butylcatechol (0.222 g, 1 mmol), 2-aminobenzonitrile (0.118 g, 1 mmol) and 0.005 mL triethylamine in n-hexane (4 mL), was refluxed for two days and yellow color solution was obtained that turned red upon stirring at room temperature (30 °C) for four days. H2LNAP, a colorless crystalline solid appeared from recrystallization of a 1:1 CH2Cl2:MeOH solvent mixture. Yield: 2.18 g (67.7%). νmax(KBr)/cm−1: 3421 (O–H), 3354 (N–H), 2222 (–CN) (Fig. S1†).
Synthesis of NiIIL2NIS
Ni(OAc)2, (0.088 g, 0.5 mmol) was added to a mixture of H2LNAP (0.162 g, 0.5 mmol) and triethylamine in acetonitrile and stirred for three hours at room temperature. Dark green needle type crystals suitable for single crystal diffraction were formed after two days. Anal. calcd (found) for C42H48N4O2Ni: C 69.88 (69.93), H 7.28 (7.41), N 7.49 (7.41). νmax(KBr)/cm−1: 1257 (C–O), 2954, 2916 and 2869 (tertbutyl groups), 2221 (–CN) (Fig. S2†). 1H NMR (700 MHz, DMSO-d6) δ 1.010 (s, 11H), 1.081 (s, 9H), 6.213–6.215 (d, 1H), 6.904–6.906 (d, 1H), 7.493–7.516 (dt, 1H), 7.446–7.453 (dt, 1H), 7.337–7.348 (d, 1H), 7.769–7.782 (dd, 1H). 15N NMR (700 MHz, CH3NO2) δ −196 ((–CN)) −315 (–CN) (Fig. S3 and S4†).
General procedure for determination the catalytic activity of NiIIL2NIS complex in homocoupling reactions
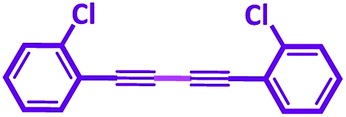
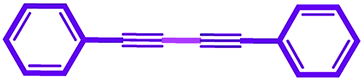
In a typical approach a mixture of phenyl acetylene (1 mmol), base (2 mmol) and NiIIL2NIS (0.017 g, 3 mol%) were stirred at room temperature in solvent (3 mL). The reaction progress was monitored by TLC and gas chromatography. After completion of the reaction, the product was extracted using the column chromatography (n-hexane/ethyl acetate (5:1)) and then characterized by 1H NMR and 13C NMR (Fig. S3–S6†). 1,4-Diphenyl buta-1,3-diyne: 1H NMR (400 MHz, DMSO-d6) δ 7.69–7.57 (m, 4H), 7.55–7.40 (m, 6H), 13C NMR (101 MHz, DMSO) δ 73.95, 82.29, 120.85, 129.40, 130.51, 132.88. 1,4-Di(pyridin-2-yl)buta-1,3-diyne: 1H NMR (400 MHz, DMSO-d6) δ 8.66 (ddd, J = 4.86, 1.76, 0.98 Hz, 2H), 7.91 (td, J = 7.75, 1.80 Hz, 2H), 7.79 (dt, J = 7.85, 1.13 Hz, 2H), 7.52 (ddd, J = 7.65, 4.82, 1.23 Hz, 2H), 13C NMR (101 MHz, DMSO) δ 72.18, 81.83, 125.38, 129.28, 137.55, 140.81, 151.11. 1,4-Bis(p-fluorophenyl)buta-1,3-diyne: 1H NMR (400 MHz, DMSO-d6) δ 7.94–7.57 (m, 4H), 7.46–7.02 (m, 4H), 13C NMR (101 MHz, DMSO) δ 73.65, 81.22, 116.71, 116.93, 135.43, 135.52, 161.95. 1,4-Bis(p-tolyl)buta-1,3-diyne: 1H NMR (400 MHz, DMSO d6) δ 7.50 (d, J = 7.68 Hz, 4H), 7.26 (d, J = 7.75 Hz, 4H), 2.35 (s, 6H), 13C NMR (101 MHz, DMSO) δ 21.65, 73.62, 82.32, 117.94, 130.03, 132.77, 140.52. 1,4-Bis(4-methoxyphenyl)buta-1,3-diyne: 1H NMR (400 MHz, DMSO-d6) δ 7.76–7.40 (m, 4H), 7.04–6.86 (m, 4H), 3.81 (s, 6H), 13C NMR (101 MHz, DMSO) δ 73.95, 82.29, 120.85, 129.40, 130.51, 132.88. 1,4-Bis(2-chlorophenyl)buta-1,3-diyne: 1H NMR (400 MHz, DMSO-d6) δ 7.79 (dd, J = 7.67, 1.67 Hz, 2H), 7.64 (dd, J = 8.16, 1.19 Hz, 2H), 7.53 (td, J = 7.75, 1.67 Hz, 2H), 7.44 (td, J = 7.59, 1.26 Hz, 2H), 13C NMR (101 MHz, DMSO) δ 55.40, 78.02, 120.56, 128.12, 130.09, 132.28, 135.26, 136.15.69–73
X-ray analysis
Crystals of NiIIL2NIS were grown from the CH3OH–CH2Cl2 solution The X-ray diffraction data were collected for a plate crystal with an Oxford Sapphire CCD diffractometer using MoKα radiation λ = 0.71073 Å, at 292(2) K, by ω–2θ method. Structure was solved by the direct methods and refined with the full-matrix least-squares method on F2 with the use of SHELX2017 program package.74,75 Analytical absorption correction applied (CrysAlisPro 1.171.38.43)76 the minimum and maximum transmission of 0.939 and 0.807. Hydrogen atoms were located from the electron density maps and their positions were constrained in the refinement. The structural data have been deposited with Cambridge Crystallographic Data Centre, the CCDC number 1883981.Computational details
Geometry optimizations were performed using the Gaussian 16 computational package.77 The density functional theory (DFT) calculations were carried out at WB97XD78 level of theory, the functional from Head-Gordon and coworkers which includes empirical dispersion, using LANL2DZ pseudo-potential basis set for Ni atom and 6-31g(d,p) for the other atoms. Frequency calculations at the same level of theory have been performed to identify the nature of all the stationary points (local minima or first-order saddle point). All structures were optimized in tetrahydrofuran (THF) solvent using the polarized continuum model (PCM)79 for modeling the solvent. Further, the energy and shape of the frontier molecular orbitals (FMOs) and electrostatic potential surfaces (ESPs) for all species involved in the catalytic cycle were calculated at the same level of theory. The intensities of the 40 lowest-energy electronic transitions of NiIIL2NIS complex were calculated by TD-DFT with a polarized continuum model (PCM) for CH2Cl2 (dielectric ε = 8.94) solvent.Conclusions
A new nickel complex of tridentate o-aminophenol ligand HLNAP was synthesized and characterized. It is evident from X-ray crystallography analysis that NiIIL2NIS exists as a square-planar geometry with bi-dentate LNIS ligand. Electrochemical studies were conducted to evaluate the redox-active behavior of the complex, and the results demonstrated two quasi reversible ligand-centered oxidation processes for this complex. The UV-Vis spectroscopy of NiIIL2NIS shows both intra-ligand and ligand-to-metal charge transfers. Finally, the catalytic activity of this nickel complex has been investigated for C–H activation and homocoupling of various terminal alkynes.We have reported the homocoupling of terminal alkynes to give rise the corresponding 1,3 diene with different substituents. The good to excellent reactivity, low catalyst amount, good reaction time under mild conditions are the most noteworthy of this project.
This is the first report of investigated metal free (e.g. Cu, Zn powder or Cu(I), Ag(I)) NiII complex without using any additive which revealed catalytic performance in this kind of coupling reaction due to the ability of ligand for switching its oxidation state from iminobenzosemiquinone to iminobenzoquinone and vice versa. In addition, our results show that electron-withdrawing substituent of cyanide on the phenol ring has good effect on none innocence character of ligand and consequently thermodynamic and kinetic control of the coupling reaction. Our theoretical results confirm the effective role of CN on the tuning of ligand redox potential and kinetic stability of intermediates in the coupling reaction.
Conflicts of interest
There is not any conflict of interest.Acknowledgements
Authors are grateful to Shiraz and Nicolaus Copernicus Universities. Special thanks to Dr Echard Bill and Prof. John F. Berry for their valuable help.References
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed.
- K. P. Butin, E. K. Beloglazkina and N. V. Zyk, Russ. Chem. Rev., 2005, 74, 531–553 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- M. J. Reinhardt, PhD thesis, The University of Edinburgh, 2013.
- B. Butschke, K. L. Fillman, T. Bendikov, L. J. Shimon, Y. Diskin-Posner, G. Leitus, S. I. Gorelsky, M. L. Neidig and D. Milstein, Inorg. Chem., 2015, 54, 4909–4926 CrossRef CAS PubMed.
- D. L. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- B. de Bruin, P. Gualco and N. D. Paul, Ligand Design in Metal Chemistry: Reactivity and Catalysis, John Wiley & Sons, 2016 Search PubMed.
- S. C. Bart, K. Chłopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901–13912 CrossRef CAS PubMed.
- K. J. Blackmore, N. Lal, J. W. Ziller and A. F. Heyduk, J. Am. Chem. Soc., 2008, 130, 2728–2729 CrossRef CAS PubMed.
- H. Grützmacher, Angew. Chem., 2008, 47, 1814–1818 CrossRef PubMed.
- C. J. Rolle III, K. I. Hardcastle and J. D. Soper, Inorg. Chem., 2008, 47, 1892–1894 CrossRef PubMed.
- P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed.
- D. Zhu, I. Thapa, I. Korobkov, S. Gambarotta and P. H. Budzelaar, Inorg. Chem., 2011, 50, 9879–9887 CrossRef CAS PubMed.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers and P. Pulay, J. Am. Chem. Soc., 2006, 128, 13175–13183 CrossRef CAS PubMed.
- S. Kokatam, The Coordination Chemistry of Redox Noninnocent O-aminophenol and Dithiolene Ligands with Transition Metal Ions, 2006 Search PubMed.
- S. E. Balaghi, E. Safaei, L. Chiang, E. W. Wong, D. Savard, R. M. Clarke and T. Storr, Dalton Trans., 2013, 42, 6829–6839 RSC.
- S. E. Balaghi, E. Safaei, M. Rafiee and M. H. Kowsari, Polyhedron, 2012, 47, 94–103 CrossRef CAS.
- Z. Alaji, E. Safaei, L. Chiang, R. M. Clarke, C. Mu and T. Storr, Eur. J. Inorg. Chem., 2014, 2014, 6066–6074 CrossRef CAS.
- E. Safaei, S. E. Balaghi, L. Chiang, R. M. Clarke, M. Webb, E. W. Y. Wong, D. Savard, C. J. Walsby and T. Storr, Dalton Trans., 2019, 48, 13326–13336 RSC.
- Z. Alaji, E. Safaei, H. Yi, H. Cong, A. Wojtczakd and A. Lei, Dalton Trans., 2018, 47, 15293–15297 RSC.
- E. Safaei, H. Bahrami, A. Pevec, B. Kozlevčar and Z. Jagličić, J. Mol. Struct., 2017, 1133, 526–533 CrossRef CAS.
- N. Leconte, J. Moutet, T. Constantin, F. Molton, C. Philouze and F. Thomas, Eur. J. Inorg. Chem., 2018, 2018, 1752–1761 CrossRef CAS.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS PubMed.
- X. H. FanandL and M. Yang, Eur. J. Org. Chem., 2011, 2011, 1467–1471 CrossRef.
- M. Wang, X. Yuan, H. Li, L. Ren, Z. Sun, Y. Hou and W. Chu, Catal. Commun., 2015, 58, 154–157 CrossRef CAS.
- M. Wang, X. Yuan, H. Li, L. Ren, Z. Sun, Y. Hou and W. Chu, Catal. Commun., 2015, 58, 154–157 CrossRef CAS.
- C. Chen and L.-M. Yang, Tetrahedron Lett., 2007, 48, 2427–2430 CrossRef CAS.
- X. H. Fan and L. M. Yang, Eur. J. Org. Chem., 2011, 8, 1467–1471 CrossRef.
- V. Percec, J.-Y. Bae and D. H. Hill, J. Org. Chem., 1995, 60, 1060–1065 CrossRef CAS.
- J. C. Galland, M. Savignac and J. P. Genêt, Tetrahedron Lett., 1999, 40, 2323–2326 CrossRef CAS.
- S. Saito, M. Sakai and N. Miyaura, Tetrahedron Lett., 1996, 37, 2993–2996 CrossRef CAS.
- S. Saito, S. Oh-tani and N. Miyaura, J. Org. Chem., 1997, 62, 8024–8030 CrossRef CAS PubMed.
- A. M. Oertel, V. Ritleng and M. J. Chetcuti, Organometallics, 2012, 31, 2829–2840 CrossRef CAS.
- T. Shimasaki, Y. Konno, M. Tobisu and N. Chatani, Org. Lett., 2009, 11, 4890–4892 CrossRef CAS PubMed.
- C. Chen and L. M. Yang, Tetrahedron Lett., 2007, 48, 2427–2430 CrossRef CAS.
- A. H. Christian, P. Müller and S. Monfette, Organometallics, 2014, 33, 2134–2137 CrossRef CAS.
- P. A. Payard, L. A. Perego, I. Ciofini and L. Grimaud, ACS Catal., 2018, 8, 4812–4823 CrossRef CAS.
- F. Alonso, P. Riente and M. Yus, Arkivoc, 2018, 4, 8–15 Search PubMed.
- S. Ghosh, A Mechanistic Insight into the Nickel-Catalyzed Homocoupling Reaction of Terminal Alkynes, 2018 Search PubMed.
- A. P. Bhat and B. R. Bhat, Appl. Organomet. Chem., 2014, 28, 383–388 CrossRef CAS.
- J. Montgomery and G. J. Sormunen, Nickel-catalyzed reductive couplings of aldehydes and alkynes, in Metal Catalyzed Reductive C–C Bond Formation, Springer, Berlin, Heidelberg, 2007 Search PubMed.
- F. Alonso and M. Yus, ACS Catal., 2012, 2, 1441–1451 CrossRef CAS.
- J. Peng, X. Liu and Y. Kishi, Tetrahedron Lett., 2011, 52, 2172–2175 CrossRef CAS.
- M. Zembayashi, K. Tamao, J. I. Yoshida and M. Kumada, Tetrahedron Lett., 1997, 18, 4089–4091 CrossRef.
- J. D. Crowley, S. M. Goldup, N. D. Gowans, D. A. Leigh, V. E. Ronaldson and A. M. Slawin, J. Am. Chem. Soc., 2010, 132, 6243–6248 CrossRef CAS PubMed.
- K. M. Miller, C. Molinaro and T. F. Jamison, Tetrahedron: Asymmetry, 2003, 14, 3619–3625 CrossRef CAS.
- Q. Chen, X. H. Fan, L. P. Zhang and L. M. Yang, Synth. Commun., 2015, 45, 824–830 CrossRef CAS.
- W. Yin, C. He, M. Chen, H. Zhang and A. Lei, Org. Lett., 2008, 11, 709–712 CrossRef PubMed.
- J. Montgomery, Angew. Chem., 2004, 43, 3890–3908 CrossRef CAS PubMed.
- E. Rajalakshmanan and V. Alexander, Synth. Commun., 2005, 35, 891–895 CrossRef CAS.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 CrossRef.
- G. Eglinton and A. R. Galbraith, Chem. Ind., 1956, 28, 736–737 Search PubMed.
- A. Hay, J. Org. Chem., 1960, 25, 1275–1276 CrossRef CAS.
- A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS.
- I. Rhee, M. Ryang and S. Tsutsumi, Tetrahedron Lett., 1969, 10, 4593–4596 CrossRef.
- S. Ghorai and C. Mukherjee, Chem. Commun., 2012, 48, 10180–10182 RSC.
- M. Nasibipour, E. Safaei, G. Wrzeszcz and A. Wojtczak, New J. Chem., 2020, 44, 4426–4439 RSC.
- J. I. van der Vlugt, Chem.–Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed.
- J. Jacquet, K. Cheaib, Y. Ren, H. Vezin, M. Orio, S. Blanchard, L. Fensterbank and M. D. El Murr, Chem.–Eur. J., 2017, 23, 15030–15034 CrossRef CAS PubMed.
- P. Chaudhuri, C. Nazari Verani, E. Bill, E. Bothe, T. Weyhermuller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS PubMed.
- A. I. Poddel’sky, V. K. Cherkasov and G. a. Abakumov, Coord. Chem. Rev., 2009, 253, 291–324 CrossRef.
- T. Karimpour, E. Safaei and B. Karimi, RSC Advances., 2019, 9, 14343–14351 RSC; T. Karimpour, E. Safaei and B. Karimi. Synfacts., 15, 0918 Search PubMed.
- A. I. Poddel’skya, I. V. Smolyaninovb, A. A. Skatovaa, A. N. Lukoyanova, G. K. Fukina, N. T. Berberovab, V. K. Cherkasova and G. A. Abakumova, Z. Anorg. Allg. Chem., 2008, 634, 1154–1160 CrossRef.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS PubMed.
- H.-F. Klein, H. Beck-Hemetsberger, L. Reitzel, B. Rodenhäuser and G. Cordier, Chem. Ber., 1989, 122, 43–51 CrossRef CAS.
- E. H. Smith and J. Whittall, Organometallics, 1994, 13, 5169–5179 CrossRef CAS.
- J. I. Aihara, J. Phys. Chem. A, 1999, 103, 7487–7495 CrossRef CAS.
- D. E. Manolopoulos, J. C. May and S. E. Down, Chem. Phys. Lett., 1991, 181, 105–111 CrossRef CAS.
- N. Devarajan, M. Karthik and P. Suresh, Org. Biomol. Chem., 2017, 15, 9191–9199 RSC.
- M. Guo, B. Chen, M. Lv, X. Zhou, Y. Wen and X. Shen, Molecules, 2016, 21, 606 CrossRef PubMed.
- L. Bettanin, G. V. Botteselle, M. Godoi and A. L. Braga, Green Chem. Lett. Rev., 2014, 7, 105–112 CrossRef.
- J. H. Li, Y. Liang and Y. X. Xie, J. Org. Chem., 2005, 70, 4393–4396 CrossRef CAS PubMed.
- S. N. Chen, W. Y. Wu and F. Y. Tsai, Green Chem., 2009, 11, 269–274 RSC.
- G. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- CrysAlisPro 1.171.38.43 software, Rigaku OD, 2015 Search PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision A. 03, 2016 Search PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem., 2008, 10, 6615–6620 CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1883981. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra04362a |
| This journal is © The Royal Society of Chemistry 2020 |