
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication and characterization of silicon oxycarbide fibre-mats via electrospinning for high temperature applications†
Zhongkan Ren a,
Christel Gervaisb and
Gurpreet Singh*a
a,
Christel Gervaisb and
Gurpreet Singh*a
aDepartement of Mechanical and Nuclear Engineering, Kansas State University, Manhattan, KS 66506, USA. E-mail: gurpreet@ksu.edu
bSorbonne Université, CNRS UMR 7574, Laboratoire de Chimie de la Matière Condensée de Paris, 75005 Paris, France
First published on 19th October 2020
Abstract
Electrospinning is an emerging technique for synthesizing micron to submicron-sized polymer fibre supports for applications in energy storage, catalysis, filtration, drug delivery and so on. However, fabrication of electrospun ceramic fibre mats for use as a reinforcement phase in ceramic matrix composites or CMCs for aerospace applications remains largely unexplored. This is mainly due to stringent operating requirements that require a combination of properties such as low mass density, high strength, and ultrahigh temperature resistance. Herein we report fabrication of molecular precursor-derived silicon oxycarbide or SiOC fibre mats via electrospinning and pyrolysis of cyclic polysiloxanes-based precursors at significantly lower weight loadings of organic co-spin agent. Ceramic fibre mats, which were free of wrapping, were prepared by a one-step spinning (in air) and post heat-treatment for crosslinking and pyrolysis (in argon at 800 °C). The pyrolyzed fibre mats were revealed to be amorphous and a few microns in diameter. Four siloxane-based pre-ceramic polymers were used to study the influence of precursor molecular structure on the compositional and morphological differences of cross-linked and pyrolyzed products. Further thermal characterization suggested the potential of electrospun ceramic mats in high temperature applications.
Introduction
Silicon-based ceramics have extraordinary potential in high-temperature applications due to their promising mechanical properties at elevated temperatures and in oxidizing environments.1 Controlled synthesis of one-dimensional non-oxide ceramic fibres, whiskers, or rods has been widely studied.2,3 Until the discovery of polymer-derived ceramics (PDCs), ceramic fibres were nearly unachievable via powder-based conventional ceramic manufacturing techniques.2 PDC, however, is beneficial for the manufacturing of complex structured ceramic components, such as fibres, films/coatings, or ceramic matrix composites (CMCs), due to its ease of handling, improved processability, decreased processing temperature, and controllable preceramic compositions, which allow the application of a variety of preforming techniques to draw fibres from preceramic dry, wet, melt spinning, and electrospinning polymers.1In our previous work,4 we prepared handspun PDC fibres from cyclic silazane using linear polymer (i.e., polyacrylic acid) as a spinning agent. This polymer acted as a template to form the fibres, while the silazane molecules were embedded or gathered on the surface of the fibre template. The silazane molecules then crosslinked and formed a rigid structure, even causing solidification if used pure. After pyrolysis, crosslinked silazanes converted into ceramics and the polyacrylic acid (PAA) decomposed, contributing free carbons to the final product. Although hand spinning produces uniform, long fibres, the high requirement of sol–gel morphology and extensive amount of human involvement required decrease its efficiency for mass production.
Electrospinning is a flexible, scalable technique that utilizes a simple setup and easy operation to draw micron to sub-micron fibres from molten polymers or polymer solutions.2,5,6 Electrospun ultra-thin fibres are desirable for applications such as electronics, photonics, sensing, filter, biomedical, or reinforcement applications.7,8 Compared to hand spinning, electrospinning is more efficient with well-controlled fibre diameter distribution. Substantial electrostatic charges, which are caused by the electrical field between the spinning needle and the collector, form the fibres.9 The fibre mats can then be heat treated to ceramic fibres using PDCs.10 The spinning solution is usually made of at least one preceramic precursor and a solvent that evaporates during spinning.10 Additionally, an organic polymer may be added as a spinning agent to optimize viscosity. Fibre diameter is controllable primarily via sol–gel properties (i.e., viscosity, conductivity, and surface tension) and spinning parameters (i.e., electrical potential and distance between the tip and collector).7,11,12 Likewise, fibre morphology is effectively tunable via spinning, crosslinking, and pyrolysis conditions.
A limited amount of research has demonstrated the electrospinning of SiOC,13–15 SiOCN,16–18 SiCN, or SiBCN19 fibres via PDCs. These fibres are usually synthesized from a mixture of linear polysiloxane (or polysilazane) and linear organic polymer, such as polyacrylonitrile or polyvinylpyrrolidone (PVP). Most fibres synthesized uniform ceramic fibres with diameters ranging from hundreds of nanometres to several micrometres. Research has also shown further implementations of electrospun fibre mats for aerogel growth,20 nanowire growth,14,19 and electrochemical applications.15,17 In the previous studies, pyrolyzed ceramic fibre surface and cross-section features were well determined with via high resolution scanning electron microscopy (SEM) or transmission electron microscope (TEM) techniques; chemical compositions were also reported by various analysis. However, characterizations of as-spun raw fibre mats and crosslinked fibre mats were often ignored. As a result, the evolution or development of chemical compositions, bonding situations and surface morphologies over each post processing stage remain incomplete. Herein, we carried out a systematic characterization of fibre mats from the raw state to crosslinked and pyrolyzed state to investigate: (1) the effect of different preceramic precursor on the final product; (2) the crosslinking efficiency of different catalyst; (3) compositional and structural development of the fibre mats; (4) the change of chemical bonding situation after crosslinking and pyrolysis of the fibre mats; (5) mechanical and thermal stability of synthesized fibre mats.
In this work, we studied the SiOC fibres that were electrospun from cyclic oligomer siloxanes and PVP hybrid precursors for the first time. Fibre mats were prepared by simple one step mixing and spinning process with isopropanol as the only solvent for siloxanes and PVP. This study utilized 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetra-siloxane (referred to as 4-TTCS), Pt-1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane complex (2% Pt-4-TTCS complex in 4-TTCS, referred to as Pt-TTCS), 1,3,5-trivinyl-1,3,5-trimethyl-cyclotrisiloxane (referred to as 3-TTCS), and 1,3-divinyl-tetramethyldisiloxane (referred to as DTDS) as SiOC precursors. These siloxanes, that have not been reported for fibre drawing purpose, are readily available and inexpensive, thereby decreasing fibre manufacturing costs compared to costly SiC fibres.1,21 The electrospinning was carried out with low PAA (spinning reagent) concentration as 20 wt%. As-spun fibre mats are crosslinked and pyrolyzed in an inert environment (ultra-high purity argon), and prepared fibre mats are free of wrapping and large enough for lab-scale CMC testing. SEM and TEM revealed the fibre morphologies. Crosslinking reactions of preceramic polymers, PVP, and polymer-to-ceramic conversion were studied via Raman spectroscopy, Fourier-transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), and nuclear magnetic resonance (NMR).
Experimental
Materials
Preceramic silicon oligomers were purchased from Gelest (Pennsylvania, USA), PVP (average Mw ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 g mol−1) and dicumyl peroxide (DCP) were obtained from Sigma-Aldrich (Missouri, USA), and isopropanol was obtained from Fisher Chemical (Kansas, USA). Matheson (Texas, USA) supplied ultra-high-purity, compressed argon gas to be used as a pyrolysis atmosphere.
300000 g mol−1) and dicumyl peroxide (DCP) were obtained from Sigma-Aldrich (Missouri, USA), and isopropanol was obtained from Fisher Chemical (Kansas, USA). Matheson (Texas, USA) supplied ultra-high-purity, compressed argon gas to be used as a pyrolysis atmosphere.
Fig. 1 shows that preceramic silicon oligomers have very similar initial structures to vinyl groups for potential crosslinking spots. Initial compositions differ slightly since 4-TTCS, Pt-TTCS, and 3-TTCS have an overall Si to O ratio of 1:1 and cyclic structures, while the ratio is 2:1 for DTDS. As shown in the following equations, crosslinking by DCP and Pt yields different mechanisms and results:
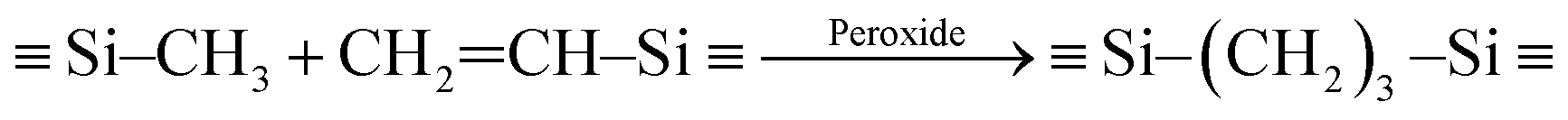 | (1) |
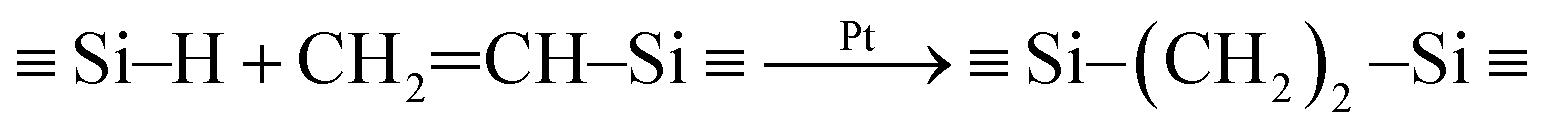 | (2) |
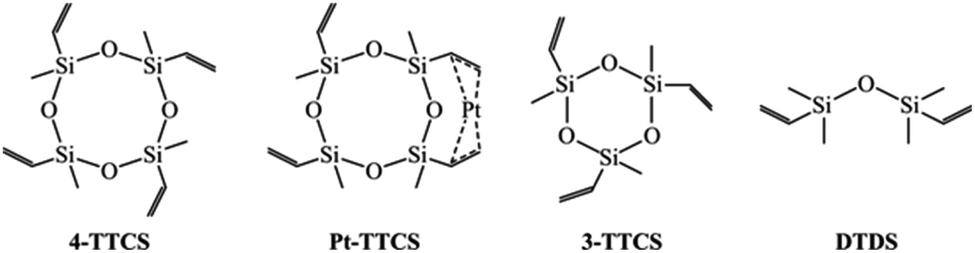 | ||
| Fig. 1 Molecular structure of preceramic silicon oligomers. | ||
Electrospinning setup
The electrospinning setup used in this work was designed and assembled within the lab. Fig. 2 shows the four major parts of the spinning setup:1 spinning solution feeder made of a syringe mount on a linear slide stroke with a stepper motor;2 fibre collector as a rotating aluminium cylinder driven by a motor;3 high-voltage power supply (30 kV maximum); and4 control system by Arduino, including sensors and motor controller.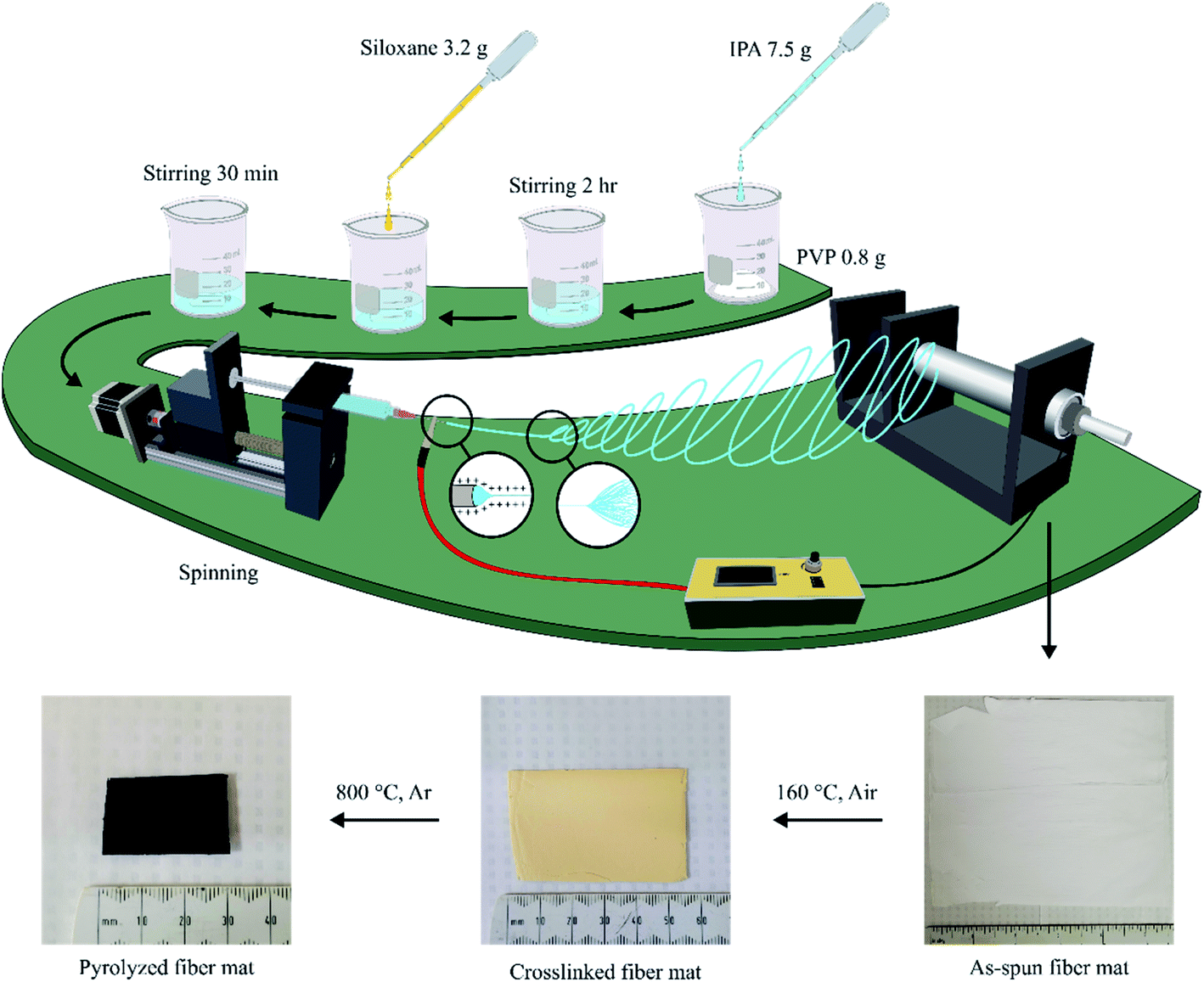 | ||
| Fig. 2 Electrospinning setup. | ||
Preparation of raw fibres
Siloxanes were initially mixed with 1% DCP as a crosslinking initiator until DCP was fully dissolved into liquid siloxanes, except for Pt-TTCS, in which Pt acted as the crosslinker. In addition, 800 mg of PVP powder was dissolved into isopropanol alcohol (IPA) with the approximate weight ratio of PVP:IPA = 1:9 via magnetic stirring at 300 rpm for 4 hours. Once a uniform solution was formed without visible particles or air bubbles, preceramic siloxanes were added into the solution with extra stirring for 1 hour. The siloxane/PVP hybrid gel was loaded into a 10 ml syringe with a flat metallic needle and fed at the speed of 5 ml h−1. The needle was subjected to the 15 kV positive output of the high-voltage power supply while the ground was connected to the fibre collector. The distance between the needle tip and the collector was set to 20 cm. A raw fibre mat measuring about 20 × 20 cm2 was produced.
Crosslinking and pyrolysis of fibre mats
The as-spun fibre mats were dried in open air for 24 hours and then transferred into the low-temperature oven for crosslinking at 160 °C for 24 hours. Crosslinked mats were cut into small squares to fit into aluminium ceramic boats for pyrolysis, which was performed at a heating rate of 2 °C min−1 and 30 min annealing at the target temperature in the tube furnace with Ar gas (at the flowrate of 5 ml min−1). The target temperature of 800 °C was selected to study polymer-to-ceramic conversion and fibre morphologies. SiOC ceramic fibres mats were synthesized after pyrolysis.Characterization
Characterization techniques were performed on raw, crosslinked, and pyrolyzed samples to determine chemical, mechanical, surface, and compositional changes of the mats at each stage. Fibre diameter and surface morphology were obtained via SEM (EVO MA10, ZEISS, Germany), and corresponding characteristics of fibre surface and cross sections were observed after focused ion beam (FIB) treatment in the SEM. XPS (K-alpha XPS/UPS photoelectron spectrometer, ThermoFisher, USA) was used for compositional analysis, and XPS survey and high-resolution scans were performed on fibre mats for a 10 s acquisition time with depth-profiling via 2 min Ar sputtering at 3 keV.Techniques such as Raman spectroscopy (ARAMIS Raman spectrometer, LabRAM HORIBA Jobin Yvon, USA), FTIR (Spectrum 400 FT-IR spectrometer, PerkinElmer, USA), and NMR spectroscopy were used to investigate the molecular structure of prepared fibre mats. Solid-state 13C CP MAS and 29Si MAS NMR spectra were recorded on a Bruker AVANCE 300 spectrometer (B0 = 7.0 T, ν0(1H) = 300.29 MHz, ν0(13C) = 75.51 MHz, ν0(29Si) = 59.66 MHz) using a 7 mm Bruker probe and spinning frequency of 5 kHz. 13C CP MAS experiments were recorded with ramped-amplitude cross-polarization in the 1H channel to transfer magnetization from 1H to 13C (i.e., recycle delay = 3 s, CP contact time = 1 ms, optimized 1H spinal-64 decoupling). Single-pulse 29Si NMR MAS spectra were recorded with recycle delays of 60 s. Chemical shift values were referenced to preceramic precursors for 13C and 29Si. Raman spectra were collected using a HeNe laser (632 nm) to analyse free carbon in the pyrolyzed system; FTIR was performed using an attenuated total reflectance (ATR) setup and collected IR spectrums ranging from 500 to 3500 nm with 64 cumulation number that provided data from functioning groups at each stage. NMR revealed detailed bonding using various elements in each sample.
Results and discussion
Microstructure
SEM images of crosslinked and pyrolyzed fibres are presented in Fig. 3a–f. The fibres were initially drawn under identical conditions (15 kV, ∼20 cm distance between needle and collector, sol–gel feeding speed of 5 ml h−1). Raw samples were uniform in diameter across the individual fibres. For 4-TTCS samples, the average fibre diameter was initially 3.5 μm, decreased to approximately 3.0 μm after crosslinking, and then further shrunk to approximately 1.5 μm (about 60% shrinkage from raw fibres) after pyrolysis. Various Si initial compounds resulted in unique features of the pyrolyzed fibres. Average fibre diameters changed significantly from 3–5 μm (for Pt-TTCS sample) to 1–3 μm (for 4-TTCS and 3-TTCS samples) and 0.2–1.0 μm (for DTDS sample). The results indicate that the crosslinking mechanism (by DCP or Pt) led to varied diameter distribution; furthermore, the large silicon compound (with higher average molecular weight) derived thick fibres for cyclic types of preceramic precursors. In addition, DTDS derived fibres were smoother (or less porous) on the fibre surface compared to the other three samples. Amorphous (nanodomain) structures of ceramic fibres were revealed by high-res TEM images (Fig. 3k–n), compare to the clear crystalline structure of nano Pt particles in Fig. 3l. | ||
| Fig. 3 SEM, TEM images, and corresponding XPS spectra of electrospun fibre mats. SEM images of (a) 4-TTCS/PVP as spun; (b) 4-TTCS/PVP crosslinked at 160 °C; (c) 4-TTCS/PVP pyrolyzed at 800 °C; (d) Pt-TTCS/PVP pyrolyzed at 800 °C; (e) 3-TTCS/PVP pyrolyzed at 800 °C; and (f) DTDS/PVP pyrolyzed at 800 °C. Further magnified images are shown in the insets of (a)–(f). TEM images of (g) 4-TTCS/PVP pyrolyzed; (h) Pt-TTCS/PVP pyrolyzed; (i) 3-TTCS/PVP pyrolyzed; and (j) DTDS/PVP pyrolyzed. Corresponding high-res TEM images of (k) 4-TTCS/PVP pyrolyzed; (l) Pt-TTCS/PVP pyrolyzed; (m) 3-TTCS/PVP pyrolyzed; and (n) DTDS/PVP pyrolyzed. | ||
Cyclic precursors derived translucent fibres, revealing the possible hollow features of the fibres, which were also captured from the cross-sectional view. Theoretically, the formation of hollow structured fibres is primarily related to the spinning process.22 Research has shown that post-heat treatment on the core–shell structure creates hollow fibre mats via electrospinning, where the core–shell structure could be formed if two different polymers were extruded simultaneously with two coaxial capillaries. However, in our case, the spinning solution was prepared by mixing preceramic silicon polymer, linear PVP polymer (as spinning agent), and solvent. The as-prepared solutions were electrospun using a single syringe needle. Hence, we suspect the core–shell structure was formed during the spinning process due to the molecular weight difference between preceramic polymers and PVP (∼1300000 g mol−1). The small, or short, molecules demonstrated increased mobility when they were applied to the same electrostatic force that can be pulled out of the rigid PVP environment and form such hollowed structures. The holes were formed during heat treatments (i.e., crosslinking and pyrolysis). Further investigation of this phenomenon is needed with other characterization techniques.
Compositional analysis
Raman spectroscopy results (Fig. 4) were used to determine carbon bonding and corresponding microstructures of 800 °C pyrolyzed systems. All samples, except Pt-TTCS/PVP, showed obvious carbon D band (around 1340 cm−1) and G band (around 1600 cm−1) absorptions in the spectra, which indicated the potential presence of a free-carbon or nanodomain structure. The curve-fitted result showed a considerable amount of disordered sp2–sp3 bonds (indicated by T band at 1204–1232 cm−1) and amorphous carbon (D′′ at 1490–1504 cm−1) in the pyrolyzed samples.23,24 The Pt-TTCS/PVP sample showed a strong fluorescence under visible (HeNe at 632 nm) and IR (diode at 785 nm) laser sources and no significant carbon D or G band in the structure. However, under a 532 nm laser source, the Pt-TTCS/PVP sample revealed carbon D and G bands, which were most obvious after the background was removed. The fluorescence may have been caused by Pt in the sample, as confirmed by XPS.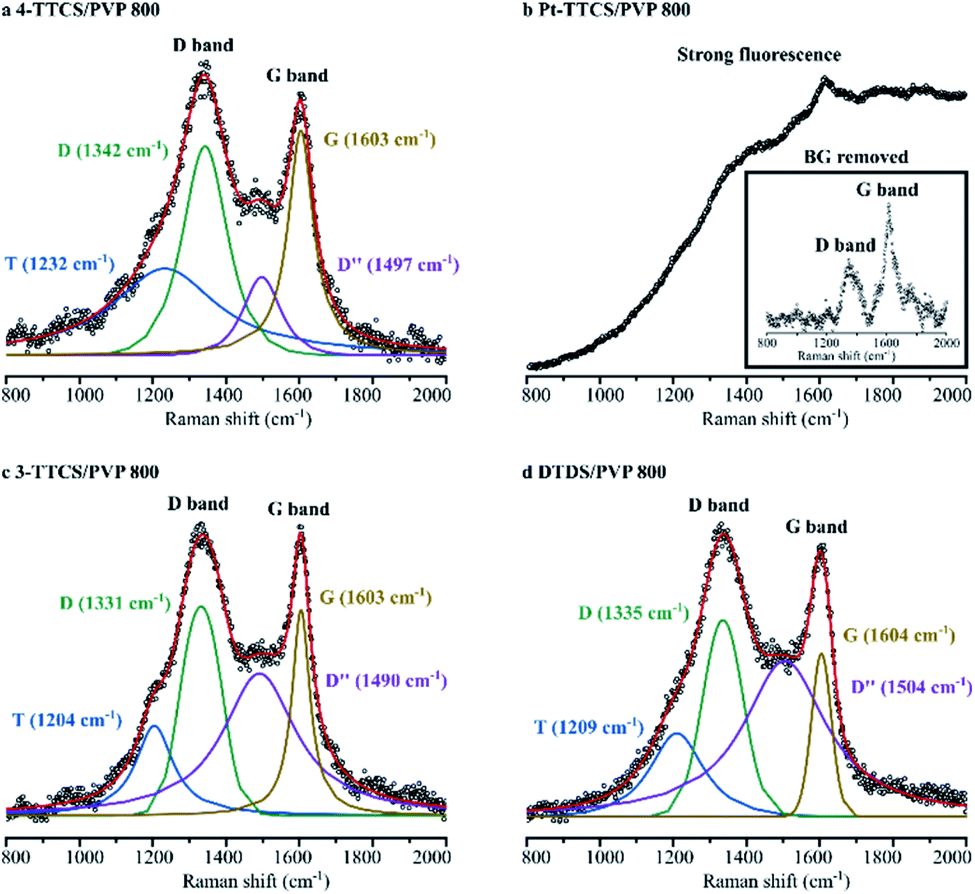 | ||
| Fig. 4 Raman spectra of pyrolyzed fibre mats with integrated peaks shown in the plot. (a) 4-TTCS/PVP; (b) Pt-TTCS/PVP sample with strong fluorescence. The spectrum with a removed background is shown in the inset: (c) 3-TTCS/PVP; (d) DTDS/PVP. | ||
FTIR spectra of raw, crosslinked, and pyrolyzed samples were collected for each type of sample (i.e., neat PVP and each PVP/preceramic silicon polymer).
Fig. 5 shows FTIR results with marked characteristic absorption bands. Corresponding characteristic peaks are summarized in Table 1 for various preceramic silicon precursors and PVP. For neat PVP samples, a comparison between raw and 160 °C heated samples (neat PVP XL) showed that all the samples retained main peaks for PVP (C–O at ∼1650 cm−1, C–H at ∼1420 cm−1 and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at ∼570 cm−1), suggesting that no obvious crosslinking reaction occurred. The high-temperature pyrolysis of heat treated neat PVP in an inert atmosphere was performed to confirm this finding. Destruction of the fibre mat suggested no or low crosslinking during the low-temperature heat treatment. When PVP was mixed with 4-TTCS or Pt-TTCS, very strong peaks, such as Si–O–Si at ∼1059 cm−1, Si–CHCH2 at ∼1000 cm−1, and Si–CH3 ∼750 cm−1, which did not exist in neat PVP samples, were observed. After crosslinking at 160 °C, decreased Si–CHCH2 and Si–CH3 peaks indicated a strong crosslinking reaction of TTCS, and after pyrolysis, the spectrum showed the decomposition of PVP organic components and the retention of Si–O in the system. Although 3-TTCS molecules are structurally resemble to 4-TTCS molecules, 3-TTCS samples showed a unique spectrum that the characteristic peaks were less intense even though similar crosslinking behaviour was observed from the decrease of Si–CHCH2 at 1662 cm−1 and Si–CH3 at 1424 and 1288 cm−1. Relatively weaker crosslinking evidence was observed in the DTDS sample with less obvious changes of Si–CH3 at 1424 and 1288 cm−1 after crosslinking. As shown in Fig. 5c, all pyrolyzed samples had strong absorptions at the Si–O (∼1050 cm−1) peak and weak absorptions at the Si–C (∼700 cm−1) peak. The relatively strong Si–O absorption indicated the presence of more Si–O than Si–C bonds in all samples.
O at ∼570 cm−1), suggesting that no obvious crosslinking reaction occurred. The high-temperature pyrolysis of heat treated neat PVP in an inert atmosphere was performed to confirm this finding. Destruction of the fibre mat suggested no or low crosslinking during the low-temperature heat treatment. When PVP was mixed with 4-TTCS or Pt-TTCS, very strong peaks, such as Si–O–Si at ∼1059 cm−1, Si–CHCH2 at ∼1000 cm−1, and Si–CH3 ∼750 cm−1, which did not exist in neat PVP samples, were observed. After crosslinking at 160 °C, decreased Si–CHCH2 and Si–CH3 peaks indicated a strong crosslinking reaction of TTCS, and after pyrolysis, the spectrum showed the decomposition of PVP organic components and the retention of Si–O in the system. Although 3-TTCS molecules are structurally resemble to 4-TTCS molecules, 3-TTCS samples showed a unique spectrum that the characteristic peaks were less intense even though similar crosslinking behaviour was observed from the decrease of Si–CHCH2 at 1662 cm−1 and Si–CH3 at 1424 and 1288 cm−1. Relatively weaker crosslinking evidence was observed in the DTDS sample with less obvious changes of Si–CH3 at 1424 and 1288 cm−1 after crosslinking. As shown in Fig. 5c, all pyrolyzed samples had strong absorptions at the Si–O (∼1050 cm−1) peak and weak absorptions at the Si–C (∼700 cm−1) peak. The relatively strong Si–O absorption indicated the presence of more Si–O than Si–C bonds in all samples.
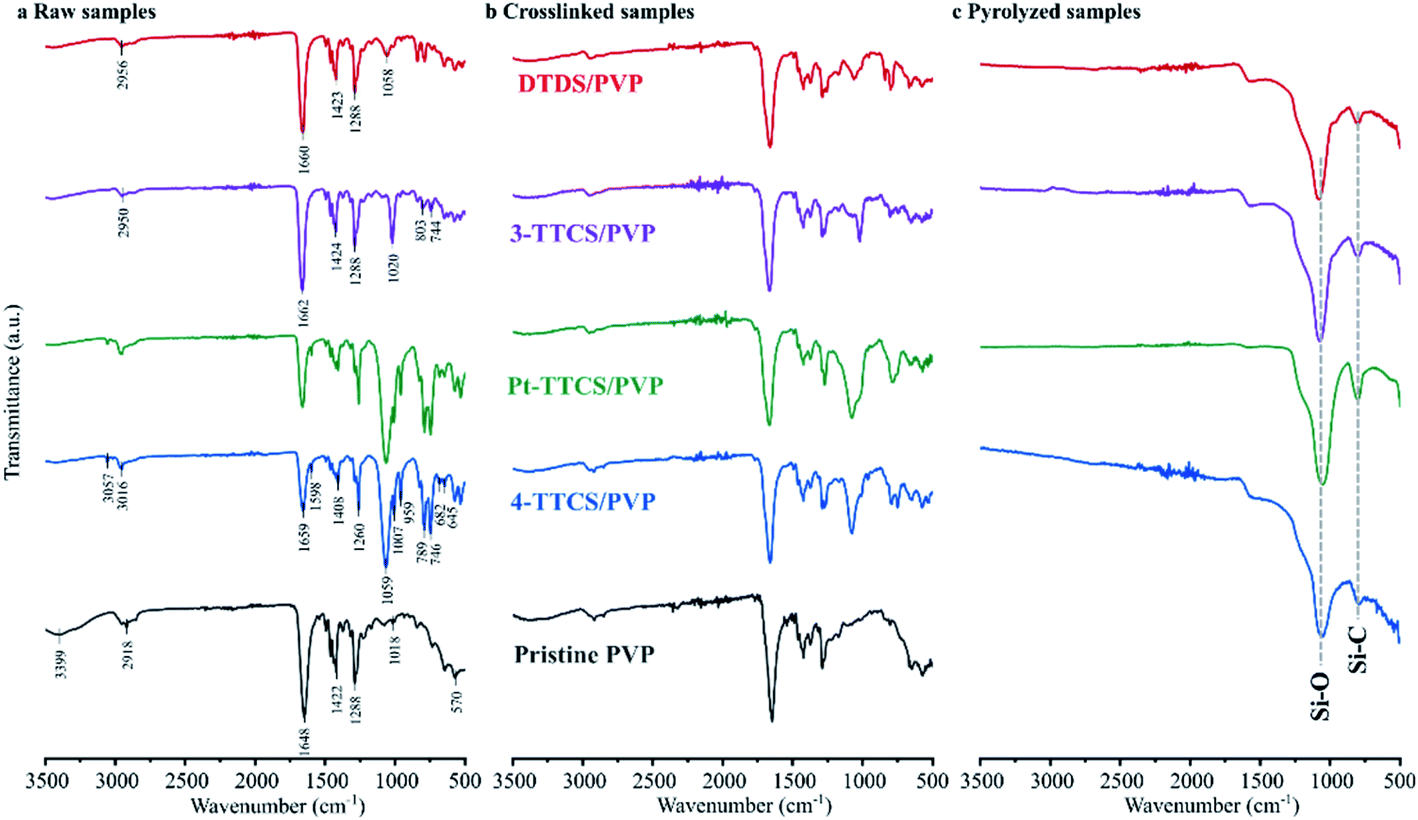 | ||
| Fig. 5 FTIR spectra of raw, crosslinked, and pyrolyzed electrospun siloxane/PVP and pristine PVP fibre mats. Major peaks of pyrolyzed samples are marked within the plot. | ||
| Wavenumber (cm−1) | Chemical group | Ref. | |
|---|---|---|---|
| PVP | 3399 | OH stretching | 25 |
| 2918 | CH2 asymmetric stretching | 25 | |
| 1648 | C–O stretching | 25 | |
| 1422 | CH bending | 25 | |
| 1288 | CH2 wagging | 25 | |
| 1018 | CH2 rock | 25 | |
| 570 | N–CO bending |
25 | |
| 4-TTCS | 3057, 3016 | Si–CHCH2 (CH asymmetric stretching) |
26 |
| 2964 | Si–CH3 (CH symmetric stretching) | 26 | |
| 1598 | Si–CHCH2 (CC stretching) |
26 | |
| 1408 | Si–CH3 (C–H asymmetric bending) | 26 | |
| 1260 | Si–CH3 (C–H symmetric bending) | 26 | |
| 1059 | Si–O–Si asymmetric stretching | 26 | |
| 1007, 959 | Si–CHCH2 (CH out-of-plane bending) |
26 | |
| 789, 746 | Si–CH3 (Si–C deformation vibration) | 27 | |
| 682, 645 | Si–CH3 (Si–C stretching) | 26 | |
| 3-TTCS | 2950 | CH2 asymmetric stretching | 28 |
| 1662 | Si–CHCH2 (CC stretching) |
28 | |
| 1424 | Si–CH3 (C–H asymmetric bending) | 28 | |
| 1288 | Si–CH3 (C–H symmetric bending) | 28 | |
| 1020 | Si–O–Si (siloxane stretch) | 28 | |
| 803, 744 | Si–CH3 (CH3 rock) | 28 | |
| DTDS | 2954 | CH2 asymmetric stretching | 29 |
| 1660 | Si–CHCH2 (CC stretching) |
29 | |
| 1423 | Si–CH3 (C–H asymmetric bending) | 29 | |
| 1288 | Si–CH3 (C–H symmetric bending) | 29 | |
| 1058 | Si–O–Si (siloxane stretch) | 29 |
XPS survey scans were performed on pyrolyzed fibre mats to investigate the sample compositions. Elemental compositions (Table 2) of pyrolyzed samples differed significantly for each preceramic polymer. The atomic weight percentages were calculated by integrating the areas under critical peaks. The Pt-TTCS/PVP sample derived the highest Si composition (41.1 at%) with the highest O (44.9 at%) and lowest C contents (13.4 at%). However, 4-TTCS/PVP, which had the same initial structure as Pt-TTCS, produced a final composition with the lowest Si (23.7 at%) and highest C (41.5 at%). Similarly structured 3-TTCS (6-ring cyclic siloxane) provided a similar composition to Pt-TTCS/PVP, while DTDS/PVP demonstrated a balanced chemical composition after pyrolysis. As mentioned, the Si and O had identical initial atomic percentages in 4-TTCS, Pt-TTCS, and 3-TTCS preceramic silicon polymers, and Si:O = 2:1 in DTDS. Therefore, the inference was made that all polymers gained oxygen during the heat treatments.
| Pyrolyzed samples | Elements (at%) | |||
|---|---|---|---|---|
| Si | C | O | Pt | |
| 4-TTCS/PVP | 23.7 | 41.5 | 34.8 | — |
| Pt-TTCS/PVP | 41.1 | 13.4 | 44.9 | 0.6 |
| 3-TTCS/PVP | 38.4 | 13.8 | 47.8 | — |
| DTDS/PVP | 29.0 | 31.8 | 39.2 | — |
Si, C, O, and high-resolution scans (shown in Fig. 6) were conducted for the samples to determine the bonding of each sample. Pt 4f high-res scan was presented in Fig. S1.† The bonding types of Si must be determined to accurately estimate the microstructures of the fibre mats. Stacked high-resolution scans showed shifting peaks according to initial compositions of preceramic polymers, revealing a variation of integrated peaks. Multi-peak fittings were performed to all high-resolution scans with marked integrated peaks:24,30 SiO4 (104.5 ± 0.1 eV), SiCO3 (103.5 ± 0.1 eV), SiC2O2 (102.9 ± 0.1 eV) for Si 2p; CO (287.1 ± 0.1 eV), C–C (284.9 ± 0.1 eV), C–Si (284.0 ± 0.1 eV) for C 1s; CO (534.4 ± 0.1 eV), O–Si (532.9 ± 0.1 eV), SiO2 (532.4 ± 0.1 eV) for O 1s. The following conclusions were drawn from the Si 2p and C 1s high-resolution scan curves: (1) low carbon content results in decreased Si–C contents, which eventually decreases SiCO3 and SiC2O2 amounts in microstructures; in fact, SiC2O2 may not exist in low C samples; (2) high C samples tend to form high C–C bonds that may lead to more free carbon in the microstructure; (3) low carbon contents in Pt-TTCS and 3-TTCS samples indicated a limited contribution of PVP after pyrolysis.
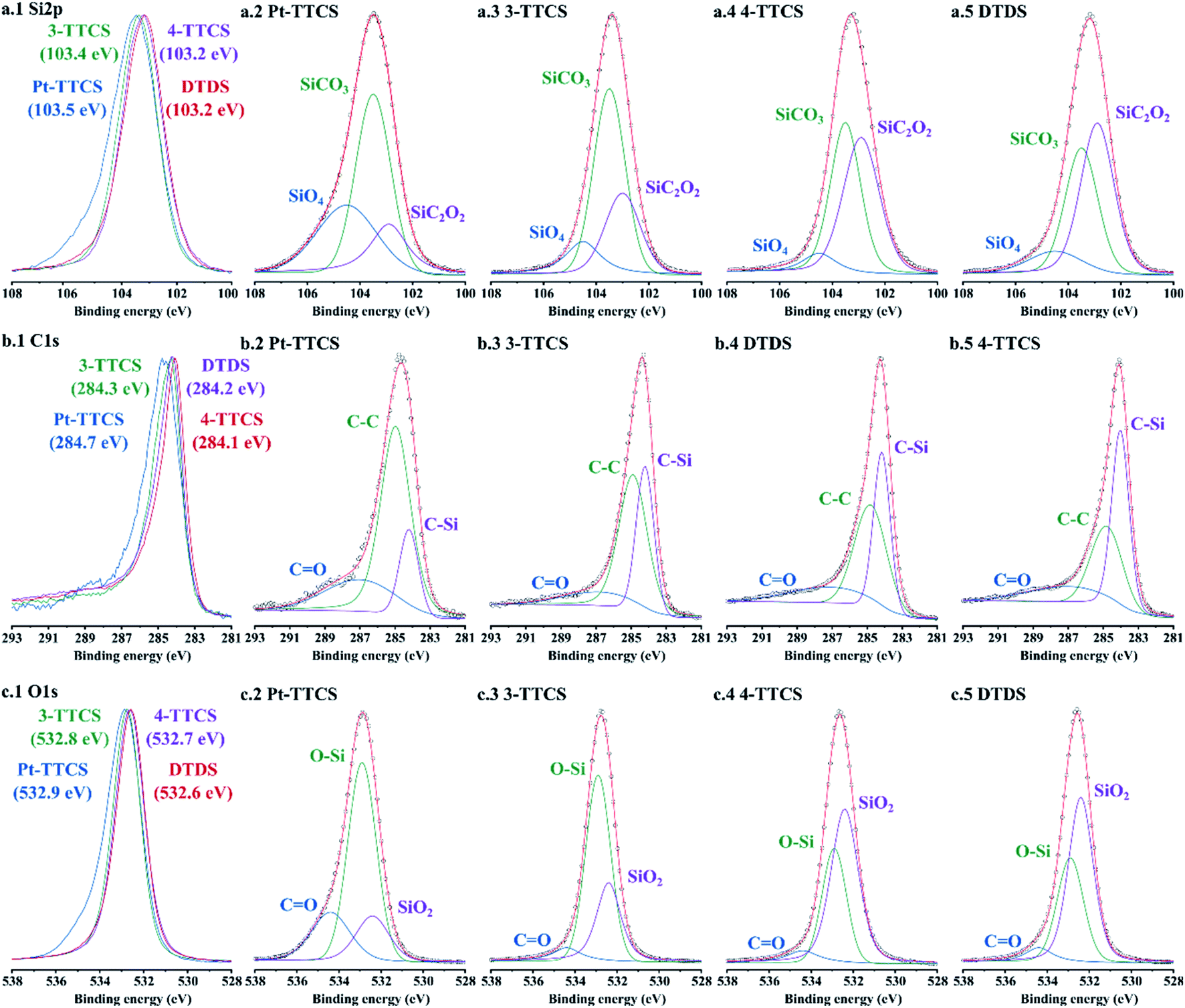 | ||
| Fig. 6 XPS spectra of all pyrolyzed fibre mat samples with various preceramic polymer precursors with plots of high-resolution scans of Si 2p, C 1s, and O 1s. The first column shows stacked high-resolution scans (normalized to 4-TTCS) of (a.1) Si 2p: (b.1) C 1s: and (c.1) O 1s, with peak centres marked. Subplots of columns 2–5 show integrated peaks with marked components. | ||
Solid-state NMR spectroscopy was performed on pristine PVP, crosslinked siloxane/PVP, and pyrolyzed samples. Bulk powder samples were used to enhance the NMR signal (Fig. 8), and corresponding NMR spectra of fibre mats were included in Fig. S3.† From the 29Si MAS analysis of crosslinked samples, ring structures of both the reserved 8- (around 32 ppm for 4-TTCS samples)31,32 and 6-units (around 23 ppm for 3-TTCS)33 were detected after crosslinking. Also, the formation of broad –CHx–MeSiO2 (for 4-TTCS and 3-TTCS samples) and –CHx–Me2SiO– (for DTDS sample)34 peaks indicated a rigid crosslinked phase by DCP after heat treatment at 160 °C. Similarly, a less-effective crosslinking reaction of Pt suggests that DCP is more efficient for this type of cyclic siloxane. 29Si MAS NMR spectra of pyrolyzed samples showed the highest amounts of SiC2O2 and SiCO3, indicating high Si–C contents in the 4-TTCS sample, and less SiCO3 in the 3-TTCS sample, as well as almost no Si–C related peaks in the Pt-TTCS sample. XPS analysis also confirmed low carbon content in the Pt-TTCS/PVP sample. The 29Si MAS spectrum was not obtainable from the pyrolyzed DTDS sample due to the formation of radicals in the free carbon phase. Additional 29Si MAS NMR spectra (Fig. S5†) of samples pyrolyzed at 1000 °C suggest more Si–C bonds might form at higher pyrolysis temperatures, however, fibre mats may also experience a severer shrinkage and even wrapping.
The significant Si–CHx characteristic peaks on the 13C CP MAS NMR spectra of crosslinked samples imply that 4-TTCS and 3-TTCS samples had the highest siloxane-to-PVP ratio after crosslinking. The remaining –CHCH2 signals suggest incomplete crosslinking of CC that may occur due to the limited mobility of siloxane molecules in the PVP environment during crosslinking. Nonetheless, the weak –CHCH2 signal in the DTDS sample demonstrated more complete crosslinking because of the improved mobility of the small molecules. This finding was consistent with the relatively high numbers of XPS-detected SiC2O2 groups.
Combining all the evidences, the compositional analyses of crosslinked and pyrolyzed fibre mats revealed the strong crosslinking reactions between preceramic precursors. Precursors, that were initially embedded in the PVP environment, crosslinked through vinyl and methyl groups. This process corresponds to the widely agreed crosslinking mechanisms of Si compounds (as shown by eqn (1) and (2) in Experimental section), and was confirmed by the decrease of Si–CHCH2 and Si–CH3 FTIR signals (Fig. 5a and b) and formation of low mobile -CHx-Me2SiO- NMR signals (Fig. 7a).
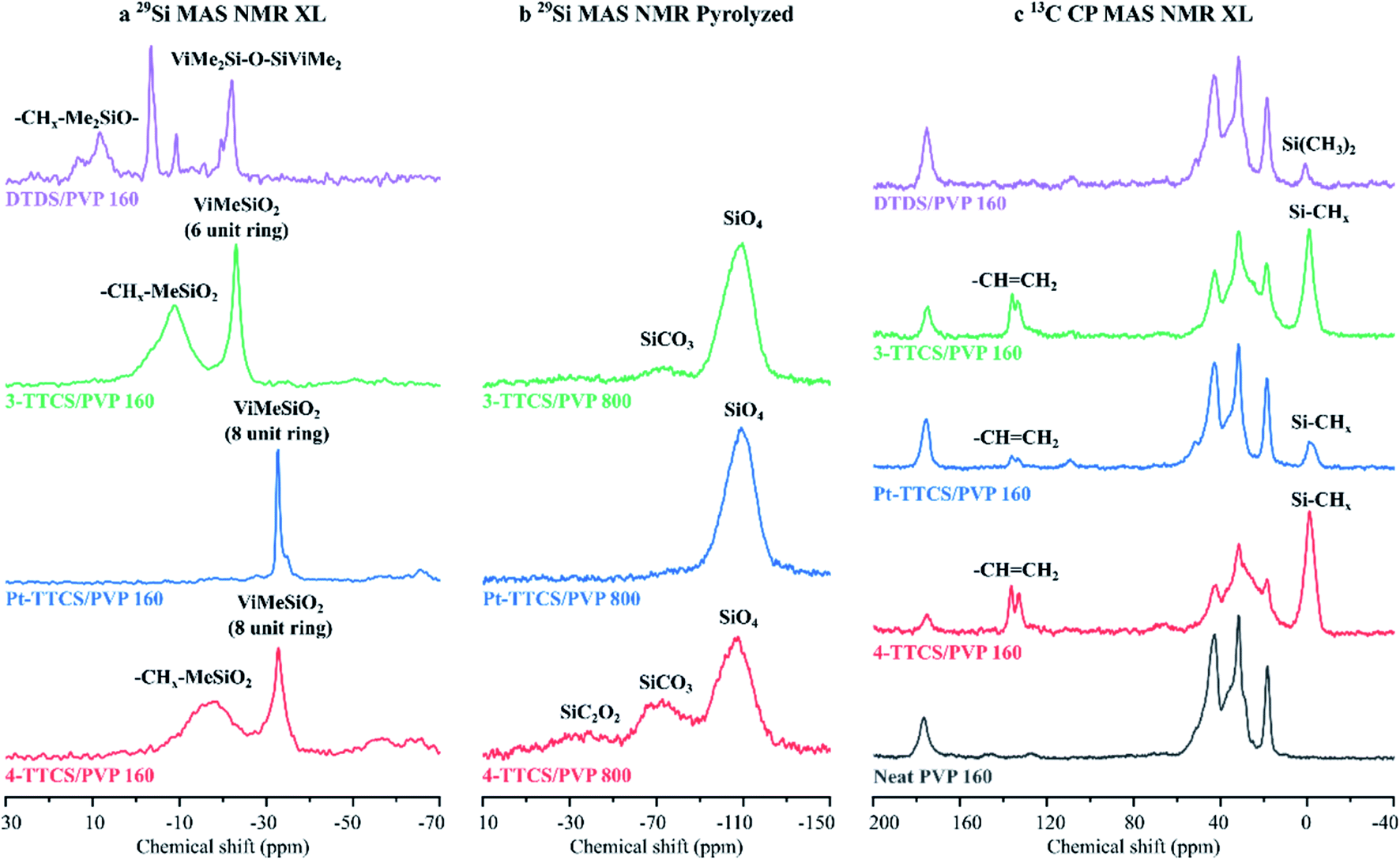 | ||
| Fig. 7 Solid-state NMR spectra crosslinked and pyrolyzed fibre mats. (a) 29Si MAS NMR XL; (b) 29Si MAS NMR pyrolyzed; and (c) 13C CP MAS NMR XL. | ||
Structurally, PVP molecules do not have a potential crosslinking site with Si compounds. Pristine PVP fibre mats also failed to survive 800 °C pyrolysis, that indicates no crosslinking behaviour of PVP molecules. In addition, FTIR and NMR of crosslinked fibre mats indicated no or weak interactions between the precursor molecules and spinner PVP molecules. FTIR spectra of pristine PVP sample showed slight decrease of C–H and CH2 signals, which might due to the thermal decomposition of the molecule. However, when precursors were added to the system, characteristic peaks of PVP did not show a further decrease. 13C CP MAS NMR spectra of PVP and hybrid samples (Fig. 7c) also showed all reserved PVP peaks. The spinner PVP molecules are expected to decompose at pyrolyzing temperature and contributes to excessive or free carbon phase.
Mechanical properties and thermal stability
Nanoindentation was performed on fibre surfaces that were embedded in epoxy resin in the pyrolyzed 4-TTCS/PVP sample. Results of the indentation revealed the fibre transverse modulus of 5.6 GPa (2.5 GPa for epoxy resin), and the plotted indentation curves were presented in Fig. S5.† In addition, all the fibre mats were directly exposed to an acetylene torch flame for 20 s (Fig. 8); the XPS analysis results are shown in Table 3, and corresponding spectra were presented in Fig. S2.† Under an oxidizing high-temperature environment, the Pt-TTCS/PVP sample did not show any changes, while the 4-TTCS/PVP and 3-TTCS/PVP mats demonstrated slight oxidation at the surface (i.e., turned grey after heat treatment). However, the DTDS/PVP samples shrunk and turned completely grey, suggesting the lowest oxidation resistance among the four tested samples. XPS analysis revealed compositional changes after exposure to the flame. The Pt-TTCS/PVP sample showed almost unchanged composition but slightly decreased Si content, while the 4-TTCS/PVP and 3-TTCS/PVP samples showed minimal changes with slightly decreased carbon content compared to previous XPS data. The DTDS/PVP sample showed critically decreased carbon content (from 31.8% to 6.8%), which resulted in shrinkage of the fibre mat. | ||
| Fig. 8 Four types of fibre mats directly exposed to acetylene flame torch. | ||
| Pyrolyzed samples | Elements (at%) | |||
|---|---|---|---|---|
| Si | C | O | Pt | |
| 4-TTCS/PVP | 24.0 | 39.5 | 36.5 | — |
| Pt-TTCS/PVP | 40.8 | 16.9 | 41.9 | 0.4 |
| 3-TTCS/PVP | 40.7 | 13.4 | 45.9 | — |
| DTDS/PVP | 40.6 | 6.8 | 52.5 | — |
Flame torch tests suggest a relation between microstructures and thermal stabilities. The best, performing Pt-TTCS sample was previously revealed with highest SiO4 bonding character (from NMR and XPS) and highest Si–O bonding (from XPS). 3-TTCS sample with slightly higher SiCO3 content showed a slight decrease in thermal stability. Consecutively, 4-TTCS, with slight SiC2O2 and significant SiCO3 structure, showed further decreased thermal stability from 3-TTCS. While, microstructure and NMR information related to DTDS was not as conclusive. It did however show the least stability to flame torch.
Conclusions
This study used electrospinning to synthesize SiOC fibre mats from four preceramic silicon oligomers and PVP hybrid spinning precursors. Electrospinning efficiently and reliably produced large fibre mats. As-spun fibre mats were subjected to two-stage heat treatment for complete polymer-to-ceramic conversion. During cross-linking and pyrolysis, the fibre surfaces changed from smooth to highly porous. Each preceramic oligomer derived unique final SiOC compositions even if the initial structures were very similar for 4-TTCS, Pt-TTCS, 3-TTCS, and DTDS, proving the controllability of final compositions.Characterization results revealed the porosity of the pyrolyzed 4-TTCS, Pt-TTCS, and 3-TTCS samples. Small DTDS molecules resulted in dense, thin fibres, while surface compositional analyses showed preserved Si composition at the fibre surfaces. Improved thermal crosslinking reactions were achieved via DCP, although Pt was shown to increase final Si composition. In the thermal stability tests, Pt-TTCS derived SiOC fibre mat demonstrated the best stability under oxidizing conditions. The Pt-TTCS, 4-TTCS, and 3-TTCS derived SiOC fibre mat samples showed slight compositional changes after heat treatment, and only the DTDS sample showed significant shrinkage and reduction in the free-carbon phase after heat treatment.
In conclusion, when fabricating PDC fibres from a preceramic polymer that is initially not spinnable, spinning agent can be added to improve viscosity and enable electrospinning, as when PVP was used in this study. The PVP showed neither a crosslinking behaviour of its own, nor crosslinking interactions with preceramic polymer. The amount of spinning agent is critical to spinnability of the hybrid precursor system, in case for PVP, 400 wt% maximum loading (precursors to PVP = 4:1) were achieved for all precursors. Different preceramic precursors and crosslinking catalysts affected the final compositions and microstructures, therefore, further influenced thermal stabilities of the final PDC fibre mats. Lower free carbon phase and higher Si–O bonding resulted in better thermal performance under acetylene flame torch.
Conflicts of interest
G. S. and Z. R. have filed for a provisional patent: U.S. Provisional Patent Application 63/011,402, – “Electrospun SiOC Fiber Mats from Polyvinylpyrrolidone/preceramic Siloxanes Hybrid Systems”; filed April 17, 2020.Acknowledgements
Financially support from National Science Foundation Partnerships for International Research and Education grant number 1743701 is gratefully acknowledged. Technical support from Nebraska Nanoscale Facility with grant number ECCS: 2025298 is gratefully acknowledged.References
- P. Colombo, G. Mera, R. Riedel and G. D. Soraru, Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics, J. Am. Ceram. Soc., 2010, 93(7), 1805–1837 CAS.
- D. Li, J. T. McCann and Y. Xia, Electrospinning: A Simple and Versatile Technique for Producing Ceramic Nanofibres and Nanotubes, J. Am. Ceram. Soc., 2006, 89(6), 1861–1869 CrossRef CAS.
- W. Sigmund, J. Yuh, H. Park, V. Maneeratana, G. Pyrgiotakis and A. Daga, et al., Processing and Structure Relationships in Electrospinning of Ceramic Fibre Systems, J. Am. Ceram. Soc., 2006, 89(2), 395–407 CrossRef CAS.
- Z. Ren, C. Gervais and G. Singh, Preparation and structure of SiOCN fibres derived from cyclic silazane/poly-acrylic acid hybrid precursor, R. Soc. Open Sci., 2019, 6(10), 190690 CrossRef CAS.
- D. Li, Y. Wang and Y. Xia, Electrospinning of Polymeric and Ceramic Nanofibres as Uniaxially Aligned Arrays, Nano Lett., 2003, 3(8), 1167–1171 CrossRef CAS.
- T. Subbiah, G. S. Bhat, R. W. Tock, S. Parameswaran and S. S. Ramkumar, Electrospinning of nanofibres, J. Appl. Polym. Sci., 2004, 96(2), 557–569 CrossRef.
- M. Bognitzki, W. Czado, T. Frese, A. Schaper, M. Hellwig and M. Steinhart, et al., Nanostructured Fibres via Electrospinning, Adv. Mater., 2001, 13(1), 70–72 CrossRef CAS.
- D. Li and Y. Xia, Fabrication of Titania Nanofibres by Electrospinning, Nano Lett., 2003, 3(4), 555–560 CrossRef CAS.
- D. H. Reneker, A. L. Yarin, H. Fong and S. Koombhongse, Bending instability of electrically charged liquid jets of polymer solutions in electrospinning, J. Appl. Phys., 2000, 87(9), 4531 CrossRef CAS.
- R. Ramaseshan, S. Sundarrajan, R. Jose and S. Ramakrishna, Nanostructured ceramics by electrospinning, J. Appl. Phys., 2007, 102(11), 111101 CrossRef.
- J. Doshi and D. H. Reneker, Electrospinning Process and Applications of Electrospun Fibres. The 1993 IEEE Industry Applications Conference Twenty-Eighth IAS Annual Meeting, IEEE, Toronto, Ontario, Canada, 1993 Search PubMed.
- A. L. Yarin, S. Koombhongse and D. H. Reneker, Bending instability in electrospinning of nanofibres, J. Appl. Phys., 2001, 89, 3018 CrossRef CAS.
- P. Lu, Q. Huang, B. Liu, Y. Bando, Y.-L. Hsieh and A. K. Mukherjee, Macroporous Silicon Oxycarbide Fibres with Luffa-like Superhydrophobic Shells, J. Am. Chem. Soc., 2009, 131(30), 10346–10347 CrossRef CAS.
- A. Guo, M. Roso, M. Modesti, J. Liu and P. Colombo, Hierarchically structured polymer-derived ceramic fibres by electrospinning and catalyst-assisted pyrolysis, J. Eur. Ceram. Soc., 2014, 34, 549–554 CrossRef CAS.
- S. B. Mujib, R. Cuccato, S. Mukherjee, G. Franchin, P. Colombo and G. Singh, Electrospun SiOC ceramic fibre mats as freestanding electrodes for electrochemical energy storage applications, Ceram. Int., 2020, 46, 3565–3573 CrossRef.
- Y. Liu, W. Yang, X. He and H. Hou, Tailored synthesis of amorphous SiCNO mesoporous fibres through combining a facile electrospinning process and microwave-assisted pyrolysis, Ceram. Int., 2019, 45, 8640–8645 CrossRef CAS.
- S. A. Smith, B. P. Williams and Y. L. Joo, Effect of polymer and ceramic morphology on the material and electrochemical properties of electrospun PAN/polymer derived ceramic composite nanofibre membranes for lithium ion battery separators, J. Membr. Sci., 2017, 526, 315–322 CrossRef CAS.
- S. Sarkar, A. Chunder, W. Fei, L. An and L. Zhai, Superhydrophobic Mats of Polymer-Derived Ceramic Fibres, J. Am. Ceram. Soc., 2008, 91(8), 2751–2755 CrossRef CAS.
- J. Wilfert, R. v. Hagen, R. Fiz, M. Jansen and S. Mathur, Electrospinning of preceramic polymers for the preparation of SiBNC felts and their modification with semiconductor nanowires, J. Mater. Chem., 2012, 22, 2099–2104 RSC.
- Y. Si, X. Wang, L. Dou, J. Yu and B. Ding, Ultralight and fire-resistant ceramic nanofibrous aerogels with temperature-invariant superelasticity, Sci. Adv., 2018, 4, eaas8925 CrossRef.
- Z. Ren and G. Singh, Nonoxide polymer-derived CMCs for “super” turbines, Am. Ceram. Soc. Bull., 2019, 98(3), 34–39 CAS.
- D. Li and Y. Xia, Electrospinning of Nanofibres: Reinventing the Wheel?, Adv. Mater., 2004, 16(14), 1151–1170 CrossRef CAS.
- G. Mera, A. Navrotsky, S. Sen, H.-J. Kleebe and R. Riedel, Polymer-derived SiCN and SiOC ceramics – structure and energetics at the nanoscale, J. Mater. Chem. A, 2013, 1(12), 3826–3836 RSC.
- L. David, R. Bhandavat, U. Barrera and G. Singh, Silicon oxycarbide glass-graphene composite paper electrode for long-cycle lithium-ion batteries, Nat. Commun., 2016, 7, 10998 CrossRef CAS.
- Kamaruddin, D. Edikresnha, I. Sriyanti, M. M. Munir and Khairurrijal, Synthesis of Polyvinylpyrrolidone (PVP)-Green Tea Extract Composite Nanostructures using Electrohydrodynamic Spraying Technique, The 4th International Conference on Advanced Materials Science and Technology, IOP Publishing, 2017 Search PubMed.
- A. Nyczyk, C. Paluszkiewicz, A. Pyda and M. Hasik, Preceramic polysiloxane networks obtained by hydrosilylation of 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane, Spectrochim. Acta, Part A, 2011, 79(4), 801–808 CrossRef CAS.
- Y. Zhang, Y. Li, J. Shao and C. Zou, Fabrication of superhydrophobic fluorine-free films on cotton fabrics through plasma-induced grafting polymerization of 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane, Surf. Coat. Technol., 2015, 276, 16–22 CrossRef CAS.
- D. D. Burkey and K. K. Gleason, Structure and mechanical properties of thin films deposited from 1,3,5-trimethyl-1,3,5-trivinylcyclotrisiloxane and water, J. Appl. Phys., 2003, 93(9), 5143 CrossRef CAS.
- P. R. Dvornic, V. V. Gerov and M. N. Govedarica, Polymerization by Hydrosilation. 2. Preparation and Characterization of High Molecular Weight Poly[(1,1,3,3-tetramethyldisiloxanyl)ethylene] from 1,3-Dihydridotetramethyldisiloxane and 1,3-Divinyltetramethyldisiloxane, Macromolecules, 1994, 27(26), 7575–7580 CrossRef CAS.
- R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Preparation and structure of silicon oxycarbide glasses derived from polysiloxane precursors, J. Sol-Gel Sci. Technol., 1997, 8, 327–330 CAS.
- C. G. Pantano, A. K. Singh and H. Zhang, Silicon Oxycarbide Glasses, J. Sol-Gel Sci. Technol., 1999, 14, 7–25 CrossRef CAS.
- G. D. Sorarù, G. D'Andrea, R. Campostrini, F. Babonneau and G. Mariotto, Structural Characterization and High-Temperature Behavior of Silicon Oxycarbide Glasses Prepared from Sol-Gel Precursors Containing Si-H Bonds, J. Am. Ceram. Soc., 1995, 78(2), 379–387 CrossRef.
- K. Rózga-Wijas, J. Chojnowski, M. Ścibiorek and W. Fortuniak, Polysiloxane–silica hybrids from novel precursors by the sol–gel process, J. Mater. Chem., 2005, 15, 2383–2392 RSC.
- D. Seyferth and J. Robison, Cyclopolymerization of metalloid-containing α,ω-dienes. 1,3-Divinyltetramethyldisiloxane, 1,3-divinyltetramethyldisilazane, and 1,3-divinylpentamethyldisilazane, Macromolecules, 1993, 26(3), 407–418 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04060f |
| This journal is © The Royal Society of Chemistry 2020 |