
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of isobemisiose, neosartose, and fischerose: three α-1,6-linked trehalose-based oligosaccharides identified from Neosartorya fischeri†
E. J. Kuenstner‡§
 ,
E. A. Palumbo,
J. Levine and
N. L. Snyder*
,
E. A. Palumbo,
J. Levine and
N. L. Snyder*
Department of Chemistry, Davidson College, Box 7120, Davidson, NC 28036, USA. E-mail: nisnyder@davidson.edu
First published on 12th June 2020
Abstract
Three complex α-1,6-linked trehalose-based oligosaccharides with unique preservation properties, isobemisiose, neosartose, and fischerose, were recently identified from the extreme stress-tolerant ascospores of Neosartorya fischeri. Herein, we report the first concise, scalable, and iterative chemical synthesis of these oligosaccharides from a differentially protected thioglycoside donor and a selectively protected, asymmetric trehalose acceptor. This work constitutes an improved synthesis of isobemisiose, and is also the first reported synthesis of neosartose, a tetrasaccharide, and fischerose, a pentasaccharide, in good yield. Importantly, in-depth studies of biological function are enabled by this synthetic platform.
Introduction
Trehalose 1, a disaccharide comprised of two glucose monomers linked in a α,α-1,1 fashion, is found in over 80 different species of plants, insects, algae, fungi, and bacteria (Fig. 1).1 The oligosaccharides and mycolic acids that contain trehalose (e.g. 2 & 3) are important biomolecules involved in a variety of functions ranging from pathogenicity and autophagy induction to membrane stability.2–4 Notably, the atypical α,α-1,1 glycosidic linkage of trehalose renders the disaccharide non-reducing, which is believed to impart significant resistance to degradation by chemical, thermal, and desiccating means.5,6 The precise biophysical mechanism by which trehalose derivatives stabilize lipid membranes is the subject of ongoing investigation, and has previously been explained by a number of different hypotheses.7–9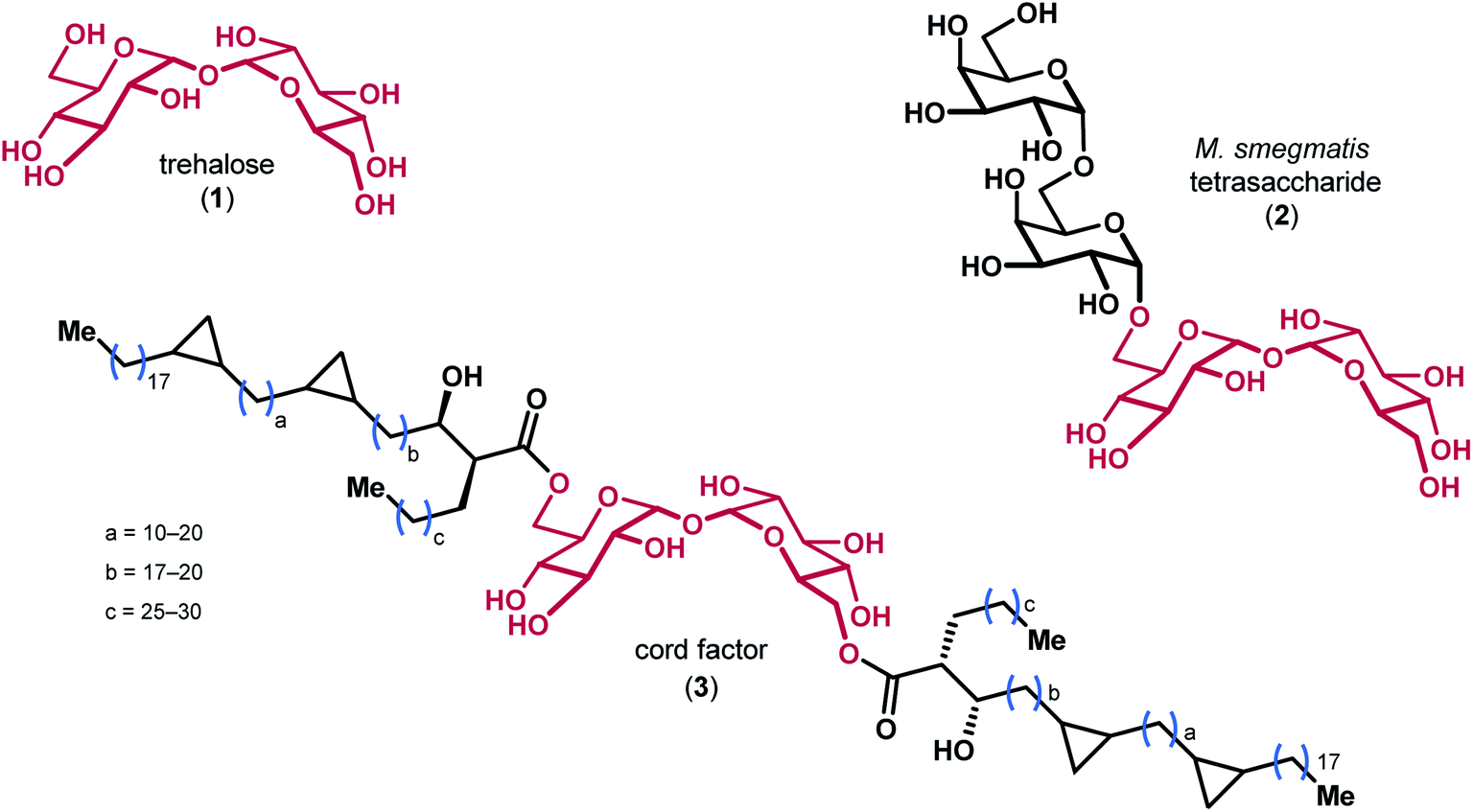 | ||
| Fig. 1 The structures of trehalose and representative derivatives of biological importance, with the trehalose core highlighted in red. | ||
In 2015, Wyatt et al. identified and characterized a series of trehalose-containing oligosaccharides responsible for the unique preservation properties in the extreme stress-resistant ascospores of Neosartorya fischeri (Aspergillus fischeri) (Fig. 2).10–12 FTIR spectroscopy was used to ascertain Tg and WTC values for combinations of isobemisiose 4, neosartose 5 and fischerose 6. In aggregate, the data suggested that the trehalose-containing oligosaccharides form a high-density glass that is postulated to serve a protective role for cytosolic biomolecules in vivo. Moreover, recent studies have indicated that sol gels embedded with bovine LDH have increased stability in the presence of neosartose 5 and fischerose 6 when compared to trehalose.13 The complexity of these compounds and their unique preservation properties ultimately led us to engage in a synthetic effort to prepare these compounds to supplement our ongoing studies to broaden our understanding of the role of these unique compounds in protein preservation. Our results are reported below.
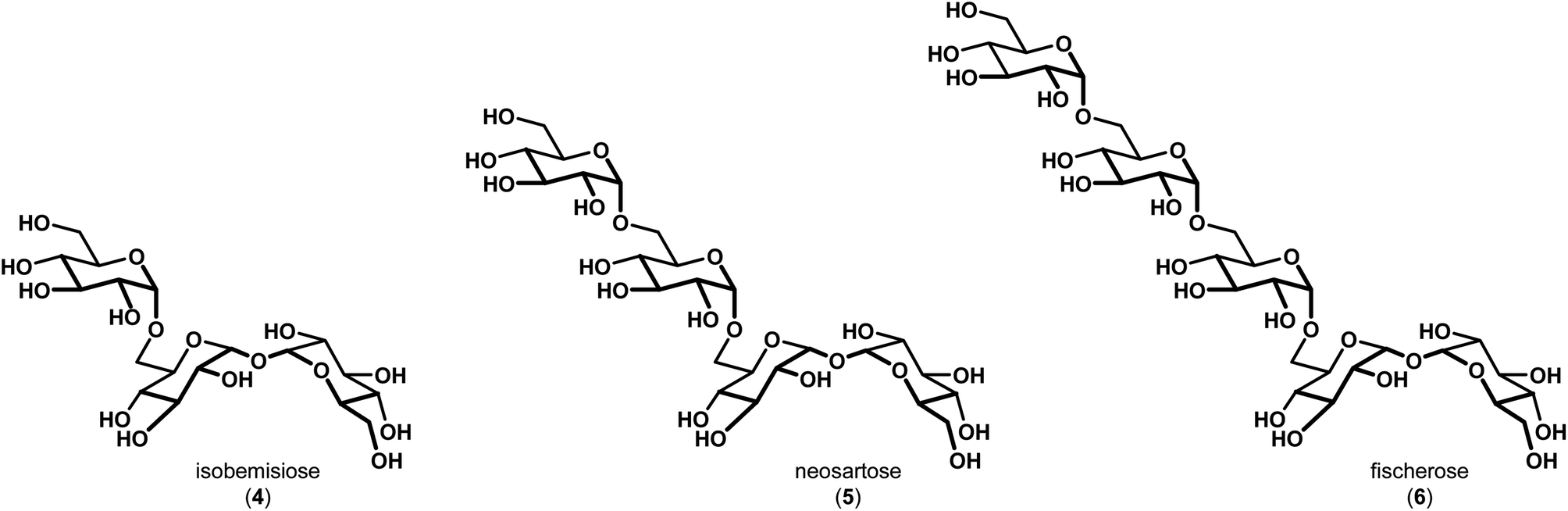 | ||
| Fig. 2 Trehalose-containing oligosaccharides with preservation capabilities identified in Neosartorya fischeri (Aspergillus fischeri). | ||
Results and discussion
We envisioned a linear assembly of the oligosaccharides 4, 5, and 6 via a series of iterative glycosylations. The crux of our strategy was the unification of a bifunctional glycosyl donor with an asymmetric trehalose acceptor. Whereas differential removal of the glucosyl C6 protecting group would allow for additional glycosylations, global deprotection would afford the requisite targets. These considerations led to the identification of readily accessible thioglycoside 7 (ref. 14) and trehalose derivative 8 (ref. 15) as suitable building blocks (Fig. 3). Thioglycoside 7 was particularly attractive because of the precedence for using this donor to generate α-glucosidic linkages with high diasteroselectivity, albeit with different nucleophiles, and for its straightforward synthesis on multigram scales.14 Both thioglycoside 7 and trehalose analog 8 were readily synthesized from commercially available building blocks.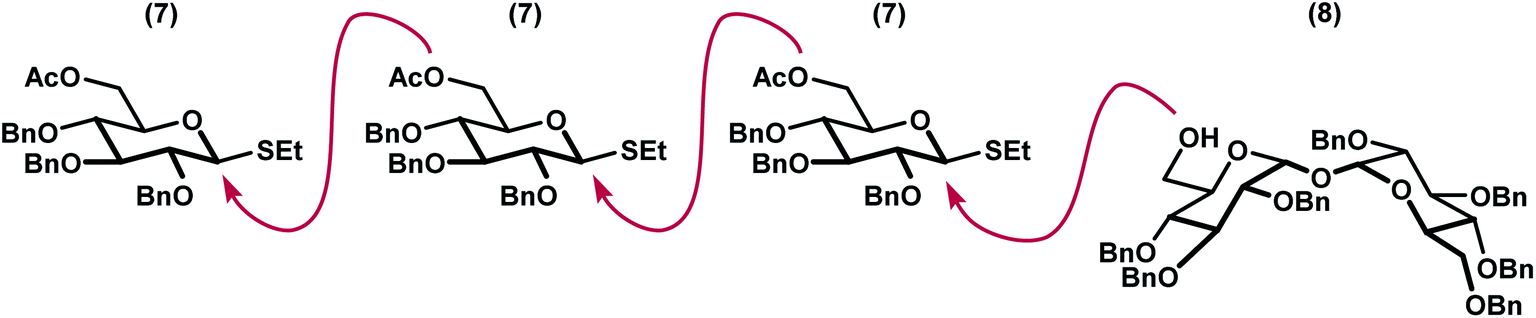 | ||
| Fig. 3 Iterative oligosaccharide synthesis by incorporation of a bifunctional glycosyl linchpin. | ||
Our synthesis began with a moderately stereoselective glycosylation of donor 7 with acceptor 8 via the use of NIS and catalytic triflic acid to form trisaccharide 9 (Scheme 1). Despite the use of a non-participating benzyl group at C2 in conjunction with a binary solvent mixture, we were unable to achieve greater than 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r. to favor the desired α anomer.16,17 Purification of these diastereomers was best achieved following saponification to alcohol 9 and the corresponding β-linked epimer (not shown). Tentative assignment of the relative stereochemistry of the newly-formed glycosidic bond was first accomplished by examination of anomeric resonances in 13C NMR, and later confirmed via comparison to a previously-synthesized sample of 9.18 Iterative glycosylations under identical conditions gave 10 and 11, which proceeded with comparable diastereoselectivity. Removal of the remaining benzyl protecting groups via hydrogenolysis required the addition of a few drops of water as a co-solvent to effect full dissolution of the partially deprotected oligosaccharides, but proceeded in near-quantitative yields. All 13C and 1H NMR spectra of the deprotected compounds, which had been presumed to be of the α configuration, fully matched the previous report, confirming our previous assignments of relative stereochemistry.10
:1 d.r. to favor the desired α anomer.16,17 Purification of these diastereomers was best achieved following saponification to alcohol 9 and the corresponding β-linked epimer (not shown). Tentative assignment of the relative stereochemistry of the newly-formed glycosidic bond was first accomplished by examination of anomeric resonances in 13C NMR, and later confirmed via comparison to a previously-synthesized sample of 9.18 Iterative glycosylations under identical conditions gave 10 and 11, which proceeded with comparable diastereoselectivity. Removal of the remaining benzyl protecting groups via hydrogenolysis required the addition of a few drops of water as a co-solvent to effect full dissolution of the partially deprotected oligosaccharides, but proceeded in near-quantitative yields. All 13C and 1H NMR spectra of the deprotected compounds, which had been presumed to be of the α configuration, fully matched the previous report, confirming our previous assignments of relative stereochemistry.10
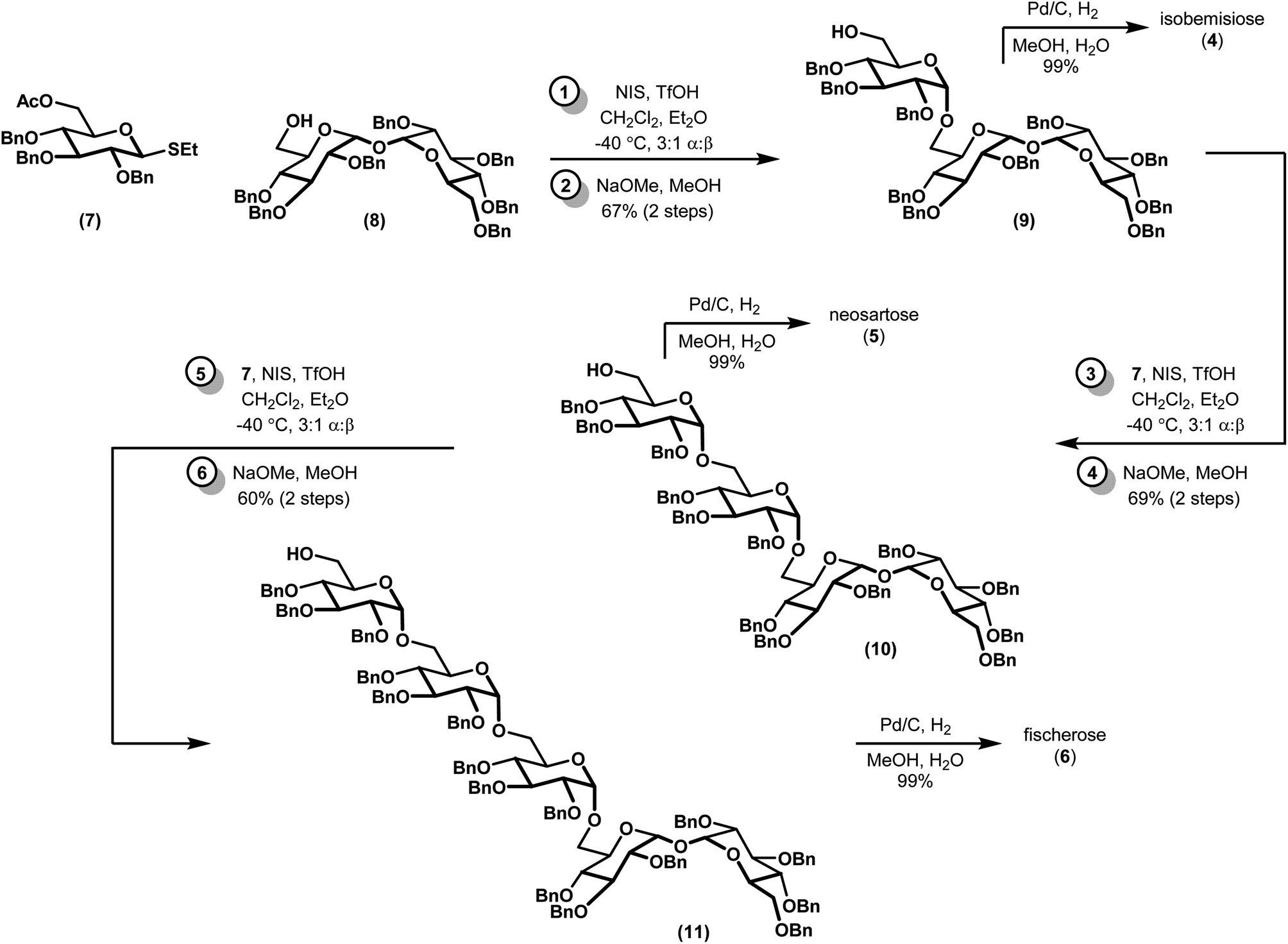 | ||
| Scheme 1 Synthesis of α-1,6-linked oligosaccharides. | ||
The stereoselectivity of our glycosylation, while modest, merits additional discussion. Indeed, the reactivity of carbohydrate building blocks is critical for forming regio- and diastereoselective glycosidic linkages.19–22 First, our results with thioglycoside donor 7 differs from an earlier report by Koto and co-workers18 that remarkably predates the isolation of isobemisiose from a natural source. Their report, which involves glycosylation of acceptor 8 with the lactol (C1 hemiacetal) analog of 7 in the presence of excess pyridine and TMS triflate, gave ca 12:1 d.r. to favor the α-anomer albeit in lower yields and on smaller scales. While the original authors did not comment on the origin of this high diastereoselectivity, other reports have speculated that it may arise via remote participation of the C6 acetyl group.23 Indeed, similar mechanisms of selectivity have been noted in the synthesis of an epimeric trehalose-containing galactoside.24 However, the presence of a C6 acetate in 7 suggests a more complex interplay between remote participation and various ion-pair species. Recent reports by Zhu25 and Kowalska,26 which were published while this manuscript was in preparation, suggest that the α-selectivity of this reaction might be improved by using trifluoroacetimidates instead of thioglycoside donors. In contrast, the role of glycosylation acceptors in promoting stereochemical outcomes is less established than that of glycosylation donors, but research in this area may still provide relevant insight to our results. Numerous studies across various reaction manifolds have correlated reactive acceptors (e.g. primary alcohols) with β-selectivity.27,28 Conversely, less reactive counterparts (e.g. secondary alcohols with adjacent electron-withdrawing groups) generally display α-selectivity.22 It might be expected, then, that replacement of the benzyl protecting groups in 8 for benzoyl groups could improve the diastereoselectivity of our glycosylation. These speculations aside, the present paper emphasizes the impact of seemingly subtle changes in stereoelectronic environments during the formation of reactive intermediates in glycosylations.
In conclusion, we have described a concise synthesis of three complex oligosaccharides: isobemisiose (4), neosartose (5), and fischerose (6). Our sequence to 6, which is the largest synthesized α-linked trehalose-derived oligosaccharide described to date, is unprecedented. Sequences to the former species compare favorably to reports of enzymatic incubations and de novo syntheses of smaller trehalose oligosaccharides.18,24,29–33 Several advantages over the single study for isobemisiose reported by Koto18 are particularly noteworthy, including the overall yield and scalability (e.g. 66% of 4 over three steps on a gram scale, as opposed to ca. 45% over three steps on a hundred milligram scale). In addition, our methodology provides us with the ability to generate theretofore unknown stereoisomers. The aforementioned compounds are the subject of ongoing biological investigations in our laboratory, the results of which will be reported in due course.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to thank Peter H. Seeberger for enabling a fruitful collaboration with Jan Dijksterhuis which inspired this work. This work was partially supported by an NSF-RUI (#1708546) award and Faculty Study and Research grants to N. L. S. E. A. P. would like to thank Davidson College for support through the Davidson Research Initiative.Notes and references
- Q. Chen and G. G. Haddad, J. Exp. Biol., 2004, 207, 3125 CrossRef CAS PubMed
.
- D. B. Kastrinsky, N. S. McBridge, K. M. Backus, J. J. LeBlanc and C. E. Barry III, Mycolic Acid/Cyclopropane Fatty Acid/Fatty Acid Biosynthesis and Health Relations, in Comprehensive Natural Products II, ed. H.-W. Liu and L. Mander, Elsevier, 2010, vol. 1.04, pp. 65–145 Search PubMed
- S. Sarkar, J. E. Davies, Z. Huang, H. Tunnacliffe and D. C. Rubinsztein, J. Biol. Chem., 2007, 282, 5641 CrossRef CAS PubMed
- M. Ohta, Y. T. Pan, R. A. Laine and A. D. Elbein, Eur. J. Biochem., 2002, 269, 3142 CrossRef CAS PubMed
- T. Higashiyama, Pure Appl. Chem., 2002, 74, 1263 CAS
- A. D. Elbein, Y. T. Pan, I. Pastuszak and D. Carrol, Glycobiology, 2003, 13, 17R CrossRef CAS PubMed
- J. F. Carpenter and J. H. Crowe, Cryobiology, 1988, 25, 459 CrossRef CAS
- H. Levine and L. Slade, BioPharm, 1992, 5, 36 CAS
- N. K. Jain and I. Roy, Protein Sci., 2008, 18, 2 Search PubMed
- T. T. Wyatt, G. J. Gerwig, J. P. Kamerling, H. A. B. Wösten and J. Dijksterhuis, Carbohydr. Res., 2015, 411, 49 CrossRef CAS PubMed
- T. T. Wyatt, M. R. van Leeuwen, E. A. Golovina, F. A. Hoekstra, E. J. Kuenstner, E. A. Palumbo, N. L. Snyder, C. Visagie, A. Verkennis, J. E. Hallsworth, H. A. B. Wösten and J. Dijksterhuis, Environ. Microbiol., 2015, 17, 395 CrossRef CAS PubMed
- Isobemisiose has previously been isolated, see: D. L. Hendrix and M. E. Salvucci, J. Insect Physiol., 2001, 47, 423 CrossRef CAS PubMed
- Unpublished data.
- J. Wang, B. Elchert, Y. Hui, J. Y. Takemoto, M. Bensaci, J. Wennergren, R. Rai and C.-W. T. Chang, Bioorg. Med. Chem. Lett., 2004, 12, 6397 CrossRef CAS PubMed
- C. McDonnell, O. López, P. Murphy, J. G. Fernández Bolaňos, T. Hazell and M. Bols, J. Am. Chem. Soc., 2004, 126, 12374 CrossRef CAS PubMed
- For a recent review of protecting groups in glycosylations, see: B. Ghosh and S. S. Kulkarni, Chem.–Asian J., 2020, 15, 450 CrossRef CAS PubMed
- K.-K. T. Mong, T. Nokami, N. T. T. Tran and P. B. Nhi, Solvent Effect on Glycosylation, in Selective Glycosylations: Synthetic Methods and Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, Bennett, C.S, 1st edn, 2017, pp. 59–77 Search PubMed
- S. Koto, K. Yago, S. Zen, F. Tomonaga and S. Shimada, Bull. Chem. Soc. Jpn., 1986, 59, 411 CrossRef CAS
- B. Fraser-Reid and J. C. López, in React. Tuning Oligosacch. Assem, ed. B. Fraser-Reid and J. C. López, Springer, Berlin, Heidelberg, 2011, pp. 1–29 Search PubMed
- For a recent review see: T. G. Frihed, M. Bols and C. M. Pedersen, Chem. Rev., 2015, 115, 4963 CrossRef CAS PubMed
- M. T. C. Walvoort, G. A. van der Marel, H. S. Overkleeft and J. D. C. Codée, Chem. Sci., 2013, 4, 897 RSC
- S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., 2018, 130, 8372 CrossRef
- A. V. Demchenko, Synlett, 2003, 9, 1225 Search PubMed
- The presence of a C4 benzoyl group enabled the formation of a [2.2.1]-trioxobicyclohexane via remote participation to impede β attack. Such intermediates would naturally be inaccessible in the case of glycosylation with 7. For details, refer to: M. A. Chaube, V. V. Sarpe, S. Jana and S. S. Kulkarni, Org. Biomol. Chem., 2016, 14, 5595 RSC
- S. Zhu, G. Samala, E. T. Sletten, J. L. Stockdill and H. M. Nguyen, Chem. Sci., 2019, 10, 10475 RSC
- K. Kowalska and C. M. Pedersen, Org. Biomol. Chem., 2020, 18, 1918 RSC
- S. Kaeothip, S. J. Akins and A. V. Demchenko, Carbohydr. Res., 2010, 345, 2146 CrossRef CAS PubMed
- J. Kalikanda and Z. Li, Carbohydr. Res., 2011, 346, 2380 CrossRef CAS PubMed
- K. Ajisaka and H. Fujimoto, Carbohydr. Res., 1990, 199, 227 CrossRef CAS PubMed
- M. Kurimoto, T. Nishimoto, T. Nakada, H. Chaen, S. Fukuda and Y. Tsujisaka, Biosci., Biotechnol., Biochem., 1997, 61, 699–703 CrossRef CAS PubMed
- N. K. Smith, S. G. Gilmour and R. A. Rastall, Enzyme and Microbiol Technology, 1997, 21, 349 CrossRef CAS
- As disclosed in a private communication with Ben Swarts, recent enzymatic methods have not been fruitful in the synthesis of neosartose and fischerose. For example: B. L. Urbanek, D. C. Wing, K. S. Haislop, C. J. Hamel, R. Kalscheuer, P. J. Woodruff and B. M. Swarts, ChemBioChem, 2014, 15, 2066 CrossRef CAS PubMed
- An epimeric α,β-(1,1) trisaccharide has also been reported via chemical synthesis: T. E. C. L. Ronnow, M. Meldal and K. Bock, Tetrahedron: Asymmetry, 1994, 5, 2109 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, in addition to 13C & 1H NMR spectra for 4, 5, 6, 9, 10, 11, and select β anomers, is available online. See DOI: 10.1039/d0ra04137h |
| ‡ Present address: Entasis Therapeutics, 35 Gatehouse Drive, Waltham, MA 02451, USA. |
| § Previous address: Department of Chemistry, Hamilton College, 198 College Hill Road, Clinton, NY 13323, USA. |
| This journal is © The Royal Society of Chemistry 2020 |