
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol–ene coupling reaction achievement and monitoring by “in situ” UV irradiation NMR spectroscopy†
Natalia Toncheva-Moncheva a,
Miroslav Dangalovb,
Nikolay G. Vassilev*b and
Christo P. Novakov*a
a,
Miroslav Dangalovb,
Nikolay G. Vassilev*b and
Christo P. Novakov*a
aInstitute of Polymers, Bulgarian Academy of Sciences, 103-A, Akad. G. Bonchev Str., 1113 Sofia, Bulgaria. E-mail: hnovakov@polymer.bas.bg
bInstitute of Organic Chemistry with Center of Phytochemistry, Bulgarian Academy of Sciences, 9, Acad. G. Bonchev Str., 1113 Sofia, Bulgaria
First published on 2nd July 2020
Abstract
In this study, the possibilities of a new “in situ” LED UV illumination NMR spectroscopic technique for performing an initiator-free thiol–ene “click” coupling reaction of an allyl-functionalized poly(allyl glycidyl ether) (PAGE) prepolymer with a number of mono- and di-oligo polyethylene glycol (PEG) thiols is demonstrated. The state-of-the-art setup constructed with LEDs as UV light sources that illuminate through optical fibers directly into an NMR testing tube at a fixed wavelength of 365 nm is appropriate for various polymeric materials and biologically active substances. The selected experimental protocol uses a series of periods of irradiation and dark periods, thus providing opportunities to conduct an effective thiol–ene “click” reaction and simultaneously study the kinetics of the photochemical reaction with the exposure time, as well as macromolecular association directly in a solution applying the whole types of NMR methods: from conventional 1H or 13C NMR to diffusion NMR spectroscopy (DOSY). In addition, the molecular mass characteristics of the prepared copolymers were studied by gel-permeation chromatography (GPC). The observed differences in the reaction rates as well as in the size of species formed (the corresponding hydrodynamic radiuses Rh of aggregates) as a result of the coupling process of parent PAGE prepolymers and model PEG thiols were thoroughly discussed and the reaction pathway proposed.
Introduction
The rise of “click” chemistry in the recent decade is a result of its favorable synthetic characteristics such as quantitative yields, regioselectivity, use of environmentally friendly solvents, mild reaction conditions and nonchromatographic purification.1 Highly efficient reactions of thiols with reactive unsaturated carbon–carbon double bonds (“enes”)2–10 in particular have gained growing interest for their advantages such as no involvement of metallic catalysts, compatibility with a range of functional groups and mild reaction conditions. Thiol–ene chemistry possesses many characteristics of “click” chemistry with an outstanding efficiency even in the presence of oxygen. Among a range of metal-free “click” reactions,11 the photoinduced thiol–ene radical addition reactions have been recognized as widely used tools for polymer synthesis and modification and as efficient bio-orthogonal reactions.12–14 Radical-mediated thiol–ene coupling reactions have become a useful approach for attaching a wide range of functional groups, i.e. biomolecules like proteins and peptides commonly bearing free thiols.15,16The free-radical addition17–21 is applicable to isolated double bonds such as allyls and can be generated by photoinitiation. An interesting example for the achievement of modular functionalization of precursor amphiphilic block copolymers with mercaptans via a single thiol–ene addition reaction with the participation of allyl groups was described by Jing et al.22 Some applications within the biomedical field of thiol–ene polymers comprising different kinds of multifunctional enes and thiols were collected and reviewed.23 It was suggested that such polymers are suitable for the preparation of nanoparticles for drug-delivery systems, as biomimetic hydrogels and as dental restorative resins.
An excellent example of potentiality of thiol–ene chemistry as a new “click” reaction that can be performed in the absence of solvents and under photochemical initiation has been demonstrated by Hawker19 and Walter et al.24 They have presented a facile and efficient synthesis procedure of poly(thioether) dendrimers using thiol–ene addition reactions for both the construction of a dendritic backbone and the functionalization of chain ends.
Irradiation, with or without added photoinitiators, is the most common method for initiating radical thiol–ene reactions. There are several examples for the thiolation reactions conducted using type I photoinitiators, i.e. DMPA,25–31 TMDPO32 and type 2 ones, i.e. BP based,32–34 TX,32 etc., performing under irradiation using a UV lamp emitting in the range of 320–400 nm.
The preparation of novel photoinitiators is still of interest. For example, Xi and coworkers reported a novel amine-catalyzed thiol-Michael reaction.35 Upon irradiation at 320–390 nm in the presence of a thiol and activated substrate, a newly prepared photolabile primary amine, NPPOC-hexylamine, serves as the initiating species for the thiol-Michael reaction with a series of thiols and methyl acrylates.
Although fewer in number, there are examples in the literature describing thiol–ene coupling reactions performed under irradiation in the absence of an initiator. In one of the pioneering works on photopolymerization carried out in the absence of photoinitiators, Bowman and co-workers have described thiol monomer copolymerization with a wide variety of vinyl monomers.36 The polymerization reactions proceed under irradiation with light centered at 365 nm in the presence of oxygen, demonstrating advantages relative to the common drawbacks associated with radical photopolymerization.
A combination of controlled radical polymerization and subsequent “click” reactions to prepare the molecular brushes with PGMA as the main chain and PEG side chains whose side chain terminals were transformed into various functionalities has been established.37 A series of thiols with different functionalities were introduced by thiol–ene reactions12,38 induced by UV light radiation using a mercury vapor lamp. A “green” approach for the synthesis and thiol–ene post-modification of alkene-functionalized poly(2-oxazoline)s has also been demonstrated.39 “Green” solvents have been chosen to perform thiol–ene reactions on PDecEnOx homopolymers with two-different thiols under irradiation with UV light without any additional photoinitiators. The preparation of thioetherfunctional biscyclocarbonates under initiator-free UV irradiation of ene functional groups cyclocarbonated with 2,2′-oxybis(ethane-1-thiol) has been reported by Benyahya et al.40 Jing X. described successful modular functionalization of precursor amphiphilic block MPEG-b-P(LA-co-MAC) and allyl-PEG-b-PLA copolymers via a radical thiol–ene addition reaction with model mercaptans initiated by UV light without adding any photoinitiators. Authors stated that the thiol–ene addition reaction is universal and any allyl-bearing polymers can be functionalized with other functionalized mercaptans as modifiers.22 The reaction is a safe approach to biopolymer functionalization. The synthesis of end- and side-chain-functionalized polyesters based on allyloxyl PEG-bl-p(L-lactide) and methoxy PED-bl-p(L-lactide-co-2-methyl-2-allyloxycarbonyl propylene carbonate) as well as their thiol–ene modifications with three common thiols accomplished under radical conditions with UV irradiation without added photoinitiator has also been accomplished.28 Liang et al. reported a method for nanoparticle modification by the conversion of carboxylic acid-functionalized Fe2O3 nanoparticles to thiol-functional ones, which subsequently reacted with vinylferrocene upon initiator-free UV exposure.41
An interesting combination of thiol–ene chemistry with polyhedral oligomeric silsesquioxanes (POSS) for the preparation of hybrid, photocrosslinked polymerosomes was described by Jiang et al.42 Upon irradiation with UV light at 364 nm, the hydrophobic domain of the polymerosomes obtained from a co-assembly of acrylate-functionalised poly(ether amine) and POSS(Sn)8 has been crosslinked via radical-mediated thiol-Michael reactions.
Light-emitting diodes (LEDs) have been used as UV sources to conduct the crosslinking polymerization of representative thiol–ene systems by Crivello back in 2004.43 The “in situ” LED illumination NMR technique has been extensively used in the recent years.44–46 Excellent examples of various photo catalytic reactions conducted using such techniques that combine both traditional and advanced NMR methods were published by Gschwind group.47–51 The review highlights new ideas, concepts and methods in the field of photocatalysis and photopolymerization and demonstrates the utility of the “in situ” LED illumination NMR method.52 Meanwhile, a full quantification of the light-mediated Gilch polymerization with “in situ” UV-irradiation NMR spectroscopy experiments has been described.53 The concentration–time profiles of the photochemical reactions have been recorded with continuous UV-irradiation applied using a broad band light source in combination with a 320–400 nm filter. A UV-driven premonomer photolysis technique was used.
The aim of the present work is to investigate the feasibility of performing initiator-free thiol–ene coupling reactions of poly(allyl glycidyl ether) macroagents with a number of model PEG-(di)thiols under UV illumination using a LED source directly into the NMR spectroscopy tube. LED sources emitting at a fixed wavelength of 365 nm are appropriate for various polymeric and biological materials. In addition, a kinetics study of “click” reaction as well as the behavior of the obtained copolymers in a solution by diffusion NMR spectroscopy and GPC was done.
Experimental
Materials
All solvents (methanol, dichloromethane, and tetrahydrofuran) as well as ethyl vinyl ether (99%, Aldrich) were purified by distillation. Deionized water was obtained from a Millipore MilliQ system and additionally filtered through a 220 nm PTFE filter. Allyl glycidyl ether (99% Fluka) was distilled under reduced pressure. KOH (Merck), 1,2-propandiol (Aldrich), benzene (Merck), p-toluenesulfonic acid (Merck), and 1,6-diphenyl-1,3,5-hexatriene (Aldrich) were used without purification. Hexa(ethylene glycol)dithiol (Sigma-Aldrich, > 97%, Mn = 314.46 g mol−1) (HEGDT), O-(2-mercaptoethyl)-O′-methyl-hexa(ethylene glycol) (Sigma-Aldrich, ≥ 95%, Mn = 356.48 g mol−1) (HEGMT) and poly(ethylene glycol)methyl ether thiol (Sigma-Aldrich, Mn = 2000 g mol−1) (PEGMET) were used as received.Synthesis
Synthesis of functionalized poly(allyl glycidyl ether) (PAGE) by anionic ring-opening polymerization (ROP) in bulk. PAGE was obtained following a procedure described in our previous publication.54 First, anionic ROP of allyl glycidyl ether (AGE) was carried out in a glass reactor. Then, 1,2-propanol (0.18 g, 2.3 mmol) was mixed with KOH (0.20 g, 3.5 mmol) and stirred at 50 °C for 2 hours in an argon atmosphere (Scheme 1). Following this, the reaction mixture was cooled to room temperature and dried two times by freeze drying with benzene (0.5 mL per portion) for 4 h at high vacuum. AGE (5.5 g, 48.0 mmol) was added in portions of 1.1 g each every 10 h. Polymerization was carried out in bulk at 80 °C for 50 h in an argon atmosphere. The reaction medium was cooled to room temperature and polymerization was stopped by addition of methanol. Methanol was removed under vacuum and the mixture was additionally treated with hydrochloric acid (HCl 27.95 mL, 40% in methanol) for neutralization. The resulting polymer was isolated by centrifugation and then was dried under vacuum. | ||
| Scheme 1 Synthesis of an allyl functional poly(allyl glycidyl ether) (PAGE) macroagent. | ||
Yield: 4.565 g, (82%); 1H-NMR (CDCl3, δ ppm): 1.18 (t, 3H, CH3-g), 1.55 (d, 3H, CH3-f′), 3.41–3.71 (m, 5H, CH2-a,c, CH-b), 3.98 (d, 2H, CH2-d), 4.38 (m, 1H, =CH, e′), 5.20 (dd, 2H, CH2-f), 5.88 (m, 1H, CH-e), 5.95 (d, 1H, –CH = , d′), MGPCn = 1300 g mol−1, Mw/Mn = 1.1, MHNMRn = 1000 g mol−1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v THF:deionized water for 2 days using a 1 or 3.5 kDa Mw cutoff membrane depending on the obtained copolymer molecular weight. The THF was removed using a rotary vacuum evaporator, and the copolymer products were recovered by freeze-drying.
:2 v/v THF:deionized water for 2 days using a 1 or 3.5 kDa Mw cutoff membrane depending on the obtained copolymer molecular weight. The THF was removed using a rotary vacuum evaporator, and the copolymer products were recovered by freeze-drying.Methods
Nuclear Magnetic Resonance (NMR) LED-NMR experiments were carried out using a self-made setup constructed with three LEDs as light sources operating at 365–470 nm, 440–460 nm or 650–670 nm coupled to an optical fiber. The setup includes a control unit connected to the IPSO computer through BNC connections. The other end of the optical fiber is inserted in a coaxial insert and illuminates the NMR sample. The usage of the coaxial insert provides uniform illumination to the whole reaction solution, thus ensuring that the irradiated volume matches the one detected by the NMR coil. In the experiments described herein, a LED model SST-10-UV with a wave length of 365–370 nm and an optical power of 130 mW at the end of the optical fiber using an LED current of 1 A was utilized.13C{1H} NMR spectra were recorded using a Bruker Avance II+ 600 NMR spectrometer, with a 5 mm direct detection dual broadband probe (BBO) and a gradient coil delivering a maximum gradient strength of 53 G cm−1. The following experimental parameters were used: spectral width of 230 ppm, 32 K time domain data points, relaxation delay of 2 s, number of scans 8k, and Waltz16 broadband 1H decoupling scheme.
1H NMR spectra and 1H DOSY spectra were recorded using a Bruker Avance II+ 600 NMR spectrometer, with a 5 mm direct detection dual probehead (BBO) probe and a 10 A gradient amplifier, providing a maximum gradient strength of 53 G cm−1. The experiments were performed at 20 °C or 40 °C. The DOSY spectra were acquired using the Diff suite package integrated in TopSpin 3.5 using a double-stimulated echo pulse sequence to compensate for possible convection during the measurements. The spectra were recorded with 16 K time domain data points in t2 dimension, 32 scans, diffusion delay (Δ) 50 ms, sine-shaped gradient pulses of (δ) 2 ms, and relaxation delay of 2.8 s. A gradient ramp of 32 linearly distributed gradient amplitude values was used. The starting and final gradient amplitudes were optimized to ensure optimal signal attenuation, typically from 2 to 53 G cm−1 for all samples. The spectra were processed with an exponential window function (line broadening factor 1), 32k data points in the F2 dimension, and 128 data points in the diffusion dimension, using the fitting routine integrated in the TopSpin 3.5 package. The diffusion coefficients were calculated by fitting the sum of the columns along the chemical shift of each signal in the DOSY spectrum with a variant of the Stejskal–Tanner equation adapted for the particular pulse sequence used.
Samples were irradiated at six-hour intervals with an LED light source operating at 365 nm. Each illumination was followed by a two-hour “dark” interval, over which several 1H as well as DOSY NMR spectra were taken in order to monitor the course of the response upon discontinued irradiation. Such a procedure was applied until the PAGE prepolymer was fully reacted.
Results and discussion
The synthesis of hydroxyl terminated bifunctional poly(allyl glycidyl ether) (PAGE) macroagent was achieved by anionic ring-opening polymerization initiated by alkali metal alkoxides carried out in bulk as described by us in a previous paper (Scheme 1).54 The alkoxide initiator has been obtained in a reaction of deprotonation of low-molecular-weight diols with KOH. For performing an initiator-free thiol–ene “click” coupling reaction, the PAGE precursor with degree of polymerization (DP = 10) was selected, to be consistent with the molecular weights of the corresponding thiol partners. The structure and molecular mass characteristics of PAGE were proved and evaluated by GPC, 1H and 13C NMR spectroscopy. In fact, a well-defined prepolymer of a narrow molar mass distribution (MHNMRn = 1000 g mol−1, MGPCn = 1300 g mol−1, PDI = 1.1, calculated towards PSt standards) was obtained (Fig. SI. 1, SI. 2, in ESI†). 1H and 13C NMR spectra peak assignment shown in the inset is presented in Fig. SI. 2, SII. 16–19 in ESI,† respectively. The presence of the signals corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in the allyl group of PAGE allows assessment of the extent of the coupling reaction with thiols under the selected experimental conditions. It should be noted that the molar mass of the PAGE determined by GPC in THF as the eluent corresponded well with the one determined by 1H NMR (Mn = 1000 g mol−1) taken in CDCl3.
C double bonds in the allyl group of PAGE allows assessment of the extent of the coupling reaction with thiols under the selected experimental conditions. It should be noted that the molar mass of the PAGE determined by GPC in THF as the eluent corresponded well with the one determined by 1H NMR (Mn = 1000 g mol−1) taken in CDCl3.
The 1H, 13C NMR, COSY, HSQC and DOSY spectra and GPC traces of the chosen three different thiol-end-capped precursors, namely, difunctional, hexa(ethylene glycol)dithiol (HEGDT) (Fig. SII. 1–5†), monofunctional, O-(2-mercaptoethyl)-O′-methyl-hexa(ethylene glycol) (HEGMT) (Fig. SII. 6–10†) and monofunctional polymers containing poly(ethylene glycol)methyl ether thiol (PEGMET) (Fig. SII. 11–15 and SIII. 2†), used in attempts to perform coupling reactions with PAGE prepolymers are also available in ESI.† The analysis of NMR spectra and GPC chromatograms and peak assignment shows that the HEGDT dithiol monomer is contaminated with dimer compounds containing a disulfide link according to the presence of signals in the 1H spectrum at 2.85 ppm and in 13C NMR one at 38.3 ppm. The existence of a shoulder peak in the GPC eluogram of PEGMET also suggests the presence of two components in the thiol reagent (Fig. SIII. 2†).
Representative figures of the used LED set-up together with detailed description of the equipment are given in the ESI and Fig. SIV. 1.† In general, the set-up includes three LEDs operating at 365–370 nm, 440–460 nm and 650–670 nm, respectively. The LED source of 365 nm was chosen because in all the groups of UV radiation, UV-A (315–400 nm) radiation is friendliest on diverse habitats, because it is not absorbed by native DNA, thus having poor efficiency in inducing its damage.55 The latter would allow the application of the methodology to a wide range of biologically active molecules.
The “click” coupling reactions with the participation of PAGE and HEGMT, HEGDT or PEGMET under UV illumination using an LED source at 365 nm wavelength were carried out “in situ” in deuterated benzene under mild conditions at 20 °C in an inert atmosphere. A reaction temperature of 40 °C was only used in the case of “click” coupling with PEGMET. A 10% excess of thiol-end-capped compounds with respect to PAGE were used in all cases to enable highly efficient macromolecular coupling and drive the reaction to completion.
The efficiency of the “click” reaction was followed by 1H NMR spectroscopy. The 1H spectra were taken regularly and the progress of coupling between PAGE and the used three thiol-end-capped compounds were evaluated by the decrease in the signal of protons of the double bonds of the allyl group. The decrease in the double bond content with the exposure time is shown in Fig. 1. Data obtained by “in situ” NMR measurements were fitted with a mono exponential function, and the standard errors in rate constant are ±2% (R2 > 0.99).
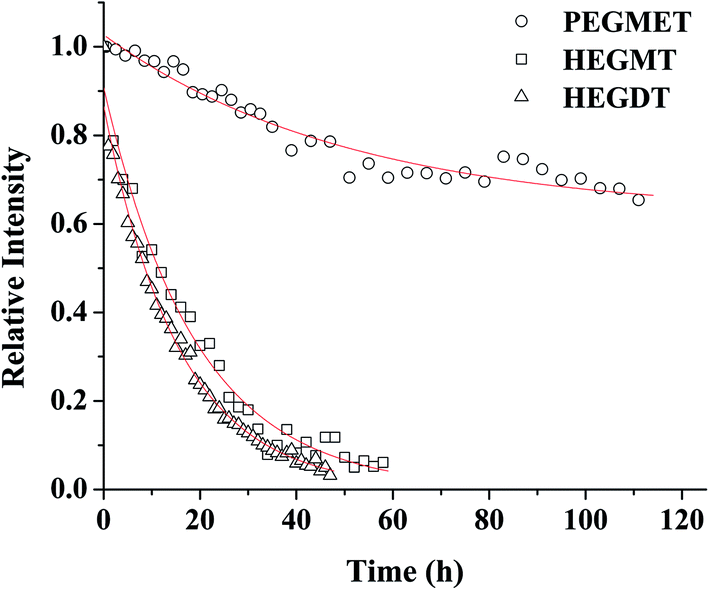 | ||
| Fig. 1 Consumption of CC of PAGE in the course of LED UV-irradiation: (△) HEGDT rate constant (1.8 × 10−5 s−1); (□) HEGMT rate constant (1.1 × 10−5 s−1), (○) PEGMET rate constant (5.4 × 10−6 s−1). | ||
Tracking the course of the curves of CC consumption presented in Fig. 1 shows the difference in the ability of different types of thiol monomers for the reaction of coupling with the PAGE prepolymer. The binding of oligomeric thiols with PAGE proceeded completely, although slowly and non-linearly with the increase in time. As expected, the presence of a second binding center in the dithiol molecule contributes for a noticeably faster, but not dramatically, coupling reaction. At the same time, thiol–ene “click” reaction between PAGE and polymer-containing thiol reagent PEGMET proceeds much more slowly and the process was not accomplished even after 120 h of irradiation. The results indicate that even conducting the UV irradiation at a higher temperature (40 °C), about 30% binding of PEGMET was reached (Fig. 1 and SII. 23†). Presumably, the dissolution of such relatively long-chain thiol molecules was accompanied with their warping, making more straitened the attack on the double bond of PAGE. One should note that PAGE macromolecules of the prepolymer are in shrink conformation, hampering double bond accessibility.
The success of the “click” coupling reaction was confirmed by 1H NMR analysis (Fig. 2, SII. 21 and SII. 23†). As evidenced from the 1H NMR spectra of PAGE and HEGDT reaction mixture (A) before and (B) after LED UV irradiation, the signals at 5.05–5.2 ppm and at 5.7–5.9 ppm assigned to the protons corresponding to the double bond in the allyl group of PAGE (Fig. 2A) entirely disappeared at the end of illumination, and new signals at 1.8 and 2.6 ppm corresponding to methylene protons adjacent to the sulfide bond appeared (Fig. 2B and SII. 21B†).
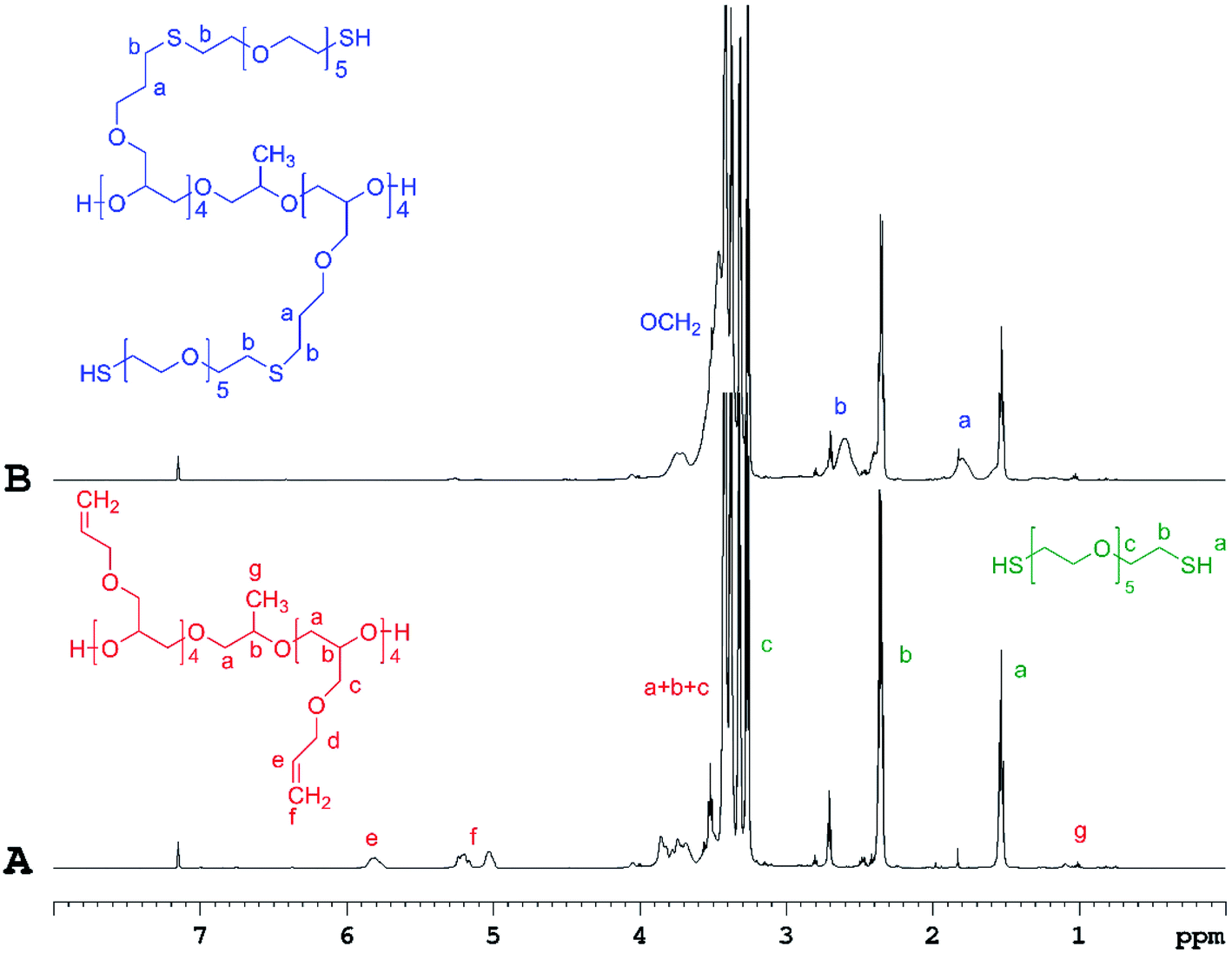 | ||
| Fig. 2 1H NMR spectra of PAGE and HEGDT reaction mixture (A) before and (B) after illumination at 365 nm for 48 h at RT, taken in C6D6 (600 MHz). | ||
After LED UV-irradiation, following the above-described procedures for the purification of the reaction mixtures from the excess of thiol–ene coupling partners, the isolated copolymers were analyzed by GPC to determine their molecular mass characteristics. The calculated values from the corresponding peaks in eluograms are summarized in Table 1 together with those obtained for starting PAGE and HEGDT, and HEGMT and PEGMET reagents. Representative GPC curves of PAGE-co-HEGDT, PAGE-co-HEGMT and PAGE-co-PEGMET copolymers are presented in Fig. SIII. 1, Fig. SIII. 2 in ESI† and Fig. 3, respectively.
| Sample | MGPCn [g mol−1] | Mw/Mn |
|---|---|---|
| PAGE | 1300 | 1.08 |
| HEGDT | — | — |
| HEGMT | 600 | 1.02 |
| PEGMET | 3600 (I) | 1.04 |
| 1600 (II) | 1.05 | |
| PAGE-co-HEGDT | 5800 | 1.25 |
| PAGE-co-HEGMT | 3100 | 1.15 |
| PAGE-co-PEGMET | 9300 (I) | 1.04 |
| 4300 (II) | 1.06 |
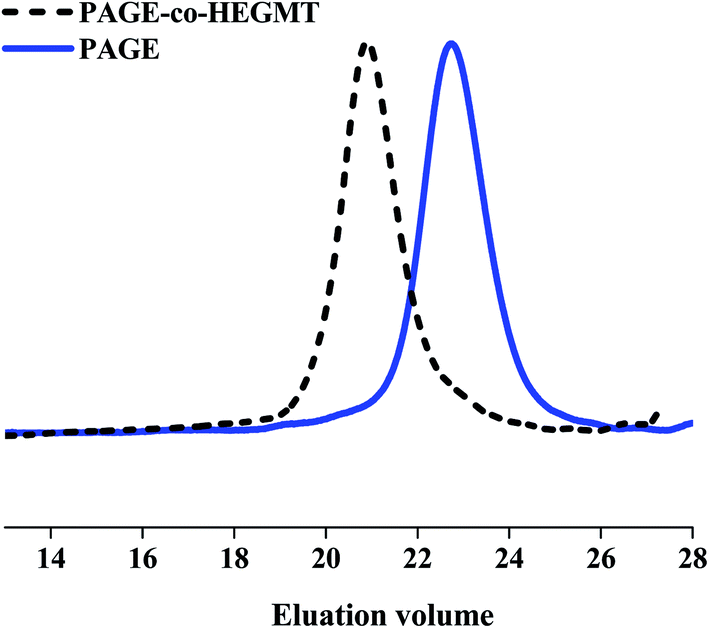 | ||
| Fig. 3 GPC traces of PAGE (solid blue line) and PAGE-co-HEGMT copolymer (short-dash black line). | ||
The course of eluograms proved the successful completion of the tested coupling reaction by LED UV-irradiation without using any initiator (Fig. 3.). The chromatogram peaks are shifted to smaller elution times, which correspond to an increase in the molecular weight of copolymers. The observed presence of a bimodal peak in GPC traces of PAGE-co-PEGMET copolymers as well as a noticeable shoulder in the eluogram of PAGE-co-HEGDT one corresponds well with the composition of the starting thiols discussed above (see. Fig. SIII. 1. and SIII. 2 in ESI†). The molecular mass of the PAGE-co-PEGMET copolymer also fits well with the theoretically calculated value, taking into account the higher molar mass and polydispersity (containing fractions that differ almost twice in molecular weight, Table 1) of starting thiols.
The effect of the number of reactive thiol–ene functional groups is also well pronounced, and the value of the molecular weight of PAGE-co-HEGDT obtained with a dithiol reagent is almost twice larger than the corresponding one of PAGE-co-HEGMT copolymer obtained with a mono thiol compound of comparable EG chain length (see. Fig. SIII. 1. and SIII. 2 in ESI†)). In addition, formation of a noticeable amount of PAGE-co-HEGDT gel-fraction was observed, suggesting occurrence of cross-linking reaction during irradiation in the presence of a dithiol reagent.
Further evidence for the changes in size and morphology of the macromolecules was provided by diffusion-ordered NMR spectroscopy (DOSY) spectra of the studied reaction mixtures in C6D6. DOSY is a valuable tool for complex mixture analysis, exploiting differences in translational diffusion coefficients and, hence, particle sizes as a means to spectroscopic discrimination between different components in a solution mixture. Therefore, simultaneously with the decrease in the double bond content with the exposure time (Fig. 1) observed in 1H NMR spectra, the hydrodynamic radius Rh of the obtained product species was calculated. After every 6 h of irradiation, a dark period was applied, during which 1H DOSY spectra of the reaction mixture were taken. It should be pointed that the integral of the residual signal of C6D6 was used as a reference for the calibration of the integrals of the allylic protons. As no noticeable change in the integrals was observed after the dark period, one can conclude that no reaction took place at that time. The Rh value was calculated according to the Stokes–Einstein equation using viscosity V = 0.6942 at T = 20 °C or V = 0.5395 at T = 40 °C.56
As it is mentioned before, the thiol partners used in the coupling reaction, namely, HEGDT and PEGMET, contain some amount of dimer compounds with a disulfide building link. Their presence in the reaction mixture reflects the molecular mass characteristics, as it is seen from the GPC results (Table 1) and is expected to affect the behavior of copolymers and the size of their species formed in a solution.
The change in Rh of PAGE-co-HEGDT and PAGE-co-HEGMT-based species with the exposure time follow an exponential increase, which trend has to be expected for a pseudo-first order kinetics, as shown on Fig. 4A. Moreover, the presence of a second thiol group in HEGDT accelerates the process and, therefore, increases both the molecular weight of the corresponding PAGE-co-HEGDT copolymer and the size of the formed species, respectively.
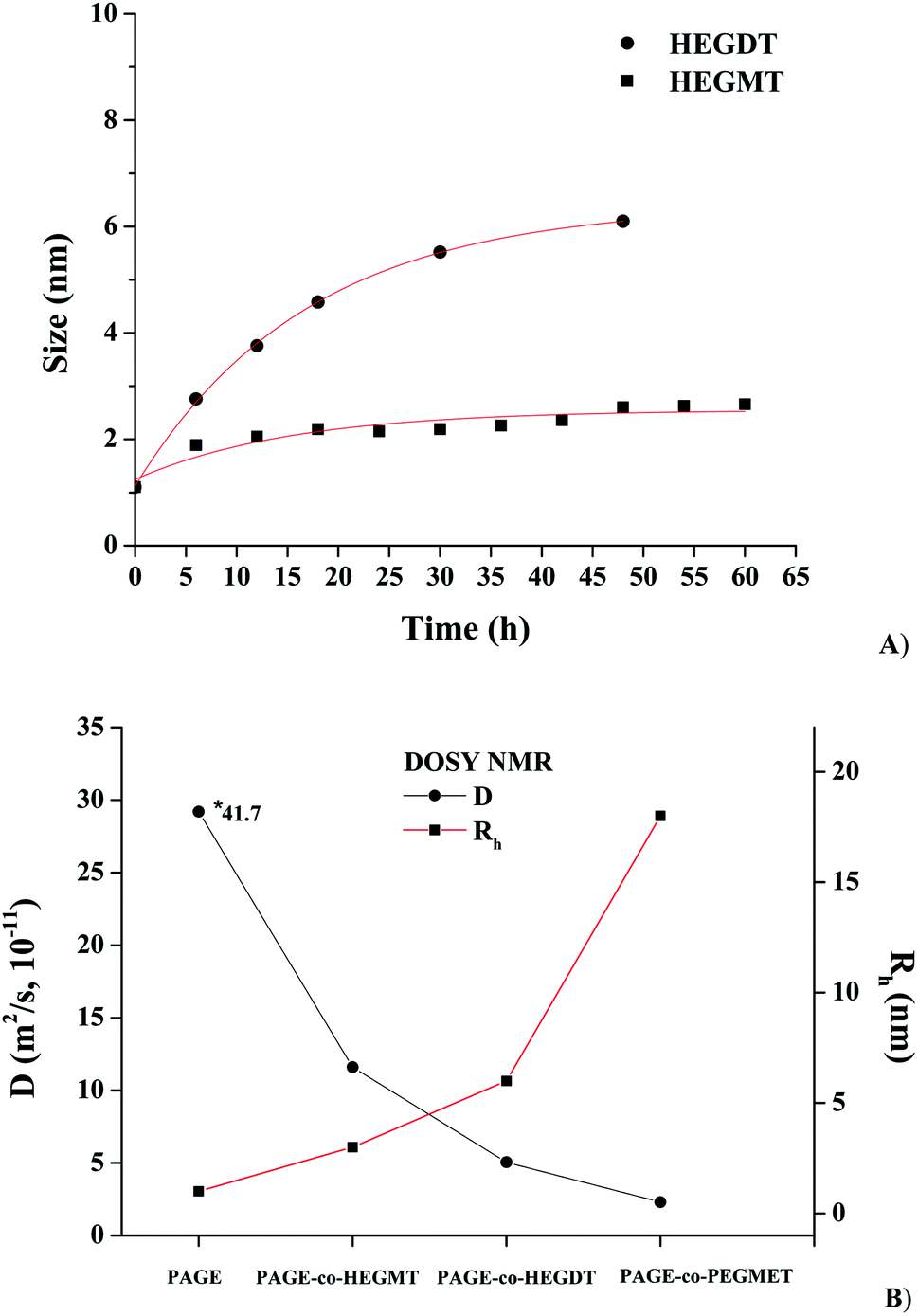 | ||
| Fig. 4 DOSY NMR: (A) Change in hydrodynamic radius of PAGE-co-HEGDT and PAGE-co-HEGMT based species with the exposure time at RT. (B) D and Rh values of PAGE prepolymer and PAGE-co-HEGDT, PAGE-co-HEGMT and PAGE-co-PEGMET-based species after the thiol–ene “click” coupling reaction, in C6D6 at RT. *The value was taken at 40 °C. | ||
The diffusion coefficients (D) and the calculated Rh values obtained from 1H DOSY NMR spectra of the starting PAGE macroagent and the synthesized copolymers taken after the LEDs UV treatment are summarized in Fig. 4B. It is noteworthy that the PAGE-co-PEGMET copolymer has the highest molecular weight and volume of the macromolecules. Hence, the particle size of its species in solution is largest (Fig. 4B).
Representative DOSY spectra of PAGE prepolymers and HEGDT thiol reaction mixtures (A) before and (B) after LED UV irradiation are shown in Fig. 5.
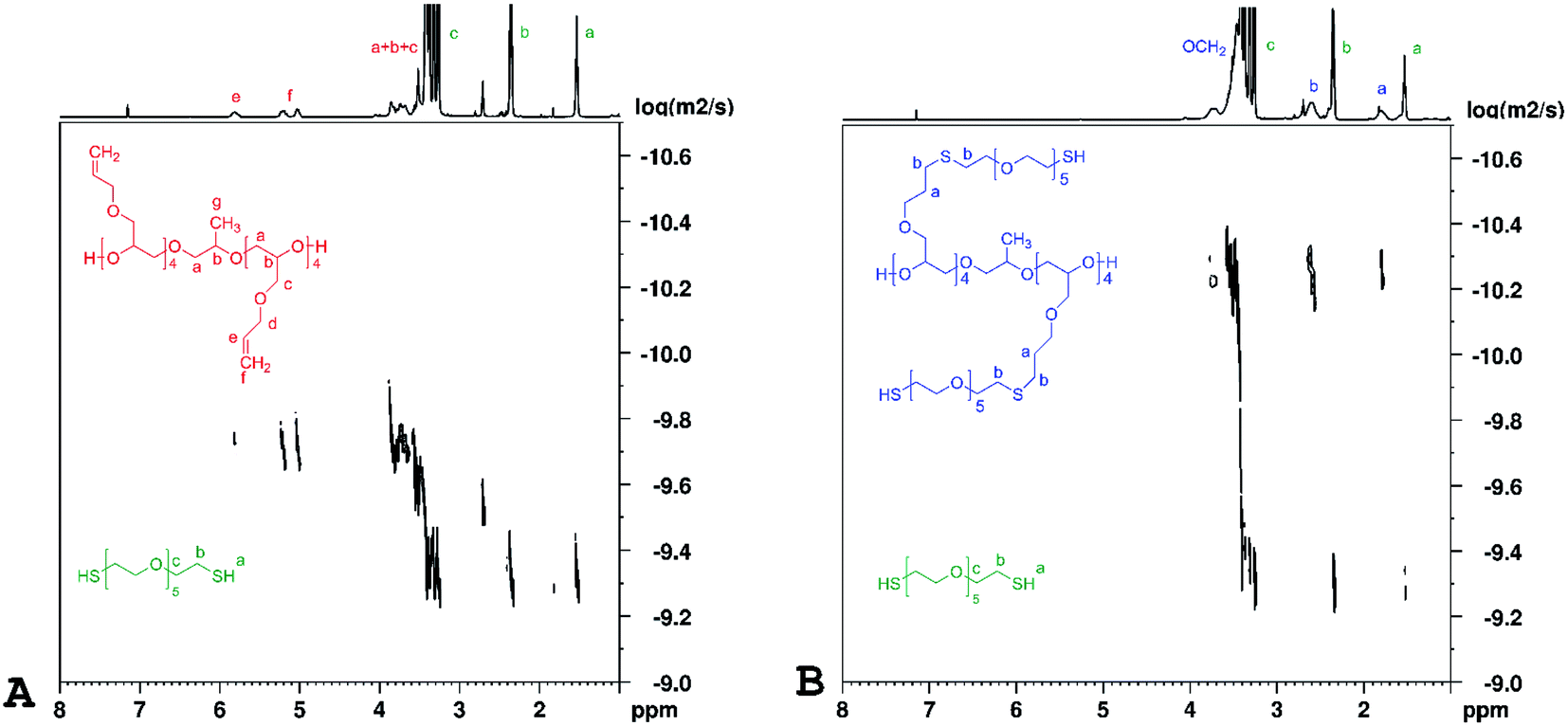 | ||
| Fig. 5 1H DOSY NMR spectra of PAGE and HEGDT reaction mixture (A) before and (B) after illumination at 365 nm for 48 h at room temperature, taken in C6D6 (600 MHz). | ||
It is clearly noticeable that the signals corresponding to reagents before illumination and the newly appeared ones with a smaller diffusion coefficient corresponding to the copolymer product were well separated. The values of diffusion coefficients and Rh of species size, calculated on their basis presented in Fig. 4, are in good agreement with the GPC data. In the DOSY spectrum measured after the irradiation (Fig. 5B), a signal corresponding to unreacted thiols is observed as its quantity is set in a slight excess relative to the amount of PAGE prepolymers.
Based on the results for the molecular weights of copolymers calculated from GPC analyses as well as on data for decrease in the content of allyl double bonds in thiol molecules taken from “in situ” NMR experiments, the thiol–ene reaction sequences for the synthesis of PAGE-thiol presented in Scheme 2 are proposed.
 | ||
| Scheme 2 General scheme of the preparation of copolymers via a thiol–ene “click” coupling reaction of PAGE and HEGDT, HEGMT or PEGMET under UV irradiation at a fixed wavelength of 365 nm without use of any photoinitiator. | ||
In conclusion it can be noted that the energy of the LED source of irradiation used in these experiments was low, which limited the useful experimental volume, and hence the concentration of the reagents and the yield of product. However, the methodology used to conduct the polymerization and its simultaneous tracking demonstrated its application power under mild conditions that are suitable to a number of partners, i.e. for various biological objects.
Moreover, the proposed strategy can be easily reproduced to large-scale experimental volumes and set-ups, thus allowing the preparation of copolymers having predefined compositions and macromolecular characteristics via an initiator-free thiol–ene “click” coupling reaction.
Conclusion
Versatile UV-irradiated NMR spectroscopy for “in situ” study of coupling reactions of PAGE and few oligo-/PEG (di)thiols without any photoinitiator has been successfully implemented. The preformed functionalized PAGE macroagent has been prepared via anionic ROP, while the thiol–ene modifications have been done via LED UV illumination of the mixture of solutions of reagents directly inside the NMR spectrometer testing tube. 1H NMR spectra and GPC traces of resulting products after LED UV illumination support the expected turnaround of “click” reaction and formation of PEGylated (co)polymers. The reaction efficiency depends on the type of thiols (mono or dithiol) at the selected PAGE molecular weight. Upon coupling with the dithiol reagent, the formation of a gel-fraction in a noticeable amount was observed, suggesting the occurrence of cross-linking reactions, which is a subject of a further study. In general, the experimental setup and protocol, which consists of a series of LED irradiation intervals with measurement of 1H NMR spectra (thus tracking the degree of photochemical reaction) and dark periods and measurement of 1H DOSY spectra (allowing estimation of the size of macromolecules in the solution), can be successfully applied for the encapsulation of biologically active substances and the design of new polymeric materials via an initiator-free thiol–ene coupling reaction mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. M. and C. N. are grateful to the Bulgarian Operational Program “Science and Education for Smart Growth” 2014–2020, co-funded by EU through the European structural and investment funds (Grant BG05M2OP001-1.002-0012-C01) for supporting the modernization of research infrastructure at IP-BAS. M. D. and N. V. thank the Bulgarian National Science Fund (Project KP-06-H 29/6) for funding the experimental work and INFRAMAT (D01-155) project by the Ministry of Education and Science for supporting the construction and delivery of the irradiation device.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- J. Justynska and H. Schlaad, Macromol. Rapid Commun., 2004, 25, 1478 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- F. Lutz and H. Schlaad, Polymer, 2008, 49, 817 CrossRef.
- C. Rissing and D. Y. Son, Organometallics, 2008, 27, 5394 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487 CrossRef CAS.
- M. Heggli, N. Tirelli, A. Zisch, J. A. Hubbell and S. Perrier, Bioconjugate Chem., 2003, 14, 967 CrossRef CAS PubMed.
- J. Rieger, K. V. Butsele, P. Lecomte, C. Detrembleur, R. Jerome and C. Jerome, Chem. Commun., 2005, 14, 274 RSC.
- L. T. T. Nguyen, M. T. Gokmen and F. E. Du Prez, Polym. Chem., 2013, 4, 5527 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- M. Bednarek, React. Funct. Polym., 2013, 73, 1130 CrossRef CAS.
- D. E. Borchmann, N. ten Brummelhuis and M. Weck, Macromolecules, 2013, 46, 4426 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573 RSC.
- L. C. You and H. Schlaad, J. Am. Chem. Soc., 2006, 128, 13336 CrossRef CAS PubMed.
- J. Justynska, Z. Hordyjewicz and H. Schlaad, Macromol. Symp., 2006, 240, 41 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS PubMed.
- Y. Geng, D. E. Discher, J. Justynska and H. Schlaad, Angew. Chem., Int. Ed., 2006, 45, 7578 CrossRef CAS PubMed.
- N. T. Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946 CrossRef.
- J. Yue, X. Li, G. Mo, R. Wang, Y. Huang and X. Jing, Macromolecules, 2010, 43, 9645 CrossRef CAS.
- T. O. Machado, C. Sayer and P. H. H. Araujo, Eur. Polym. J., 2017, 86, 200 CrossRef CAS.
- M. V. Walter, P. Lundberg, A. Hult and M. Malkoch, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2990 CrossRef CAS.
- S. Das, M. Kar and S. S. Gupta, Polym. Chem., 2013, 4, 4087 RSC.
- C. Lluch, J. C. Ronda, M. Galia, G. Lligadas and V. Cadiz, Biomacromolecules, 2010, 11, 1646 CrossRef CAS PubMed.
- M. Stemmelen, C. Travelet, V. Lapinte, R. Borsali and J.-J. Robin, Polym. Chem., 2013, 4, 1445 RSC.
- D. Esquivel, O. van den Berg, F. J. Romero-Salguero, F. E. Du Prez and P. Van Der Voort, Chem. Commun., 2013, 49, 2344 RSC.
- Y. Li, W.-B. Zhang, J. E. Janoski, X. Li, X. Dong, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Macromolecules, 2011, 44, 3328 CrossRef CAS.
- Y.-J. Wang and C.-M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1645 CrossRef CAS.
- B. Schulte, C. A. Dannenberg, H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1243 CrossRef CAS.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103 CrossRef CAS.
- G. Temel, N. Karaca and N. Arsu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5306 CrossRef CAS.
- H. Xie, L. Hu, Y. Zhang and W. Shi, Prog. Org. Coat., 2011, 72, 572 CrossRef CAS.
- W. Xi, M. Krieger, C. J. Kloxin and C. N. Bowman, Chem. Commun., 2013, 49, 4504 RSC.
- N. B. Cramer, J. Paul Scott and Ch. N. Bowman, Macromolecules, 2002, 35, 5361 CrossRef CAS.
- L. Zou, W. Zhu, Y. Chen and F. Xi, Polymer, 2013, 54, 481 CrossRef CAS.
- C. Hoyle, A. Lowe and C. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- K. Kempe, R. Hoogenboom and U. Schubert, Macromol. Rapid Commun., 2011, 32, 1484 CrossRef CAS PubMed.
- S. Benyahya, M. Desroches, R. Auvergne, S. Carlotti, S. Caillol and B. Boutevin, Polym. Chem., 2011, 2, 2661 RSC.
- C. Liang, L. Jing, X. Shi, Y. Zhang and Y. Xian, Electrochim. Acta, 2012, 69, 167 CrossRef CAS.
- B. Yu, X. Jiang, N. Qin and J. Yin, Chem. Commun., 2011, 47, 12110 RSC.
- M. R. Zonca, B. Falk and J. V. Crivello, J. Macromol. Sci., Part A: Pure Appl. Chem., 2004, A41, 741 CrossRef CAS.
- C. Feldmeier, H. Bartling, E. Riedle and R. M. Gshwind, J. Magn. Reson., 2013, 232, 39 CrossRef CAS PubMed.
- H. R. Ward and R. G. Lawler, J. Am. Chem. Soc., 1967, 89, 5518 CrossRef CAS.
- J. Bargon and H. Fischer, Z. Naturforsch., A: Phys. Sci., 1967, 22, 1556–1562 CAS.
- C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347 CrossRef CAS PubMed.
- H. Bartling, A. Eisenhofer, B. Konig and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860 CrossRef CAS PubMed.
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444 CrossRef CAS PubMed.
- D. Petzold, P. Nitschke, F. Brandl, V. Scheidler, B. Dick, R. M. Gschwind and B. König, Chem. - Eur. J., 2019, 25, 361 CrossRef CAS PubMed.
- S. Wang, L. Nanjundappa, J. Hioe, R. M. Gschwind and B. Koenig, Chem. Sci., 2019, 10, 4580 RSC.
- Ph. Nitschke, N. Lokesh and R. M. Gschwind, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 86 CrossRef CAS PubMed.
- A.-K. Schoenbein, J. Kind, C. M. Tiele and J. Michels, Macromolecules, 2018, 51, 4678 CrossRef CAS.
- B. Stoyanova, Ch. Novakov, Ch. Tsvetanov and S. Rangelov, Macromol. Chem. Phys., 2016, 217, 2380 CrossRef CAS.
- R. G. Alscher, J. L. Donahue and C. L. Cramer, Physiol. Plant., 1997, 100, 224 CrossRef CAS.
- J. A. Dixon and R. W. Schiessler, J. Phys. Chem., 1954, 58(5), 430 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03902k |
| This journal is © The Royal Society of Chemistry 2020 |