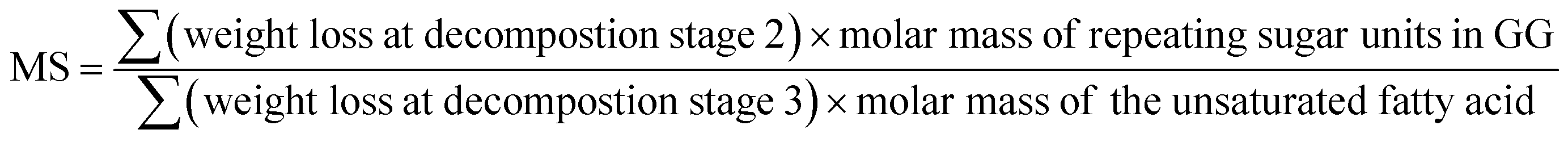DOI:
10.1039/D0RA04322B
(Paper)
RSC Adv., 2020,
10, 32050-32057
Rheological behaviours of guar gum derivatives with hydrophobic unsaturated long-chains†
Received
14th May 2020
, Accepted 13th August 2020
First published on 28th August 2020
Abstract
Three guar gum derivatives with hydrophobic unsaturated long-chains were successfully synthesized, including oleic guar gum (OGG), linoleic guar gum (LGG) and erucic guar gum (EGG). The guar gum derivatives were characterized by Fourier transform infrared (FTIR) spectroscopy and thermogravimetric analysis (TGA). Moreover, comparative studies on the rheological behaviours of guar gum (GG) and three guar gum derivatives were performed. Regardless of their hydrophobic side groups, all sample solutions showed the properties of non-Newtonian shear-thinning fluids, and their complex viscosities (η*) increased significantly with increasing concentration. In contrast to GG, OGG or EGG, LGG exhibited the strongest shear-thinning property when the concentration is 1 wt%. It was found that the frequency-dependent viscous (G′′) and elastic modulus (G′) curves of GG and its derivatives aqueous solutions cross at a specific frequency. This crossover frequency decreases with increasing sample concentration, indicating that the viscoelasticity is highly dependent on polymer concentration. In addition, when the molar substitution (MS) of the repeating sugar units in the guar gum derivatives is less than 0.08, the LGG solution showed more obvious solid-like rheological properties than the OGG and EGG solutions at the same concentration.
1 Introduction
The hydrophobic associated water-soluble polymers are a class of water-soluble polymers modified with a small molar percentage of hydrophobic groups. Due to their practical and fundamental importance, they are receiving increasing attention in academic and industrial laboratories.1–9 In an aqueous environment, the hydrophobic groups of these polymers aggregate to minimize their surface area exposed to the solvent, which leads to intermolecular or intramolecular association. Above a certain polymer concentration, the hydrophobic interactions between these molecules tend to form a three-dimensional network of polymer chains, resulting in a rapid increase in the hydrodynamic size of the polymer, thereby increasing the apparent viscosity.3,9–11 In addition, the hydrophobically associated water-soluble polymers exhibit interesting shear behaviour to avoid irreversible degradation of high molecular polymers under high shear stress, and also show high surface and interface activities. The combination of rheological behaviour, surface and interface activities of these polymers is of great significance, especially in a range of important commercial applications, such as super absorbency, enhanced oil recovery, flocculation, latex paints, industrial thickeners, controlled drug release, protein separations and biological/medical purposes.
Therefore, the synthesis and solution properties of hydrophobically associated water-soluble polymers have attracted more and more attention in the past few decades. In particular, natural polysaccharides have been widely used as the hydrophilic part of amphiphilic macromolecules because of their unique properties, for example, the ability to form polymeric liquid crystals or weak gel systems. Among them, guar gum is one of the important natural polysaccharides, in which the galactose side chains are connected to the mannose main chains with an average molecular ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.12 The solubility in cold and hot water and other unique characteristics, such as stabilizing, thickening, binding, emulsification, gelation, etc. make it suitable for various applications. And, these properties can be further adjusted through chemical modification to make it most suitable for specific applications.13–16 Central to many applications of both guar gum and its derivatives is the viscosity and rheological properties exhibited by its aqueous solution. Therefore, mastering the viscosity and rheological behaviour of guar gum and its various derivatives in aqueous solution is very important to meet the requirements of users.
:2.12 The solubility in cold and hot water and other unique characteristics, such as stabilizing, thickening, binding, emulsification, gelation, etc. make it suitable for various applications. And, these properties can be further adjusted through chemical modification to make it most suitable for specific applications.13–16 Central to many applications of both guar gum and its derivatives is the viscosity and rheological properties exhibited by its aqueous solution. Therefore, mastering the viscosity and rheological behaviour of guar gum and its various derivatives in aqueous solution is very important to meet the requirements of users.
So far, some studies have been performed to understand the viscosity characteristics of GG or its derivatives in solution.17,18 However, despite important industrial applications and related scientific interest, there is still a lack of research on the rheological behaviour of hydrophobized guar derivatives. In this work, three guar gum derivatives with hydrophobic unsaturated long-chains were successfully synthesized, including oleic guar gum (OGG), linoleic guar gum (LGG) and erucic guar gum (EGG). Furthermore, the rheological behaviour of three hydrophobic guar gum derivatives was systematically tested, in order to evaluate the influence of hydrophobic substituent on these hydrophobic guar gum derivatives under different polymer concentration and molar substitution of repeating sugar units.
2 Materials and methods
2.1. Materials
Guar gum (GG) was provided by the Jingkun Oilfield Chemistry Company, Jiangsu, China, which was purified by dissolving in water and precipitated from the solution by adding ethanol. Afterward, GG was extracted with ethanol using a Soxhlet extractor for one-week, and dried in a vacuum oven with phosphorus pentoxide for 24 h (25 °C). The Mw value of GG is 2.6 × 106 g mol−1, which is determined by gel permeation chromatography (GPC) using Shimadzu Instrument (ESI Fig. S1†). All other reagents were analytical pure and used without further purification.
2.2. Synthesis of oleic acyl chloride, linoleic acyl chloride and erucic acyl chloride
Oleic acid (8.46 g, 0.03 mol) was added into the flask with constant stirring at 35 °C and phosphorous trichloride (1.64 g, 0.012 mol) was continuously added dropwise into the reaction vessel with the dropping time controlled at 1.5–2 h. After the addition was completed, the mixture was heated to 65 °C and stirred at a stirring speed of 300 rpm for 6 h, using aqueous sodium hydroxide as the exhaust gas absorbent. When the reaction was completed, the reaction system was heated to 80 °C, and excess phosphorous trichloride was absorbed with an aqueous solution of sodium hydroxide until no bubbles appeared in the flask. The final yellow liquid was the mixture of the target oleic acyl chloride and a small amount of phosphorus trichloride, which was stored in a well-sealed dryer. Since oleic acyl chloride is easily deteriorated when the temperature is high, and a small amount of phosphorus trichloride can delay the above process, so phosphorus trichloride is retained when oleic acyl chloride is stored. If further purification is necessary, the mixture of oleic acyl chloride and phosphorus trichloride can be depressurized by vacuum pump for 10 min to remove phosphorus trichloride. The yield of the product is high as 99% verified by weighting method. Finally, the resulting product was characterized by infrared spectroscopy (FT-IR) to verify whether the reaction was successful.
The synthesis of linoleic acyl chloride and erucic acyl chloride was very similar to that of oleic acyl chloride, using linoleic acid and erucic acid as a substitute for oleic acid. In addition, the yields of the linoleic acyl chloride and erucic acyl chloride under such conditions were up to 99% and 85% (calculated by weighting method), respectively, implying this acid chlorination method was also suitable for other unsaturated fatty acids.
2.3. Synthesis of oleic guar gum (OGG), linoleic guar gum (LGG) and erucic guar gum (EGG)
A series of unsaturated fatty acid modified guar gum with different degrees of substitution were prepared by esterification. Take OGG as an example, the preparation process of unsaturated fatty acid modified guar gum was as follows: freshly distilled acetone (100 mL) and purified guar gum (10 g) were added into a four-necked flask equipped with a thermometer, mechanical stirrer, condenser and constant pressure funnel. A solution of oleic acyl chloride (0.1 g) in acetone (10 mL) was added dropwise to the above flask with the dropping time controlled at 1 h. During this process, a pH meter was used to monitor the reaction system, and the HCl generated by the reaction was timely neutralized with NaOH aqueous solution (1 M) to ensure the smooth progress of the reaction. The mixture was stirred at a constant speed (about 300 rpm using a magnet stirrer) at 50 °C for 12 h. After cooling, the mixture was filtered under reduced pressure and washed six times with acetone. The product was naturally dried for several hours and placed in a vacuum oven at room temperature for 24 h to obtain OGG.
The synthesis of LGG and EGG was very similar to that of OGG, using linoleic acyl chloride and erucic acyl chloride as a substitute for oleic acyl chloride.
2.4. Fourier transform infrared spectrometer analysis
The Fourier transform infrared (FTIR) spectra of the samples in KBr pellets were measured on a Thermo Nicolet 670 FT-IR spectrophotometer (USA). In 32 scans, the spectrum of each sample from 4000 to 400 cm−1 was collected at a resolution of 4 cm−1.
2.5. 1H NMR analysis
The purified guar gum, OGG, LGG and EGG samples (30 mg) were dissolved separately in D2O solution containing 10% DCl and hydrolyzed in an oil bath at 100 °C for 20 min. All 1H NMR analyses of samples were performed on a Bruker Avance III 600 MHz NMR spectrometer at 25 °C.
2.6. Thermogravimetric analysis
The thermogravimetric analysis (TGA) was carried out on a TA Instruments TGA Q50 thermal analyzer at a heating rate of 10 °C min−1 from 50 to 500 °C. All samples (approximately 3–8 mg) were examined under a nitrogen flow.
2.7. Rheological measurements
The rheological performance was measured on a HAAKE RS600 rheometer (Germany) with a coaxial cylinder sensor at 25 °C. After selecting the appropriate stress (5 Pa), an oscillating rheological measurement of the dynamic shear modulus was performed. The measurement result is a function of frequency (0.01 to 100 Hz).
3 Results and discussion
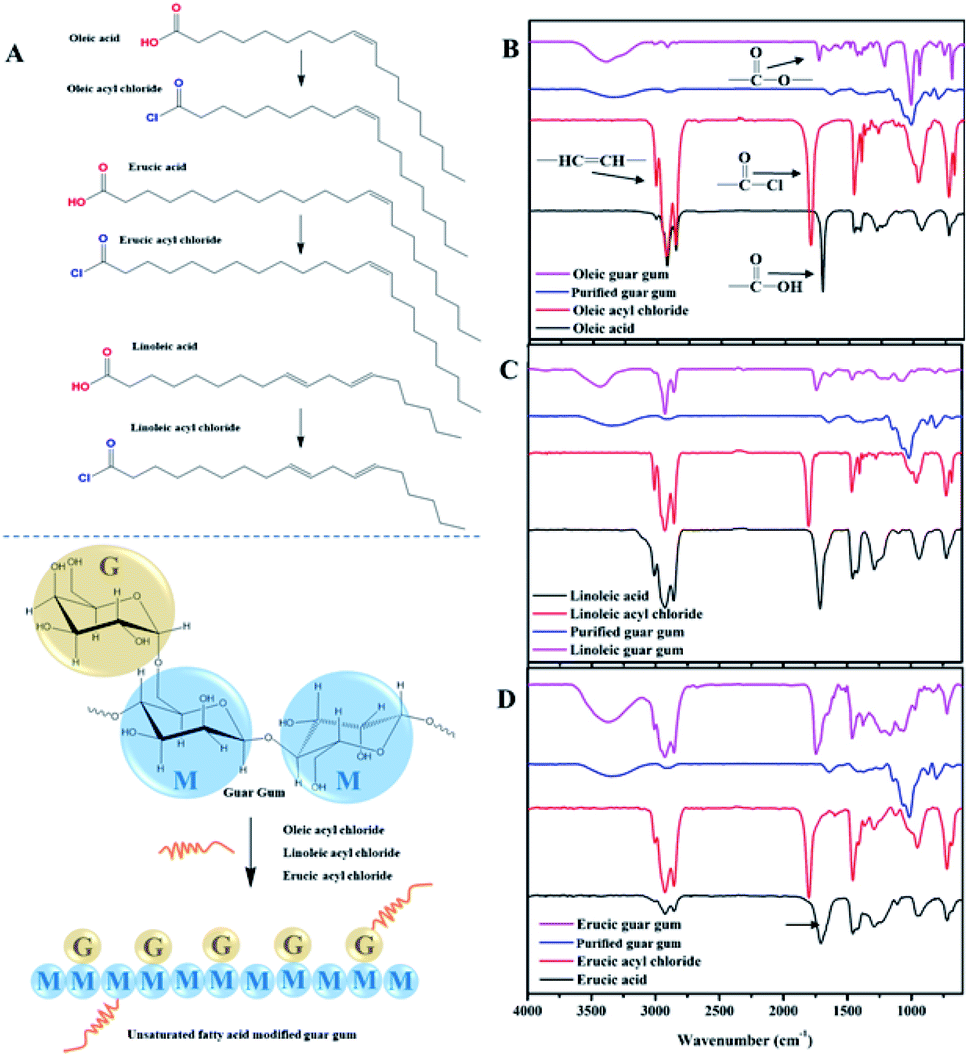
Three kinds of guar gum derivatives with hydrophobic unsaturated long-chains as shown in Fig. 1A have been prepared with the purpose to systematically study the rheology properties of hydrophobically modified guar gum.
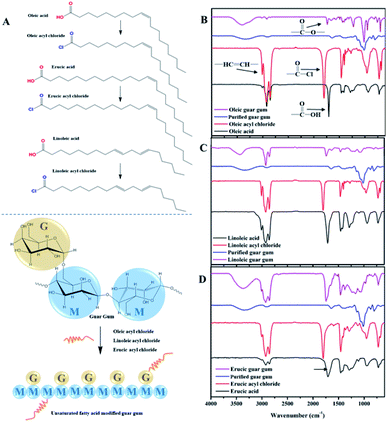 |
| | Fig. 1 (A) The synthetic route of guar gum derivatives with hydrophobic unsaturated long-chains (M is the abbreviation of Mannan; G is the abbreviation of Galactose); the infrared spectra of (B) oleic acid, oleic acyl chloride, purified guar gum, oleic guar gum, (C) linoleic acid, linoleic acyl chloride, purified guar gum, linoleic guar gum, (D) erucic acid, erucic acyl chloride, purified guar gum and erucic guar gum. | |
3.1. Characterization of the guar gum derivatives by FTIR
Fig. 1B shows the FT-IR spectra of oleic acid, oleic acid chloride and OGG. The spectrum of oleic acid displays the stretching vibration of unsaturated double bond at 3008 cm−1, the stretching vibration of methylene at 2926 and 2858 cm−1, the stretching vibration of carbonyl group at 1708 cm−1, the bending vibration of methylene at 1458 cm−1, and the in-plane oscillating vibration of methylene at 721 cm−1. For oleic acid chloride, the peak shape and intensity are very similar to those of oleic acid, with only significant differences in the carbonyl characteristic peaks. The carbonyl absorption peak of oleic acid chloride shows a large red shift from 1708 to 1801 cm−1 due to the strong electron-attracting effect of chlorine atoms, which can be used to characterize the successful synthesis of oleic acid chloride. In addition, FTIR spectra of purified GG and OGG were recorded to compare the change in their chemical structure. The absorption bands of purified GG at 1017, 1082 and 1158 cm−1 are due to the stretching vibrations of C–O(H) and the absorption band at 1648 cm−1 is due to the bending vibration of –OH groups. However, after grafting copolymerization with oleic acid, the O–H absorption peak of modified GG was significantly reduced, and new peaks appeared at 1738 and 1224 cm−1 (corresponding to the strong asymmetric stretching and weak symmetric stretching in COO groups), indicating that oleic acid has been successfully grafted onto the GG backbone.
As shown in Fig. 1C and D, the FT-IR spectra of linoleic acyl chloride and erucic acyl chloride are very similar to that of oleic acyl chloride. Due to the strong electron attraction of the chlorine atoms, the carbonyl absorption peaks of linoleic acyl chloride and erucic acyl chloride both showed a large red shift from 1711 to 1801 cm−1, demonstrating the successful synthesis of linoleic acyl chloride and erucic acyl chloride. Moreover, compared with purified GG, new peaks of about 1738 and 1224 cm−1 (strong asymmetric stretching and weak symmetric stretching in COO groups) appeared in the spectra of the modified GG after grafting copolymerization with linoleic acid and erucic acid, indicating that the linoleic acid and erucic acid have been successfully grafted onto the GG backbone, respectively.
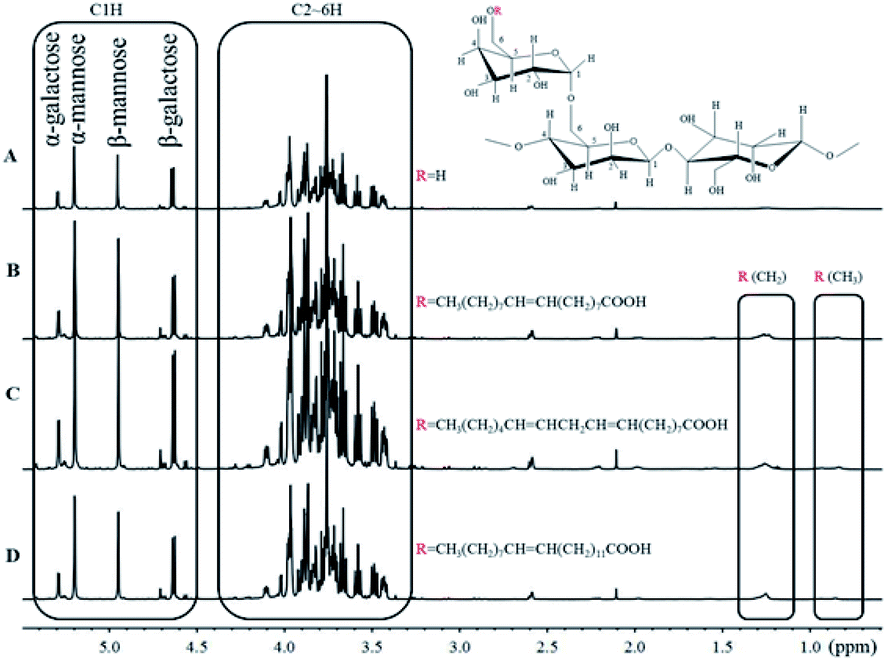
3.2. Characterization of the guar gum derivatives by 1H NMR
1H NMR spectra of purified guar gum, OGG, LGG and EGG dissolved in D2O solution containing 10% DCl are shown in Fig. 2. As we previously reported,19 the monosaccharide composition of guar gum can be analyzed by 1H NMR. The peaks at 5.20 ppm (α conformation) and 4.95 ppm (β conformation) are attributed to C1H in mannose, and the peaks at 5.29 ppm (α conformation) and 4.63 ppm (β conformation) are attributed to C1H in galactose. From the integrated area of C1H characteristic peaks of mannose and galactose, it can be seen that the ratio of galactose to mannose in guar gum is 1:1.6. Moreover, the peak at 0.85 ppm is usually attributed to the methyl groups of the guar gum derivatives, but there is no methyl group in the pure guar gum molecule. Therefore, the appearance of the methyl peak indicates that the oleic guar gum, linoleic guar gum and erucic guar gum have been successfully formed through the grafting reaction of oleic acid, linoleic acid and erucic acid, respectively. The molar substitution (MS) of repeating sugar units in OGG, LGG and EGG is obtained by the comparison between the integrated area of the peak of methyl group and that of the C1 hydrogen atom on the GG units. Table 1 shows the MS of the repeating sugar units in the guar gum derivatives calculated from the 1H NMR spectra.
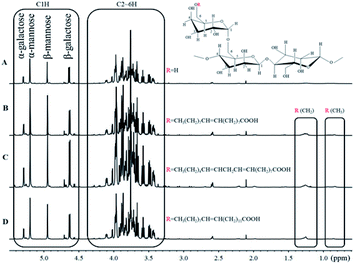 |
| | Fig. 2 1H NMR spectra of (A) purified guar gum, (B) OGG, (C) LGG and (D) EGG. | |
Table 1 Molar substitution (MS) of repeating sugar units in guar gum derivatives and the yield of prepared guar gum derivatives
| Polymer |
MS obtained by 1H NMR |
MS obtained by TGA |
Yield (%) |
| OGG-0.06 |
0.05 |
0.06 |
92.5 |
| OGG-0.01 |
0.01 |
0.01 |
90.4 |
| LGG-0.08 |
0.08 |
0.08 |
90.8 |
| LGG-0.05 |
0.06 |
0.05 |
92.1 |
| LGG-0.03 |
0.03 |
0.03 |
91.8 |
| LGG-0.02 |
0.02 |
0.02 |
94.4 |
| EGG-0.08 |
0.06 |
0.08 |
90.2 |
| EGG-0.04 |
0.04 |
0.04 |
92.9 |
| EGG-0.03 |
0.02 |
0.03 |
93.5 |
3.3. Determination of molar substitution (MS) of repeating sugar units in guar gum derivatives
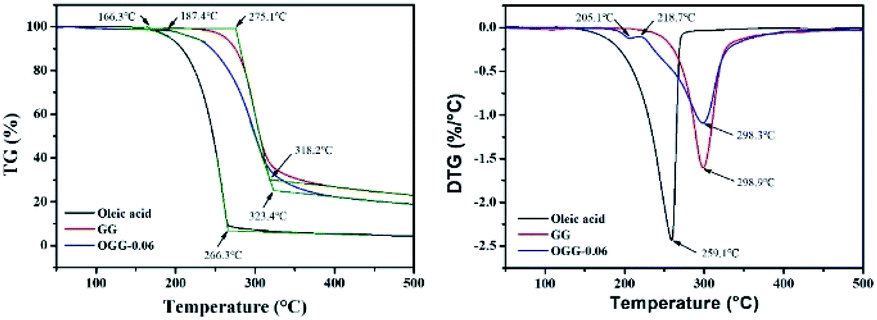
Thermogravimetric analysis (TGA) technique was used to characterize the thermal properties of the graft copolymers and revealed the percentage weight loss of purified GG and unsaturated fatty acid (oleic acid, linoleic acid, and erucic acid) at the decomposition temperature. Thus, the weight loss can be used to characterize grafting characteristic, expressed as MS of repeating sugar units, which is defined as the molar ratio of grafted unsaturated fatty acids to repeating sugar units in GG. Fig. 3 compares the typical TGA curves for purified GG, oleic acid and OGG. Notably, there are three major degradation steps in OGG, which further confirms the successful grafting of hydrophobic oleic acid on GG. In the initial stage, when the temperature increases from ambient temperature to 120 °C, the weight loss is caused by the dehydration of water contained in OGG. In the second stage of 120 to 266 °C, OGG shows a decomposition peak of hydrophobic oleic acid, and the decomposition interval is the same as that of pure oleic acid, leaving behind the relatively thermally stable GG. In the third stage from 250 to 350 °C, there is a degradation interval of GG. Comparing the degradation characteristics, only a small part of the thermal decomposition curves of purified GG and oleic acid overlap each other, which has minimal effect on the MS calculation of repeating sugar units in OGG after peak processing. Similarly, the TGA method has been used to characterize the grafting ratio of cassava starch-g-acrylamide/itaconic acid copolymer.20 The MS of repeating sugar units in OGG is calculated by the following formula:
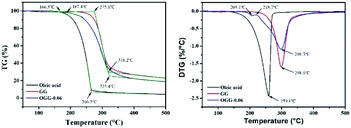 |
| | Fig. 3 Thermogravimetric analysis of GG, oleic acid and OGG under nitrogen at 10 °C min−1. | |
The MS of repeating sugar units in LGG and EGG was calculated using the same method and the results are shown in Table 1. It was found that although the final products are not exactly consistent with the feed compositions, the MS increases with the increase of the unsaturated fatty acids added, which is expected to tune the rheological properties of the modified guar gum. Furthermore, the MS of repeating sugar units in GG provides information on the grafting degree of GG and is therefore an important parameter for studying the thermal stability of graft copolymer. The study found that the initial decomposition temperature of GG derivatives with different hydrophobic unsaturated long-chains is lower than that of GG, and decreases slightly with the increase of unsaturated fatty acid content because of the early degradation of unsaturated fatty acids.
3.4. Rheological behaviour under dynamic shear
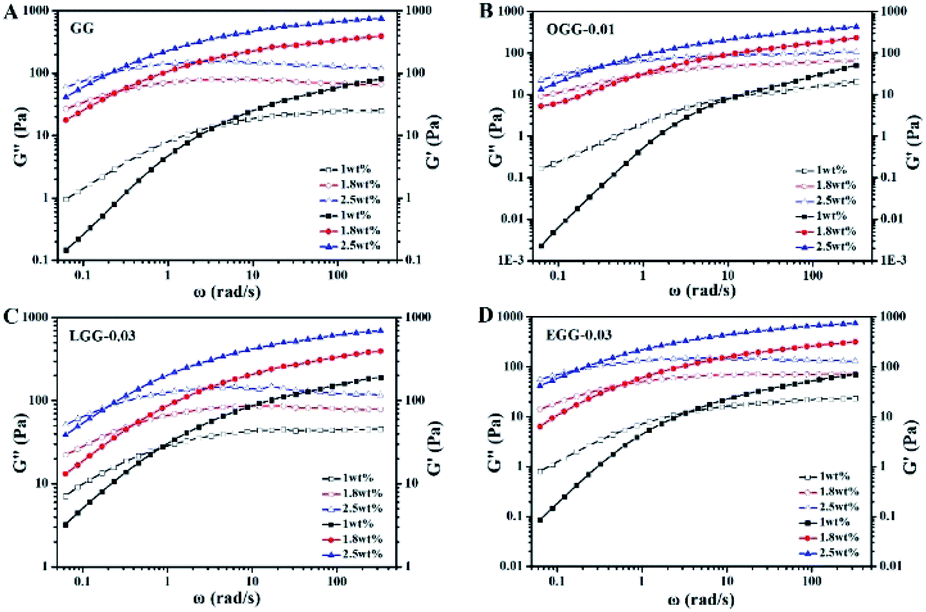
3.4.1. Effect of polymer concentration. The changes in dynamic elastic (G′) and viscous modulus (G′′) of guar gum and guar gum derivatives solutions with different polymer concentrations as a function of the angular frequency are shown in Fig. 4 (25 °C). Depending on the frequency, all polymer solutions investigated could exhibit liquid-like property (when G′ value was less than G′′ value) or solid-like property (when G′ value was greater than G′′ value). At a low frequency state, G′ value is less than G′′ value, and the response to external strain is caused by the relative movement of the polymer chains. With the increase of frequency, the increase rate of the elastic component is faster than that of the viscous component. In this case, the solution changes from liquid-like behaviour to solid-like behaviour. It is observed that the G′ and G′′ curves cross at a specific frequency, at which G′ and G′′ values are equal. For frequency above the intersection, the elastic modulus is greater than the viscous modulus, indicating that intermolecular chain crosslinks are widespread. At higher polymer concentration, the crossover frequency shifts. As the polymer concentration increases, the frequency at which the moduli are equal decreases. At higher concentrations, the polymer is less viscous due to restricted intermolecular motions, which increases the relaxation time. The crossover frequency is inversely related to this relaxation time. When G′ = G′′, the dependence of the crossover frequency on polymer concentration is consistent with the literature.4,21
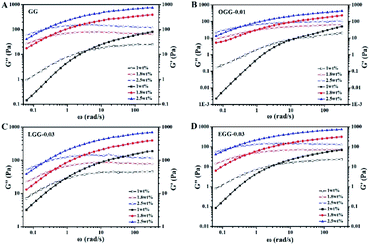 |
| | Fig. 4 Elastic (solid) and viscous modulus (hollow) as a function of angular frequency for the aqueous solutions of (A) GG, (B) OGG-0.01, (C) LGG-0.03, and (D) EGG-0.03 with different concentrations (temperature = 25 °C; pH = 7.0). | |
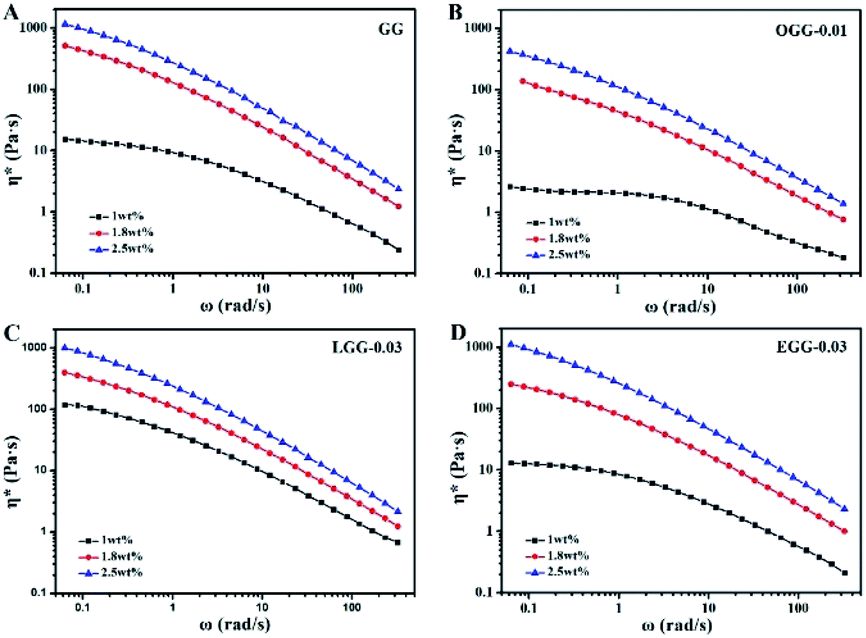
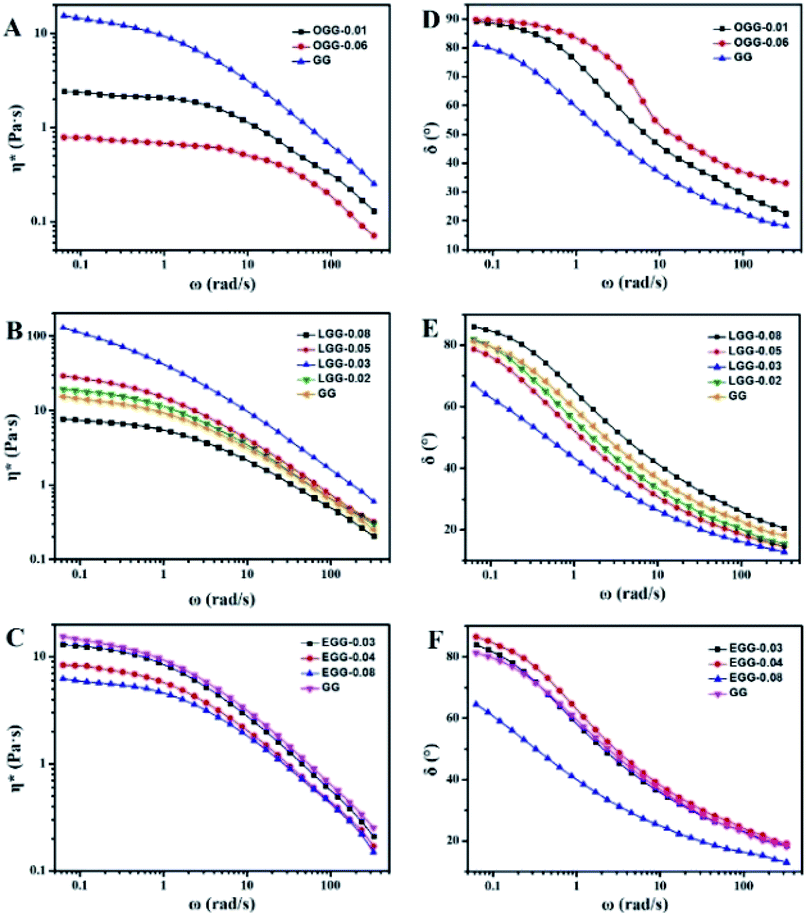
Complex viscosity (η*) is the viscosity measured with a rheometer in dynamic mode. The complex viscosities (η*) of aqueous solutions of GG and guar gum derivatives with different polymer concentrations as a function of angular frequency are shown in Fig. 5 (25 °C). The η* values were found to increase with increasing polymer concentrations at a given frequency. When all concentrations are equal, the viscosity of OGG-0.01 and EGG-0.03 solutions is lower than that of GG and LGG-0.03 solutions, indicating the difference in intrinsic viscosity (η). This phenomenon may be due to the fact that strong hydrophobic associations limit the extension of the chain conformation in OGG-0.01 and EGG-0.03, which leads to a decrease in dispersion and viscosity. In particular, the η* value decreases with increasing frequency, and the difference in the η* values is more pronounced at lower shear rates, and less so at higher shear rates, exhibiting non-Newtonian shear-shinning behaviour. Based on Graessley's entanglement theory,22 the phenomenon that viscosity decreases with increasing shear frequency is considered to be the result of a net decrease in entanglement density. Here, the net reduction in entanglement density may mainly derive from two effects, namely, the reduced number of hydrophobic associations and the reduced lifetimes of these associations, both of which are caused by the shear field. Simulation studies of associative polymers4,23–27 showed that the dissociation rate of crosslinking increases with increasing the shear rate and shear force may cause severe structural reorganizations of the polymer network.28–30 Furthermore, it is clear from the flow curves that the solution exhibits Newtonian behaviour in the low shear rate range, and this shear rate range is related to the polymer concentration.31 By fitting the frequency-dependent complex viscosity (η*) to a power–law relationship (only applicable to the non-Newtonian fluids), the shear-shinning exponent (n) can be obtained semi-qualitatively:32
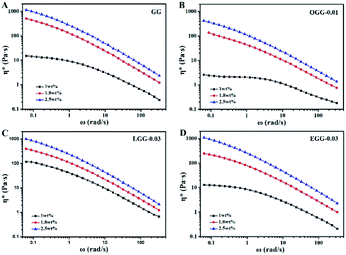 |
| | Fig. 5 Dynamic complex viscosity (η*) as a function of angular frequency (ω) for the aqueous solutions of (A) GG, (B) OGG-0.01, (C) LGG-0.03, and (D) EGG-0.03 with different concentrations (pH = 7.0; temperature = 25 °C). | |
Among them, n is the flow behaviour index. As we all known, shear-thinning exponent of an ideal Newtonian liquid should be equal to 0, because its viscosity is independent of the external shear frequency. Therefore, the shear thinning degree of the guar gum derivatives aqueous solutions can be measured by the n value, which increases with the increase of polymer concentration. As shown in Table 2, the aqueous solutions of GG, OGG-0.01 LGG-0.03 and EGG-0.03 with a higher concentration have stronger non-Newtonian shear-thinning behaviour (R2 represents a goodness-of-fit). In contrast to GG, OGG-0.01 or EGG-0.03, LGG-0.03 have an apparently higher flow behaviour index n when the concentration is 1 wt%, exhibiting the strongest shear-thinning property.
Table 2 Rheological parameters of power–law model at 25 °C: the effect of different concentrations (R2 represents a goodness-of-fit)
| Concentration (w/v) |
GG |
OGG-0.01 |
LGG-0.03 |
EGG-0.03 |
| n |
R2 |
n |
R2 |
n |
R2 |
n |
R2 |
| 1 wt% |
0.49 |
0.94 |
0.31 |
0.88 |
0.63 |
0.98 |
0.49 |
0.94 |
| 1.8 wt% |
0.72 |
0.99 |
0.64 |
0.99 |
0.69 |
0.99 |
0.66 |
0.98 |
| 2.5 wt% |
0.74 |
0.99 |
0.68 |
0.99 |
0.73 |
0.99 |
0.73 |
0.99 |
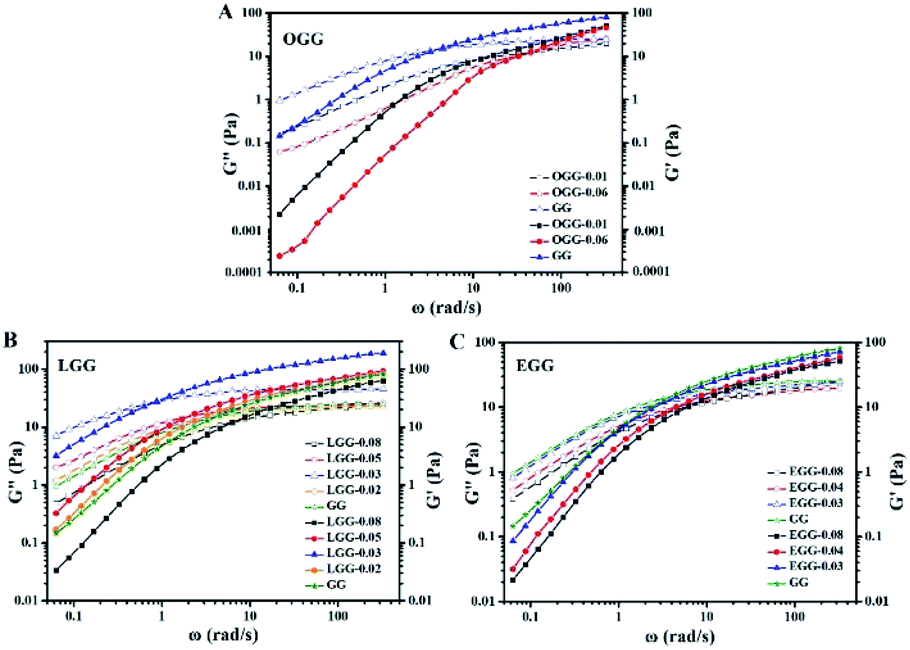
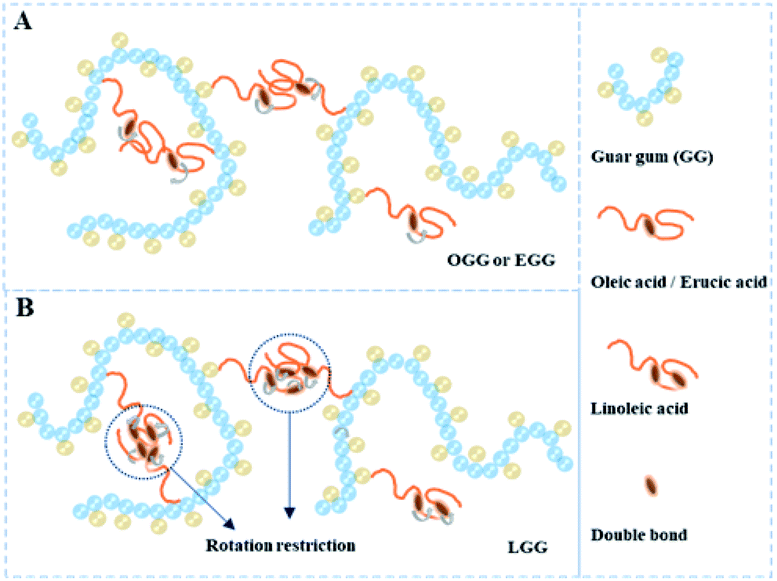
3.4.2. Effect of hydrophobic group and molar substitution degree. As shown in Fig. 6, it is the dynamic elastic (G′) and viscous modulus (G′′) for three guar gum derivatives solutions (1 wt%) with different MS as a function of the angular frequency (25 °C). Here, the reciprocal of the crossover frequency can be regarded as the characteristic time (expressed as τG′=G′′). The value of G′G′=G′′ is an indicator of the characteristic intensity of the relaxation spectrum, which is positively correlated with the high-frequency storage modulus without Rouse relaxations. According to the reported literature,4 when G′ = G′′, G′ is a function of the mass of the aggregates, and ω is proportional to the polydispersity of aggregates. As shown in Fig. 6, we can speculate that the molar mass of the aggregate is in the following order: LGG > GG > EGG > OGG, and the polydispersity is in the following order: LGG < GG < EGG < OGG (due to the higher degree of molar substitution, LGG-0.08 does not fit this order). As shown in the ESI Fig. S2,† the order of the molar mass and polydispersity of the aggregates are the same at 1.8%, which are LGG > GG > EGG > OGG and LGG < GG < EGG < OGG, respectively (LGG-0.08 and OGG-0.01 do not fit this order). These signs confirmed that the addition of oleic and erucic groups onto the GG chains could prevent the GG polymers from tending to aggregation. This may be due to the fact that the OGG and EGG configurations are less expanded (smaller hydrodynamic volume33) than the GG configuration, hence they have less intermolecular associations. In contrast, LGG has a strong hydrophobic association and can maintain a relatively solid three-dimensional crosslinking network under shear condition. The main reason for this phenomenon may be that the hydrophobic chain of linoleic acid contains two double bonds, which makes the flexibility of the LGG molecular chain is relatively lower than that of the OGG and EGG molecular chains due to the rotation restriction (the possible schematic diagrams of hydrophobic association in OGG, EGG and LGG aqueous solutions are shown in Fig. 7). Therefore, compared with OGG and EGG, LGG is easier to maintain the state of chain entanglement caused by hydrophobic association. As the MS of LGG increases, the intersection point between the two moduli moves first to a lower shear rate direction, and then moves to a higher shear rate direction, and reaches the minimum value of the shear rate when MS = 0.03. Therefore, the viscoelasticity of LGG can be adjusted by adjusting MS to meet different usage requirements. From the viewpoint of industrial application as a fracturing fluid, the relatively good viscoelasticity of the LGG aqueous solution seems to be advantageous.
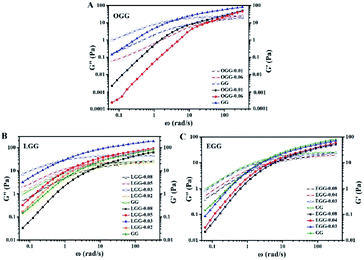 |
| | Fig. 6 Elastic (solid) and viscous modulus (hollow) as a function of angular frequency for the aqueous solutions of (A) OGG, (B) LGG, and (C) EGG with different MS (concentration = 1 wt%; temperature = 25 °C; pH = 7.0). | |
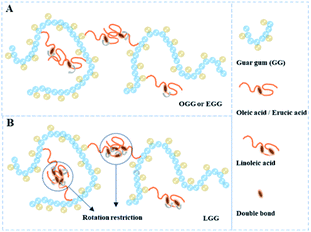 |
| | Fig. 7 Schematic diagrams of hydrophobic association in (A) OGG/EGG and (B) LGG aqueous solutions. | |
The effect of hydrophobic group and molar substitution degree on the complex viscosity (η*) for guar gum derivatives solutions (1 wt%) was also investigated as a function of angular frequency (ω) at 25 °C (Fig. 8A–C). Here, some of these curves in Fig. 8A–C are identical to those in Fig. 5. Obviously, the complex viscosities of both OGG and EGG solutions are lower than GG, and decrease with increasing MS. This probably due to the introduction of hydrophobic groups leading to a decrease in the chain extension of OGG and EGG, and it becomes more apparent as the MS increases. In comparison, when MS is less than 0.08, the complex viscosities of LGG solutions are obviously higher than that of GG, which is possibly due to the appropriate hydrophobic association of the LGG chains and the increase of intramolecular frictional force, ultimately leading to an increase in viscosity. When MS increases to 0.08, the complex viscosity of the LGG solution becomes lower than that of GG, which is mainly due to the difficulty of chain extension caused by excessive hydrophobic groups, eventually leading to the decrease of the complex viscosity.
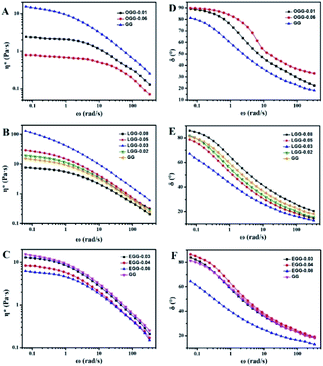 |
| | Fig. 8 Dynamic complex viscosity (η*) (A–C) and dynamic loss angle (δ) (D–F) as a function of angular frequency (ω) for the aqueous solutions of (A and D) OGG, (B and E) LGG, and (C and F) EGG with different MS (concentration = 1 wt%; pH = 7.0; temperature = 25 °C). | |
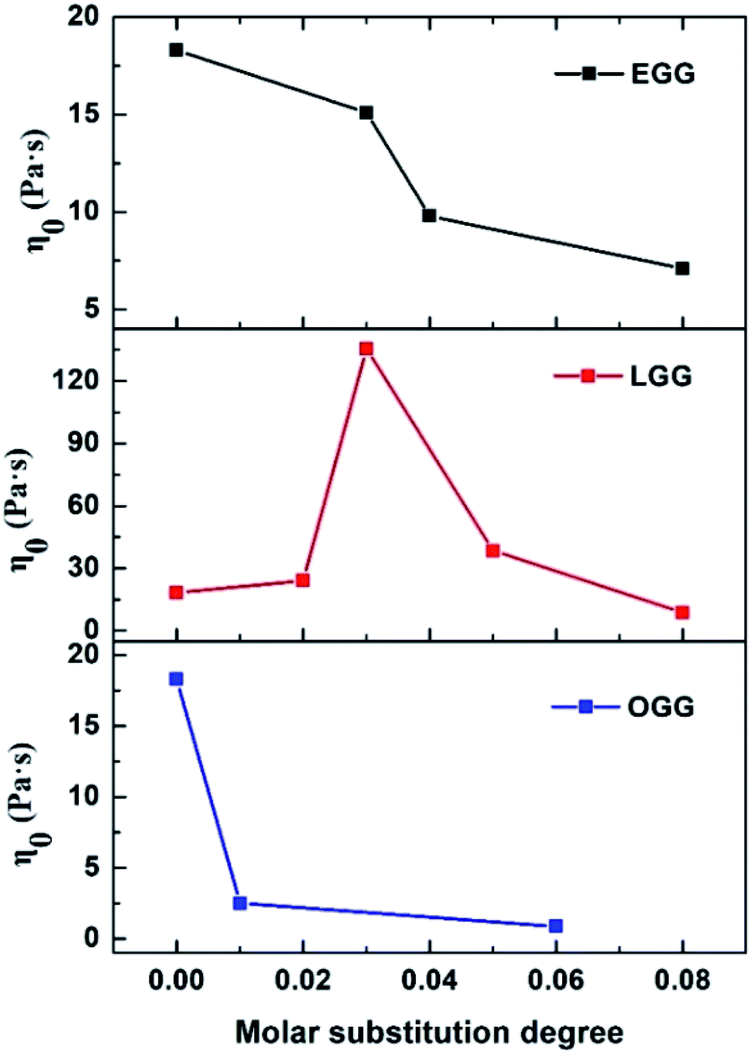
The frequency dependence of the dynamic loss angle δ (tanδ = G′′(ω)/G′(ω)) decreases as the angular frequency ω increases. This relationship is used to measure the ratio of energy dissipated as heat to energy stored during cyclic deformation.4 As shown in Fig. 8D–F, specific examples of guar gum derivatives solutions with different MS are given. The observed phenomenon can be demonstrated by the enhanced effect of elastic components to viscous components at higher frequencies. Over the entire angular frequencies (ω) range studied, the values of tanδ at the same concentration follow the sequence: LGG < GG < EGG < OGG (due to the higher degree of MS, LGG-0.08 and EGG-0.08 do not fit this order). As shown in the ESI Fig. S3,† the sequence for tanδ is different at 1.8% where it is LGG < EGG < GG < OGG (LGG-0.08 and OGG-0.01 do not fit this order). A low value of tanδ indicates that the polymer solution has a solid-like solution structure. With the increase of tanδ, the polymer solution behaves as Newtonian fluid. Thus, when MS is less than 0.08, the LGG solution seems to be dominated by appropriate hydrophobic interactions to organize itself into a relatively stable network structure (linoleic acid contains two double bonds, which limits the flexibility of the LGG chains to some extent), which confers to the LGG system more obvious solid-like rheological properties than the OGG and EGG systems. As shown in Fig. 9, LGG showed higher zero-shear viscosities (η0) than OGG and EGG (due to the higher degree of MS, LGG-0.08 does not fit this rule), especially LGG-0.03 exhibited the highest zero-shear viscosity, which again confirmed the appropriate and strong hydrophobic association in the LGG system.
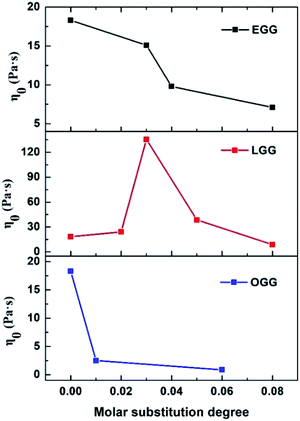 |
| | Fig. 9 Zero-shear viscosities (η0) of OGG, LGG and EGG as a function of MS (concentration = 1 wt%; pH = 7.0; temperature = 25 °C). | |
4 Conclusions
In this work, three guar gum derivatives with hydrophobic unsaturated long-chains were successfully synthesized, including OGG, LGG and EGG, and the dynamic viscoelastic properties of these polymers in aqueous system were investigated compared with GG. For all samples, with the increase of polymer concentration, the intersection point between the elastic and viscous moduli moves towards a low angular frequency, caused by an increase in intermolecular associations that hinder motion. The polymer solutions investigated all exhibited typical non-Newtonian shear-thinning fluids. Among them, when the concentration is 1 wt%, LGG-0.03 has an apparently higher flow behaviour index n, exhibiting the strongest shear-thinning property. Within the entire range of angular frequencies ω studied (the concentration of all samples is 1 wt%), it can be speculated that the molar mass of the aggregates is arranged in the following order: LGG > GG > EGG > OGG, and the polydispersity is in the following order: LGG < GG < EGG < OGG, and the values of tanδ follows the sequence: LGG < GG < EGG < OGG (due to the higher degree of MS, LGG-0.08 does not fit this order). As shown in the ESI Fig. S3,† the sequence for tanδ is different at 1.8% where it is LGG < EGG < GG < OGG (LGG-0.08 and OGG-0.01 do not fit this order). To some extent, the introduction of oleic and erucic groups onto the GG chains causes a decrease in the extended configuration of the chains due to the strong hydrophobic associations, while LGG can form a solid three-dimensional crosslinking network probably due to the appropriate hydrophobic association. Moreover, the hydrophobic chain of linoleic acid contains two double bonds. Due to the rotation restriction, the flexibility of the LGG molecular chain is relatively lower than that of the OGG and EGG molecular chains. Therefore, compared with OGG and EGG, LGG is easier to maintain the state of chain entanglement caused by hydrophobic association under shear flow. In summary, from the viewpoint of industrial application, it seems to be an effective method to achieve satisfactory application of guar gum derivatives by adjusting the hydrophobic groups and the degree of substitution.
Funding
This research was funded by China Postdoctoral Science Foundation (Grant no. 2017M623030).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge Jingkun Oilfield Chemistry Company of china for financial and material support.
References
- G. Bastiat, B. Grassl and J. François, Polym. Int., 2002, 51, 958–965 CrossRef CAS.
- B. Grassl, J. Francois and L. Billon, Polym. Int., 2001, 50, 1162–1169 CrossRef CAS.
- Z. L. Yang, B. Y. Gao, C. X. Li, Q. Y. Yue and B. Liu, Chem. Eng. J., 2010, 161, 27–33 CrossRef CAS.
- D. Risica, A. Barbetta, L. Vischetti, C. Cametti and M. Dentini, Polymer, 2010, 51, 1972–1982 CrossRef CAS.
- Y. J. Zhu, J. Zhang, B. Jing and Y. B. Tan, Adv. Mater. Res., 2013, 781–784, 431–435 Search PubMed.
- Z. Zhan, B. Du, S. Peng, J. He, M. Deng, J. Zhou and K. Wang, Carbohydr. Polym., 2013, 93, 686–690 CrossRef CAS PubMed.
- D. Szopinski, W. M. Kulicke and G. A. Luinstra, Carbohydr. Polym., 2015, 119, 159–166 CrossRef CAS PubMed.
- Y. Feng, L. Billon, B. Grassl, A. Khoukh and J. François, Polymer, 2002, 43, 2055–2064 CrossRef CAS.
- Y. Feng, L. Billon, B. Grassl, G. Bastiat, O. Borisov and J. François, Polymer, 2005, 46, 9283–9295 CrossRef CAS.
- G. O. Yahaya, A. A. Ahdab, S. A. Ali, B. F. Abu-Sharkh and E. Z. Hamad, Polymer, 2001, 42, 3363–3372 CrossRef CAS.
- J. T. Ma, P. Cui, L. Zhao and R. H. Huang, Eur. Polym. J., 2002, 38, 1627–1633 CrossRef CAS.
- D. Mudgil, S. Barak and B. S. Khatkar, J. Food Sci. Technol., 2014, 51, 409–418 CrossRef CAS PubMed.
- A. M. A. Hasan and M. E. Abdel-Raouf, Egypt. J. Pet., 2018, 27, 1043–1050 CrossRef.
- N. Thombare, U. Jha, S. Mishra and M. Z. Siddiqui, Int. J. Biol. Macromol., 2016, 88, 361–372 CrossRef CAS PubMed.
- S. Thakur, B. Sharma, A. Verma, J. Chaudhary, S. Tamulevicius and V. K. Thakur, Int. J. Polym. Anal. Charact., 2018, 23, 621–632 CrossRef CAS.
- G. Sharma, S. Sharma, A. Kumar, A. H. Al-Muhtaseb, M. Naushad, A. A. Ghfar, G. T. Mola and F. J. Stadler, Carbohydr. Polym., 2018, 199, 534–545 CrossRef CAS PubMed.
- L.-M. Zhang, J.-F. Zhou and P. S. Hui, J. Sci. Food Agric., 2005, 85, 2638–2644 CrossRef CAS.
- R. Lapasin, L. De Lorenzi, S. Pricl and G. Torriano, Carbohydr. Polym., 1995, 28, 195–202 CrossRef CAS.
- P. X. Gong, S. H. Peng, J. P. He, M. Y. Deng, B. Jiang and K. Wang, Carbohydr. Res., 2011, 13, 1973–1977 CrossRef PubMed.
- P. Lanthong, R. Nuisin and S. Kiatkamjornwong, Carbohydr. Polym., 2006, 66, 229–245 CrossRef CAS.
- L. Wang and L.-M. Zhang, Ind. Crops Prod., 2009, 29, 524–529 CrossRef CAS.
- M. M. Cross, J. Colloid Sci., 1965, 20, 417–437 CrossRef CAS.
- D. Bedrov, G. Smith and J. F. Douglas, Polymer, 2004, 45, 3961–3966 CrossRef CAS.
- R. D. Groot and W. G. Agterof, Macromolecules, 1995, 28, 6284–6295 CrossRef CAS.
- M. Wilson, A. Rabinovitch and A. R. C. Baljon, Macromolecules, 2015, 48, 6313–6320 CrossRef CAS.
- D. Bedrov, G. D. Smith and J. F. Douglas, Europhys. Lett., 2002, 59, 384–390 CrossRef CAS.
- J. Billen, M. Wilson and A. R. C. Baljon, Chem. Phys., 2015, 446, 7–12 CrossRef CAS.
- R. Lund, R. A. Lauten, B. Nyström and B. Lindman, Langmuir, 2001, 17, 8001–8009 CrossRef CAS.
- P. Khalatur, A. Khokhlov and D. Mologin, J. Chem. Phys., 1998, 109, 9602–9613 CrossRef CAS.
- P. G. Khalatur, A. R. Khokhlov, J. N. Kovalenko and D. A. Mologin, J. Chem. Phys., 1999, 110, 6039–6049 CrossRef CAS.
- S. Venkataiah and E. Mahadevan, J. Appl. Polym. Sci., 1982, 27, 1533–1548 CrossRef CAS.
- R. G. Larson, The structure and rheology of complex fluids, Oxford University Press, New York, 1999 Search PubMed.
- W.-M. Kulicke and R. Kniewske, Rheol. Acta, 1984, 23, 75–83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04322b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Minmin Fan
c,
Minmin Fan