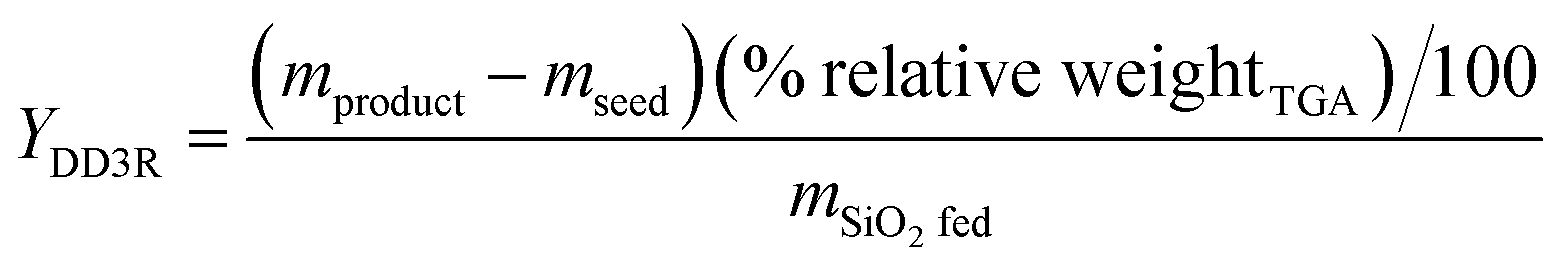DOI:
10.1039/D0RA04293E
(Paper)
RSC Adv., 2020,
10, 27523-27530
Comparative study on different strategies for synthesizing all-silica DD3R zeolite crystals with a uniform morphology and size†
Received
14th May 2020
, Accepted 9th July 2020
First published on 23rd July 2020
Abstract
In the last three decades, the all-silica deca-dodecasil 3R (DD3R) zeolite has been extensively studied as a significant potential class of porous materials in adsorptive separations. However, the use of most existing synthesis methods is unable to produce pure DD3R crystals with a uniform morphology and size. The present research, is therefore intended to provide a facile protocol to synthesize pure DD3R crystals with a controllable morphology and size and with a high reproducibility and productivity. Special attention was focused on investigating the effects of the type of seeds and the mineralizing reagent on the phase-purity, morphology, and crystal size of the resultant DD3R crystals. Various techniques, such as X-ray diffraction (XRD), scanning electron microscopy (SEM), N2 adsorption–desorption at 77 K, and thermogravimetric analysis (TGA) were then used to characterize the synthesized samples. The results show that by adding a small amount of “amorphous” DD3R or “amorphous” ZSM-58 seeds, the pure DD3R crystals with a uniform morphology and size can be synthesized using 1-adamantanamine (1-ADA) as a structure-directing agent (SDA), KF was used as a mineralizing reagent, and LUDOX AS-30 as a silicon source at 443 K for 1 d. In addition, the pure, large and uniform hexahedron DD3R crystals can be prepared using fumed silica as seeds, although the crystallization time takes a longer period of 3 d. The present work could stimulate fundamental research and industrial applications of the all-silica DD3R zeolite.
Introduction
Deca-dodecasil 3R (DD3R) is a member of the clathrasil family that possesses topologically different frameworks. Gies1,2 performed pioneering work on the synthesis and structural identification of a clathrasil DD3R. For the thermal treatment of the synthesized DD3R sample, the guest molecules were decomposed and the fragments were driven out of the 19-hedron cages, transforming the clathrasil into a phase possessing zeolitic properties.2
In the last three decades, zeolites with DDR topology have been extensively studied as a significant potential class of industrial porous materials for use in different fields such as adsorptive separations3–35 and catalysis.28,36–42 The development of new efficient gas separation techniques in order to improve the current expensive and high-energy-demanding industrial separation processes is a matter of interest.28 In comparison with the critical diameters of propane/propylene and unsaturated linear C4 hydrocarbons, the eight-ring windows of DD3R are accessible to propylene, buta-1,3-diene, and trans-but-2-ene molecules, but they exclude propane, but-1-ene, and cis-but-2-ene molecules, implying that DD3R might be effective as an adsorbent for the separation and purification of propylene/propane and unsaturated linear C4 hydrocarbon mixtures.5–10 Compared with other zeolites, the all-silica DD3R, which is highly hydrophobic and stable up to high temperatures,5 displays good prospects for application in the aforementioned adsorptive separations. Additionally, DD3R membranes have an excellent separation performance for CO2/CH4 and CO2/N2 mixtures.11–21 Most recently, Wang et al.22 reported that a DD3R membrane showed great potential for on-stream CO2 removal from the Xe-based closed-circuit anesthesia system.
In addition, zeolites with DDR topology are the most efficient catalysts used to produce light olefins such as ethylene, propylene, and butylenes in the methanol-to-olefin (MTO) process.36–42
Unfortunately, the applications of DD3R are largely limited owing to the difficulties of synthesis. The first work on the synthesis of DD3R crystals was reported by Gies1 in 1984, and later den Exter et al.3 optimized and scaled up the synthesis. However, their syntheses took 25–42 d, and additionally, the synthesized crystals were not at all uniform in morphology and size. Although Gascon et al.8 reduced the synthesis time to 2 d by adding DD3R seeds, their synthesis was hardly reproducible, and more importantly, the synthesized DD3R phase was contaminated with some undesirable byproducts such as Sigma-2, leading to low productivity. Yang et al.43 introduced a fluoride route into the synthesis of DD3R crystals in 2009, which was later optimized by Gücüyener et al.,9 reducing the synthesis time to 1 d. However, the synthesized crystals were not uniform in morphology and size, and more importantly, the DD3R phase was not pure. Recently, several research groups have tried to develop different methods to synthesize DD3R crystals with a short synthesis time, high purity, low cost, and controllable morphology and size. Zheng et al.44 used the ball-milled Sigma-1 as seeds to induce the crystallization of DD3R crystals and successfully synthesized a pure DD3R zeolite with a crystallization time of ca. 9 h. Yang et al.45 adjusted the pH of the Sigma-1-seeded precursor solution and hydrothermal crystallization to control the morphology of the resulting DD3R crystals. Sen et al.46 developed a sonochemical method to synthesize pure DD3R crystals at room temperature over 5 d. A microwave-aided heating method was used to significantly reduce the DD3R-crystal synthesis time from 25 to 3 d without seeding and to 6 h with seeding.47 Liu et al.48 used (NH4)2SiF6 as a silica source to synthesize DD3R crystals over 12 h without seeding, because (NH4)2SiF6 is very reactive and can enhance the nucleation and crystal growth of DD3R significantly. Bai et al.49 developed an environment-friendly synthesis method to prepare DD3R crystals, in which only the silica source, a small amount of template, and a trace amount of water were used in the synthesis, without adding a toxic solvent such as ethylenediamine or the mineralizing reagent fluoride. Without seeding, Kajihara et al.50 used the essential reagents, that is, a silica source, water, and 1-ADA, hydrothermally to synthesize DD3R crystals and found that the synthesis time could be significantly reduced from 15–20 d at a hydrothermal temperature of 433 K to 5 d at a hydrothermal temperature of 473 K. Most recently, Wang et al.51 took advantage of the synergy of microwave heating and seeding to significantly reduce the synthesis time of DD3R crystals to a few hours, and in addition, an inorganic base, such as KOH, NaOH, or LiOH, was used as a mineralizing reagent instead of ethylenediamine and fluoride, which is comparatively more environment-friendly and economical. Although significant progress in the synthesis of DD3R crystals, in terms of the synthesis time, cost, phase purity and so forth, has been made in recent years, unfortunately, most of the synthesized crystals were not uniform in morphology and size. Therefore, the synthesis of pure DD3R crystals with a uniform morphology and size is still a challengeable subject.
Herein, a comparative study on the different synthesis methods for DD3R crystals was carried out and the effects of the type and amount of seeds, and the amount of mineralizing reagent KF and so forth, on the phase purity, morphology, and size of the resultant DD3R crystals were investigated in detail. The purpose of the current research was to provide a facile protocol to synthesize DD3R crystals with a pure and controllable morphology and size and with a high reproducibility and productivity.
Experimental
Chemicals
All chemical reagents were used as received without further purification. The details of the chemical reagents used, including their providers and specifications, are summarized in Table 1.
Table 1 Providers and specifications of the chemical reagents used
| Reagents |
Providers |
Specification |
| Guaranteed reagent. Analytic reagent. Electronic grade. |
| 1-Adamantanamine |
Sigma-Aldrich |
GRa (≥97%) |
| Ethylenediamine |
Sigma-Aldrich |
GR (≥99%) |
| Tetramethoxysilane |
Shanghai Jiachen |
99% |
| Tropine |
Aladdin |
GR (≥99%) |
| Methyl iodide |
Shanghai Bangcheng |
ARb |
| LUDOX |
Sigma-Aldrich |
HS-40, AS-30 |
| Potassium fluoride |
Aladdin |
EGc (≥99.95%) |
| Potassium hydroxide |
Aladdin |
GR (≥99%) |
| Sodium hydroxide |
Aladdin |
GR (≥99%) |
| Ethanol |
Shanghai Lianshi |
98% |
| Deionized water |
Homemade |
— |
| Fumed silica |
Sigma-Aldrich |
AR |
Synthesis procedures
“Amorphous” DD3R seeds. Based on the recipe and synthesis procedure reported in the literature,4 the “amorphous” DD3R seeds were prepared with a much shorter hydrothermal reaction time than that reported in the literature. They were prepared as follows, the structure directing agent (SDA) 1-adamantanamine (1-ADA) was dissolved in ethylenediamine (EN) at room temperature, followed by adding deionized water, then the mixture was sonicated for 1 h. After heating to 368 K and being maintained at this temperature for 1 h under magnetic stirring, the mixture was cooled to 273 K in ice and ice-cooled tetramethoxysilane (TMOS) was then added dropwise under vigorous stirring. The mixture, with a molar ratio of 47 1-ADA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 SiO2:404 EN:11240 H2O, was heated up to 368 K again and maintained at this temperature under stirring until the mixture became clear. Finally, the solution was transferred into a 50 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 433 K for 2 d under static conditions, then cooled to room temperature naturally. The white product was washed with distilled water and then dried at 333 K overnight. The synthesized sample was named “amorphous” DD3R seeds.
:100 SiO2:404 EN:11240 H2O, was heated up to 368 K again and maintained at this temperature under stirring until the mixture became clear. Finally, the solution was transferred into a 50 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 433 K for 2 d under static conditions, then cooled to room temperature naturally. The white product was washed with distilled water and then dried at 333 K overnight. The synthesized sample was named “amorphous” DD3R seeds.
“Amorphous” ZSM-58 seeds. Based on the recipe and the synthesis procedure reported in the literature,52 the “amorphous” ZSM-58 seeds were prepared with a much shorter hydrothermal reaction time than that reported in the literature. They were prepared as follows, first, the SDA methyltropinium iodide (MTI) was prepared by adding methyl iodide dropwise into a solution of tropine in ethanol at 273 K under stirring and then the suspension was kept under reflux for 72 h. After cooling and filtration, the resultant solid product MTI was washed with ethanol and dried at 353 K. Afterwards, LUDOX HS-40 as a silica source was added into a solution of MTI in deionized water and the mixture was then stirred overnight, followed by the addition of NaOH solution. The resultant solution, with a molar ratio of 17.5 MTI:70 SiO2:11.5 Na2O:2800 H2O, was stirred for 0.5 h. Finally, the solution was transferred into a 50 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 433 K for 1 d under stirring, then cooled to room temperature naturally. The white product was washed with distilled water and then dried at 333 K overnight. The synthesized sample was named “amorphous” ZSM-58 seeds.
Sigma-2 seeds. The SDA 1-ADA was completely dissolved in the aqueous solution of LUDOX AS-30 as the silica source, followed by adding the mineralizing reagent KF. Then, the mixture was subsequently stirred at room temperature for 2 h. Finally, the aged gel, with a molar ratio of 47 1-ADA:100 SiO2:50 KF:8000 H2O, was transferred to a 50 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 433 K for 1 d under static conditions, then cooled to room temperature naturally. The white product was washed with distilled water and then dried at 383 K overnight. The synthesized sample was named Sigma-2 seeds.
DD3R seeds. The SDA 1-ADA was completely dissolved in the aqueous solution of LUDOX AS-30 as the silica source, followed by addition of the mineralizing reagent KF. After the mixture was stirred at room temperature for a while, the pre-prepared “amorphous” DD3R seeds (0.1 wt%) were added into the above mentioned mixture. Then, the mixture was subsequently stirred for 2 h. Finally, the aged gel, with a molar ratio of 47 1-ADA:100 SiO2:100 KF:8000 H2O, was transferred to a 50 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 433 K for 1 d under static conditions, then cooled to room temperature naturally. The white product was washed with distilled water and then dried at 383 K overnight, and calcined at 823 K for 2 h and 973 K for 8 h. The synthesized sample was named DD3R seeds.
DD3R crystals. The synthesis procedure was very similar to that described for preparing the DD3R seeds, except (i) the pre-prepared seeds with amounts varying from 0.1 to 0.3 wt% were added into the synthesis solutions; (ii) the synthesis solutions with different molar ratios of 1-ADA:SiO2:KF:H2O were used; and (iii) the hydrothermal crystallization time varied from 1 to 3 d.
Characterization
The powder X-ray diffractometry (XRD) patterns were collected on a Philips PW 3040/60 diffractometer using Cu Kα radiation (λ = 0.1541 nm) in the scanning range of 2θ of 5–50° at 2.40° min−1. Scanning electron microscopy (SEM) was carried out on an S-4800 apparatus (Hitachi) equipped with a field emission gun. The thermogravimetric analysis (TGA) was performed on a NETZSCH STA 449C thermogravimetric analyzer at a temperature ramp of 5 K min−1 up to 1173 K in flowing air with a rate of 20 mL min−1. The textural properties were determined by N2 adsorption–desorption at 77 K using an ASAP 2020 apparatus (Micromeritics Instrument Corp.). The samples were degassed under a vacuum at 573 K for 8 h prior to the adsorption measurements. The specific surface areas, SBET, were calculated using the multiple-point Brunauer–Emmett–Teller (BET) method in the relative pressure range of p/p0 = 0.05–0.15. The micropore volumes, Vmicro, were calculated using the t-plot method.
Results and discussion
Effects of seeds
In a conventional synthesis for the all-silica DD3R, the crystallization times of up to 25 to 48 d undoubtedly hinder their in-depth research and development. It is known that adding seeds into a hydrothermal synthesis batch can significantly accelerate the rate of zeolite crystallization and reduce the synthesis time.53–55 Indeed, the addition of DD3R crystals as seeds can reduce the synthesis time to 1–2 d.8,9 Here, a systematic investigation was conducted on the effects of the addition of different previously prepared seeds on the phase purity, morphology, and size of the resultant zeolite crystals.
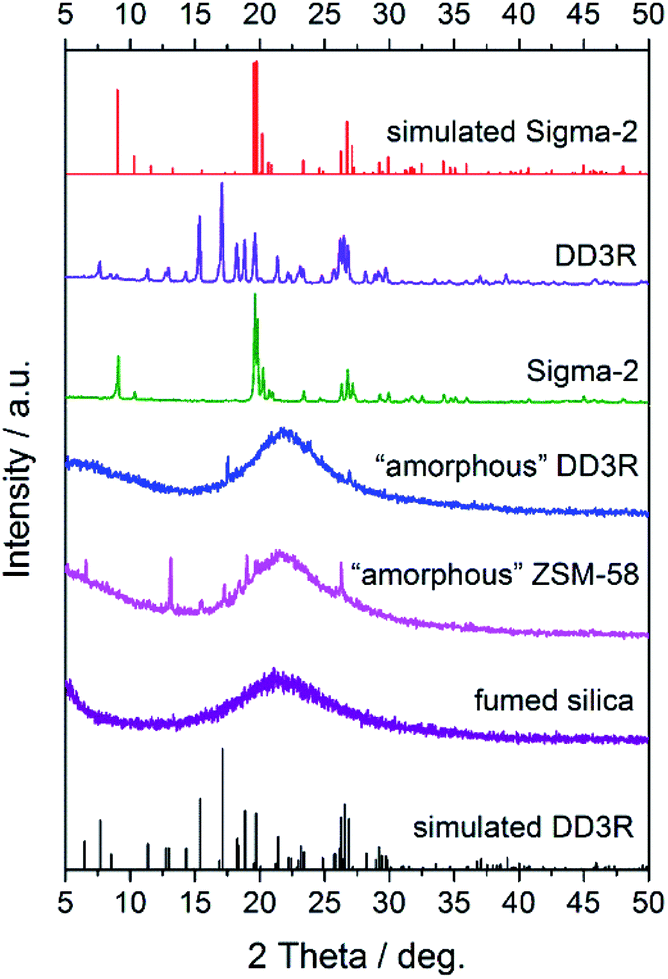
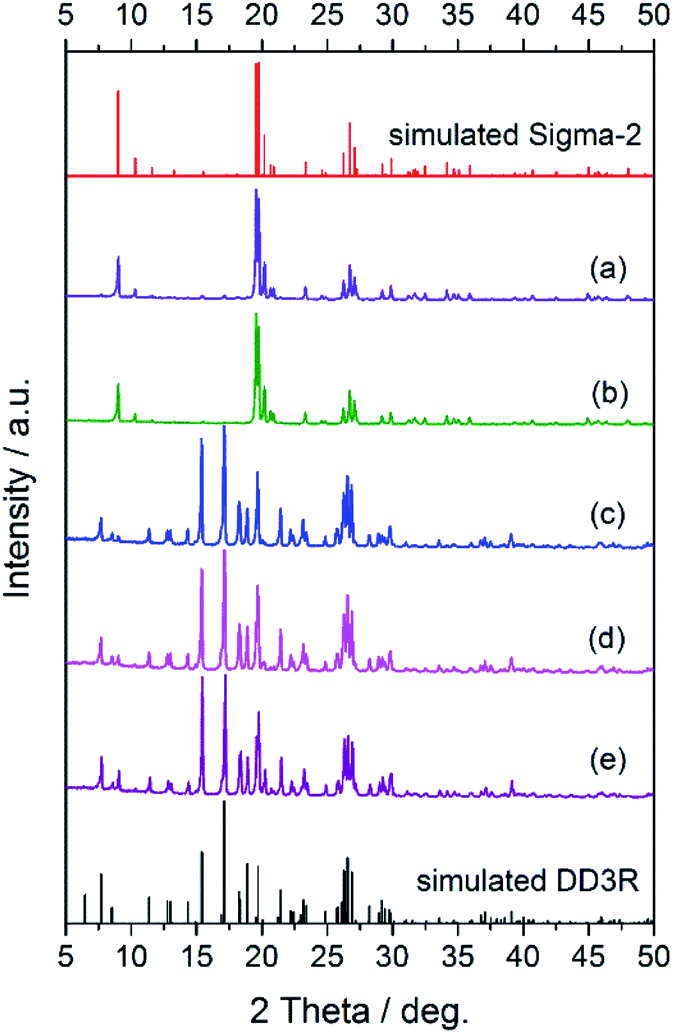
Fig. 1 shows the XRD patterns of the synthesized seeds, from which it can be clearly seen that the characteristic peaks of the synthesized DD3R and Sigma-2 seeds match their corresponding simulated ones very well, indicating that the pure DD3R and Sigma-2 phases are obtained. On the other hand, based on the recipe and synthesis procedure for DD3R and ZSM-58 reported in the literature,4,52 only their amorphous phases were obtained or the synthesized zeolite crystals may be too small to be detectable using the XRD technique, owing to the much shorter crystallization times used compared to those reported in the literature. These prepared seeds with an amount of 0.1 wt% of the total mass were individually added into the synthesis solution, the aged gel composition had a molar ratio of 47 1-ADA:100 SiO2:100 KF:8000 H2O, and the hydrothermal crystallization time was 1 d to prepare the DD3R crystals. As using target crystals as seeds has been widely acknowledged, synthesis with seeding of the pure DD3R crystals was first conducted. Unexpectedly, the resulting synthesized product was Sigma-2 crystals mixed with a few DD3R crystals, as evidenced by the XRD pattern and the SEM image, shown in Fig. 2 and 3, respectively, although Sigma-2 and DD3R are different types of zeolites. Similarly, if Sigma-2 seeds are added into the synthesis solution, the main product of Sigma-2 crystals mixed with a few DD3R crystals is obtained, as shown in Fig. 3b. In contrast, by adding either “amorphous” DD3R or “amorphous” ZSM-58 seeds, pure DD3R crystals with a uniform shape and size were successfully synthesized, as evidenced by the XRD pattern and the SEM image, shown in Fig. 2 and 3, respectively. Thus, the type of seeds plays an important role in the DD3R synthesis, although the reasons cannot be clearly given at this stage. Plausibly, either the “amorphous” DD3R or “amorphous” ZSM-58 seeds, with tiny particles and/or crystallites (see SEM images in Figs. S1 and S2 in the ESI†) but a high surface energy, are able to remain suspended throughout the solution, giving a more homogeneous reaction condition with further nucleation or crystallization sites. Therefore, the yielded products were uniform and did not contain any hybrid crystals. On the other hand, it is not conducive to obtain pure DD3R using relatively larger Sigma-2 or DD3R crystals as seeds. In general, in zeolite synthesis, the added seeds can provide “sites” for nucleation or crystallization, reduce the supersaturation of the synthesis solution to promote the crystallization process, and consume the raw materials rapidly and complete the crystallization with the secondary nucleation of seeds.53–55
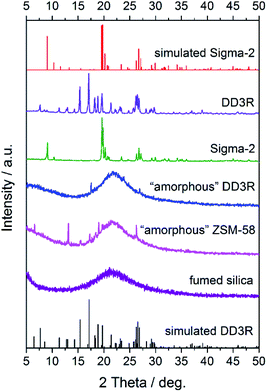 |
| | Fig. 1 XRD patterns of the prepared seeds and commercial fumed silica. | |
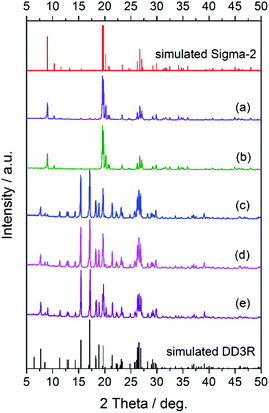 |
| | Fig. 2 XRD patterns of the zeolite samples synthesized using DD3R seeds (a), Sigma-2 seeds (b), “amorphous” DD3R seeds (c), “amorphous” ZSM-58 seeds (d), and fumed silica seeds (e), in comparison with the simulated DD3R and Sigma-2 patterns. | |
 |
| | Fig. 3 SEM images of the samples synthesized by adding different pre-prepared DD3R seeds (a), Sigma-2 seeds (b), “amorphous” DD3R seeds (c), and “amorphous” ZSM-58 seeds (d). | |
To understand the role of “amorphous” DD3R or “amorphous” ZSM-58 seeds in the formation of DD3R crystals with a uniform shape and size, commercial fumed silica particles (0.1 wt%) were used as seeds in the synthesis, in which the aged gel composition was the same as that described in the last paragraph, but the crystallization time was 3 d. Interestingly, pure and large, but uniform, DD3R crystals were successfully synthesized, as evidenced by the XRD (Fig. 2) and SEM (Fig. 4) characterization results. This phenomenon strongly suggests that the seeds added in the synthesis solution mainly promote zeolite crystallization by providing an additional surface area for the dissolved material to grow onto, rather than crystal nucleation by providing “secondary building units” (secondary nucleation).53 In summary, the added seeds not only reduced the crystallization time, but also significantly affected the phase composition of the resulting product. As the amount of added seeds is very small, the effects of the seeds as an SDA, providing secondary building units to the synthesis mixture, should be very limited. In addition, because the crystallization is completed in a static environment, the secondary nucleation caused by friction, fluid shear, and other mechanisms is not expected in the current case. Therefore, the role of the seeds in the synthesis could be dominated by reducing the supersaturation of the synthesis mixture and thus inhibiting the heterocrystalline nuclei. Consequently, the pure DD3R crystals can be obtained using “amorphous” DD3R, “amorphous” ZSM-58 or fumed silica as seeds.
 |
| | Fig. 4 SEM images of the DD3R crystals synthesized using the commercially available fumed silica particles as seeds. | |
Effects of the amount of seeds
As already demonstrated above, the addition of seeds to the zeolite synthesis batch can significantly accelerate the rate of crystallization. Meanwhile, the addition of the seeds can also induce the formation of more nuclei so that the resultant zeolite crystals with a controllable morphology and size can be obtained by varying the amount of the seeds added.53,56 As an example, different amounts of “amorphous” DD3R seeds of 0.1, 0.2, and 0.3 wt% were individually added into the synthesis solution, the aged gel had a molar composition of 47 1-ADA:100 SiO2:100 KF:8000 H2O, and the crystallization time was 1 d to investigate the effects of the amount of seeds on the morphology and size of the resultant crystals. As shown in Fig. 5, when the amount of seeds increases from 0.1 to 0.2 wt%, the resultant crystal morphology remains unchanged but the size decreases. By further increasing the amount of the seeds to 0.3 wt%, the crystal size increases again. This indicates that the crystal size of the resultant zeolite can be decreased by increasing the number of nuclei added to the system or induced. On the other hand, increasing the amount of the seeds also enhances the surface area of the nuclei for the crystal growth, resulting in an increase in the crystal size of DD3R. The XRD patterns of the synthesized DD3R crystals are shown in Fig. S3 in the ESI.†
 |
| | Fig. 5 SEM images of the DD3R crystals synthesized using different amounts of “amorphous” DD3R seeds: (a) 0.1 wt%, (b) 0.2 wt%, and (c) 0.3 wt%. | |
Effects of the mineralizing reagent
Mineralizing reagents are the compounds (or ionic salts) needed to form new phases from a metastable phase through precipitation, dissolution and so forth.54,57,58 For the synthesis of silica-based zeolites that are normally prepared in an alkaline medium, the replacement of the hydroxide anions by fluoride anions as mineralizers makes it possible to obtain zeolites under near-neutral conditions.59,60 KF, instead of the conventionally applied ethylenediamine, has a positive influence on the formation of highly pure DD3R crystals.43 Therefore, the different molar ratios of KF to SiO2 in the synthesis, with 0.1 wt% of “amorphous” DD3R seeds, a molar ratio of 47 1-ADA:100 SiO2:x KF:8000 H2O (x varied from 50 via 100 to 150) in the gel, and a crystallization time of 1 d, were used to investigate the effects of the added amount of KF on the phase-purity and size of the resultant DD3R crystals. The SEM images shown in Fig. 6 indicate that the crystal size slightly increases with an increase in the dosed amount of KF. On the other hand, when the molar ratio of KF to SiO2 is reduced to 0.5, not only are a few Sigma-2 crystals formed, but also the morphology and size of the resultant DD3R crystals is not uniformly distributed anymore. However, this phase-impurity cannot be detected using the XRD technique (see the XRD pattern in Fig. S4 in the ESI†) owing to the very small number of Sigma-2 crystals formed in the sample. These results confirm that pure and uniform DD3R crystals are obtained only if the dosage of KF used is sufficient. It is well known that the added fluoride plays a decisive role in the zeolite synthesis.60–62 When fluoride is used as an SDA, the fluorine ion is generally believed to exist in the gaps of the zeolite structure, which may form a fluorosilicate ion and neutralize the positive charge of the SDA. As a mineralizing reagent, fluoride is considered to accelerate the formation of Si–OH, thus strengthening the crystallization of the zeolite and eliminating the connection defect of Si–O–Si. In the synthesis of the all-silica DD3R, KF mainly acts as a mineralizing reagent. At a low KF concentration, the rate of crystallization is lower than that of nucleation, leading to phase-impurity of the resultant product and an uneven crystal size distribution. In contrast, when the amount of KF used is sufficient, pure and uniform DD3R crystals can be obtained.
 |
| | Fig. 6 SEM images of the DD3R crystals synthesized using 0.1 wt% of “amorphous” DD3R seeds with different KF/SiO2 molar ratios of (a) 0.5, (b) 1.0, and (c) 1.5 in the gel. | |
Effects of the H2O and KOH dosages
The amount of added H2O should affect the concentration of the zeolite synthesis solution. If too little H2O is added, the synthesis solution is concentrated, leading to significant restrictions on mass and heat transfer. On the other hand, when too much H2O is added, the diluted synthesis solution will reduce the supersaturation for nucleation. Therefore, the dosage of H2O also influences the morphology and size of the resultant DD3R crystals. By decreasing the amount of H2O in the synthesis, that is, a gel molar composition of 47 1-ADA:100 SiO2:100 KF:6000 H2O, DD3R crystals with a uniform octahedron morphology and size were obtained, as shown in Fig. 7a, and the corresponding XRD pattern is shown in Fig. S5a in the ESI.† It was observed that when the KF concentration in the synthesis solution is increased to a certain extent, the morphology of the resulting DD3R crystals tends to transition from hexahedron to octahedron, see Fig. 5b and c. In the current case, although the tendency is not to investigate the effects of the added KF amount on the DD3R crystal morphology, the reduction of the amount of H2O used also increases the KF concentration, resulting in a transition in the morphology from hexahedron to octahedron.
 |
| | Fig. 7 SEM images of the DD3R crystals synthesized using 0.1 wt% of “amorphous” DD3R seeds with different gel molar compositions: 47 1-ADA:100 SiO2:100 KF:6000 H2O (a), and 47 1-ADA:100 SiO2:100 KF:8000 H2O:2 KOH (b). | |
In the synthesis of L-zeolite (LTL topology), the crystal morphology can be modulated by the K+ concentration.63 Therefore, it is speculated that the K+ concentration in the synthesis of DD3R also has an effect on its crystal morphology. For this purpose, a small dosage of KOH was added into the synthesis solution with a molar composition of 47 1-ADA:100 SiO2:100 KF:8000 H2O:2 KOH. Indeed, the morphology of the resultant DD3R crystals changed from hexahedron to diamond-like, as evidenced by the SEM image shown in Fig. 7b. The corresponding XRD pattern is shown in Fig. S5b in the ESI.†
Thermal stability and textural properties
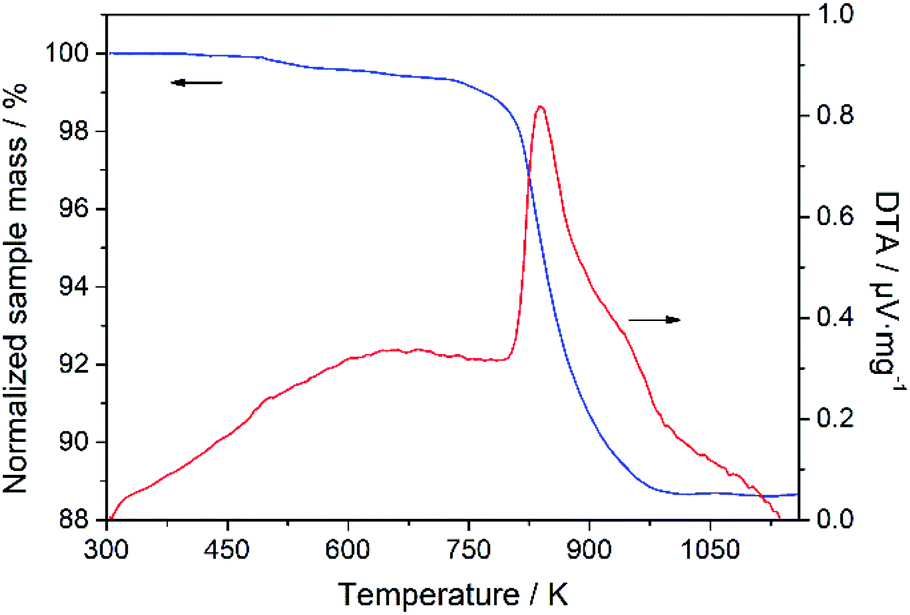
Thermal gravimetric analysis experiments were carried out, and the representative measured TGA curves are shown in Fig. S6 in the ESI.† As an example, Fig. 8 shows the TGA and differential thermal analysis (DTA) curves of Sample 1 represented in Table 2, in which the TGA curve indicates a small weight loss in a low temperature range, caused by the desorption of some of the adsorbed impurities on the sample, and a significant weight loss at about 823 K, corresponding to the exothermic peak in the DTA curve, owing to the decomposition of the SDA 1-ADA. There is no obvious change in the mass from 973 to 1173 K, indicating the complete decomposition of the SDA and the high thermal stability of the DD3R zeolite. The total weight loss is about 11 wt%, consistent with that reported in the literature.4
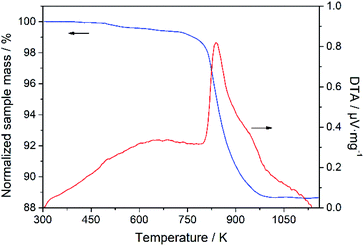 |
| | Fig. 8 TGA and DTA curves of sample 1 represented in Table 2. | |
Table 2 Details of the synthesis of some representative DD3R samples
| Sample code |
(1-ADA):(SiO2):(KF):(H2O) |
Seeds |
Synth. time/d |
| Type |
Amount/wt% |
| 1 |
47:100:100:8000 |
“Amorphous” DD3R |
0.2 |
1 |
| 2 |
47:100:100:8000 |
“Amorphous” DD3R |
0.1 |
1 |
| 3 |
47:100:150:8000 |
“Amorphous” DD3R |
0.1 |
1 |
| 4 |
47:100:100:6000 |
“Amorphous” DD3R |
0.1 |
1 |
| 5 |
47:100:100:8000 |
Fumed SiO2 |
0.1 |
3 |
Additionally, the yield of the DD3R crystals from each synthetic procedure was calculated using the following formula:9
| |
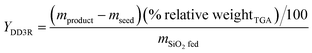 | (1) |
in which
mproduct,
mseed, and
mSiO2 fed are the masses of the product, added seeds, and the fed SiO
2, respectively, and % relative weight
TGA represents the percentage of the total weight loss of the product characterized by TGA. The synthesis details of several representative samples are shown in
Table 2 and their corresponding characterization results, including the crystal morphology and size, yield, and textural properties, are summarized in
Table 3. All of the yields shown in
Table 3 are slightly higher than those reported in the literature,
9 owing to the fact that the pure DD3R crystals were synthesized in the representative samples. The determined
SBET and
Vmicro of samples 1 and 2, shown in
Table 3, are almost identical to the theoretic ones,
1 indicating the high phase-purity and perfection of the synthesized DD3R crystals. In addition, the measured
SBET and
Vmicro decreased with the increasing DD3R crystal size, because the trace amount of the 1-ADA molecules remaining inside the crystals blocks the zeolite cavities for the adsorption of N
2, and this effect will be enhanced when the crystal size becomes larger.
Table 3 Properties of the synthesized representative DD3R samples
| Sample code |
Morphology and size |
SBET/m2 g−1 |
Vmicro/cm3 g−1 |
Weight loss/wt% |
Yield/% |
| 1 |
Hexahedral, ∼3 μm |
366 |
0.14 |
11 |
91 |
| 2 |
Hexahedral, ∼5 μm |
352 |
0.14 |
11 |
91 |
| 3 |
Hexahedral, ∼8 μm |
339 |
0.14 |
11 |
90 |
| 4 |
Octahedral, ∼8 μm |
283 |
0.13 |
11 |
88 |
| 5 |
Hexahedral, ∼15 μm |
276 |
0.12 |
11 |
89 |
Conclusions
By adding a small amount of “amorphous” DD3R or “amorphous” ZSM-58 seeds, pure DD3R crystals with a uniform morphology and size were successfully synthesized using 1-ADA as an SDA, KF as a mineralizing reagent, and LUDOX AS-30 as a silicon source at 443 K for 1 d. For the first time, the pure and uniform hexahedral DD3R crystals with a large size of ca. 15 μm were prepared using fumed silica as seeds, although the crystallization time took a longer period of 3 d. In addition to the seed effects, other effects, such as the mineralizing reagent, K+ and F− concentrations in the synthesis mixture, have been investigated as well. The results show that the role of the added seeds is dominated by reducing the supersaturation of the synthesis mixture and thus inhibiting the heterocrystalline nuclei. If the KF used in the synthesis is sufficient, pure DD3R crystals with a uniform morphology and size can be obtained. The current research, therefore, provides a facile protocol to synthesize pure and DD3R crystals with a controllable morphology and size and with a high reproducibility and productivity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support from the National Key R&D Program of the People's Republic of China (2017YFB0405802).
Notes and references
- H. Gies, J. Inclusion Phenom. Macrocyclic Chem., 1984, 2, 275–278 CrossRef CAS.
- H. Gies, Z. Kristallogr., 1986, 175, 93–104 CAS.
- M. J. den Exter, J. C. Jansen and H. van Bekkum, Stud. Surf. Sci. Catal., 1994, 84, 1159–1166 CrossRef CAS.
- M. J. den Exter, J. C. Jansen, H. van Bekkum and A. Zikánova, Zeolites, 1997, 19, 353–358 CrossRef CAS.
- W. Zhu, F. Kapteijn and J. A. Moulijn, Chem. Commun., 1999, 2453–2454 RSC.
- W. Zhu, F. Kapteijn, J. A. Moulijn and J. C. Jansen, Phys. Chem. Chem. Phys., 2000, 2, 1773–1779 RSC.
- W. Zhu, F. Kapteijn, J. A. Moulijn, M. C. den Exter and J. C. Jansen, Langmuir, 2000, 16, 3322–3329 CrossRef CAS.
- J. Gascon, W. Blom, A. van Miltenburg, A. Ferreira, R. Berger and F. Kapteijn, Microporous Mesoporous Mater., 2008, 115, 585–593 CrossRef CAS.
- C. Gücüyener, J. van den Bergh, A. M. Joaristi, P. C. M. M. Magusin, E. J. M. Hensen, J. Gascon and F. Kapteijn, J. Mater. Chem., 2011, 21, 18386–18397 RSC.
- H. I. Mahdia and O. Muraza, Sep. Purif. Technol., 2019, 221, 126–151 CrossRef.
- T. Tomita, K. Nakayama and H. Sakai, Microporous Mesoporous Mater., 2004, 68, 71–75 CrossRef CAS.
- S. Himeno, T. Tomita, K. Suzuki and S. Yoshida, Microporous Mesoporous Mater., 2007, 98, 62–69 CrossRef CAS.
- S. Himeno, T. Tomita, K. Suzuki, K. Nakayama, K. Yajima and S. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 6989–6997 CrossRef CAS.
- J. van den Bergh, W. Zhu, F. Kapteijn, J. A. Moulijn, K. Yajima, K. Nakayama, T. Tomita and S. Yoshida, Res. Chem. Intermed., 2008, 34, 467–474 CrossRef CAS.
- J. van den Bergh, W. Zhu, J. Gascon, J. A. Moulijn and F. Kapteijn, J. Membr. Sci., 2008, 316, 35–45 CrossRef CAS.
- E. Kim, S. Hong, E. Jang, J. H. Lee, J. C. Kim, N. Choi, C. H. Cho, J. Nam, S. K. Kwak, A. C. K. Yip and J. Choi, J. Mater. Chem. A, 2017, 5, 11246–11254 RSC.
- E. Hayakawa and S. Himeno, Sep. Purif. Technol., 2019, 218, 89–96 CrossRef CAS.
- Y. Jeong, S. Hong, E. Jang, E. Kim, H. Baik, N. Choi, A. C. K. Yip and J. Choi, Angew. Chem., Int. Ed., 2019, 58, 18654–18662 CrossRef CAS PubMed.
- M. Q. Wang, L. Bai, M. Li, L. Y. Gao, M. X. Wang, P. H. Rao and Y. F. Zhang, J. Membr. Sci., 2019, 572, 567–579 CrossRef CAS.
- J. Okazaki, H. Hasegawa, N. Chikamatsu, K. Yajima, K. Shimizu and M. Niino, Sep. Purif. Technol., 2019, 218, 200–205 CrossRef CAS.
- E. Hayakawa and S. Himeno, Microporous Mesoporous Mater., 2020, 291, 109695 CrossRef CAS.
- X. R. Wang, Y. T. Zhang, X. Y. Wang, E. Andres-Garcia, P. Du, L. Giordano, L. Wang, Z. Hong, X. H. Gu, S. Murad and F. Kapteijn, Angew. Chem., Int. Ed., 2019, 58, 15518–15525 CrossRef CAS PubMed.
- J. Kuhn, K. Yajima, T. Tomita, J. Gross and F. Kapteijn, J. Membr. Sci., 2008, 321, 344–349 CrossRef CAS.
- J. Kuhn, J. M. Castillo-Sanchez, J. Gascon, S. Calero, D. Dubbeldam, T. J. H. Vlugt, F. Kapteijn and J. Gross, J. Phys. Chem. C, 2009, 113, 14290–14301 CrossRef CAS.
- A. Vidoni and D. M. Ruthven, Ind. Eng. Chem. Res., 2012, 51, 1383–1390 CrossRef CAS.
- H. Maghsoudi, M. Soltanieh, H. Bozorgzadeh and A. Mohamadalizadeh, Adsorption, 2013, 19, 1045–1053 CrossRef CAS.
- T. Binder, C. Chmelik, J. Kärger, A. Martinez-Joaristi, J. Gascon, F. Kapteijn and D. Ruthven, Microporous Mesoporous Mater., 2013, 180, 219–228 CrossRef CAS.
- M. Moliner, C. Martínez and A. Corma, Chem. Mater., 2014, 26, 246–258 CrossRef CAS.
- A. Caravella, P. F. Zito, A. Brunetti, E. Drioli and G. Barbieri, J. Chem. Eng. Data, 2015, 60, 2343–2355 CrossRef CAS.
- A. Lauerer, T. Binder, J. Haase, J. Kärger and D. M. Ruthven, Chem. Eng. Sci., 2015, 138, 110–117 CrossRef CAS.
- T. Binder, A. Lauerer, C. Chmelik, J. Haase, J. Kärger and D. M. Ruthven, Ind. Eng. Chem. Res., 2015, 54, 8997–9004 CrossRef CAS.
- H. Maghsoudi, Adsorption, 2015, 21, 547–556 CrossRef CAS.
- E. Kim, K. Lim, T. Lee, K. Ha, D. Han, J. Nam, N. Choi, I. Cho, A. C. K. Yip and J. Choi, Chem. Eng. J., 2016, 306, 876–888 CrossRef CAS.
- Y. Zhang, S. Chen, R. Shi, P. Du, X. Qiu and X. Gu, Sep. Purif. Technol., 2018, 204, 234–242 CrossRef CAS.
- Y. Zhang, X. Qiu, Z. Hong, P. Du, Q. Song and X. Gu, J. Membr. Sci., 2019, 581, 236–242 CrossRef CAS.
- Y. Kumita, J. Gascon, E. Stavitski, J. A. Moulijn and F. Kapteijn, Appl. Catal., A, 2011, 391, 234–243 CrossRef CAS.
- J. van den Bergh, C. Gücüyener, J. Gascon and F. Kapteijn, Chem. Eng. J., 2011, 166, 368–377 CrossRef CAS.
- I. Yarulina, J. Goetze, C. Gücüyener, L. van Thiel, A. Dikhtiarenko, J. Ruiz-Martinez, B. M. Weckhuysen, J. Gascon and F. Kapteijn, Catal. Sci. Technol., 2016, 6, 2663–2678 RSC.
- I. Yarulina, A. Dikhtiarenko, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2017, 7, 300–309 RSC.
- J. Goetze, F. Meirer, I. Yarulina, J. Gascon, F. Kapteijn, J. Ruiz-Martínez and B. M. Weckhuysen, ACS Catal., 2017, 7, 4033–4046 CrossRef CAS PubMed.
- J. Goetze, I. Yarulina, J. Gascon, F. Kapteijn and B. M. Weckhuysen, ACS Catal., 2018, 8, 2060–2070 CrossRef CAS PubMed.
- J. H. Kang, F. H. Alshafei, S. I. Zones and M. E. Davis, ACS Catal., 2019, 9, 6012–6019 CrossRef CAS.
- Q. Yang, S. L. Zhong and X. Lin, Chin. J. Inorg. Chem., 2009, 25, 191–194 CAS.
- F. Zheng, W. Jing, X. Gu, N. Xu and J. Dong, J. Mater. Sci., 2013, 48, 6286–6292 CrossRef CAS.
- S. W. Yang, J. Provenzano, A. Arvanitis, W. H. Jing and J. H. Dong, J. Porous Mater., 2014, 21, 1001–1007 CrossRef CAS.
- M. Sen, A. Bose, P. Pal, J. K. Das, N. Das and H. Richter, J. Am. Ceram. Soc., 2014, 97, 52–55 CrossRef CAS.
- J. Zhang, M. Li, Y. Lin, C. Liu, X. Liu, L. Bai, D. Hu, G. Zeng, Y. Zhang, W. Wei and Y. Sun, Microporous Mesoporous Mater., 2016, 219, 103–111 CrossRef CAS.
- C. Liu, L. Bai, J. M. Zhang, D. Hu, M. Li, G. F. Zeng, Y. F. Zhang, W. Wei and Y. H. Sun, Microporous Mesoporous Mater., 2016, 225, 312–322 CrossRef CAS.
- L. Bai, C. Liu, M. Li, Y. H. Wang, G. Z. Nan, D. Hu, Y. F. Zhang, G. F. Zeng, W. Wei and Y. H. Sun, Microporous Mesoporous Mater., 2017, 239, 34–39 CrossRef CAS.
- K. Kajihara, R. Takahashi, H. Kato and K. Kanamura, J. Ceram. Soc. Jpn., 2018, 126, 221–229 CrossRef CAS.
- M. X. Wang, M. Li, M. Q. Wang, Y. Zhang, L. Bai and Y. F. Zhang, Microporous Mesoporous Mater., 2019, 288, 109596 CrossRef CAS.
- J. Kuhn, J. Gascon, J. Gross and F. Kapteijn, Microporous Mesoporous Mater., 2009, 120, 12–18 CrossRef CAS.
- S. Gonthier and R. W. Thompson, Stud. Surf. Sci. Catal., 1994, 85, 43–73 CrossRef CAS.
- M. T. Weller and S. E. Dann, Curr. Opin. Solid State Mater. Sci., 1998, 3, 137–143 CrossRef CAS.
- E. J. P. Feijen, J. A. Martens, P. A. Jacobs, J. Weitkamp and W. Hölderich, Stud. Surf. Sci. Catal., 1994, 85, 3–21 CrossRef.
- U. Díaz, V. Fornés and A. Corma, Microporous Mesoporous Mater., 2006, 90, 73–80 CrossRef.
- R. M. Barrer, Zeolites, 1981, 1, 130–140 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- H. Kessler, J. Patarin and C. Schott-Darie, Stud. Surf. Sci. Catal., 1994, 85, 75–113 CrossRef CAS.
- M. A. Camblor, L. A. Villaescusa and M. J. Díaz-Cabañas, Top. Catal., 1999, 9, 59–76 CrossRef CAS.
- S. I. Zones, R. J. Darton, R. Morris and S.-J. Hwang, J. Phys. Chem. B, 2004, 109, 652–661 CrossRef PubMed.
- Y. Yang, J. Pinkas, M. Schäfer and H. W. Roesky, Angew. Chem., Int. Ed., 1998, 37, 2650–2653 CrossRef CAS PubMed.
- A. G. Gomez, G. d. Silveira, H. Doan and C.-H. Cheng, Chem. Commun., 2011, 47, 5876–5878 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04293e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Shuhua Wangc and
Weidong Zhu
ab,
Shuhua Wangc and
Weidong Zhu