
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Metal-free [2+2+1] cycloaddition polymerization of alkynes, nitriles, and oxygen atoms to functional polyoxazoles†
Lichao Dong *,
Tian Lan,
Yin Liang,
Shifeng Guo and
Hao Zhang
*,
Tian Lan,
Yin Liang,
Shifeng Guo and
Hao Zhang
The 306th Institute of China Aerospace Science & Industry Corp., Beijing, 100074, China. E-mail: 610557405@qq.com
First published on 25th June 2020
Abstract
The metal-free [2+2+1] cycloaddition polymerization of alkynes, nitriles, and O-atoms for the regioselective assembly of highly substituted oxazole compounds has been achieved by the use of iodosobenzene (PhIO) with trifluoromethanesulfonic acid (TfOH). The present reaction could be applied to a facile synthesis of polyoxazoles. In this work, the cycloaddition polymerization of 4-cyano-4′-ethynylbiphenyl and PhIO was developed and modified polyoxazole was prepared. All experimental conditions such as polymerization solvent, temperature, catalyst and time were systematically studied. The structure of the obtained polyoxazole was characterized by GPC and NMR, and its thermal properties were studied by TGA. In addition, the good thermal stability of polyoxazoles with unreacted terminal alkynes and cyano groups makes them potentially useful for modifying resins.
Introduction
Multicomponent cycloaddition reactions (MCCA), which allow for multiple bond formation in a single operation, can provide more atom-, step-, and time-economical conversion of simple starting materials to complex and multifunctional cyclic compounds. Based on these reactions, mild and efficient approaches to substituted pyridines, furans, pyrroles, and some important azoles using heteroatom-containing unsaturated compounds as starting materials have been developed.1–6In recent decades, MCCA have drawn great attention and have been exploited to synthesize new functional polymers because of their high atom efficiency, shorter reaction time and higher overall yields than multiple-step syntheses.7–9 Furthermore, MCCA can introduce some special functional groups which largely enrich the functions and diversities of compounds. And a series of polymers with new main chains, side groups, and topologies have therefore been successfully prepared, indicating the vitality of MCCA in polymer science.10–12
Since oxazole derivatives have found widespread applications not only as biologically active compounds but also as synthetic intermediates,13–15 much effort has been directed toward devising methods for the synthesis of substituted oxazoles.16–18 However, the research on oxazoles mainly concentrated in the small molecules at present. The polyoxazoles may have new features and functions different from small molecules. In 2000, Gong's group reported the synthesis of a hyperbranched poly(aryletheroxazole) with terminal phenolic groups, an ABB′ monomer containing a pair of phenolic groups and an aryl fluoride which is activated toward displacement by the attached oxazole ring was prepared. The nucleophilic substitution of the fluoride with the phenolic group leads to the formation of ether linkage and subsequently the hyperbranched poly(aryletheroxazole).19 In 2001, Peter Wipf et al. reported a novel approach for polyoxazole synthesis, which was prepared by a Chan-type rearrangement of a tertiary amide. But this method requires multiple steps of reaction, and the operation is complicated, which limits its application.20 In 2005, Jeffery M. Atkins et al. reported a two-stage iterative process for the synthesis of polyoxazoles, which begins with C2-chlorination of a lithiated oxazole using hexachloroethane. However, the method also requires multiple steps and the obtained polymer is an oligomer.21 Although there are some related reports, there is still few research on polyoxazoles. Following the daily increasing requirement on molecular functionality, developing simple and efficient polymerization and providing novel or unprecedented structures has become an important issue.
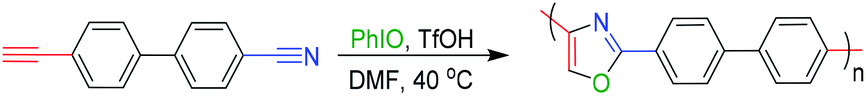
In 2013 and 2017, Akio Saito et al. reported that the multicomponent cycloaddition of oxazole could be achieved by employing alkynes, nitriles, and O-atoms (Scheme 1).22,23 Inspired by the above-mentioned small molecule's reaction, we have successfully designed and developed a facile and efficient polymerization to produce functional polyoxazoles in this work (Scheme 2). The reaction conditions were optimized systematically, and polyoxazoles with satisfied solubility, thermal stability and molecular weights (up to 3300 g mol−1) were obtained in good yield (up to 85%). Polyoxazoles' structures were well characterized by GPC and NMR. Polymerization mechanism is proposed according small molecule's reaction. Moreover, the thermal properties were investigated and the good thermal stability of the polyoxazoles with unreacted terminal alkynes and nitriles endows them potential applications for modified resins.
 | ||
| Scheme 1 Syntheses of oxazoles by one-pot MCR. | ||
 | ||
| Scheme 2 Synthetic route for the polyoxazoles. | ||
Experimental
Materials
Unless stated otherwise, all chemicals were obtained from commercial suppliers and used without further purification. The monomers 4-cyano-4′-ethynylbiphenyl were synthesized according to the commonly used synthetic routes (see Scheme 3). 1-Bromo-4-ethynylbenzene and 4-cyanophenylboronic acid were bought from Energy Chemical. PhIO and TfOH were bought from Innochem and used without further purification. Chloroform-d was bought from Innochem. All monomers put into vacuum oven to drying.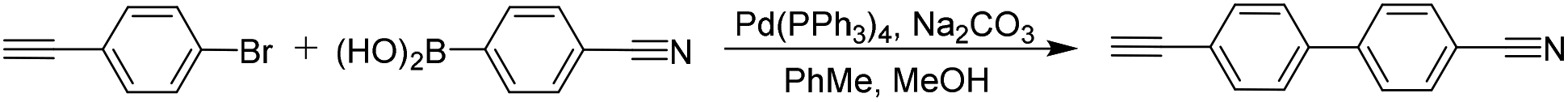 | ||
| Scheme 3 The synthetic route of monomer 4-cyano-4′-ethynylbiphenyl. | ||
Instruments
Weight-average molecular weights (Mw) and polydispersity indices (Mw/Mn) of the polymers were estimated on a Waters gel permeation chromatography (GPC) system equipped with a Waters 1515 isocratic HPLC pump and Waters 2414 refractive index detector. Polystyrene standards were utilized, and THF was used as the eluent at a flow rate of 1.0 mL min−1. 1H NMR and 13C NMR spectra were measured on a Bruker AV 400 spectrometer.Polymerization
All the polymerization reactions were conducted using standard techniques. A typical polymerization procedure is given below by employing the conditions in Table 1, entry 2 as an example. Into a 10 mL tube equipped with a magnetic stir bar were added 4-cyano-4′-ethynylbiphenyl (0.49 mmol, 100 mg), PhIO (0.098 mmol, 21.7 mg), TfOH (0.082 mmol, 7.25 μL) and 3 mL of DMF. The reaction mixture was stirred under N2 atmosphere at 80 °C for 24 h. Then the solution was added into 200 mL of methanol with vigorous stirring. The precipitate was filtered and washed with hexane and dried in a vacuum at room temperature to a constant weight. Other polymerization reactions also have the same procedure in different conditions.| Entries | Solvent | Yield (%) | M n b | M w b | Đ c |
|---|---|---|---|---|---|
| a Carried out at 80 °C for 24 h, concentration: 0.16 M. PhIO (1.8 equiv.), TfOH (1.5 equiv.). b Determined by GPC in THF using a linear polystyrene as calibration standard; Mw = weight-average molecular weight. c Đ = Mw/Mn, where Mn = number-average molecular weight. | |||||
| 1 | DMSO | 50 | 3200 | 3400 | 1.06 |
| 2 | DMF | 80 | 2400 | 2900 | 1.2 |
| 3 | DCE | 12 | 1000 | 1400 | 1.4 |
| 4 | 1,4-Dioxane | 20 | 4400 | 5500 | 1.25 |
| 5 | Toluene | 10 | 1700 | 2300 | 1.35 |
Synthesis of 4-cyano-4′-ethynylbiphenyl
The monomers 4-cyano-4′-ethynylbiphenyl were prepared by the Suzuki reaction24 and the synthetic route was shown in Scheme 3. The structural information was characterized by spectroscopic techniques, and the results agreed with its target structures (Fig. S1 and S2†).Under a nitrogen atmosphere, methanol (60 mL) and toluene (150 mL) were injected into a 500 mL round-bottom flask containing 1 g (5.5 mmol) of 1-bromo-4-ethynylbenzene, 0.81 g (5.5 mmol) of 4-cyanophenylboronic acid, 0.16 g (0.14 mmol) of Pd(PPh3)4 and 2.01 g (18.97 mmol) of Na2CO3. The solution was stirred at 90 °C for 24 h and cooled down to room temperature. After solvent was evaporated by reduced pressure distillation, the solid residues were dissolved in proper amount of dichloromethane, and the obtained solution was washed with dilute hydrochloric acid and deionized water, respectively. The organic phase was dried over anhydrous MgSO4 and concentrated, the crude product obtained was purified by silica gel column chromatography using dichloromethane/petroleum ether (1/4, v/v) as eluent. The 4-cyano-4′-ethynylbiphenyl was obtained as a white solid in 73.0% yield. 1H NMR (400 MHz, CDCl3) δ 7.76–7.72 (d, 1.8 Hz, 2H), 7.68 (d, J = 8.4, 1.8 Hz, 2H), 7.60 (d, J = 8.4, 1.8 Hz, 2H), 7.55 (d, J = 8.3, 1.8 Hz, 2H), 3.18 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 144.63, 139.39, 132.85, 132.71, 127.67, 127.14, 122.58, 118.80, 111.39, 83.06, 78.80.
Results and discussion
Polymerization
Generally, the optimized conditions for small molecule reactions are not necessarily suitable for polymerizations. Thus, the polymerization conditions were first systematically investigated.Firstly, we investigated the solvent effect on the polymerization; the results are summarized in Table 1. The property of solvent is significant for the specific occurrence of polymerization. Five different solvents including dimethyl sulfoxide (DMSO, aprotic solvent), N,N-dimethylformamide (DMF, aprotic solvent), 1,2-dichloroethane (DCE, haloalkane solvent), 1,4-dioxane (aprotic solvent) and toluene (aromatic hydrocarbon solvent) were tested. The highest yield (80%), as well as the modest Mw (2900 g mol−1), was achieved in DMF-a suitable solvent for the synthesis of polyoxazoles via cycloaddition reaction. Although the polymerization also occurred in other solvents, Mw or yields of the polymer products was lower significantly. Therefore, DMF was used as the polymerization solvent in the following experiments.
Secondly, the effects of catalyst concentration on the polymerization efficiency were determined. According to the mechanism of small molecule's reaction, we fixed the ratio of PhIO and TfOH. Then decreasing the total amount of the two catalysts to 0.2 equiv. and 0.167 equiv. and produced a polymer with the desired Mw of 3300 g mol−1 in the moderate isolated yield of 85% (Table 2, entry 6). There was no signification change in molecular weight and yields in the process, which was mainly due to the increased viscosity of the polymerization system and rigidity of polymer structure. This result shown that polymerization is naturally terminated during the later period of polymerization because of the more difficult diffusion of chain segments. Therefore, considering the economy of catalyst, PhIO (0.2 equiv.) and TfOH (0.167 equiv.) were used as the catalyst in the following experiments.
| Entries | PhIO (equiv.) | TfOH (equiv.) | Yield (%) | M n | M w | Đ |
|---|---|---|---|---|---|---|
| a Carried out in DMF for 24 h, concentration: 0.16 M. | ||||||
| 1 | 1.2 | 1 | 78 | 2500 | 3000 | 1.2 |
| 2 | 0.9 | 0.75 | 84 | 2500 | 3100 | 1.24 |
| 3 | 0.6 | 0.5 | 82 | 2600 | 3200 | 1.23 |
| 4 | 0.45 | 0.375 | 80 | 2600 | 3200 | 1.23 |
| 5 | 0.3 | 0.25 | 81 | 2600 | 3300 | 1.27 |
| 6 | 0.2 | 0.167 | 85 | 2700 | 3300 | 1.22 |
The effects of temperature on the polymerization are summarized in Table 3. The yield of polymer product increased with the increase of temperature from 40 to 120 °C. However, the molecular weight almost unchanged. This result further supports the above inference: polymerization is naturally terminated because of the more difficult diffusion of chain segments during the later period of polymerization. Therefore, considering the simplicity of the operation, the polymerization temperature was optimized as 40 °C.
| Entries | Temperature (°C) | Yield (%) | M n | M w | Đ |
|---|---|---|---|---|---|
| a Carried out in DMF for 24 h, concentration: 0.16 M. PhIO (0.2 equiv.), TfOH (0.167 equiv.). | |||||
| 1 | 40 | 75 | 2400 | 2900 | 1.2 |
| 2 | 60 | 77 | 2400 | 2900 | 1.2 |
| 3 | 80 | 80 | 2400 | 2900 | 1.2 |
| 4 | 90 | 82 | 2500 | 3000 | 1.2 |
| 5 | 100 | 81 | 2500 | 3000 | 1.2 |
| 6 | 110 | 84 | 2400 | 2900 | 1.2 |
| 7 | 120 | 85 | 2500 | 3100 | 1.24 |
The effects of time course on the polymerization were evaluated, as shown in Table 4. The yield of the polymer product gradually increased with the polymerization time increased from 2 h to 24 h. However, further prolonging the reaction time to 36 h resulted in almost unchanged Mw, suggesting that the polymer chain length growth became difficult after a certain time. One reason is the increased viscosity of the polymerization system and the other is the more difficult diffusion of chain segments during the later period of polymerization. Based on these results, take the yield into consideration, the reaction time was optimized as 24 h.
| Entries | Time (h) | Yield (%) | M n | M w | Đ |
|---|---|---|---|---|---|
| a Carried out in DMF at 40 °C, concentration: 0.16 M. PhIO (0.2 equiv.), TfOH (0.167 equiv.). | |||||
| 1 | 2 | 60 | 2500 | 3000 | 1.2 |
| 2 | 6 | 66 | 2600 | 3100 | 1.19 |
| 3 | 12 | 74 | 2500 | 3000 | 1.2 |
| 4 | 24 | 80 | 2400 | 2900 | 1.2 |
| 5 | 36 | 82 | 2500 | 3000 | 1.2 |
To sum up, the poor solubility caused the more losses of polymer during the post-treatment process, which resulted in a lower yield. Considering the environmental friendliness, economy and simple experimental operation, we chose N,N-dimethylformamide (DMF) as solvent, PhIO (0.2 equiv.) and TfOH (0.167 equiv.) as the dosage of catalyst, reaction time of 24 h, and polymerization temperature of 40 °C as the optimal polymerization conditions for further investigation.
Structural characterization
As above mentioned, the resultant polymers are all soluble in highly polar organic solvents, which enable us to characterize their structures spectroscopically. Satisfactory analysis data corresponding to their expected molecular structures were obtained from 1H NMR spectroscopy, which could offer detailed information about the polymer structures.In the 1H NMR spectra (Fig. 1), the chemical shift of 4-cyano-4′-ethynylbiphenyl mainly assembled at δ = 3.1 ppm and 7.4–8.0 ppm, which corresponding to the proton resonances of alkynyl and benzene ring group. After the polymerization, the chemical shift of newly formed oxazole ring still resonance at δ = 7.4–8.0 ppm, which was overlapped with the peak of benzene ring. The overall peak shape appears as a broad peak. These results suggest that the cyano groups and alkyne groups have been converted into oxazole group by the polymerization. Moreover, the terminal alkyne and cyano groups has the ability to continue with the electrophilic addition process according to the small molecule reaction, because there is still alkyne or cyano groups in the obtained polymer from Fig. 1B. However, due to the higher viscosity achieved during the later period of polymerization, the diffusion of macromolecular terminal alkynes becomes more difficult, and then, polymerization is naturally terminated.
 | ||
| Fig. 1 1H NMR spectra of (A) 4-cyano-4′-ethynylbiphenyl, (B) polyoxazoles in CDCl3. The solvent peaks are marked with asterisks. | ||
The 13C NMR spectra further substantiate the conclusion drawn from their 1H NMR spectral analysis (Fig. 2). In the 13C NMR spectra, the peak at δ = 80, 85 and 118 ppm, which were attributed to the resonance of the Ca, Cb and Cc in 4-cyano-4′-ethynylbiphenyl, completely disappeared in the polyoxazole spectra, as shown in Fig. 2. Meanwhile, the new peak corresponding to the oxazole carbons appeared at δ = 146 ppm in the polyoxazole spectra. All the results abovementioned indicated that the polymerization reacted in a manner similar to small molecules reaction and the target polymer was obtained as we expected.
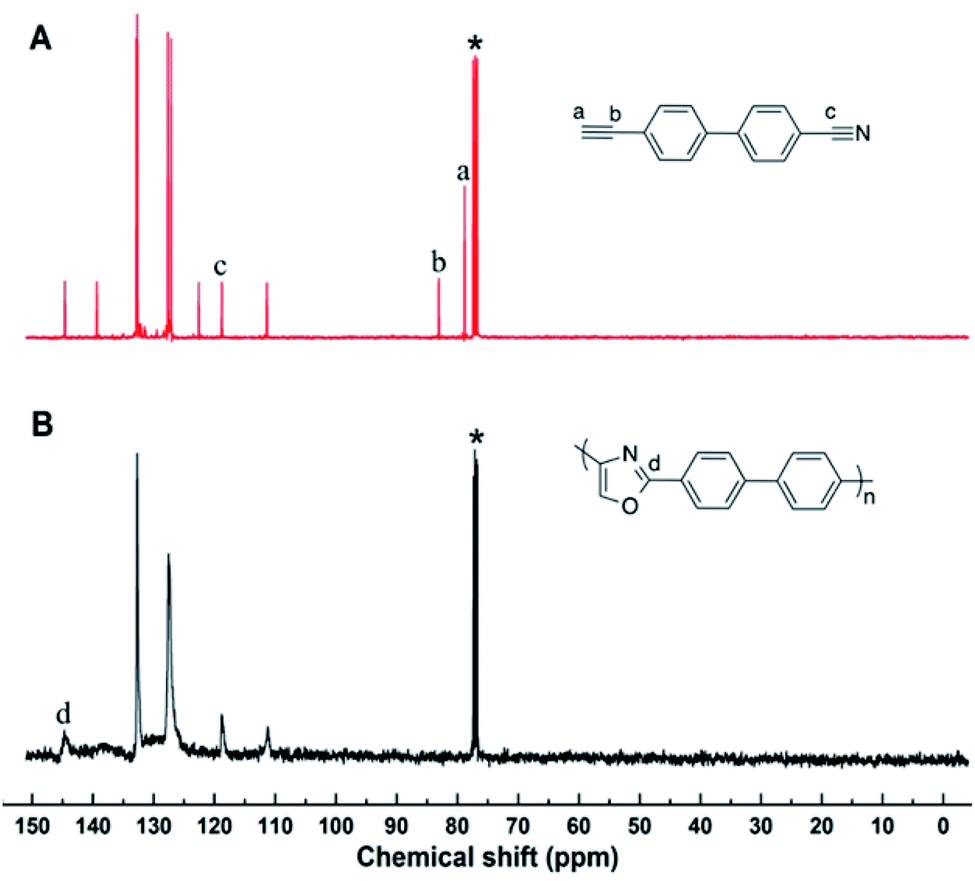 | ||
| Fig. 2 13C NMR spectra of (A) 4-cyano-4′-ethynylbiphenyl, (B) polyoxazoles in CDCl3. | ||
Based on the obtained polymer is a conjugated structure, we measured its UV absorption, the Fig. S3† shows the UV absorption spectra of the polymer (Table 2, entry 6) in THF. The maximum absorption peaks was located at 290 nm, respectively, which belonged to the π–π* transition of the phenyl. Meanwhile, this absorption peak ranges from 290 nm to 400 nm, indicating that the conjugate absorption of the structure (π–π* transition of the phenyl and oxazole group) is continuous. This result shows that the polymers synthesized are conjugated, and further proof of the occurrence of polymerization.
Polymerization mechanism
To study the polymerization mechanism, we referred to the literature studying small molecule's reaction and proposed a tentative mechanism of this polymerization, as shown in Scheme 4. At first, the ion pair of Ph-I+(OH)[OTf]− is formed by the addition reaction of PhIO and TfOH. Then the formed Ph-I+(OH)[OTf]− proceeds the electrophilic addition reaction with alkynes to form the OTf and I(OH)(Ph) substituted olefin derivatives Int-A. Int-B is formed through the subsequent ligand exchange in Int-A. And then, the nucleophilic vinylic substitution of Int-B with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CRCN, which contributes to the Michael-acceptor ability of Int-B, leads to Int-C. Subsequently, Int-C reacts with H2O to generate Int-D and/or Int-E, which are converted to Int-F. Finally, the reductive elimination of Int-F gives the target oxazoles G. The G continue to repeat the whole process from Int-A to G by the same catalytic mechanism. Ultimately, the polyoxazoles are obtained by repeating this process until the polymerization process is over. Due to the higher viscosity achieved during the later period of polymerization, the diffusion of chain segments becomes more difficult, and then, polymerization is naturally terminated.
CRCN, which contributes to the Michael-acceptor ability of Int-B, leads to Int-C. Subsequently, Int-C reacts with H2O to generate Int-D and/or Int-E, which are converted to Int-F. Finally, the reductive elimination of Int-F gives the target oxazoles G. The G continue to repeat the whole process from Int-A to G by the same catalytic mechanism. Ultimately, the polyoxazoles are obtained by repeating this process until the polymerization process is over. Due to the higher viscosity achieved during the later period of polymerization, the diffusion of chain segments becomes more difficult, and then, polymerization is naturally terminated.
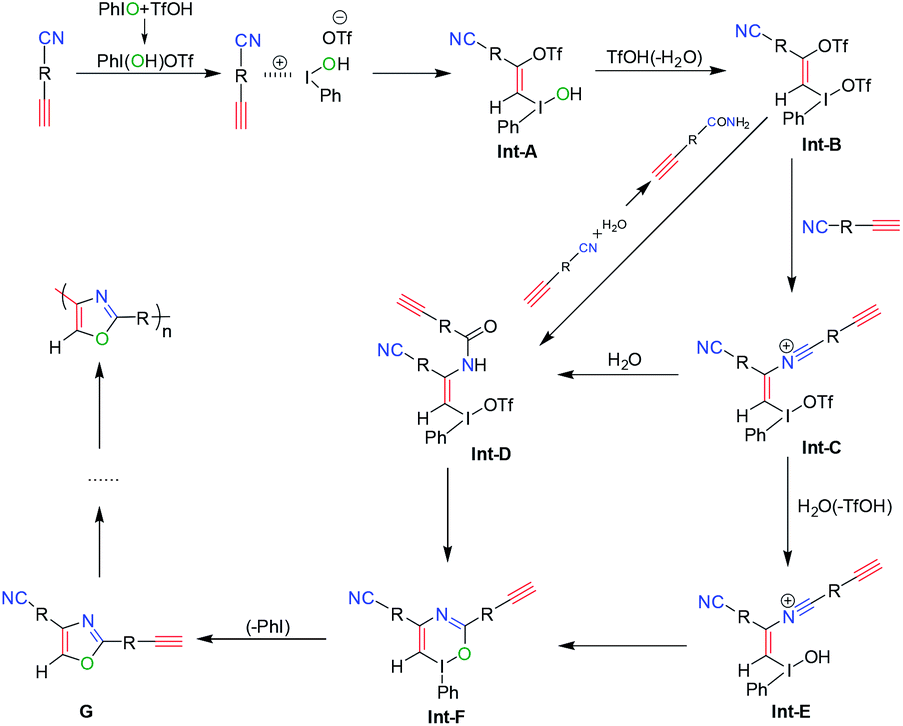 | ||
| Scheme 4 The proposed polymerization process of polyoxazoles. | ||
Thermal properties and applications
The thermal stability of polymer was evaluated by thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC). As shown in Fig. 3, the polymer lost only 5% weight as heated at 340 °C under a nitrogen atmosphere due to the degradation of the unstable 6-substituent group, suggesting that they were highly thermal-stable. Fig. S4† shows the differential scanning calorimetry (DSC) curves of the polymer (Table 2, entry 6). The glass transition temperature (Tg) was 208 °C. The results indicated that the polyoxazoles had the good thermal stability, which further confirms the analysis of TGA. Therefore, the good thermal stability of the polyoxazoles with unreacted terminal alkynes and nitriles endows them application potentials for modified resins materials.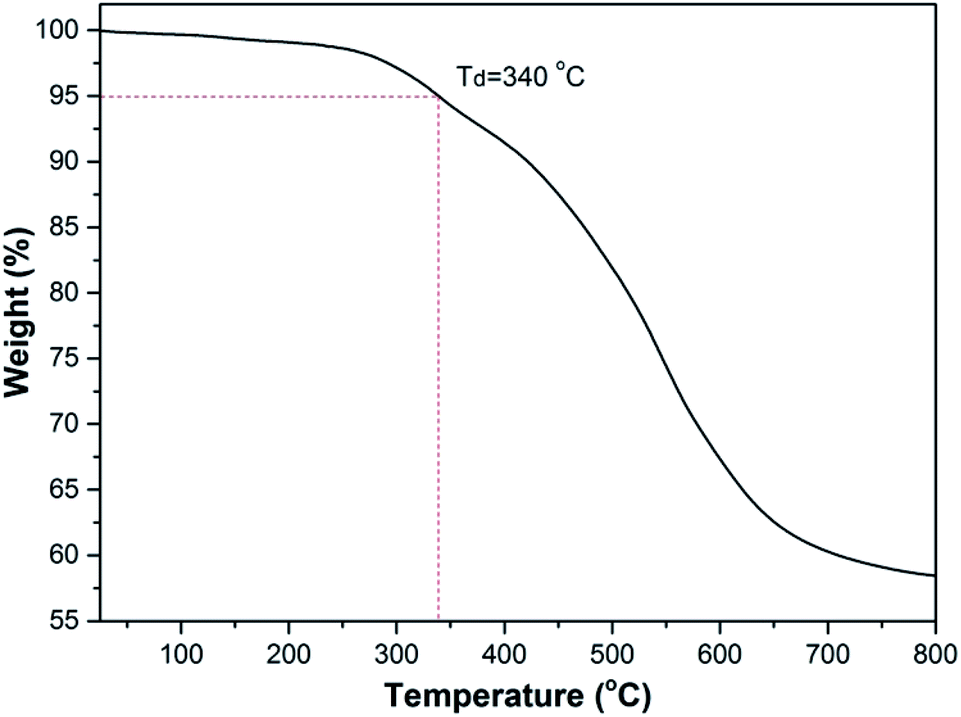 | ||
| Fig. 3 Thermograms of the polyoxazoles. The measurements were performed under N2 atmosphere at the heating rate of 10 °C min−1. | ||
The polyoxazoles can modify the polyarylacetylene or cyanate ester resins by binary copolymer, which can endow the modified resin with high temperature resistance performance and mechanical property. The detailed methods are as following (Scheme 5).
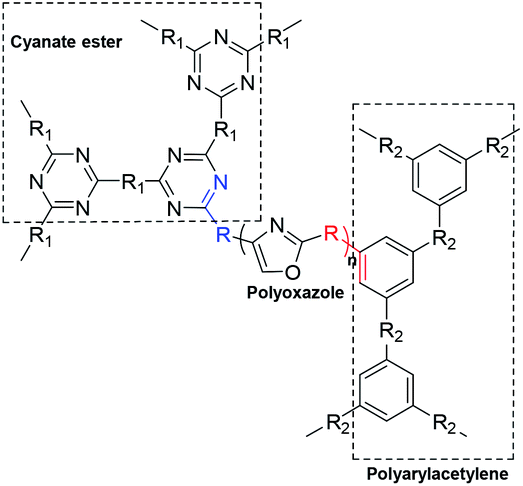 | ||
| Scheme 5 Cyanate ester or polyarylacetylene modified by the polyoxazoles. | ||
In addition, we also studied the effect of molecular weight on the thermodynamic stability of the resulting polymer, as shown in Fig. S5,† with the molecular weight gradually increasing, the thermal decomposition temperature (Tg) of the resulting polymer also gradually increases, and the thermal stability is improved. This is mainly due to the entanglement of the polymer segments, which increased the interaction between the molecular chains and improved the thermal stability of the polymer.
Conclusions
In this paper, the cycloaddition polymerization of 4-cyano-4′-ethynylbiphenyl and PhIO was developed to prepare modified polyoxazoles with metal-free reactions. The structure of polyoxazoles was well characterized by GPC and NMR and the thermal properties were investigated. Furthermore, the good thermal stability of the polyoxazoles with unreacted terminal alkynes and nitriles endows them potential applications for modified resins materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants 21490574, 21875019, 51673024, and 51803009).Notes and references
- J. C. Wu, W. B. Xu, Z. X. Yu and J. Wang, J. Am. Chem. Soc., 2015, 137, 9489–9496 CrossRef CAS.
- V. Nair, A. U. Vinod, N. Abhilash, R. S. Menon, V. Santhi, R. L. Varma, S. Viji, S. Mathew and R. Srinivas, Tetrahedron, 2003, 59, 10279–10286 CrossRef CAS.
- H. C. Chiu and I. A. Tonks, Angew. Chem., Int. Ed., 2018, 57, 6090–6094 CrossRef CAS.
- I. V. Efimov, Y. M. Shafran, N. N. Volkova, N. A. Beliaev, P. A. Slepukhin and V. A. Bakulev, Chem. Heterocycl. Compd., 2016, 52, 743–749 CrossRef CAS.
- H. M. Sheldrake, T. W. Wallace and C. P. Wilson, Org. Lett., 2005, 7, 4233–4236 CrossRef CAS.
- B. Song, K. Hu, A. J. Qin and B. Z. Tang, Macromolecules, 2018, 51, 7013–7018 CrossRef CAS.
- Y. Z. Huang, P. Chen, B. Wei, R. R. Hu and B. Z. Tang, Chin. J. Polym. Sci., 2019, 37, 428–436 CrossRef CAS.
- Y. Li and B. J. Cheng, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1645–1650 CrossRef CAS.
- B. Wei, W. Z. Li, Z. J. Zhao, A. J. Qin, R. R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 5075–5084 CrossRef CAS.
- D. J. Dibble, M. J. Umerani, A. Mazaheripour, Y. S. Park, J. W. Ziller and A. A. Gorodetsky, Macromolecules, 2015, 48, 557–561 CrossRef CAS.
- H. Q. Deng, R. R. Hu, E. G. Zhao, C. Y. K. Chan, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2014, 47, 4920–4929 CrossRef CAS.
- W. Q. Fu, L. C. Dong, J. B. Shi, B. Tong, Z. X. Cai, J. G. Zhi and Y. P. Dong, Polym. Chem., 2018, 9, 5543–5550 RSC.
- H. Z. Zhang, Z. L. Zhao and C. H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS.
- X. H. Li, J. M. Li, Y. H. Tao, Z. X. Peng, P. Lu and Y. G. Wang, Sens. Actuators, B, 2017, 247, 609–616 CrossRef CAS.
- Y. S. Li, D. K. Hua, D. S. Zhao, X. Y. Liu, H. W. Jin, G. P. Song, Z. N. Cui and L. H. Zhang, Bioorg. Med. Chem., 2017, 25, 1852–1859 CrossRef CAS.
- M. Bakherad, R. Doosti, M. Mirzaee and K. Jadidi, Tetrahedron, 2017, 73, 3281–3287 CrossRef CAS.
- A. Ohura, T. Itoh, H. Ishida, A. Saito and K. Yamamoto, Asian J. Org. Chem., 2017, 6, 673–676 CrossRef CAS.
- T. Soeta, A. Matsumoto, Y. Sakata and Y. Ukaji, J. Org. Chem., 2017, 82, 4930–4935 CrossRef CAS.
- Z. H. Gong, C. M. Leu, F. I. Wu and C. F. Shu, Macromolecules, 2000, 33, 8527–8533 CrossRef CAS.
- P. Wipf and J. L. Methot, Org. Lett., 2001, 3, 1261–1264 CrossRef CAS.
- J. M. Atkins and E. Vedejs, Org. Lett., 2005, 15, 23351–23354 Search PubMed.
- T. Yagyu, Y. Takemoto, A. Yoshimura, V. V. Zhdankin and A. Saito, Org. Lett., 2017, 19, 2506–2509 CrossRef CAS.
- A. Saito, A. Taniguchi, Y. Kambara and Y. Hanzawa, Org. Lett., 2013, 15, 2672–2675 CrossRef CAS.
- E. E. Pocock, R. J. Mandle and J. W. Goodby, Soft Matter, 2018, 14, 2508–2514 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04249h |
| This journal is © The Royal Society of Chemistry 2020 |