
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arenium cation or radical cation? An insight into the cyclodehydrogenation reaction of 2-substituted binaphthyls mediated by Lewis acids†
Patricia Camargo Solórzano,
María T. Baumgartner ,
Marcelo Puiatti* and
Liliana B. Jimenez*
,
Marcelo Puiatti* and
Liliana B. Jimenez*
INFIQC, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, X5000HUA, Córdoba, Argentina. E-mail: mpuiatti@fcq.unc.edu.ar; ljimenez@fcq.unc.edu.ar; Tel: +54-351-5353867 int. 53330
First published on 9th June 2020
Abstract
Perylene and its derivatives are some of the most interesting chromophores in the field of molecular design. One of the most employed methodologies for their synthesis is the cyclodehydrogenation of binaphthyls mediated by Lewis acids. In this article, we investigated the cyclodehydrogenation reaction of 2-substituted binaphthyls to afford the bay-substituted perylene. By using AlCl3 as a Lewis acid and high temperatures (the Scholl reaction), two new products bearing NH2 and N(CH3)2 groups at position 2 of the perylene ring were synthesized. Under these conditions, we were also able to obtain terrylene from ternaphthalene in 38% yield after two cyclodehydrogenation reactions in a single step. The attempts to promote the formation of a radical cation (necessary intermediary for the oxidative aromatic coupling mechanism) by using FeCl3 or a strong oxidant like 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) did not yield the expected products. DFT calculations suggested that the lack of reaction for oxidative aromatic coupling is caused by the difference between the oxidation potentials of the donor/acceptor couple. In the case of the Scholl reaction, the regiochemistry involved in the formation of the σ-complex together with the activation energy of the C–C coupling reaction helped to explain the differences in the reactivity of the different substrates studied.
Introduction
In the last decades, there has been special interest in the design of specific chromophoric structures due to their possible application in molecular devices such as light-collecting antennas and organic light emitting diodes (OLEDs), among other applications.1 Among these structures, perylene and its derivatives are particularly interesting, since they exhibit excellent electronic and optical properties.2 Perylene shows characteristic fluorescence with high quantum yield, which varies depending on both the nature of the substituents attached to the polycycle and their positions (e.g. the axial or equatorial regions).3 Recent applications of perylene derivatives (mostly perylene diimides, PDIs),4 include organic field effect transistors,5 organic photovoltaic cells,6 optical switches7 and molecular wires.8Because of the importance of perylene and its derivatives, different strategies have been developed for their synthesis (Scheme 1, I–V). Among these routes are intramolecular condensation of quinone derivatives (I),9 base-induced dimerization of benzoisoquinoline-diones (II),10 the Ullmann reaction (III),11 decarboxylation of the perylene-3,4,9,10-tetracarboxylic dianhydride mediated by basic conditions (IV),12 and the cyclodehydrogenation of the binaphthalene (V).13 In general, the yields of these reactions are good to low. Perhaps, the best option to obtain large quantities of perylene is route IV, but it usually requires high temperatures (more than 350 °C). On the other hand, route V is an attractive option, since it can take place through up to three different mechanisms depending on the reactants and the experimental conditions used. For example, Rickhaus et al.13d were able to obtain quantitative yields of perylene by using an excess of electrons (route V), however no bay-substituted perylene was reported following this last one. Despite these recent advances, the synthetic procedures to generate perylene are currently limited. Furthermore, only routes I and II can yield substituted perylenes; peri-substituted (axial region) by route II and ortho- and bay-substituted by route I.
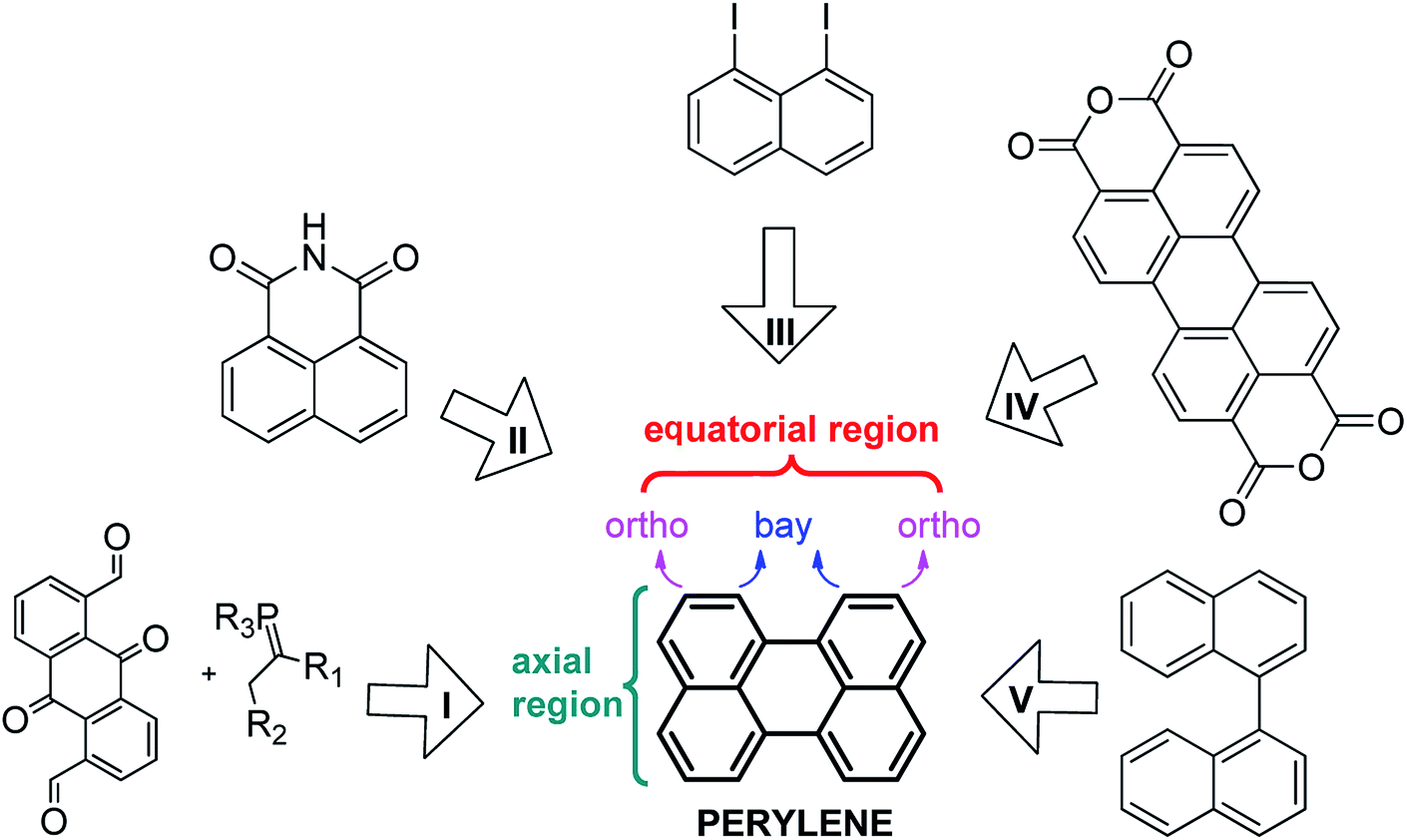 | ||
| Scheme 1 Retrosynthesis of perylene. Route: (I) intramolecular condensation of quinone derivatives; (II) dimerization of benzoisoquinoline-diones in basic media; (III) Ullmann reaction; (IV) decarboxylation of the perylene tetracarboxylic dianhydride mediated by basic conditions and high temperatures and (V) cyclodehydrogenation of the binaphthalene. | ||
Although the Lewis acid-mediated cyclodehydrogenation (another way by which route V can occur) is one of the oldest methodologies used to obtain polycyclic aromatic hydrocarbons,14 i.e. perylene,13b,15 terrylene,16 and dibenzotetraphenylperiflanthene,17 the operating mechanism presents some ambiguities. In front of this discussion, Gryko et al.18 proposed the distinction between two possible mechanisms through which polycyclic aromatic products can be generated. One of the postulated mechanisms is known as the Scholl reaction, that proceeds through the formation of an electrophilic complex or σ-complex (shown as H+ for simplicity in Scheme 2, but it could also be a σ-complex formed with a Lewis acid).18 Usually, methodologies related to this mechanism involve the use of strong Lewis acids such as AlCl3 dissolved in chlorobenzene,19 complexes of fused salts such as AlCl3/NaCl20 or AlCl3/SO2,21 among others.15
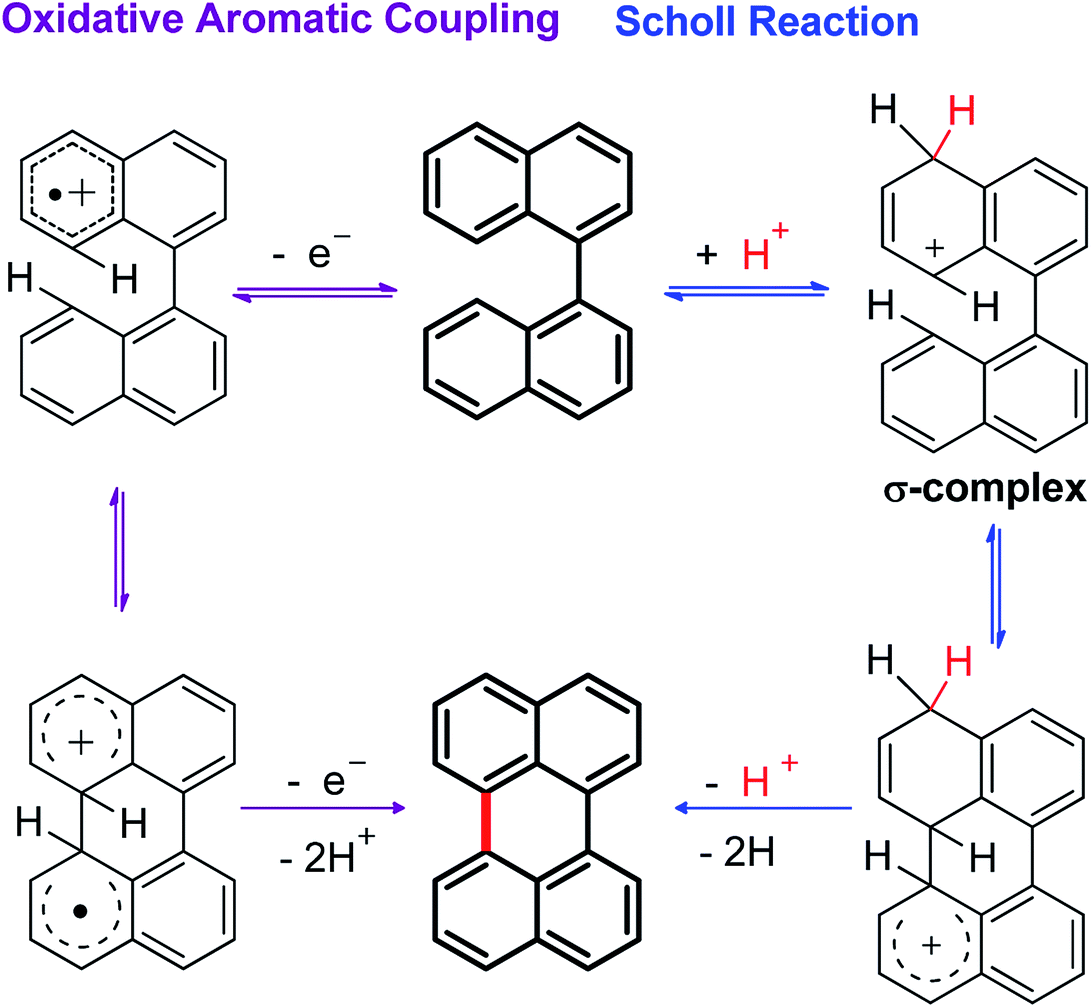 | ||
| Scheme 2 General mechanisms proposed for the cyclodehydrogenation reaction in oxidizing media (mediated by Lewis acids). | ||
In contrast, the other proposed mechanism for the reactions mediated by Lewis acids is the oxidative aromatic coupling. This mechanism involves the formation of a radical cation, followed by an intramolecular cyclization and rearomatization to give the polycyclic product (Scheme 2, left side).18 The experimental conditions to carry out this reaction involve the use of either Lewis acids such as FeCl3 (ref. 22) or strong oxidants like 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).23
The ambiguity of these reactions relies on the fact that sometimes there is no clear evidence of the operating mechanism, and they are said to occur through the Scholl mechanism without any discrimination. One of the reasons contributing to this confusion is the fact that most of the Lewis acids used in the Scholl reaction are also mild oxidants commonly employed in oxidative aromatic coupling reactions.22,24 In this regard, in some cases DFT calculations have been used to help to solve this issue.25
A prominent example in which it is possible to distinguish between oxidative aromatic coupling and Scholl mechanism is the reaction of 2-naphthol in the presence of an oxidant such as FeCl3 at 50 °C.26 Under these conditions, 2,2′-dihydroxy-1,1′-binaphthalene is formed by an oxidative aromatic coupling reaction (Scheme 3). However, it is not possible to obtain the perylene-1,12-diol compound, even when additional portions of FeCl3 were added to the system.18 In contrast, the C–C bond formation (C-8 with C-8′) to give the perylene-1,12-diol can be achieved by heating 2,2′-dihydroxy-1,1′-binaphthalene with AlCl3 at 150 °C (Scholl conditions).27
 | ||
| Scheme 3 Formation of perylene-1,12-diol. | ||
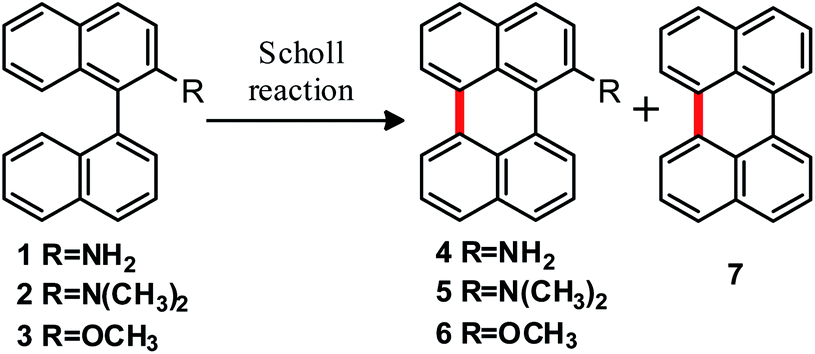
A similar behavior was also observed with naphthyl isoquinolines.13e The compound 1-(naphthalen-1-yl)-isoquinoline reacts with a melted mixture of AlCl3/NaCl at 160 °C to provide 1-azaperylene in 68% yield, but cyclization does not occur when 8-(naphthalen-1-yl)-isoquinoline is exposed to the same conditions. Moreover, neither of the two isomeric naphthyl isoquinoline substrates react in the presence of FeCl3 at 25 °C and 80 °C.18
Considering the aforementioned antecedents, 2-substituted binaphthalenes28 were synthesized and used as substrates for the preparation of equatorially functionalized perylenes. The study of the reactivity of these binaphthyl compounds can generate important contributions to the understanding of mechanisms associated with cyclodehydrogenation reactions mediated by Lewis acids. A comparison between the arenium cation (σ-complex or Scholl) mechanism and the cyclodehydrogenation initiated by radical cations (oxidative aromatic coupling) is presented for all synthesized compounds. The discussion is complemented with DFT computational studies. Furthermore, the peculiar formation of two C–C bonds in a single reaction step is presented in the synthesis of terrylene (tribenzo[de,kl,rst]pentaphene) from 1,1′:4′,1′′-ternaphthalene.
Results and discussion
Scholl and oxidative aromatic coupling experimental reactions
In an initial approach, the intramolecular cyclization of the substrates [1,1′-binaphthalen]-2-amine (1), N,N-dimethyl-[1,1′-binaphthalen]-2-amine (2) and 2-methoxy-1,1′-binaphthalene (3) was experimentally evaluated following both reaction conditions, Scholl (Table 1) and oxidative aromatic coupling.
|
||||
|---|---|---|---|---|
| Entry | Scholl reactiona | Productsb (%) | Remaining substratec (%) | |
a AlCl3/NaCl (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 eq.), substrate (0.2 eq., 0.1 mmol).b Isolated yields.c Quantified by CG-FID using the internal standard method.d Substrate (0.2 mmol), AlCl3 (1.8 mmol), C6H5Cl (10 mL). :1 eq.), substrate (0.2 eq., 0.1 mmol).b Isolated yields.c Quantified by CG-FID using the internal standard method.d Substrate (0.2 mmol), AlCl3 (1.8 mmol), C6H5Cl (10 mL). |
||||
| 1 | AlCl3/NaCl, 170 °C, 15 min | 4 (27)c | 7 (21) | 1 (12) |
| 2 | AlCl3/NaCl, 170 °C, 30 min | 4 (12)c | 7 (39) | 1 (<5) |
| 3 | AlCl3/NaCl, 170 °C, 10 min | 5 (50) | 7 (12) | 2 (<5) |
| 4 | AlCl3/NaCl, 170 °C, 20 min | 5 (40) | 7 (20) | 2 (<5) |
| 5d | AlCl3/C6H5Cl, 80 °C, 6 h | 6 (—) | 7 (—) | 3 (<5) |
The substrates were allowed to react at 170 °C within the salt mixture of AlCl3/NaCl during different reaction times (Table 1, entries 1–4). Compounds 1 and 2 afforded the cyclized products 4 and 5, respectively. To the best of our knowledge, this work describes a synthetic protocol for obtaining products 4 and 5 for the first time, since although compound 4 is mentioned in some patents, no data on the synthetic procedure or the reaction performance are known. Compound 4 decomposes under air and during purification in column chromatography (on silica gel). On the other hand, compound 5 has never been reported in literature and showed high fluorescence (ε5 (455 nm) = 6869 M−1 cm−1; Φf (5) = 0.92 in dichloromethane).30 In all cases, perylene (7) was found to be the main side product, in addition to substrate that did not react.
It is known that 1,1′-binaphthalene isomerizes to 1,2′-binaphthalene under acid-catalyzed conditions, as a consequence of the formation of an ipso-arenium ion. This process competes with the cyclodehydrogenation to form perylene;31 for example, formation of benzo[j]fluoranthene and related compounds is observed when isomerization occurs.31a In our case, the C-2′ position is blocked by the amino substituent, precluding isomerization process. In consequence, no isomerization-derived products were observed in the assayed reactions.
On the other hand, we assume that the formation of 7 could come from deamination of the products 4 or 5 due to the strongly acidic and high temperature conditions. This hypothesis is supported by the fact that an increase of reaction time leads to an increase in perylene (7) yield together with a decrease of the amino-perylene product yield (entries 2 and 4, Table 1). Although there exist several possible mechanisms for deamination of different amino compounds,32 at this stage we cannot unequivocally choose the mechanism involved in our case.
Substrate 3 has a melting point of 109–110 °C and decomposes at 125–127 °C.33 For this reason, milder Scholl reaction conditions were assayed with this substrate.19 A solution of AlCl3 in dry chlorobenzene at 80 °C was used to promote the reaction (entry 5, Table 1); however, no intramolecular cyclization product was observed and 2-methoxynaphthalene was obtained as major product, due to the C1–C1′ bond cleavage of the substrate. Some minor products from coupling between naphthyl group and chlorobenzene, homocoupling of chlorobenzene and other chlorinated compounds were detected by GC-MS.
The intramolecular cyclization by oxidative aromatic coupling conditions was evaluated by carrying out the reaction of substrates 1–3 with triflic acid (TfOH) and DDQ in dry dichloromethane for 1 hour at 0 °C. Under these conditions, the formation of cyclized products was not observed and the starting materials were fully recovered. Johnson et al. reported that 1,1′-binaphthalene in presence of TfOH at room temperature mostly produces the 2,2′-binaphthyl as a consequence of rearrangements;31a nevertheless, this isomerization reaction is not possible for our 2-substituted compounds. As an alternative methodology, solutions of 1–3 in dry dichloromethane were treated with a solution of FeCl3 in CH3NO2,16a but there was not substrate conversion at all.
To sum up, substrates 1 and 2 reacted in the Scholl reaction conditions, to produce the cyclized products of interest. Substrate 3 reacts by the Scholl pathway, but leading to no cyclodehydrogenation compounds. None of these substrates would react by the radical cation pathway (oxidative aromatic coupling).
Computational modeling
In order to find the nature of the difference in reactivity of the substrates, in both the Scholl and oxidative aromatic coupling conditions, molecular modeling studies based on Density Functional Theory (DFT)34 were done. All calculations reported here were carried out at the B3PW91/6-31+G* level of theory.35 The polarizable continuum model (IEFPCM) was employed to assess solvation using two media with different dielectric constant: dichloromethane (ε ∼ 8.93) and N-methylformamide (ε ∼ 181.5). Both solvent models afforded similar results, for simplicity the ones obtained in the latter solvent are included in the main text.36 The two possible mechanisms were evaluated, using also reference compounds selected from bibliographic data, which react, or not, within the same conditions as those employed within this work.Scholl reaction
Firstly, we address the following question, is it possible that – under Scholl reaction conditions – the substrates 1, 2 and 3 may react via the formation of the σ-complex? Two reference compounds were now included, 1-(naphthalen-1-yl)isoquinoline (8) and 8-(naphthalen-1-yl)isoquinoline (9).18 From these compounds, 8 reacted by the Scholl experimental condition whereas 9 could not form the cyclized product; they should work as positive and negative tests, respectively.Since the reactions were carried out in acidic media, the formation of the arenium cation was the first evaluated process. To study the regiochemistry of the protonation of each substrate, the relative energy difference required to form the σ-complex at different positions of the aromatic rings was calculated. A Boltzmann population analysis for the formation of the σ-complex was estimated at 170 °C (see ESI†), the most favored positions are shown in Table 2.29 It should be noted that the amino group must be protonated due to the acidic media used in the reactions. In this way, the cation of the substrate 1 (1a) was used,37 as well as the isoquinolinium cations (8a and 9a) which come from the protonation of the reference compounds 8 and 9.
| Substrates | Reference compounds | ||
|---|---|---|---|
|
|
|
|
| 5′ (72%) | 1 (98%) | 5′ (31%) | 5′ (46%) |
| 8′ (18%) | 6 (1%) | 8′ (66%) | 8′ (39%) |
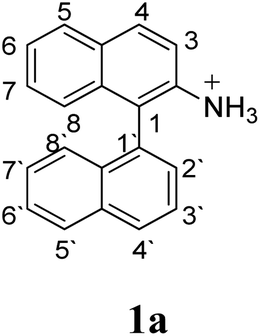
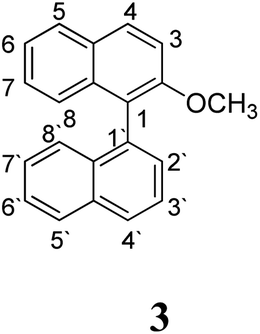
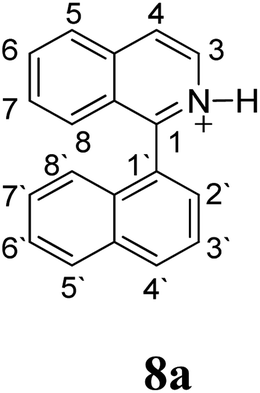
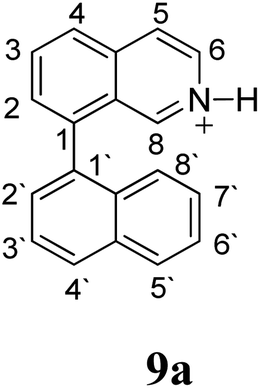
The protonation at positions 5′ and 8′ was found to be the energetically favored for 1a, 8a and 9a. When the σ-complex is generated on position 5′, the redistribution of the charge within the ring leads to an intramolecular electrophilic attack, which would generate a new C–C bond between C-8 and C-8′ (Scheme 2). On the other hand, if the σ-complex is generated in position 8′, the cyclization does not take place because a C-sp3 is formed at this position. For compound 3 the position 1 is the favored one for the protonation; however, this position does not favor the cyclization reaction.
To gain a deeper understanding of these reactions, the calculations of two different stages of the proposed Scholl mechanism (Scheme 4) were carried out. First, the free energy values obtained for the formation of the σ-complex (ΔΔGσ) were computed, using the reaction of 8a as reference.38 Secondly, the activation and reaction free energies (ΔG# and ΔGr respectively) involved in the intramolecular cyclization were evaluated; the results are presented in Table 3. The last stage of the total reaction is an oxidation with re-aromatization of the formed cycle, which should provide an important driving force.
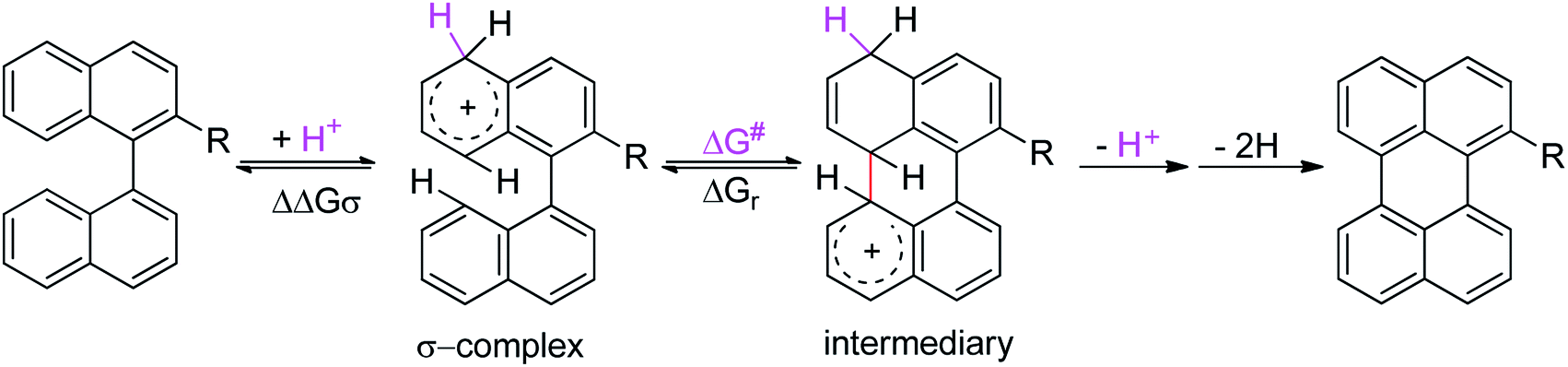 | ||
| Scheme 4 Scholl mechanism proposed for the formation of 2-R-perylene. | ||
| σ-Complex | ΔΔGr (kcal mol−1) | ΔGr (kcal mol−1) | ΔG# (kcal mol−1) | Intermediary | |
|---|---|---|---|---|---|
| a All the energies were calculated at B3PW91/6-31+G* using N-methylformamide as solvent, IEFPCM model. The energies correspond to the sum of electronic energy, solvation, and thermal free energies. | |||||
| 1σ | 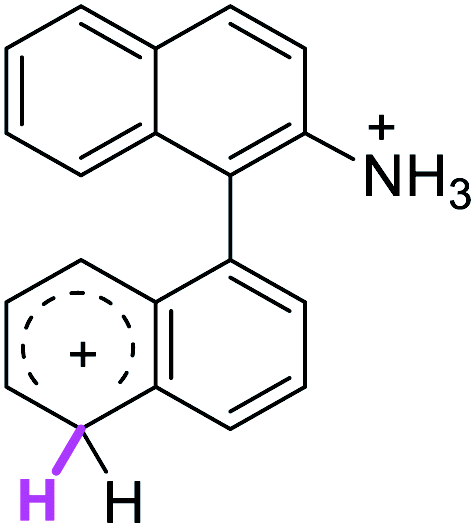 |
2.0 | 19.1 | 24.5 | 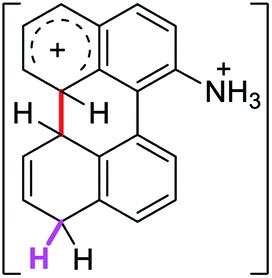 |
| 2σ | 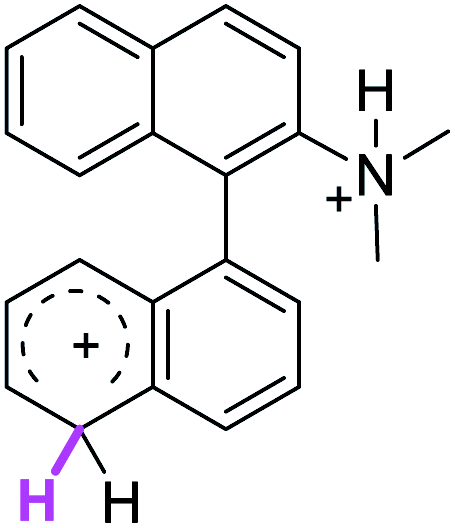 |
6.7 | 18.3 | 23.1 | 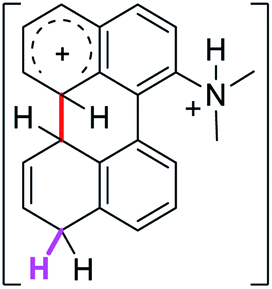 |
| 8σ | 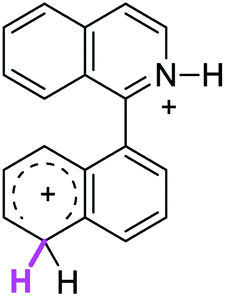 |
0.0 | 23.2 | 26.1 | 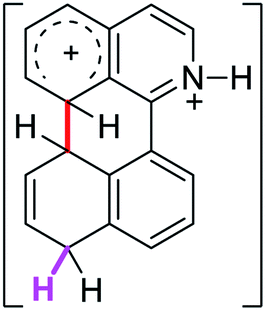 |
| 9σ | 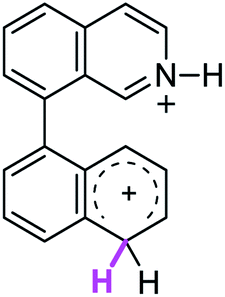 |
3.0 | 39.9 | 40.3 | 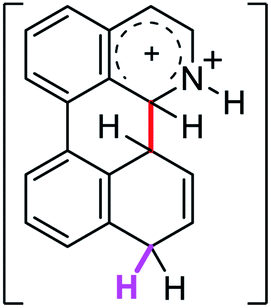 |
The ΔΔGσ values for the formation of complexes 1σ and 2σ to promote the Scholl reaction compared to that of 8σ, that is used as a reference of a compound that effectively reacts by the Scholl mechanism, indicate that it might be a possible step, even though it would entail a higher energy cost (2.0 kcal mol−1 for 1σ and 6.7 kcal mol−1 for 2σ). According to the energy of formation of the 9σ complex (∼3.0 kcal mol−1 higher than that of 8σ), compound 9 could also lead to the cyclization product. However, this product was not found when the reactions were carried out, hence the discrimination could take place after that step.
When the ΔG# energies for the intramolecular coupling were studied, it was found that the ΔG# for arenium cations 1σ and 2σ were lower than 8σ. Particularly, the 9σ complex presented values of ΔG# and ΔGr for ring closure higher than the other compounds. The difference exceeds 15 kcal mol−1, which would indicate that, in this case, the C–C coupling stage does discriminate between substrates that cyclize and those that do not generate intramolecular cyclization by this mechanism. This difference in the ΔG# and ΔGr, compared with the values for 8σ, are ascribed to the differences in the electronic distributions of 1σ, 8σ and 9σ, as presented in Fig. 1. For 1σ and 8σ the charges at position 5 and 8′ are negative and positive respectively, whereas in the case of 9σ the negative charge is closer to zero due to the protonated nitrogen adjacent to the 8′ position. The electrostatic potential help to explain how the charge is distributed after the formation of the σ complex, and which positions are nucleophilic and/or electrophilic, favoring (or not) the cyclization by an electrophilic attack.
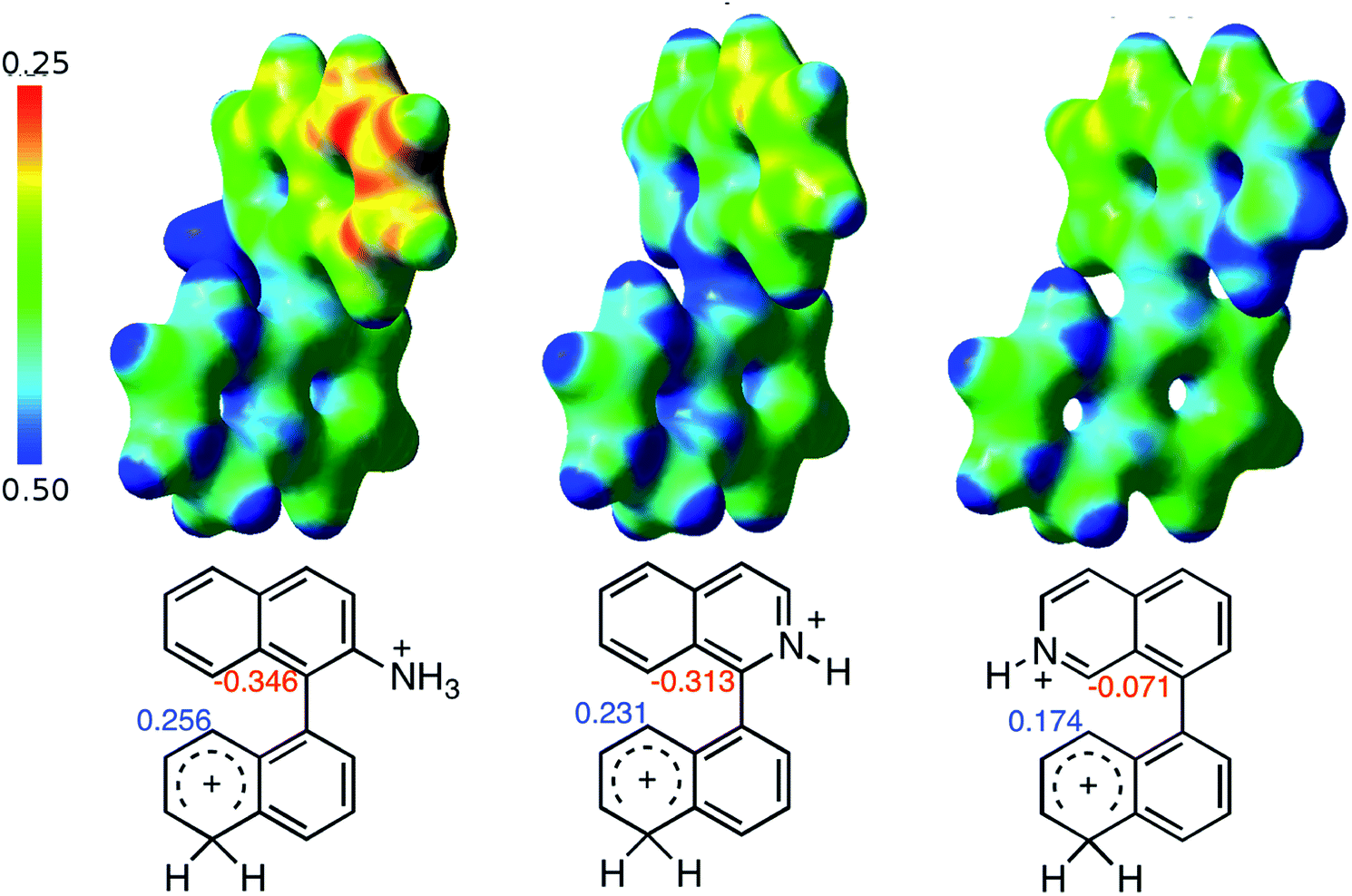 | ||
| Fig. 1 Representation of the electrostatic potential of the σ-complex of 1σ, 8σ and 9σ. In the small figure are represented the point charges that fit the electrostatic potential, according to the Merz–Singh–Kollman40 scheme, for C-5 and C-8′. | ||
As it was mentioned before, the cyclized product 6 derived from compound 3 by Scholl reaction was not observed (entry 5, Table 1). According to the analysis of the electrostatic potential of 3σ (with H+ in position 1), C-5′ and C-8 do not possess the expected charges that favor the coupling (see ESI†). In addition, the free energy for the intramolecular ring closure of 3σ is higher than the one found for compound 9σ which was used as reference of a nonreactive compound.39
Oxidative aromatic coupling
Finally, the reactivity of substrates 1–3 within oxidative aromatic coupling mechanism was also examined with computational studies. The feasibility of initiating the reaction through this mechanistic pathway (Scheme 2) was first evaluated by comparing the computed oxidation potentials.41 They were calculated for both, substrates and a group of reference molecules, the results are represented in Fig. 2. The set of reference compounds was split into two groups (Fig. 2): Group 1 (blue box) includes those compounds reported as useful for generating intramolecular cyclization mediated by an intermediary radical cation (using different oxidant agents, i. e. FeCl3, DDQ/H+, MoCl5, etc.); Group 2 (red box) includes those compounds that do not produce cyclization under oxidative aromatic coupling experimental conditions.42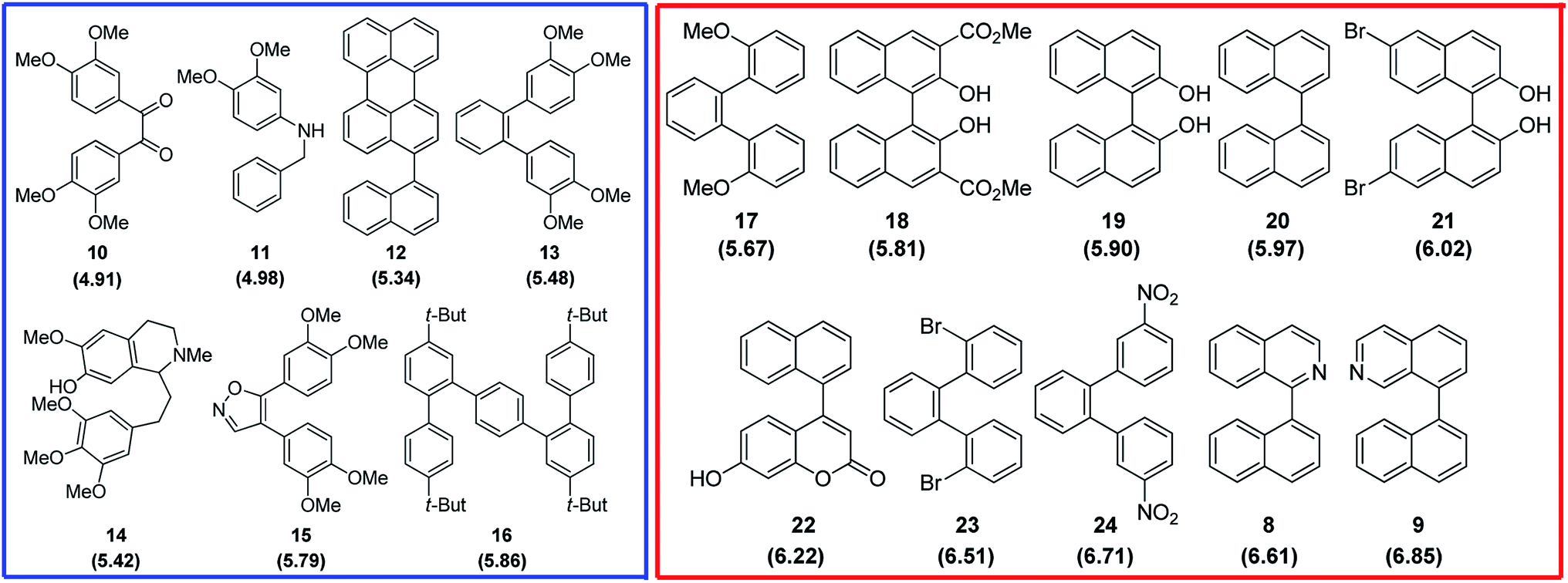 | ||
| Fig. 2 Structure of the reference compounds for oxidative aromatic coupling mechanism. Blue square: Group 1 (reactive); red square: Group 2 (non reactive). Calculated oxidation potential (eV) in parenthesis.42 | ||
As expected, the results suggest that compounds from Group 1 have lower oxidation potentials. Meanwhile, the compounds from Group 2 have higher oxidation potentials (Fig. 3).
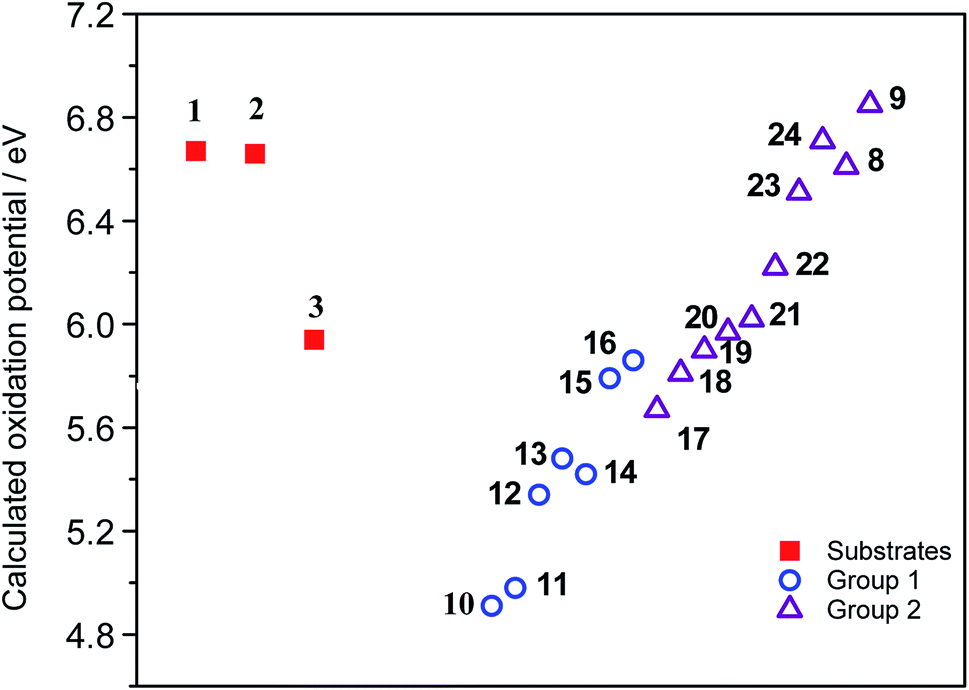 | ||
| Fig. 3 Calculated oxidation potential of the studied compounds. | ||
The oxidation potential values found for substrates 1–3 are in the region of the compounds that would hardly generate the necessary radical cation which initiates the reaction via the oxidative aromatic coupling mechanism. This could explain the lack of reactivity of these substrates experimentally observed. These results are in concordance with the previously proposed oxidation potential limit by Rathore et al. when DDQ/H+ is used as oxidant agent for cyclization of electron-rich molecules.18,23 It should be noted that this estimation of the oxidation potential, could help to predict the reactivity of new compounds toward these oxidative aromatic coupling reactions.
Cyclization of 1,1′:4′,1′′-ternaphthalene
In order to explore the scope of these reactions, we propose the synthesis of terrylene from 1,1′:4′,1′′-ternaphthalene (25), Scheme 5. In this reaction there is also another challenge, two new C–C bonds have to be formed to yield the expected product. Reaction of 1,1′:4′,1′′-ternaphthalene with a mixture of AlCl3/NaCl (5:1) was carried out during 20 min at 170 °C, the corresponding double cycled product terrylene (26) was obtained in 38% isolated yield. It is important to stand out that two new C–C bonds were generated within the same procedure. Perylene 7 (23%) was also obtained as a side product and substrate 25 (20%) was recovered.
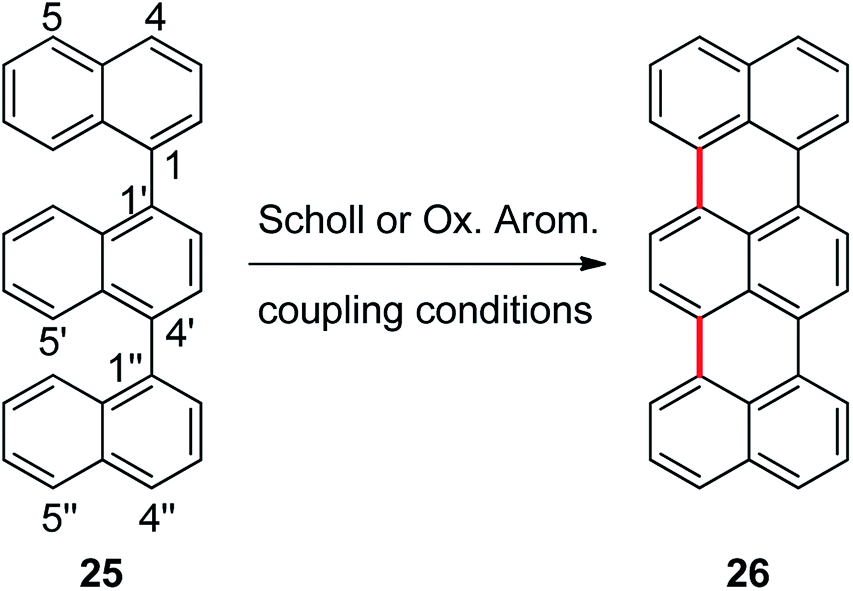 | ||
| Scheme 5 Synthesis of terrylene (26) from 1,1′:4′,1′′-ternaphthalene (25) by Scholl or oxidative aromatic coupling experimental conditions. | ||
The molecular modeling studies of compound 25 indicated that the positions 4 (64%) and 5 (11%) are the ones that protonate mostly, but the ΔG# and ΔGr for the coupling are markedly favored for the σ-complex generated in position 5 (see ESI†). The electrostatic potential of both intermediary σ-complexes are really different and help to explain this energetic difference observed for the coupling at these positions. Finally, starting form the σ-complex protonated at position 5, the energies involved in two consecutive C–C couplings of 25σ, are slightly lower than the energy values found for the other binaphthyl derivatives previously studied (1, 2, and 8).
As it was found for substrates 1–3, when substrate 25 was also exposed to oxidative aromatic coupling conditions using DDQ as the oxidant agent, the formation of the product 26 was not detected. Again, 25 was also treated with a solution of FeCl3 in CH3NO2. The reaction led to the formation of an inseparable mixture of products that could not be characterized. Moreover, none of them showed the typical fluorescence or UV-absorption profile of the terrylene.16a The computed oxidation potential found for 25 was 6.15 eV. Considering Fig. 2, the compound 25 is classified within the group of compounds that do not react by the methodology of oxidative aromatic coupling.
In brief, the results obtained by computational modeling help to explain the observed reactivity of the tested ternaphthyl compound, under both reaction conditions. Moreover, it was possible to obtain terrylene, going through two consecutive C–C coupling reactions, in a simple experimental step with Scholl conditions.
Conclusions
To summarize, three compounds derived from perylene, two of them functionalized in the equatorial region, were synthesized by a cyclodehydrogenation reaction mediated by Lewis acids, from their respective 2-substituted binaphthalene compounds through a mechanistic pathway that involves the formation of the σ-complex. Regarding the studies performed by computational methods, the regiochemistry for the formation of the σ-complex should be studied. An analysis of the electrostatic potential isosurface of the σ-complexes was very useful to predict the outcome of the cyclization reaction. According to these analyses, within this experimental condition (AlCl3/NaCl, 170 °C), substrates 1, 2 and 25 can generate the σ-complex to initiate the reaction and consequently, produce an intramolecular cyclization. For substrate 3, the results suggest that the formation of the initial σ-complex is possible on position 1, but the ring closure from the σ-complex is not energetically favored, consequently the product 6 was not observed.On the other hand, the formation of the 2-substituted perylene products by cyclodehydrogenation reactions via oxidative aromatic coupling (FeCl3 or DDQ as oxidants), e.g. radical cations as intermediates, was not detected. The studies carried out with DTF methods support that substrates 1, 2, 3, and 25 could not be experimentally oxidized and therefore, could not generate the intermediary radical cation, which is necessary to initiate the reaction and produce the cycled perylene derivative. In summary, we introduce an experimental system where it can be raised the difference between both mechanisms mediated by Lewis acids, and again, molecular modeling is a powerful tool which, on this occasion, helped us to deeply understand the key points to be considered for achieving a successful reaction.
As Gryko proposed,18 there is a clear mechanistic difference when any substrate is exposed to Lewis acids to generate a dehydrogenative aromatic coupling product by Scholl reaction or by oxidative aromatic coupling. According to the analysis of both reactions by using molecular modeling methods, it could be possible to explain differences in reactivity and to predict the outcome of a reaction with a new substrate.
Experimental section
General methods
Sodium chloride (NaCl), nitromethane (CH3NO2), 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), and trifluoromethanesulfonic acid (TFMSA) were obtained from Sigma-Aldrich and used as received. Iron trichloride (FeCl3) was obtained from Merck Millipore and used as received. Aluminum trichloride (AlCl3) was obtained commercially and sublimated before used. Dichloromethane was obtained commercially, distilled, and stored under molecular sieves (4 Å). Gas chromatographic analysis was performed on a Varian GC with a flame ionization detector and equipped with a VF-5 ms, 30 m × 0.25 mm × 0.25 μm column. Gas chromatographic/mass spectrometer analysis was carried out on a Shimadzu GC-MS QP 5050 spectrometer equipped with a VF-5 ms, 30 m × 0.25 mm × 0.25 μm column. HRMS were recorded on a Bruker, MicroTOF Q II equipment, operated with an ESI source in (positive/negative) mode, using nitrogen as nebulizing and drying gas and sodium formate 10 mM as an internal standard. HPLC analysis was carried out on a Waters 1525 Binary HPLC Pump connected to a Waters 2998 Photodiode Array Detector, and employing an Agilent Rx-Sil Analytical column (4.6 × 150 mm, 5 μm). Elemental analysis was determined on a Perkin Elmer 2400 Series II CHNS/O with a thermal conductivity detector (TCD). 1H NMR and 13C NMR were recorded on a 400 MHz Bruker nuclear magnetic resonance spectrometer.The substrates 1,28a,28c 3,43 and 25 (ref. 44) were synthesized and purified by previously published methods, their NMR spectra are shown in ESI.† The new compound N,N-dimethyl-[1,1′-binaphthyl]-2-amine (2) was obtained by the methylation of amine 1 (see ESI†).
When milder conditions were employed in the Scholl reaction, 2-methoxy-1,1′-binaphthalene (3) (0.2 mmol, 1 eq.) was dissolved in dry dichloromethane (10 mL). After stirring for 20 min under N2 atmosphere, anhydrous aluminum chloride (1.8 mmol, 9 eq.) was added. The solution was stirred at 80 °C for 6 h. For the extraction process, CH2Cl2 and H2O (3 × 20 mL) were used.
N,N-Dimethyl-[1,1′-binaphthalen]-2-amine (2)
The compound was purified as a pale yellow solid by column chromatography using as eluent a solvent gradient of pentane/ethyl acetate (100:0 → 90:10). Mp: 193–195 °C. Yield: 47% (0.17 mmol, 52 mg). 1H NMR (400 MHz, CDCl3) δ: 7.92 (t, 1J = 8.0 Hz, 3H); 7.82 (d, 1J = 8.0 Hz, 1H); 7.61 (m, 1H); 7.50 (d, 1J = 8.0 Hz, 1H); 7.48–7.44 (m, 2H); 7.37 (d, 1J = 8.0 Hz, 1H); 7.31–7.25 (m, 2H); 7.18–7.14 (m, 1H); 7.09 (d, 1J = 8.5 Hz, 1H); 2.51 (s, 6H). 13C NMR (100 MHz, CDCl3) δ = 149.9 (Cq, CAr–N); 137.2 (Cq); 134.4 (Cq); 133.9 (Cq); 133.2 (Cq); 129.7 (Cq); 129.1 (CAr–H); 128.9 (CAr–H); 128.3 (CAr–H); 127.8 (Cq), 127.8 (CAr–H); 127.6 (CAr–H); 126.7 (CAr–H); 126.1 (CAr–H); 125.9 (CAr–H); 125.8 (CAr–H); 125.8 (2 × CAr–H); 123.7 (CAr–H); 119.8 (CAr–H); 44.0 (2 × CH3). HRMS (ESI-TOF) m/z [M + Na]+ calcd for C22H19NNa: 320.1410; found: 320.1424.
Perylene-1-amine (4)
The product was purified as a yellow oily solid by semipreparative TLC using as eluent pentane/ethyl acetate (90:10). In this specific case, a second consecutive semipreparative TLC was made. The product decomposes when it is exposed to air. Isolated yield: 14% (4 mg). 1H, COSY, HMBC and HSQC-DEPT NMR spectra are showed in ESI.† 1H NMR (400 MHz, (CD3)2CO) δ = 7.97 (d, 1J = 7.6 Hz, 1H); 7.73–7.69 (m, 2H); 7.23 (d, 1J = 7.7, 1H); 7.17–7.10 (m, 3H); 7.06–6.99 (m, 2H); 6.81 (t, 1J = 7.6 Hz, 1H); 6.72 (d, 1J = 8.8 Hz, 1H); 5.58 (s, 2H, NH2). 13C NMR (100 MHz, (CD3)2CO) δ = 145.6 (Cq, C–N); 134.6 (Cq); 132.3 (Cq); 131.6 (Cq); 130.5 (Cq); 129.7 (Cq); 129.1 (Cq); 128.9 (CAr–H); 128.2 (CAr–H); 127.9 (Cq); 126.9 (CAr–H); 126.7 (2C, CAr–H); 124.8 (CAr–H); 122.9 (CAr–H); 122.2 (CAr–H); 121.7 (CAr–H); 121.5 (CAr–H); 118.9 (CAr–H); 109.7 (Cq). 1H–1H COSY NMR ((CD3)2CO): δH/δH 7.97/7.01; 7.71/6.81; 7.69/7.04; 7.23/7.04; 7.16/6.81; 7.11/6.72; 7.13/7.01. 1H–13C HSQC NMR ((CD3)2CO): δH/δC 7.97/121.5; 7.71/121.7; 7.69/118.9; 7.23/126.7; 7.16/128.2; 7.13/124.8; 7.11/128.9; 7.04/126.7; 7.01/126.9; 6.81/122.9; 6.72/122.2. 1H–13C HMBC NMR ((CD3)2CO): δH/δC 7.97/109.7; 7.97/124.8; 7.97/129.7; 7.71/128.2; 7.71/130.5; 7.71/131.6; 7.69/126.7; 7.69/129.1; 7.69/129.7; 7.23/118.9; 7.23/124.8; 7.23/129.7; 7.16/121.7; 7.16/128.9; 7.16/130.5; 7.13/121.5; 7.13/126.7; 7.13/129.7; 7.11/128.2; 7.11/130.5; 7.11/145.6; 7.04/131.6; 7.04/134.6; 7.01/132.3; 7.01/134.6; 6.81/127.9; 6.81/129.1; 6.72/109.7; 6.72/127.9; 5.58/109.7; 5.58/122.2. MS (EI): m/z 267 (100.0%), 133 (36.0%), 118 (19%).
N,N-Dimethylperylene-1-amine (5)
The compound was purified as an orange oily solid by semipreparative TLC using as eluent a solvent gradient of pentane/ethyl acetate (100:0 → 90:10). Isolated yield: 50% (15 mg). 1H, 13C, COSY, HMBC and HSQC-DEPT NMR spectra are showed in ESI.† 1H NMR (400 MHz, (CD3)2CO) δ: 9.21 (dd, 1J = 7.8, 2J = 1.0 Hz, 1H); 8.21 (dd, 1J = 7.5, 2J = 1.1 Hz, 2H); 7.73–7.64 (m, 4H); 7.54–7.46 (m, 3H); 7.38 (t, 1J = 7.8 Hz, 1H); 2.85 (s, 6H). 13C NMR (100 MHz, (CD3)2CO) δ: 151.0 (Cq, C–N); 135.6 (Cq); 132.5 (Cq); 132.4 (Cq); 131.9 (Cq); 131.6 (Cq); 131.6 (Cq); 130.8 (Cq); 129.5 (CAr–H); 128.5 (CAr–H); 128.2 (CAr–H); 127.5 (CAr–H); 127.1 (CAr–H); 127.0 (CAr–H); 125.2 (CAr–H); 124.5 (CAr–H); 122.1 (CAr–H); 121.5 (CAr–H); 120.1 (CAr–H); 119.6 (Cq); 43.4 (2C, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3). HRMS (ESI-TOF) m/z [M + Na]+ calcd for C22H17NNa: 318.1253; found: 318.1261.
H3). HRMS (ESI-TOF) m/z [M + Na]+ calcd for C22H17NNa: 318.1253; found: 318.1261.
Perylene (7)
The product was purified as a pale yellow solid by semipreparative TLC using as eluent pentane/ethyl acetate (90:10). 1H and 13C NMR spectra are showed in ESI.† 1H NMR (400 MHz, CDCl3) δ: 8.19 (d, 1J = 7.8 Hz, 4H); 7.68 (d, 1J = 8.0 Hz, 4H); 7.48 (t, 1J = 7.7 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ: 134.9 (2 × Cq); 131.4 (2 × Cq); 129.0 (4 × Cq); 128.0 (4 × CAr–H); 126.7 (4 × CAr–H); 120.4 (4 × CAr–H). MS (EI): m/z 252 (100%), 125 (18%), 113 (7%).
Terrylene (26)16a
After reaction the crude was extracted in a Soxhlet apparatus, using solvents in increasing polarity starting from ethyl acetate (100 mL, refluxing for 24 h) and then toluene (100 mL, refluxing for 24 h). The product 26 was isolated as a dark solid. Isolated yield: 38% (29 mg). UV-visible spectrum is showed in ESI.† UV-vis: λmax in dichloromethane/nm: 550.4, 508.9, 476.1. Elemental analysis: calc. for C30H16: C 95.7%, H 4.28%; found: C 95.82%, H 4.18%.Computational procedure
All the calculations were performed with the Gaussian09 program.34 Based on previous results,45 the B3PW91 DFT functional46 and the 6-31+G(p) basis set were selected. Calculations were performed with full geometry optimization including in all cases the effect of the solvent through the Tomasi's polarized continuum model (IEFPCM)36a as implemented in the Gaussian package. After refinement, the characterization of stationary points was done by Hessian matrix calculations, with all positive eigenvalues for a minimum and only one negative eigenvalue for the TSs. The xyz coordinates of all the obtained geometries are included in the ESI,† together with the SCF energy, in the case of TSs, the value of the negative frequency is also included.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Secretaría de Ciencia y Tecnología, Universidad Nacional de Córdoba (SECyT), and the Agencia Nacional de Promoción Científica y Técnica (ANPCyT). P. C. S. gratefully acknowledges a fellowship from CONICET.References
- (a) Molecular Devices for Solar Energy Conversion and Storage, ed. H. Tian, G. Boschloo and A. Hagfeldt, Springer Nature, Singapore, 2018 Search PubMed; (b) 2D Materials for Photonic and Optoelectronic Applications, ed. Q. Bao and H. Y. Hoh, Woodhead Publishing, Elsevier, Duxford, 2020 Search PubMed; (c) V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics. Concepts, Research, Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2014 Search PubMed.
- (a) J. A. Mikroyannidis, M. M. Stylianakis, M. S. Roy, P. Suresh and G. D. Sharma, Synthesis, photophysics of two new perylene bisimides and their photovoltaic performances in quasi solid state dye sensitized solar cells, J. Power Sources, 2009, 194, 1171–1179 Search PubMed; (b) J. T. Markiewicz and F. Wudl, Perylene, Oligorylenes and Aza-Analogs, ACS Appl. Mater. Interfaces, 2015, 7, 28063–28084 Search PubMed; (c) E. Kozma, W. Mróz, F. Villafiorita-Monteleone, F. Galeotti, A. Andicsová-Eckstein, M. Catellania and C. Botta, Perylene diimide derivatives as red and deep red-emitters for fully solution processable OLEDs, RSC Adv., 2016, 6, 61175–61179 Search PubMed; (d) É. Torres, R. Bogel-Lukasik, M. N. Berberan-Santos, S. Höfle, A. Colsmannd and M. J. Brites, N,N′-Diaryl-perylene-3,9-diamine derivatives: synthesis, characterization and electroluminescence properties, RSC Adv., 2016, 6, 107180–107188 Search PubMed; (e) R. K. Gupta and A. A. Sudhakar, Perylene-Based Liquid Crystals as Materials for Organic Electronics Applications, Langmuir, 2019, 35, 2455–2479 Search PubMed.
- (a) R. K. Dubey, D. Inan, S. Sengupta, E. J. R. Sudhölter, F. C. Grozema and W. F. Jager, Tunable and highly efficient light-harvesting antenna systems based on 1,7-perylene-3,4,9,10-tetracarboxylic acid derivatives, Chem. Sci., 2016, 7, 3517–3533 Search PubMed; (b) C. Huang, S. Barlow and S. R. Marder, Perylene-3,4,9,10-tetracarboxylic Acid Diimides: Synthesis, Physical Properties, and Use in Organic Electronics, J. Org. Chem., 2011, 76, 2386–2407 Search PubMed.
- (a) M. Sun, K. Müllen and M. Yin, Water-soluble perylenediimides: design concepts and biological applications, Chem. Soc. Rev., 2016, 45, 1513–1528 Search PubMed; (b) A. Venkateswararao, S.-W. Liu and K.-T. Wong, Organic polymeric and small molecular electron acceptors for organic solar cells, Mater. Sci. Eng., R, 2018, 124, 1–57 Search PubMed; (c) F. Feng, W. Jiang and Z. Wang, Synthesis and Application of Rylene Imide Dyes as Organic Semiconducting Materials, Chem.–Asian J., 2018, 13, 20–30 Search PubMed; (d) G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Nonfullerene Acceptor Molecules for Bulk Heterojunction Organic Solar Cells, Chem. Rev., 2018, 118, 3447–3508 Search PubMed.
- (a) B. A. Jones, M. J. Ahrens, M. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, High-Mobility Air-Stable n-Type Semiconductors with Processing Versatility: Dicyanoperylene-3,4:9,10-bis(dicarboximides), Angew. Chem., Int. Ed., 2004, 43, 6363–6366 Search PubMed; (b) M. Gsanger, J. H. Oh, M. Konemann, H. W. Hoffken, A.-M. Krause, Z. Bao and F. Wurthner, A Crystal-Engineered Hydrogen-Bonded Octachloroperylene Diimide with a Twisted Core: An n-Channel Organic Semiconductor, Angew. Chem., Int. Ed., 2010, 49, 740–743 Search PubMed; (c) L. Wang, X. Zhang, G. Dai, W. Deng, J. Jie and X. Zhang, High-mobility air-stable n-type field-effect transistors based on large-area solution-processed organic single-crystal arrays, Nano Res., 2018, 11, 882–891 Search PubMed; (d) L. Zonga, Y. Gonga, Y. Yua, Y. Xiea, X. Guohua, Q. Peng, Q. Li and Z. Li, New perylene diimide derivatives: stable red emission, adjustable property from ACQ to AIE, and good device performance with an EQE value of 4.93%, Sci. Bull., 2018, 63, 108–116 Search PubMed.
- (a) L. Schmidt-Mende, A. Fechtenkotter, K. Müllen, E. Moons, R. H. Friend and J. D. Mackenzie, Self-organized discotic liquid crystals for high-efficiency organic photovoltaics, Science, 2001, 293, 1119–1122 Search PubMed; (b) C. Li and H. Wonneberger, Perylene Imides for Organic Photovoltaics: Yesterday, Today, and Tomorrow, Adv. Mater., 2012, 24, 613–636 Search PubMed; (c) E. Kozma and M. Catellani, Perylene diimides based materials for organic solar cells, Dyes Pigm., 2013, 98, 160–179 Search PubMed; (d) S. Kumar, D. Panigrahi and A. Dhar, Role of organic interfacial modifiers in inverted polymers solar cells: an in-depth analysis of perylene vs. fullerene organic modifiers, Appl. Surf. Sci., 2018, 435, 855–862 Search PubMed; (e) C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3, 18003 Search PubMed.
- M. Pärs, K. Gräf, P. Bauer, M. Thelakkat and J. Köhler, Optical gating of perylene bisimide fluorescence using dithienylcyclopentene photochromic switches, Appl. Phys. Lett., 2013, 103, 221115 Search PubMed.
- (a) H. Wang, L. Chen, Z. Zhao and Y. Xiao, Aryl-bisalkynyl bridged perylene diimides dimers: efficient synthesis, properties and improved electron mobilities, Dyes Pigm., 2017, 144, 184–189 Search PubMed; (b) X. Chena, Y.-N. Wanga, R.-X. Ronga, C.-M. Zhaoa, X.-L. Lia and K.-R. Wanga, Synthesis, thermo-responsive behavior of cyclodextrin modified bi-perylene monoimide derivative, Dyes Pigm., 2019, 160, 779–786 Search PubMed.
- (a) F. Ilhan, D. S. Tyson, D. J. Stasko, K. Kirschbaum and M. A. Meador, Twisted, Z-Shaped Perylene Bisimide, J. Am. Chem. Soc., 2006, 128, 702–703 Search PubMed; (b) B. B. Rao, J.-R. Wei and C.-H. Lin, New Synthetic Routes to Z-Shape Functionalized Perylenes, Org. Lett., 2012, 14, 3640–3643 Search PubMed.
- (a) H. Langhals, Cyclic carboxylic imide structures as structure elements of high stability. Novel developments in perylene dye chemistry, Heterocycles, 1995, 40, 477–500 Search PubMed; (b) C. S. Sample, E. Goto, N. V. Handa, Z. A. Page, Y. Luo and C. J. Hawker, Modular synthesis of asymmetric rylene derivatives, J. Mater. Chem. C, 2017, 5, 1052–1056 Search PubMed; (c) T. Sakamoto and C. Pac, A “green” route to perylene dyes: direct coupling reactions of 1,8-naphthalimide and related compounds under mild conditions using a “new” base complex reagent, t-BuOK/DBN, J. Org. Chem., 2001, 66, 94–98 Search PubMed.
- G. Dyker, Transition metal catalyzed annulation reactions. Part 3. Palladium-catalyzed annulation reactions of 1,8-diiodonaphthalene, J. Org. Chem., 1993, 58, 234–238 Search PubMed.
- (a) H. Langhals and S. Grundner, Fluoreszenzfarbstoffe mit Fünfring-Carbonsäureimid-Strukturen, Chem. Ber., 1986, 119, 2373–2376 Search PubMed; (b) J. T. M. V. Dijk, A. Hartwijk, A. C. Bleeker, J. Lugtenburg and J. Cornelisse, Gram Scale Synthesis of Benzo[ghi]perylene and Coronene, J. Org. Chem., 1996, 61, 1136–1139 Search PubMed.
- (a) A. Homer, XCVI.—The Friedel-Crafts' reaction applied to naphthalene. The action of di-, tri-, and tetra-alkyl halides. Preparation of αβα′β′-dinaphthanthracene, J. Chem. Soc., 1910, 97, 1141–1154 Search PubMed; (b) R. Scholl, C. Seer and R. Weitzenböck, Perylen, ein hoch kondensierter aromatischer Kohlenwasserstoff C20H12, Ber. Dtsch. Chem. Ges., 1910, 43, 2202–2209 Search PubMed; (c) R. Weitzenböck and C. Seer, Zur Kenntnis des Perylens und seiner Derivate. (2. Mitteilung), Ber. Dtsch. Chem. Ges., 1913, 43, 1994–2000 Search PubMed; (d) M. Rickhaus, A. P. Belanger, H. A. Wegner and L. T. Scott, An Oxidation Induced by Potassium Metal. Studies on the Anionic Cyclodehydrogenation of 1,1′-Binaphthyl to Perylene, J. Org. Chem., 2010, 21, 7358–7365 Search PubMed; (e) D. T. Gryko, J. Piechowska and M. Gałęzowski, Strongly Emitting Fluorophores Based on 1-Azaperylene Scaffold, J. Org. Chem., 2010, 75, 1297–1300 Search PubMed; (f) P. Schlichting, U. Rohr and K. Müllen, Easy synthesis of liquid crystalline perylene derivatives, J. Mater. Chem., 1998, 8, 2651–2655 Search PubMed.
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Synthetic Applications of Oxidative Aromatic Coupling-From Biphenols to Nanographenes, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 Search PubMed.
- F. A. Vingiello and L. Ojakaar, New polycyclic aromatic hydrocarbons with seven fused rings, Tetrahedron, 1966, 22, 847–860 Search PubMed.
- (a) Y. Avlasevich, C. Kohl and K. Müllen, Facile synthesis of terrylene and its isomer benzoindenoperylene, J. Mater. Chem., 2006, 16, 1053–1057 Search PubMed; (b) E. Buchta and J. Bösche, Polycyclische Verbindungen, XII. Ter-a-naphthyl und terrylen, Liebigs Ann. Chem., 1962, 660, 33–40 Search PubMed.
- M. Wehmeier, M. Wagner and K. Müllen, Novel Perylene Chromophores Obtained by a Facile Oxidative Cyclodehydrogenation Route, Chem.–Eur. J., 2001, 7, 2197–2205 Search PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Comparison of oxidative aromatic coupling and the Scholl reaction, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 Search PubMed.
- S. Nagarajan, C. Barthes, N. K. Girdhar, T. T. Dang and A. Gourdon, Methylterrylene isomers, Tetrahedron, 2012, 68, 9371–9375 Search PubMed.
- (a) E. Clar, W. Kelly, D. G. Stewart and J. W. Wright, Benzo[a]perylene and some of its derivatives, J. Chem. Soc., 1956, 2652–2656 Search PubMed; (b) I. Agranat, Y. N. Oded, T. Mala′bi, S. Pogodin and S. Cohen, The linkage between reversible Friedel–Crafts acyl rearrangements and the Scholl reaction, Struct. Chem., 2019, 30, 1579–1611 Search PubMed.
- H. Z. Lecher, M. Scalera and W. S. Forster, US Pat., 2416931, Mar. 4, 1947, C. A., 1947, 41, 5151.
- T. Horibe, S. Ohmura and K. Ishihara, Structure and Reactivity of Aromatic Radical Cations Generated by FeCl3, J. Am. Chem. Soc., 2019, 141, 1877–1881 Search PubMed.
- L. Zhai, R. Shukla, S. H. Wadumethrige and R. Rathore, Probing the Arenium-Ion (Proton Transfer) versus the Cation-Radical (Electron Transfer) Mechanism of Scholl Reaction Using DDQ as Oxidant, J. Org. Chem., 2010, 75, 4748–4760 Search PubMed.
- A. A. O. Sarhanw and C. Bolm, Iron(III) chloride in oxidative C–C coupling reactions, Chem. Soc. Rev., 2009, 38, 2730–2744 Search PubMed.
- (a) B. T. King, J. Kroulík, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, Controlling the Scholl Reaction, J. Org. Chem., 2007, 72, 2279–2288 Search PubMed; (b) S. Nobusue, K. Fujita and Y. Tobe, Skeletal Rearrangement of Twisted Polycyclic Aromatic Hydrocarbons under Scholl Reaction Conditions, Org. Lett., 2017, 19, 3227–3230 Search PubMed; (c) J. Liu, A. Narita, S. Osella, W. Zhang, D. Schollmeyer, D. Beljonne, X. Feng and K. Müllen, Unexpected Scholl Reaction of 6,7,13,14-Tetraarylbenzo[k]tetraphene: Selective Formation of Five-Membered Rings in Polycyclic Aromatic Hydrocarbons, J. Am. Chem. Soc., 2016, 138, 2602–2608 Search PubMed.
- (a) K. Ding, Q. Xu, Y. Wang, J. Liu, Z. Yu, B. Du, Y. Wu, H. Koshima and T. Matsuura, Novel two-phase oxidative cross-coupling of the two-component molecular crystal of 2-naphthol and 2-naphthylamine, Chem. Commun., 1997, 693–694 Search PubMed; (b) K. Ding, Y. Wang, L. Zhang, Y. Wu and T. Matsuura, A novel two-phase oxidative coupling of 2-naphthols suspended in aqueous Fe3+ solutions, Tetrahedron, 1996, 52, 1005–1010 Search PubMed.
- A. Zinke and R. Dengg, Eine Synthese des Perylens über das 1,12-Dioxyperylen, Monatsh. Chem., 1922, 43, 125–128 Search PubMed.
- (a) A. B. Pierini, M. T. Baumgartner and R. A. Rossi, Regiochemistry of the coupling of aryl radicals with nucleophiles derived from the naphthyl system. Experimental and theoretical studies, J. Org. Chem., 1991, 56, 580–586 Search PubMed; (b) L. B. Jimenez, N. V. Torres, J. L. Borioni and A. B. Pierini, Synthesis of triaryls: hydroxy and amine dinaphthyl and diphenanthryl aryls by one-pot electron-transfer nucleophilic substitution reactions, Tetrahedron, 2014, 70, 3614–3620 Search PubMed; (c) A. B. Pierini, M. T. Baumgartner and R. A. Rossi, Photostimulated reactions of haloarenes with 2-naphtylamide ions. A facile synthesis of 1-aryl-2-naphthylamines, Tetrahedron Lett., 1987, 28, 4653–4656 Search PubMed.
- Information about the relative ΔGσ of formation of the arenium cation at all positions is included in the ESI.†.
- UV-vis and fluorescence spectra are shown in ESI.†.
- (a) S. L. Skraba-Joiner, E. C. McLaughlin, A. Ajaz, R. Thamatam and R. P. Johnson, Scholl Cyclizations of Aryl Naphthalenes: Rearrangement Precedes Cyclization, J. Org. Chem., 2015, 80, 9578–9583 Search PubMed; (b) P. Kovacic and F. W. Koch, Coupling of Naphthalene Nuclei by Lewis Acid Catalyst-Oxidant, J. Org. Chem., 1965, 30, 3176–3281 Search PubMed.
- R. J. Baumgarten and V. A. Curtis, in Supplement F. The Chemistry of Amino, Nitroso and Nitro Compounds and their Derivatives. Part 2., ed. S. Patai, John Wiley & Sons Ltd., Jerusalem, 1982, p. 929 Search PubMed.
- O. M. Demchuk, K. Kapłon, L. Mazur, D. Strzelecka and K. M. Pietrusiewicz, Readily available catalysts for demanding Suzuki–Miyaura couplings under mild conditions, Tetrahedron, 2016, 72, 6668–6677 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) J. P. Perdew, Electronic Structure of Solids, Akademie Verlag, Berlin, 1991 Search PubMed; (b) K. Burke, J. P. Perdew and Y. Wang, Electronic Density Functional Theory: Recent Progress and New Directions, Plenum Press, New York, 1998 Search PubMed.
- (a) J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 Search PubMed; (b) Since the properties of the mixture of AlCl3 and NaCl as solvent are not known, we carried out the calculations within these too different media, even though the results were the same, within less than 1 kcal mol−1 of difference in the ΔG computed in both solvents models.
- The distribution in the formation of the σ-complex of 1a (R
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH3+) and 2a (RN(CH3)3+) could be considered as similar, hence the calculations on 2a were not carried out.
NH3+) and 2a (RN(CH3)3+) could be considered as similar, hence the calculations on 2a were not carried out. - Photochromism: Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier Science, 2003 Search PubMed.
- A value of 86.1 kcal mol−1 was found for the ΔGr of the formation of the product coupling (intermediary).
- (a) U. C. Singh and P. A. Kollman, An approach to computing electrostatic charges for molecules, J. Comput. Chem., 1984, 5, 129–145 Search PubMed; (b) B. H. Besler, K. M. Merz Jr and P. A. Kollman, Atomic charges derived from semiempirical methods, J. Comp†ut. Chem., 1990, 11, 431–439 Search PubMed.
- It should be noted that this potential represents the absolute value of half of the redox reaction, a hemi-couple, whereas the experimental values are relative to a reference hemi-couple where the reduction takes place. See ref. 43.
- References of experimental studies of compounds presented in Fig. 2, used for the calculations of oxidation potential are included in ESI,† section 7, p. 28.
- (a) A. I. Vogel, Vogel's Textbook of Practical Organic Chemistry, ed. B. S. Furnis, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, John Wiley & Sons, Inc., New York, 5th edn, 1989 Search PubMed; (b) E. Guillen, J. Hierrezuelo, R. Martínez-Mallorquín, J. M. López-Romero and R. Rico, Suzuki–Miyaura monocouplings of p-dibromobiphenyl and substituted p-dibromo(penta-p-phenylenes), Tetrahedron, 2011, 67, 2555–2561 Search PubMed.
- W. Guo, E. Faggi, R. M. Sebastian, A. Vallribera, R. Pleixats and A. Shafir, Direct Arylation of Oligonaphthalenes Using PIFA/BF3·Et2O: From Double Arylation to Larger Oligoarene Products, J. Org. Chem., 2013, 78, 8169–8175 Search PubMed.
- J. L. Borioni, M. Puiatti, D. M. A. Vera and A. B. Pierini, In search of the best DFT functional for dealing with organic anionic species, Phys. Chem. Chem. Phys., 2017, 19, 9189–9198 Search PubMed.
- (a) J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 Search PubMed . Erratum: Phys. Rev. B, 2018, 98, 079904(E); (b) J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 Search PubMed . Erratum: Phys. Rev. B Condes. Matter Mater. Phys., 1993, 48, 4978; (c) J. P. Perdew, K. Burke and Y. Wang, Generalized gradient approximation for the exchange-correlation hole of a many-electron system, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 Search PubMed; (d) A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and computational information. See DOI: 10.1039/d0ra04213g |
| This journal is © The Royal Society of Chemistry 2020 |