
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOnium salts improve the kinetics of photopolymerization of acrylate activated with visible light
Janina Kabatc *a,
Katarzyna Iwińskaa,
Alicja Balceraka,
Dominika Kwiatkowskaa,
Agnieszka Skotnickaa,
Zbigniew Czechb and
Marcin Bartkowiakb
*a,
Katarzyna Iwińskaa,
Alicja Balceraka,
Dominika Kwiatkowskaa,
Agnieszka Skotnickaa,
Zbigniew Czechb and
Marcin Bartkowiakb
aUniversity of Science and Technology, Faculty of Chemical Technology and Engineering, Seminaryjna 3, 85-326 Bydgoszcz, Poland. E-mail: nina@utp.edu.pl; Fax: +48 52 374 9005; Tel: +48 52 374 9112
bWest Pomeranian University of Technology, Institute of Chemical Organic Technology, Pułaskiego 10, 70-322 Szczecin, Poland
First published on 1st July 2020
Abstract
The aim was study the influence of onium salts on the kinetics of photopolymerization in the visible light region. Trimethylolpropane triacrylate TMPTA was selected as a monomer, and activated by 1,3-bis(phenylamino)squaraine (SQ) used as a photosensitizer in addition to tetramethylammonium n-butyltriphenylborate (B2). The iodonium salt [A–I–B]+X− functioned as a second radical initiator, bearing a different substitution pattern for the cation. The ternary system was formulated with different concentrations of both borate and diphenyliodonium salts. Differential scanning calorimetry was used to investigate the polymerization reaction over the photoactivation time carried out at 300 nm < λ < 500 nm irradiation. When the squaraine dye/borate salt was used as photoinitiator, a slow polymerization reaction was observed and a lower monomer conversion. The addition of a third component (onium salt) increased the polymerization rate and conversion. Ternary photoinitiator systems showed improvement in the polymerization rate of triacrylate leading to high conversion in a short photoactivation time. The photoinitiating ability of bi- and tri-component photoinitiators acting in the UV-Vis region for initiation polymerization of triacrylate was compared with those of some commercially used photoinitiating systems. It was also found, that, the parallel electron transfer from an excited state of the sensitizer to [A–I–B]+X−, and an electron transfer from a ground state of R(Ph)3B−N(CH3)4+ to an excited state of the sensitizer results in two types of initiating radical.
Introduction
One of the modern and rapidly developing technologies is production of various types of polymeric materials in polymerization reaction. That process may be initiated, for example photochemically or thermally. The light activated polymerization is called photopolymerization. This process is characterized by: (i) low energy consumption, (ii) the possibility of using non-solvent compositions, (iii) high efficiency, and others.1,2 The photopolymerization occurs via radical (radical polymerization RP) or ionic mechanisms (cationic/anionic polymerization CP/AP). A photoinitiator plays a key role in radical initiated chain reaction. Because of this, many research groups still design new or improve known photoinitiators, especially those operating in the visible region of the spectrum.3–5 For example, diaryliodonium salts are efficient photoinitiators for cationically initiated polymerization. The very low energy of the C–I bond allows the decomposition of iodonium salt after irradiation with ultraviolet light.As a result, an iodophenyl radical cation and a reactive phenyl radical are formed.6,7 On the other hand, alkyltriphenylborate salts may be other example of onium salts used in activation of polymerization reaction.8 The active radicals are also formed as a result of bimolecular electron transfer quenching of photoexcited sensitizer. For an activation of these photoinitiators UV light sources are very often used for paints and coatings. It is well known, that the use of UV light is not recommended in the biological field. In that case, it is possible to apply dye, that absorbs in the visible light region as sensitizer, that allow a reaction with onium salt, promoting its decomposition. Therefore, the onium salts can also act in the radical polymerization of acrylates.6,9,10 Many different organic dyes have been investigated as the visible light sensitizers, for example: camphorquinone,11–13 indoles,14 coumarine,15 ketocoumarin and derivatives,16 xanthenic dyes,17,18 polymethines,19 hemicyanines,20 squaraines,21–23 pyrromethenes,24 2-amino-4,6-diphenyl-benzene-1,3-dicarbonitrile derivatives,4 2-(diethylamino)-4,6-diphenyl-benzene-1,3-dicarbonitrile derivatives,5 resazurin, flavins, safranine and others.8,25–29
An electron transfer process builds the main driving source to generate initiating radicals in UV-Vis photopolymerization.19 The first excited singlet state of dye (sensitizer) transfers/accepts an electron to/from coinitiator resulting in formation of initiating radicals.
In our previous papers it was shown that, squaraine dyes act as photosensitizers for alkyltriphenylborate salts, N-alkoxypyridinium salts and diphenyliodonium salts, which do not absorb light in the visible region.8,30–32 It was hypothesized, that squaraine may act as photosensitizer simultaneously for two different onium salts. Such approach can improve the reactivity of photoinitiator system. The aim of this study was to evaluate an influence of onium salts on the kinetics of polymerization of triacrylate (TMPTA) during irradiation with the visible light.
Generally, the squarylium dyes have been rarely used in dyeing photoinitiating systems. There are only few papers described their photoinitiating ability of polymerization of acrylates, epoxides, vinyl ether. In such photoinitiators, various 2,3,3-trimethylindolenine-based or 2-methylbenzothiazole-based squaraine dyes were applied.33,34
It should be also noted, that trimethylindolenine-based squaraines (green/red light photosensitizers) absorb at longer wavelength than dye studied by us (blue light photosensitizer). A characteristic feature of bis-phenylamino squaraine is a presence of nitrogen atom in the conjugated double bonds system. There are not any amine and iminium terminals in chromophore structure in contrast to mentioned above photosensitizers. Therefore, in order to improve the knowledge on the ability of bis-phenylamino squaraine to initiation of radical polymerization of triacrylate was described in our article.
Moreover, bis-phenylaminosquaraine dyes have not been studied in the multicomponent photoinitiating systems, yet.
The novelty of the present work is the enhancement of the photoinitiating efficiency of blue-light sensitizer-based initiator by simple modification of chemical composition of photoinitiator. It was shown here, that an introduction of a second onium salt results in better kinetic parameters of blue-light activated polymerization of triacrylate.
In this paper, we look for some iodonium and/or alkyltriphenyl borate salts, that in a presence of suitable photosensitizer are able to initiate the radical polymerization of 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate under irradiation with blue light.
Experimental part
Materials
Diphenyliodonium chloride (I1), diphenyliodonium hexafluorophosphate (I2), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), 1-methyl-2-pyrrolidinone (MP) were supplied by Aldrich Chemical Co. (Poland) and used without further purification. 1,3-Bis(phenylamino)squaraine (SQ) and tetramethylammonium n-butyltriphenylborate (B2) were synthesized in our laboratory, as it was described previously.35,36 Onium salts, such as: (4-methoxyphenyl)phenyliodonium p-toluenesulfonate (I77), (4-methoxyphenyl)-(4-nitrophenyl)iodonium p-toluenesulfonate (I81), (3-bromophenyl)-(4-methoxyphenyl)iodonium p-toluenesulfonate (I84), (4-fluorophenyl)-(4-methoxyphenyl)iodonium p-toluenesulfonate (I93) were synthesized by PhD J. Ortyl from Cracow University of Technology, as described in the literature.37 All substrates and solvents necessary for the preparation of coinitiators were purchased from Aldrich Chemical Co. (Poland) and used without further purification.The fluorescence quenching measurements
The fluorescence quenching measurements were performed using a single-photon counting system UV-VIS-NIR Fluorolog 3 Spectrofluorimeter (Horiba Jobin Yvon). The apparatus uses a picosecond diode laser (370 nm) generating pulses of about 50 ps for the excitation. Short laser pulses in combination with a fast microchannel plate photodetector and ultrafast electronics make a successful analysis of fluorescence decay signals in the range of single picoseconds possible. The dye was studied at a concentration able to provide equivalent absorbance at 370 nm (0.2 in the 10 mm cell). The rate constant for fluorescence quenching was determined in 1-methyl-2-pyrrolidinone as a solvent. The concentration of dye was 2 × 10−5 M and that of quenchers was in the range from 1 × 10−4 M to 1 × 10−3 M. The fluorescence quenching at 440 nm was measured in deaerated solution by bubbling with argon.Kinetics of polymerization by photo-differential scanning calorimetry
A regular photo-DSC setup was used to determine the photoinitiation efficiency of the UV-Vis photoinitiator systems in acrylate. For this purpose, the kinetic parameters of free radical polymerization of 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate photoinitiated by bi- and tri-component photoinitiating systems composed of 1,3-bis(phenylamino)squaraine in the presence of diphenyliodonium salt or/and borate salt were determined using a Differential Scanning Calorimeter TA DSC Q2000 Instrument equipped with a high-pressure mercury lamp (Photo-DSC). The heat evolved during reaction was registered for radiation range 300–500 nm and at a constant intensity of 30 mW cm−2. The measurements were performed at a sampling interval of 0.05 s per point in isothermal conditions under a nitrogen flow of 50 mL min−1. Samples weighing 30 ± 0.1 mg were placed into an open aluminum liquid DSC pan. The measurements were carried out under identical conditions. The sample was maintained at a prescribed temperature for 2 min before each measurement run began. The polymerization mixture was composed of 1.8 mL of monomer, 0.2 mL of 1-methyl-2-pyrrolidinone, the sensitizer and an appropriate coinitiator. The concentration of photoinitiators was in the range from 1 × 10−3 to 3 × 10−3 M. 1-Methyl-2-pyrrolidinone was necessary due to the poor solubility of sensitizer in monomer. The polymerizing solution without a coinitiator was used as the reference sample. The degree of conversion (C%) is directly proportional to the number of reactive groups (double bonds) in the monomer molecule. This parameter was calculated using eqn (1):
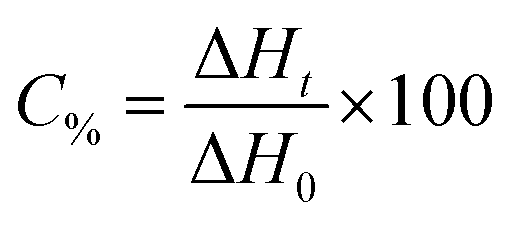 | (1) |
The rate of polymerization (Rp) is derived from the amount of heat released during the process, which is expressed by eqn (2):
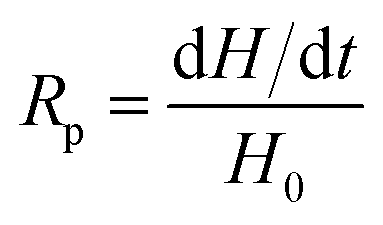 | (2) |
The overall ability to the initiation reaction was calculated using eqn (3):
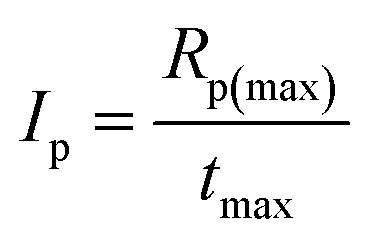 | (3) |
Cyclic voltammetry measurements
The electrochemical measurements were evaluated by Cyclic Voltammetry (CV) using the ER466 Integrated Potentiostat System (eDAQ, Poland) in a three-electrode configuration. The electrolyte was 0.1 M tetrabutylammonium perchlorate in dry acetonitrile. 1 mm platinum disk electrode was used as a working electrode, platinum and Ag/AgCl were used as auxiliary and references electrodes, respectively. All solutions were deoxygenated with N2 for at least 15 min prior to measurements. The computer-controlled potentiostat was equipped with EChem Software.Nanosecond laser flash photolysis experiment
Transient absorption spectra and decay kinetics were carried out using the nanosecond laser flash photolysis method. The experiments were performed using a LKS.60 Laser Flash Photolysis apparatus (Applied Photophysics). Laser irradiation at 355 nm from the third harmonic of the Q-switched Nd:YAG laser from a Lambda Physik/model LPY 150 operating at 65 mJ per pulse (pulse width about 4–5 ns) was used for the excitation. Transient absorbances at preselected wavelengths were monitored by a detection system consisting of a monochromator, a photomultiplier tube (Hamamatsu R955) and a pulsed xenon lamp (150 W) as a monitoring source. The signal from the photomultiplier was processed by a Hewlett–Packard/Agilent an Agilent Infiniium 54810A digital storage oscilloscope and an Acron compatible computer.Results and discussion
2-Ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate as multifunctional monomer was chosen to study the reactivity of UV-Vis photoinitiator systems. A combination of squarylium dye (1,3-bis(phenylamino)squaraine (SQ)) and a radical initiator, an onium salt (iodonium or/and borate), was selected as photoinitiator system (see Chart 1).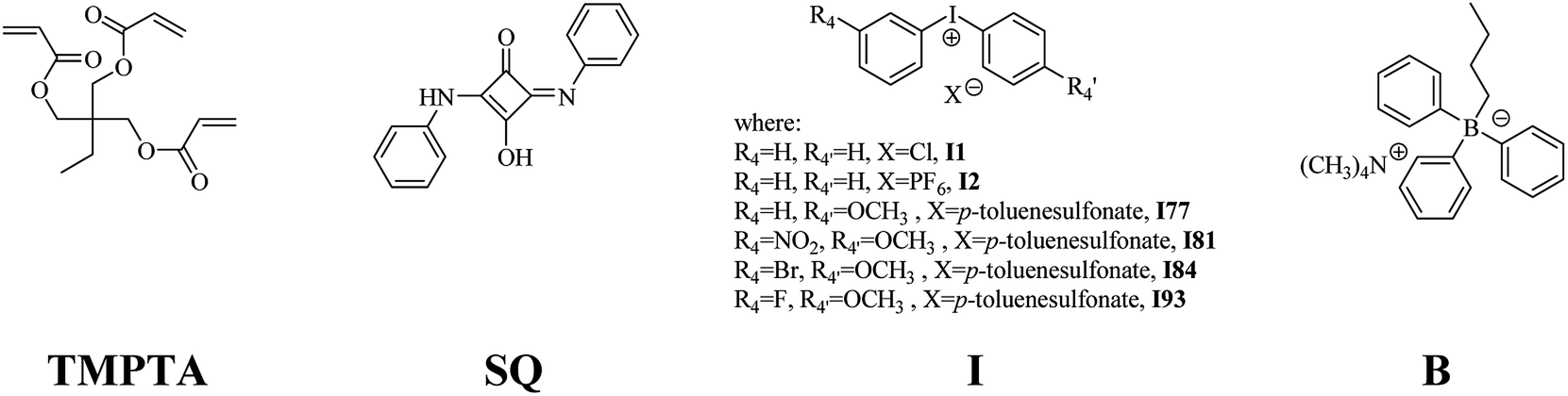 | ||
| Chart 1 The composition of polymerizing mixture composed of: monomer, photosensitizer and selected coinitiator or coinitiators. | ||
Following bi- and tri-component photoinitiators: SQ/I, SQ/B2 and SQ/I/B2 were studied.
The sensitizer absorbs light from 320 nm to 450 nm with the maximum at 400 nm in 1-methyl-2-pyrrolidinone used as a solvent.30 All coinitiators studied absorb below 300 nm. In this case, only squaraine dye is excited by light source used, because an almost satisfied overlap exists between absorption of sensitizer and emission of the high-pressure mercury lamp (Omnicure 2000) used in photo-DSC studies.
Scheme 1 describes the necessary pathways to generate initiating radicals in the redox system comprising the dye (sensitizer) as an electron donating moiety and the iodonium salt with the general cation structure [A–I–B]+ as electron acceptor, functioning as radical initiator. On the other hand, the pathways to generate initiating radicals in redox system comprising squaraine dye as electron acceptor and an alkyltriphenylborate salt as electron donating moiety as radical initiator is different.
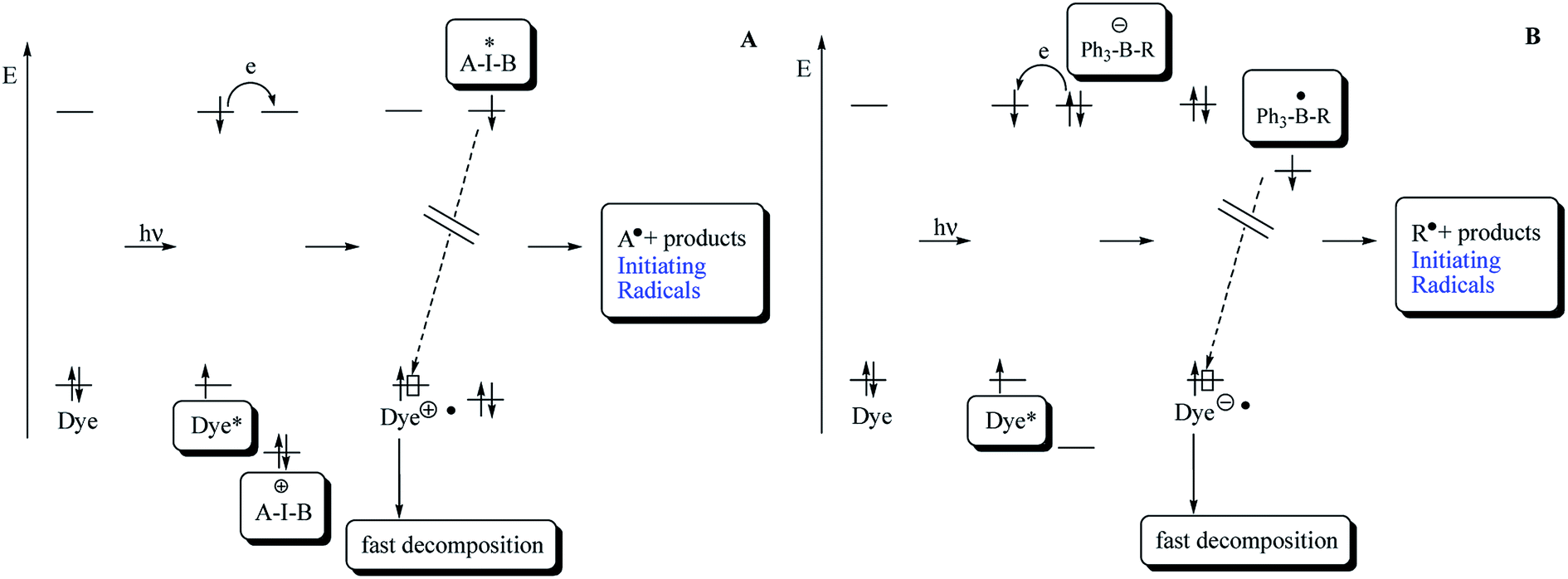 | ||
| Scheme 1 Reactions occurring during UV-Vis-initiated radical polymerization based on electron transfer. | ||
Excitation of dye results in formation of first excited singlet state Dye*. Electrochemical data allow a rough estimate of the HOMO and LUMO energies of Dye and iodonium and borate salts, respectively. Electron transfer from Dye* to [A–I–B]+ results in reduction of the iodonium salt and yields the iodyl radical [A–I–B]˙. The short-living intermediate cleavages with a high efficiency and yields initiating aryl radicals A˙. In the case of alkyltriphenylborate salt, an electron transfer from [Ph3–B–R]− to Dye* results in reduction of dye, and yields the boranyl radical [Ph3–B–R]˙. The short-living intermediate cleavages with a high efficiency and yields initiating alkyl radicals R˙. The fast decomposition of [A–I–B]˙, [Ph3–B–R]˙, Dye˙+ and Dye˙− results in decrease of the efficiency of back electron transfer.
It was found, that the irradiation of a photosensitizer alone with visible light does not induce polymerization reaction.
The main factor that controls the rate constant of electron transfer is the exothermicity of the reaction. This value may be estimated using the oxidation potential of an electron donor and the reduction potential of an electron acceptor. The possibility of electron-transfer from donor to acceptor molecules was determined on the basis of electrochemical properties of all components of photoinitiating systems. For this purpose, the redox potentials were measured using cyclic voltammetry. The values of free energy change ΔGel for electron transfer were calculated according to Rehm–Weller equation.30,38 In this study, diphenyliodonium salts were used as the electron acceptors and squaraine dye as electron donor, but borate salt as electron donor with sensitizer acting as electron acceptor.
These iodonium salts with various substituents in 4- or 4′-positions possess reduction potentials (Ered) in the range from −1.00 eV to −0.175 eV,20 while squaraine dye (SQ) exhibits an oxidation potential (Eox) of 1.13 eV and reduction potential of −0.128 eV. The oxidation potential of borate salt is 1.153 eV. SQ possesses an excitation energy (E00) of 2.938 eV derived from the absorption and fluorescence spectrum. These electrochemical data (Eox, Ered, E00) allow calculating the free enthalpy of electron transfer ΔGel, eqn (4), resulting in the range form −159.9 kJ mol−1 to −72 kJ mol−1. The negative values of ΔGet demonstrated that as a result of an electron transfer reaction between photosensitizer and coinitiators for all initiating systems under study, the radicals initiating polymerization process are formed.
The thermodynamic data presented in Table 1 confirmed, that these reactions are possible from thermodynamic point of view, that should lead to similar reactivity (expect to salt I2) between excited dye and [A–I–B] and Dye* and [Ph3–B–R]−.
| ΔGel = Eox − Ered − E00 | (4) |
| Electron acceptor | Ered [eV] | ΔGeta [kJ mol−1] | kbl × 104 [s−1] | kq × 10−10 [M−1 s−1] |
|---|---|---|---|---|
| a Calculated using the singlet state energy 0 → 0 for SQ E00 = 2.938 eV. | ||||
| I1 | −0.494 | −126.8 | 5.64 | 6.26 |
| I2 | −1.00 | −72.01 | 0.946 | 4.0 |
| I77 | −0.206 | −154.6 | 0.7 | 8.12 |
| I81 | −0.554 | −121.06 | 0.159 | 6.1 |
| I84 | −0.175 | −157.6 | ||
| I93 | −0.302 | −145.4 | 53.1 | |
| Electron donor | Eox [eV] | ΔGeta [kJ mol−1] | kbl × 104 [s−1] | kq × 10−10 [M−1 s−1] |
|---|---|---|---|---|
| B2 | 1.153 | −159.9 | 1.3 | 2.69 |
However, the kinetic results obtained (Fig. 1–3 and Table 2) show that the thermodynamic data do not fit with this hypothesis.
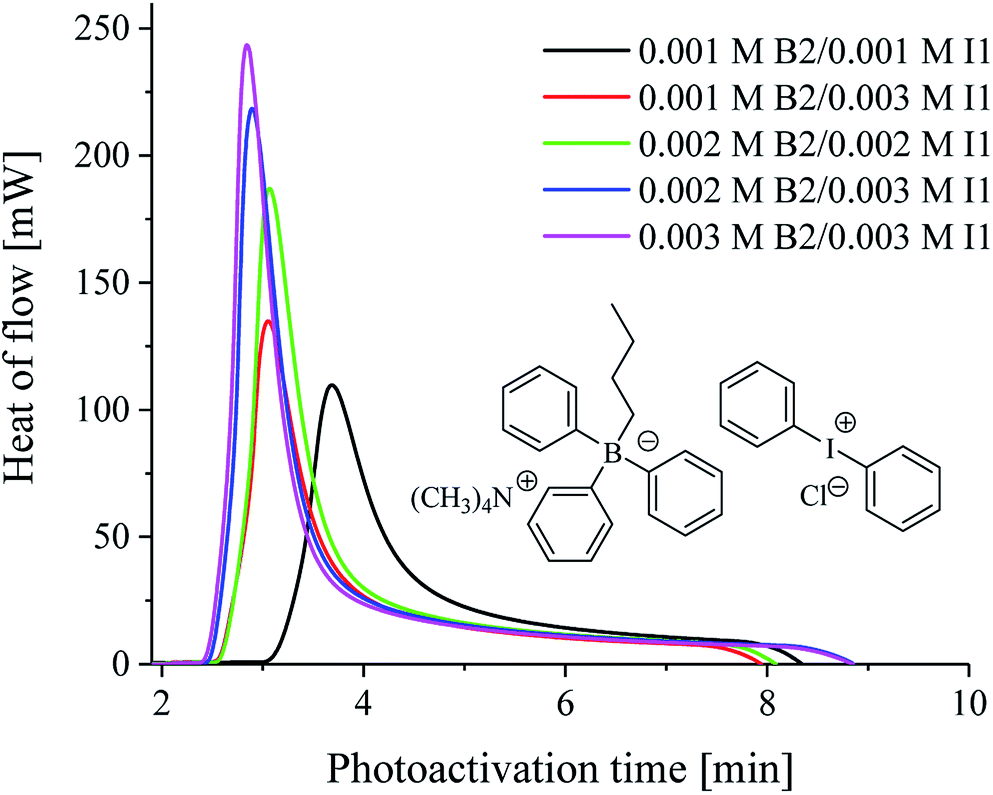 | ||
| Fig. 1 The kinetic curves recorded during radical polymerization of TMPTA initiated by 1,3-bis(phenylamino)squaraine in a presence of two coinitiators marked in the figure. The concentration of sensitizer was 1 × 10−3 M, the concentration of borate and iodonium salts is marked in the figure. The irradiation range was from 300 nm to 500 nm with intensity 30 mW cm−2. | ||
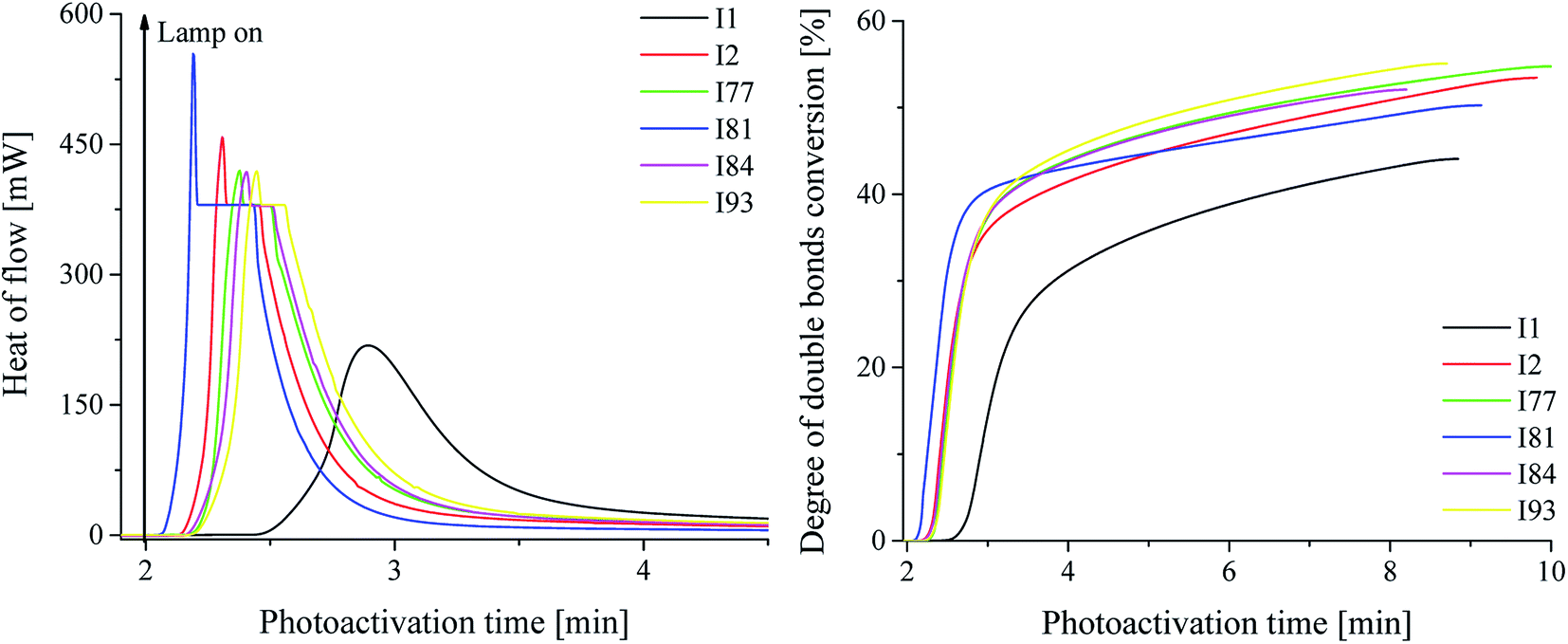 | ||
| Fig. 2 L: the kinetic curves recorded during radical polymerization of TMPTA initiated by 1,3-bis(phenylamino)squaraine in a presence of two coinitiators marked in the figure. P: degree of double bonds conversion versus irradiation time. The concentration of sensitizer was 1 × 10−3 M, and concentration of borate and iodonium salts was 2 × 10−3 M and 3 × 10−3 M, respectively. The irradiation range was from 300 nm to 500 nm with intensity 30 mW cm−2. | ||
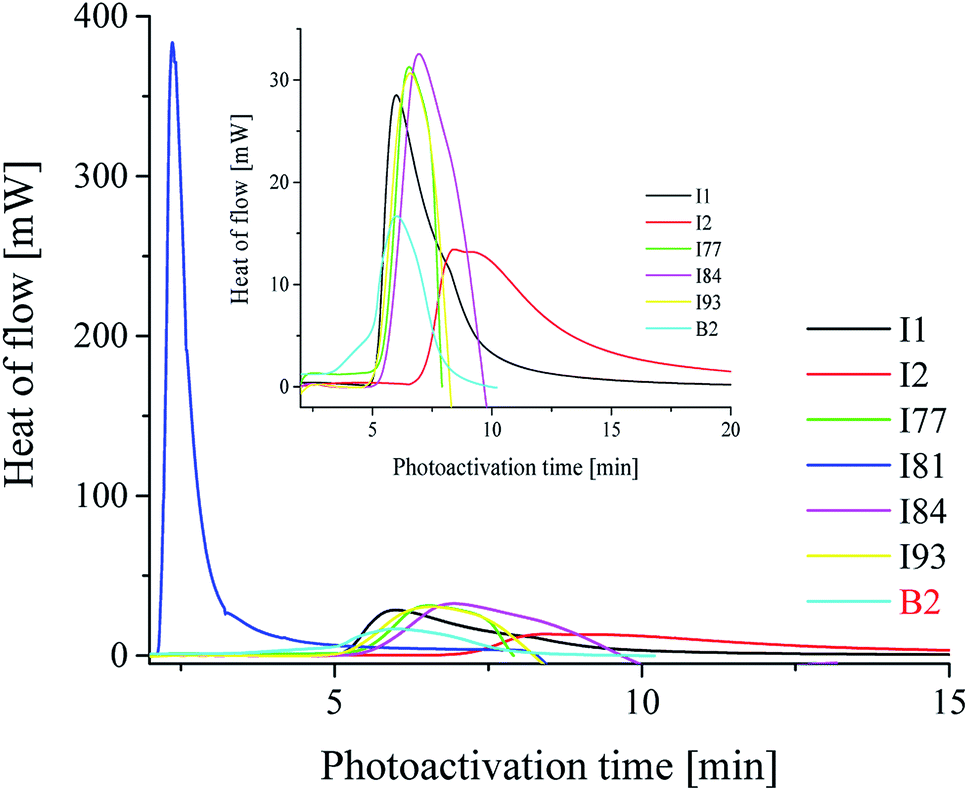 | ||
| Fig. 3 The kinetic curves recorded during radical polymerization of TMPTA initiated by 1,3-bis(phenylamino)squaraine in a presence of coinitiator marked in the figure. The concentration of sensitizer and coinitiator was 5 × 10−3 M. The irradiation range was from 300 nm to 500 nm with intensity 30 mW cm−2. | ||
| Coinitiator | Qmax [mW] | tmax [min] | Rp × 103 [s−1] | Ip × 103 | Rp/Rp(B2) | Cp [%] |
|---|---|---|---|---|---|---|
| a tmax is the time needed for reach of the maximum value of heat flow. | ||||||
| I1 | 28.5 | 6.01 | 1.12 | 3.11 | 2.12 | 16.9 |
| I2 | 13.4 | 8.46 | 0.53 | 1.10 | 1 | 6 |
| I77 | 31.3 | 6.55 | 1.23 | 3.41 | 2.33 | |
| I81 | 383.7 | 2.36 | 15.15 | 126 | 28.57 | 48 |
| I84 | 32.6 | 6.97 | 1.28 | 3.54 | 2.42 | |
| I93 | 30.7 | 6.6 | 1.21 | 3.35 | 2.28 | |
| B2 | 16.7 | 6.01 | 0.66 | 1.83 | 1 | 10.5 |
| I1/B2 | 218.44 | 2.9 | 8.62 | 68.7 | 13.09 | 44.1 |
| I2/B2 | 457.9 | 2.3 | 18.07 | 148 | 27.44 | 54 |
| I77/B2 | 419.86 | 2.37 | 16.57 | 138 | 25.16 | 54.7 |
| I81/B2 | 554.09 | 2.19 | 21.87 | 166 | 33.20 | 50.3 |
| I84/B2 | 417.93 | 2.4 | 16.50 | 135 | 25.04 | 52 |
| I93/B2 | 419.12 | 2.44 | 16.54 | 113 | 25.11 | 55.1 |
The representative polymerization curves and conversions of monomer during polymerization at different times of irradiation are shown in Fig. 1–3.
In Fig. 1, the typical kinetic curves of a photoinitiated polymerization, show the heat flow as a function of the photoactivation time, as well as the influence of borate salt and diphenyliodonium salt on the polymerization rate for the tertiary systems.
The influence of different initiator systems on the kinetics of photopolymerization is presented in Fig. 2, as well as the degree of monomer conversion as a function of the photoactivation time, using a ternary initiator systems.
For the comparison the photoinitiating ability of bi-component photoinitiators on the kinetic of polymerization of TMPTA is shown in Fig. 3.
The kinetic parameters of polymerization observed for different compositions of photoinitiators are shown in Table 2.
The maximum of polymerization heat roughly describes the reactivity of a photopolymer system19,39 because the system crosslinks during exposure. It is a characteristic point where auto-acceleration and verification are equivalent. Thus, diffusion processes control the reactivity and reaction rate constants change with conversion. Furthermore, termination kinetics often changes from bimolecular termination to termination by initiator radicals at higher conversion degrees.19,39 Nevertheless, choosing of maximum heat flow forms a reasonable compromise to describe the reactivity, because this quantity relates somehow to the initiation efficiency. In other words, the initiator efficiency is defined as the ability of the investigated photoinitiators to initiate free radical polymerization and is expressed as its rate (Rp) (see the data in Table 2).
Barner-Kowollik et al. determined the initiation efficiency of a photoinitiator via a trifold combination of pulsed laser polymerization and subsequent ionization mass spectrometry and femtosecond transient absorption spectroscopy (PLP-ESI-MS).30,40 This method is suitable only for initiators forming identical radical fragments originating from disparate source molecules. The photoinitiators studied dissociate into different radicals (e.g. phenyl, p-methoxyphenyl, p-nitrophenyl, p-bromophenyl, p-fluorophenyl and n-butyl). The quantitative initiation evaluation of different fragments originating from disparate sources molecules requires additional information regarding the radicals' reactivity towards vinyl bonds and/or stability of initiating radicals or other factors.30 It is well known, that the stability of radicals decreases from primary alkyl radical (n-butyl) to phenyl radicals. Radicals possess single occupied molecular orbital which is quite higher in energy. Any factors, that decrease energy of SOMO result in increase of radical stability and decrease its reactivity. Type of substituent attached may influence on the stability of radical formed. An electron-donating and an electron-withdrawing properties depend on the energy of SOMO (see Scheme 2).
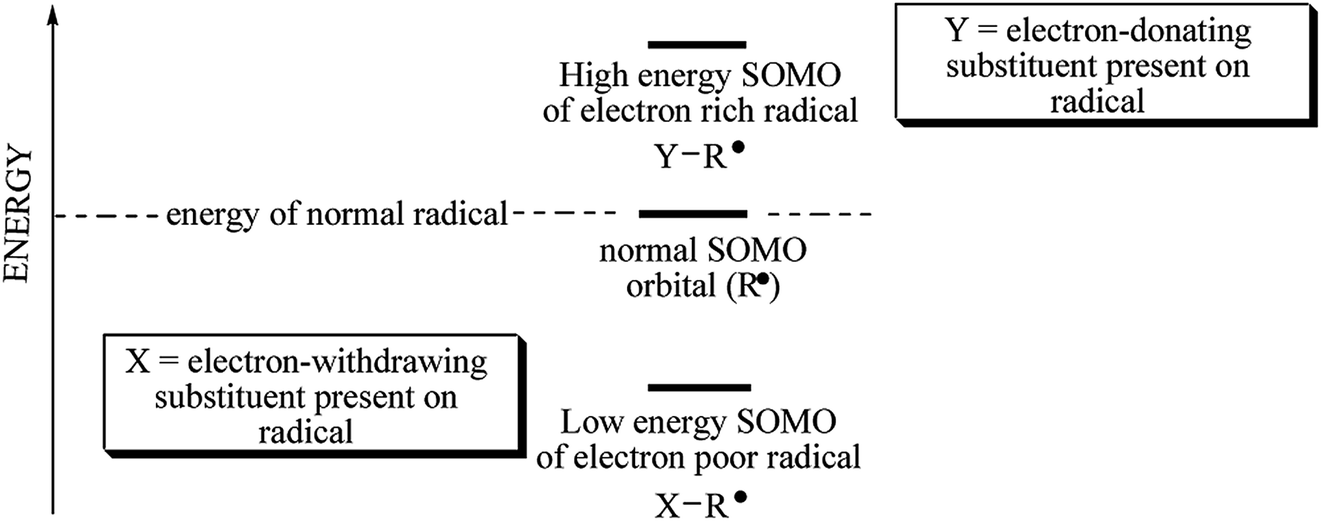 | ||
| Scheme 2 Illustrative diagram of energy of radicals. | ||
As it was mentioned above, several different p-substituted phenyl radicals are formed as a result of carbon–iodide bond cleavage in diphenyliodyl radical. The substitution in para position of phenyl ring may stabilize a radical formed. Generally, the increased substitution improves the stability of radical formed, and less energy is required for its formation. When both electron-donating and electron-withdrawing groups are present in the same molecule, with a suitable separation between them, an additional captodative stabilization of radical occurs.41 The methoxy-, nitro-, bromo- and fluoro-substituted diphenyliodonium salts were chosen for the study. Substituent effects on radical stabilization energy (RSE) values can be interpreted as a combination of three different molecular orbital interactions: (a) resonance stabilization through interaction of the radical center with p-systems; (b) stabilization through hyperconjugation of the radical center with adjacent C–H bonds; and (c) stabilization through interaction of the radical center with high lying orbitals describing lone pair electrons.42 The RSE value for sigma radical derived from n-butyltriphenylboranyl radical is about 12.2 kJ mol−1.42 The substituent effect on RSE values may be observed for the p-substituted diphenyliodonium salts under study. The substituent attached at the para position of phenyl ring may stabilize a radical. The methoxy group with unshared electron pairs stabilizes the radical by means of three electron bonding. Basing on Coote et al.,42 the radical stabilization energies theoretically calculated at G3(MP2)-RAD level, for phenyl, p-methoxyphenyl and p-nitrophenyl are −37 kJ mol−1, −41.6 kJ mol−1 and −40.4 kJ mol−1, respectively. Taking this into account, more stable p-nitro, p-bromo and p-fluorophenyl radicals are formed in the case of iodonium salt, than n-butyl radical.
Basing on the Scheme 2 and literature focused on the estimation of radical stabilization energies (RSEs) one can conclude, that coinitiator-based radical possessing of electron-withdrawing substituent in para position of phenyl ring decomposes faster than diphenyliodonyl with electron-donating group.41 This fact may be a reason of an excellent photoinitiating ability of (4-methoxyphenyl)-(4-nitrophenyl)iodonium p-toluenesulfonate (I81).
From the data presented in Table 2 and Fig. 3, it is seen that most of the iodonium salts (I1, I2, I77, I84, I93) exhibit no large differences regarding the reactivity in triacrylate (TMPTA). On the other hand, the effect of diphenyliodonium salt as second coinitiator dramatically reduced the photoactivation time required to reach a higher rate of polymerization and conversion of monomer when compared with the system without iodonium salt. It should be mentioned, that the concentration of coinitiator in bimolecular systems was higher than in three-component ones and equal 5 × 10−3 M. It was found, that when only 5 × 10−3 M of iodonium salt or borate salt was used, the maximum rate of polymerization (Rp(max) (s−1)) values equal 15 × 10−3 s−1 was observed for diphenyliodonium salt (I81) possessing nitro (NO2) and methoxy (CH3O) groups at para position in both phenyl rings. Similar results were observed for 2-(p-N,N-dimethylaminostyryl)benzoxazole used as a photosensitizer and described in earlier paper.20 Other bi-component photoinitiators initiate radical polymerization of triacrylate with very low and comparable rates, e.g. about 1.2 × 10−3 M. The worst photoinitiating ability possesses photoinitiator composed of squaraine dye and tetramethylammonium n-butyltriphenylborate salt (SQ/B2). However, for the ternary photoinitiator systems, the Rp(max) was about 20–30-times higher than for SQ/B2 system. When bis(p-substituted)diphenyliodonium salt was used as a coinitiator, the Rp(max) values were: 0.009, 0.018, 0.017, 0.022, 0.016 and 0.017 for 3 × 10−3 M iodonium salt. When equimolar concentration of borate salt and iodonium salt, e.g. 2 × 10−3 M was used, a slight inhibitory was observed and the Rp(max) decreased about 20% (see Fig. 1).
The ternary photoinitiator system, formed by squarylium dye, borate salt and diphenyliodonium salt, showed an expressively higher Rp(max) on the photoactivated polymerization. As presented in Table 1, the Rp(max) for SQ/B2/I systems reached from 8.62 × 10−3 s−1 to 21.87 × 10−3 s−1, while the binary system SQ/B2, at the same reaction conditions reached 0.66 × 10−3 s−1. After 60 s of photoactivation using ternary system, the monomer conversion was about 40% (except for salt I1), the total degree of double bonds conversion reached about 55% and for the binary system the total monomer conversion was achieved about 17, 6, and 48% for salts I1, I2 and I81, respectively after 4–5 min of irradiation. For the binary systems composed of sensitizer and borate salt the total degree of double bonds conversion was reached 10.5% after 8 min of photoactivation. It should be noted, that in binary systems the concentration of coinitiators was 2–2.5-times higher than in ternary photoinitiators.
The values of photoinitiation index (Ip) expressed as a quotient of maximum rate of polymerization and time required to reach the maximum rate of heat release, obtained from the exothermal curve,31 show that all of the compounds used as coinitiators in the polymerization, possess different effectiveness to the generation of active centers. The photoinitiation index is ranging from 0.001 to 0.17 (see data in Table 2). The highest values were achieved for bi-component photoinitiator: SQ/I81 of about 0.126, and tri-component: I2/B2 and I81/B2 of about 0.148 and 0.166, respectively.
The spectral range (300–500 nm) used under study is classical for photopolymerization. Therefore, the photoinitiating ability of new initiators was compared with that of commercially available initiators. It should be also mentioned, that the ability to initiation of polymerization of triacrylate by tertiary systems is better than that observed for commercially available initiators. For the comparison, camphorquinone (CQ) as a popular sensitizer was used in presence of following coinitiators: ethyl p-(N,N-dimethylamino)benzoate (EDMAB), diphenyliodonium chloride (I1), N-methyldiethanolamine (MDEA), and N-phenylglycine. The kinetic curves are shown in Fig. 4.
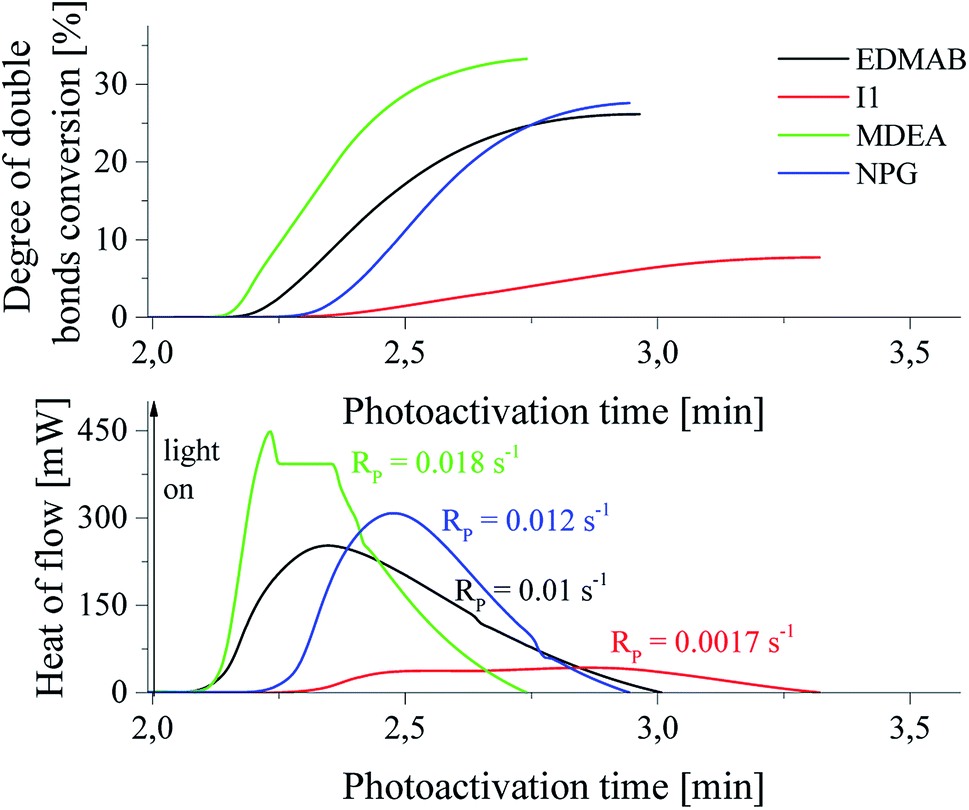 | ||
| Fig. 4 The photoinitiating ability of commercially available photoinitiators: CQ/ethyl p-(N,N-dimethylamino)benzoate (EDMAB), CQ/diphenyliodonium chloride (I1), CQ/N-methyl-diethanolamine (MDEA), CQ/N-phenylglycine (NPG) for initiation of polymerization of TMPTA under irradiation with light 300 nm-500 nm, Ia = 30 mW cm−2. The photoinitiator concentration was 5 × 10−3 M. | ||
The rates of polymerization initiated by commercial photoinitiators under experimental conditions are in the range from 0.0017 s−1 to 0.018 s−1. The best photoinitiating ability shows initiator composed of camphorquinone and N-methyldiethanolamine (MDEA), that is similar to photoinitiating efficiency of system: squarine dye/p-substituted diphenyliodonium salt (I81). The initiation efficiency of camphorquinone and diphenyliodide (I1) is comparable with that observed for bimolecular systems: squarine dye and diphenyliodonium salts possessing: hydrogen, bromine, fluorine atoms in para position of phenyl ring. Ternary initiators are similarly effective in initiating of TMPTA polymerization as the most effective commercially available system: CQ/MDEA. The rate of polymerization observed in a case of initiator: squaraine/(4-methoxyphenyl)-(4-nitrophenyl)iodonium p-toluenesulfonate (I81) is about 20% higher than that observed for the best commercial initiator studied. It should be also noted, that the degree of TMPTA conversion achieved for commercial photoinitiators is relatively low and reaches values ranging from 12.4 to 33.5%. Polymerization initiated by the best photoinitiators under study leads to higher values of double bond conversion in TMPTA and ranging from 44% to 55%.
It should be also noted, that for all tested bimolecular photoinitiators a long inhibition time was observed. An introduction of iodonium salt to bimolecular system SQ/B2 distinctly decreases the inhibition time and leads immediately to a start of the photopolymerization process. It was found, that the maximum rates of polymerization were observed below 60 s of irradiation, in the most cases after 10 or 20 seconds.
The diphenyliodonium salt concentration has little effect on Rp(max), indicating that even at low concentrations, this salt participate efficiently in the monomer polymerization. The beneficial role of the diphenyliodonium salt in photoinitiated free radical polymerization has been well recognized. Iodonium salts are very effective in reacting with an excited Dye*, accepting an electron and generating free radicals, that can start the polymerization, as viewed in Scheme 3.32
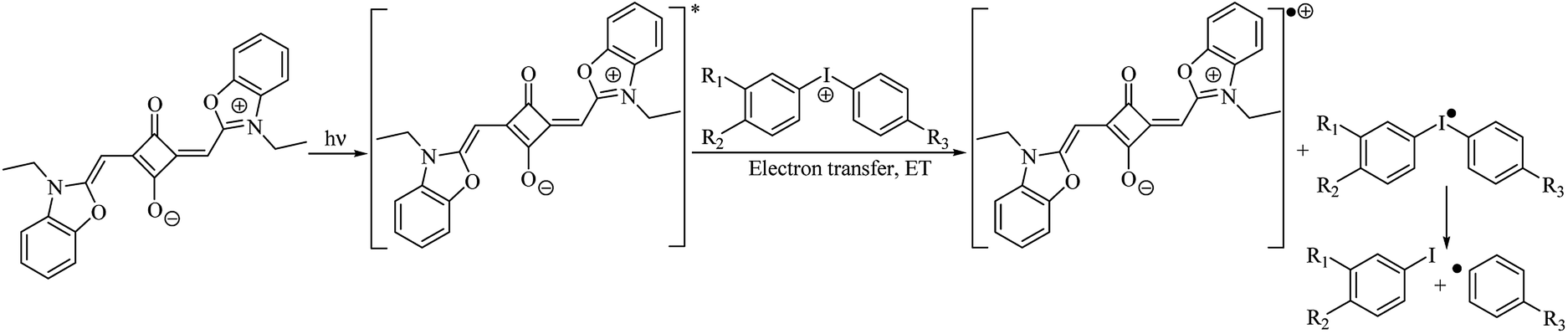 | ||
| Scheme 3 Mechanism of free radicals formation in bimolecular photoinitiating system. | ||
Additionally formed, phenyl radicals are highly reactive toward vinyl bonds and demonstrated high addition rate constants to double bonds of acrylate monomers in free radical polymerization, thus resulting in the shortened induction period (Fig. 2). On the other hand, the free volume effect also could contribute to the high polymerization efficiency. Namely, the polymerization rate is faster than the volume shrinkage, leading to an increase in free-volume formation.43,44 This phenomenon increases the mobility of the residual double bond and resulting in an increase in the final conversion.
The values of Rp/Rp(B2) given in Table 2 represent the ratio between the rate observed for given photoinitiating system and that with the lowest polymerization rate, e.g. SQ/borate salt (B2). As it is seen, the following order in terms of the efficiency of polymerization is observed:
| B2 ≈ I2 < I1 < I77 ≈ I84 ≈ I93 < I1/B2 < I2/B2 ≈ I77/B2 ≈ I84/B2 ≈ I93/B2 < I81 < I81/B2. |
It was also found, that photoinitiators used were bleaching during radical polymerization process. This suggests that the photoinduced decomposition of sensitizer and photoinitiators is caused by photoinduced intermolecular electron transfer.30,45 The changes of absorption spectra of squarine dye in a presence of selected coinitiators are shown in Fig. 5.
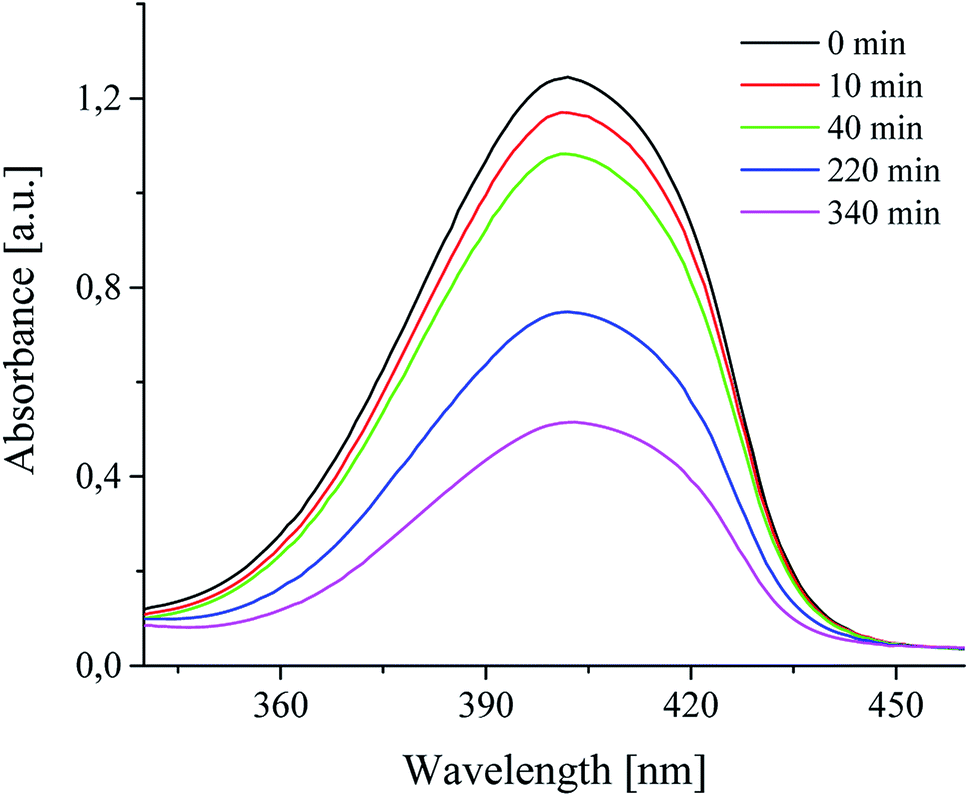 | ||
| Fig. 5 The changes of absorption spectra of squaraine dye in 1-methyl-2-pyrrolidinone as a solvent, in presence of diphenyliodonium hexafluorophosphate (I2) observed during irradiation (300 nm < λ < 500 nm) in selected periods of time. The light intensity was 30 mW cm−2. | ||
The photobleaching ratio may be calculated using eqn (5):
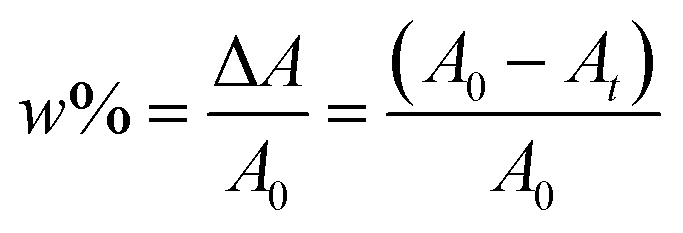 | (5) |
Fig. 6 shows a decrease of absorbance of photosensitizer in presence of selected coinitiators during irradiation with visible light.
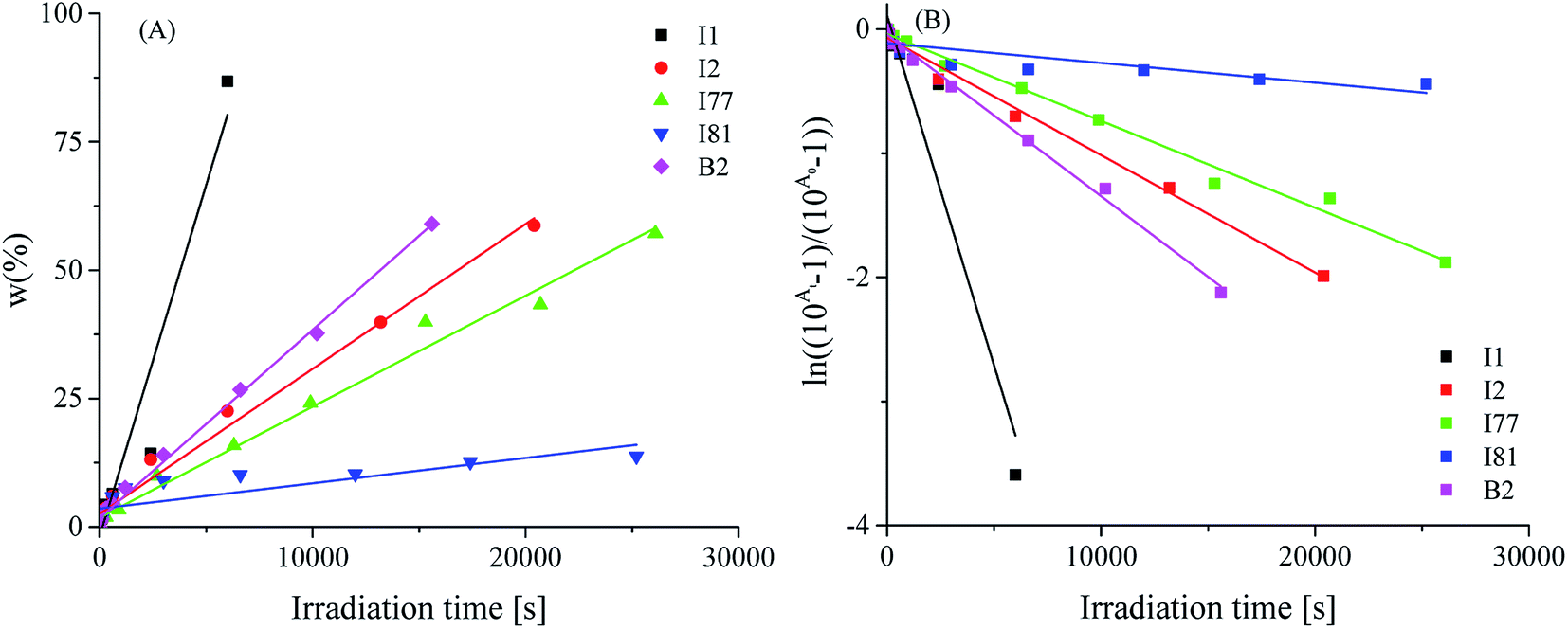 | ||
| Fig. 6 The changes of photobleaching ratios at 402 nm under various visible light irradiation time. | ||
The photobleaching process may also be presented as a relationship expressed by eqn (6).
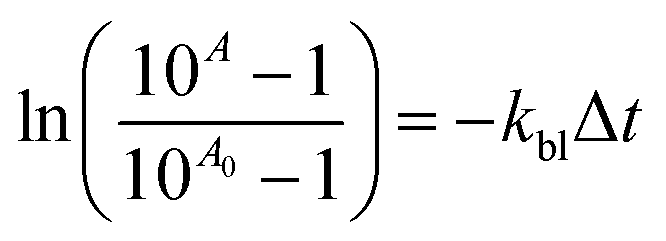 | (6) |
For almost all coinitiators, the linear relationship between the changes of absorbance and irradiation time is observed, expect to coinitiator I81 for which the relationship is exponential. The linear fitting coefficients are in the range from 0.985 to 0.996. The values of R2 for linear fitting for coinitiator I81 is about 0.66. Better result is achieved for exponential fitting. The fitting coefficient equals 0.95. Basing on the eqn (6) the photobleaching rate constants were calculated and summarized in Table 1. They are ranging from 1.59 × 10−5 s−1 to 5.64 × 10−4 s−1. Based on the kinetics of photobleaching, it is seen that the squaraine dye faster photobleaching in presence of diphenyliodonium chloride and tetramethylammonium n-butyltriphenylborate. Therefore, this may be a reason of very low ability to initiation of radical polymerization by systems composed of I1 and B2 salts. Taking this into account, one can conclude, that the phenyl and butyl radicals faster react in other way, than the addition to monomer molecule. In that case the photobleaching is faster than photoinitiation. In other words, the photobleaching process competes with the initiation of polymerization. It was found earlier,30 that in presence of coinitiator I81, the sensitizer studied undergoes photobleaching very slow, and the relationship between the decrease of absorbance and irradiation time is not linear. The system composed of this coinitiator is the best bimolecular initiator for radical polymerization of TMPTA.
In the next step, an effect of concentration of coinitiator on the quenching of the excited singlet state of sensitizer was studied. The fluorescence quenching experiment was carried out to determine the interactions between photoexcited sensitizer and coinitiators. Tetramethylammonium n-butyltriphenylborate and diphenyliodonium p-toluenesulphonates were used are not fluorescent compounds. For this purpose, both spectroscopic methods were used: time-resolved fluorescence spectroscopy and nanosecond laser flash photolysis. The results obtained as the Stern–Volmer plots are presented in Fig. 7.
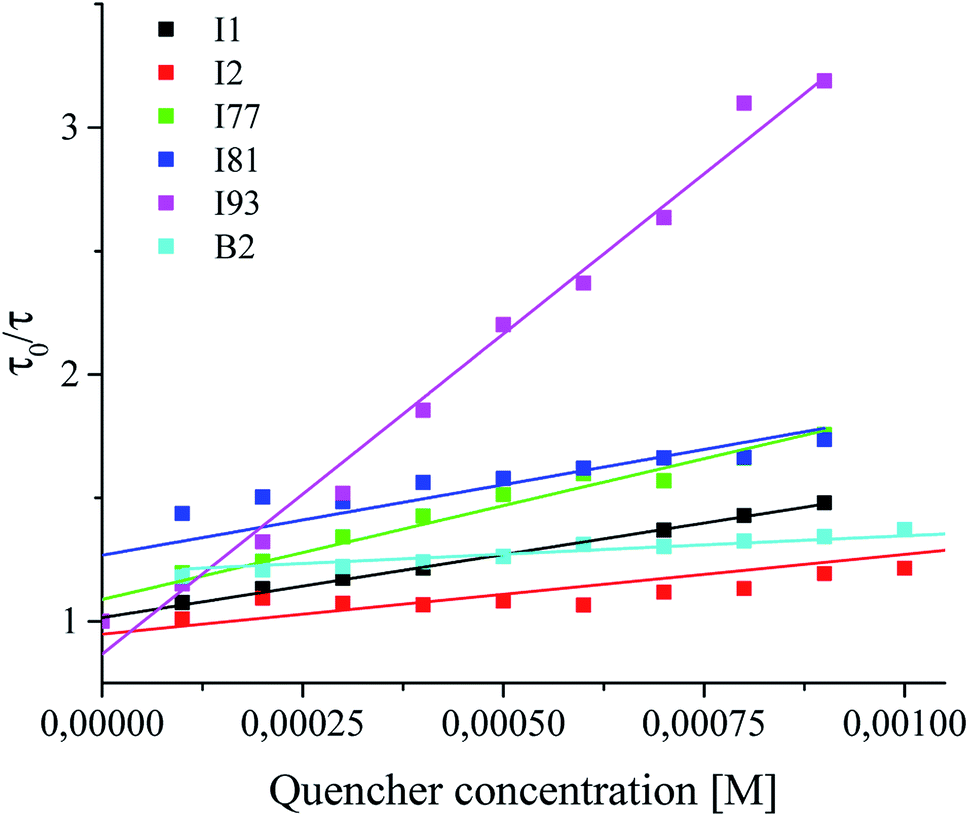 | ||
| Fig. 7 An effect of type and concentration of quencher on the fluorescence of photosensitizer. | ||
Basing on the Stern–Volmer equation,30 the fluorescence quenching rate constants kq were calculated and summarized in Table 1. The kinetics of quenching changes from 2.69 × 1010 M−1 s−1 to 53.1 × 1010 M−1 s−1 and is nearly one order of magnitude higher than that of the diffusion controlled bimolecular reaction constant (about 2 × 109 M−1 s−1). It was found here, that the fluorescence quenching rate constants depend on a type of coinitiator. These results confirmed that coinitiators studied are effective fluorescence quenchers for photoexcited sensitizer, due to strong interactions with photosensitizer in excited singlet state. Therefore, it might be conclude, that the quenching occurs predominantly through the intramolecular ion-pair pathway, e.g. electron transfer process. The highest values of kq is observed for coinitiator I93, but the lowest for borate salt B2. There is not any correlation between the rate of polymerization and the rate of fluorescence quenching. It can be seen that in the case of coinitiators (I2 and B2), which cause very fast photobleaching of sensitizer, the lowest values of the fluorescence quenching rate constants and at the same time the lowest rates of initiation of free radical polymerization of TMPTA are also observed.
The mechanism of free radicals formation in a ternary photoinitiator system is more complicated, than that for bimolecular ones. Therefore, for the explanation of a role of both onium salts as coinitiators, when squarylium dye is used as a photosensitizer, the nanosecond laser flash photolysis experiments were done. From our previous studies it is known, that the electron-transfer photosensitization involves the absorption of light by squarylium dye and formation of photoexcited sensitizer (SQ)*. In presence of coinitiator, the excited complex is formed, and two parallel redox processes occur. They are reduction of iodonium salt and/or oxidation of borate salt, via electron transfer. Next, diphenyliodonyl radical and alkyltriphenyl boranyl radicals decompose rapidly into iodobenzene and phenyl radical, and/or triphenylborate and alkyl radical, making the reaction irreversible. The phenyl and alkyl reactive species are effective in initiating of the polymerization reaction. Radicals generated in the polymerization propagation are effective in cleaving both C–I (path 1) and C–B (path 2) bonds, as viewed in Scheme 4, releasing another phenyl radical and/or alkyl radical and allowing the polymerization reaction occur.6,8
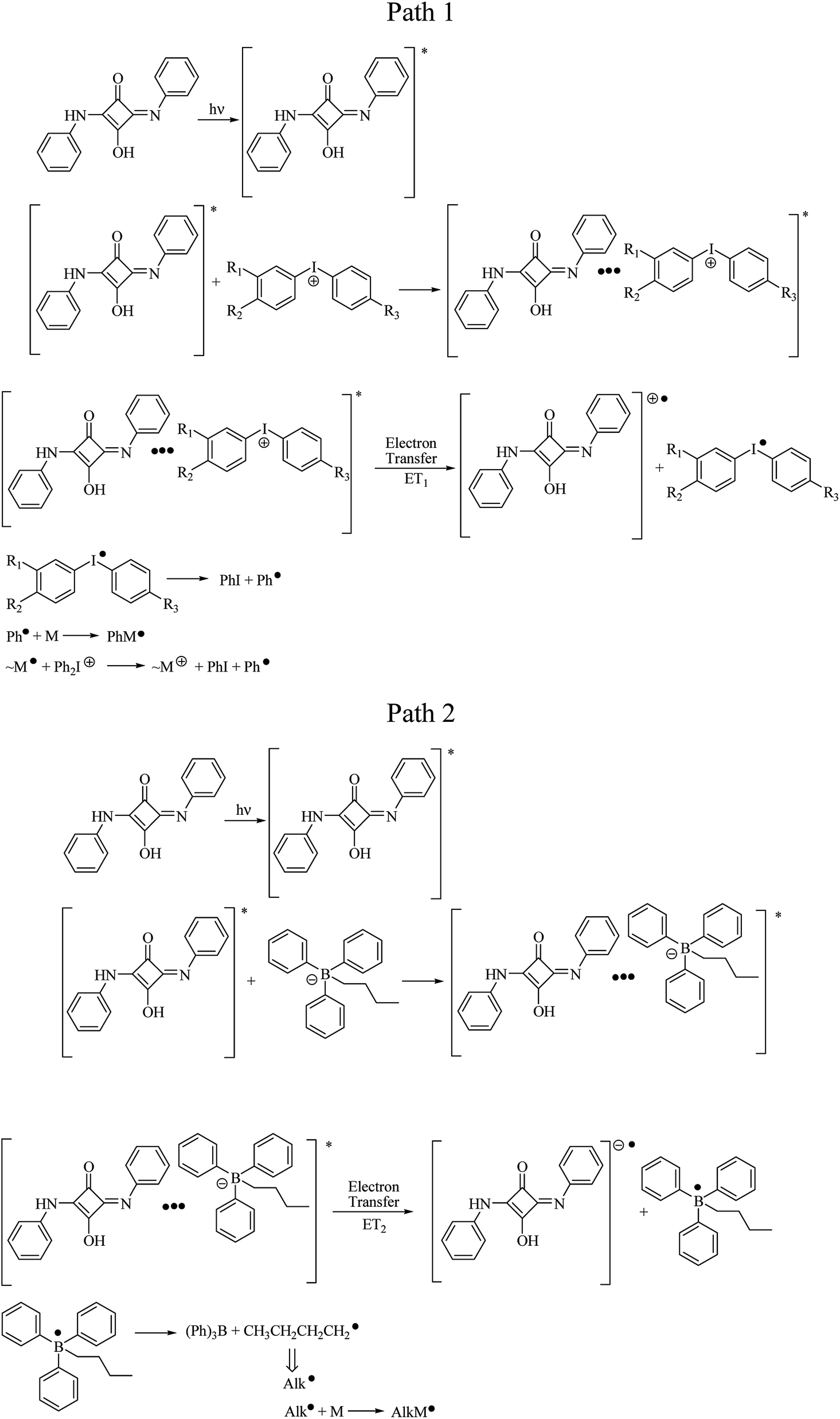 | ||
| Scheme 4 Mechanism of initiating radicals formation in bi- and tri-component photoinitiating systems. | ||
It is seen, that the coinitiator plays a role of an electron donor or an electron acceptor. The electrochemical properties of sensitizer influence on its behavior insight photoexcited complex.
This hypothesis is supported by a nanosecond laser flash photolysis experiment.
It was found, that sensitizer in deaerated solution in acetonitrile undergoes excitation with 355 nm laser pulse. One transient absorption band about 360 nm is observed in nanoseconds time scale, as a result of excited state of squaraine formation (see Fig. 8).
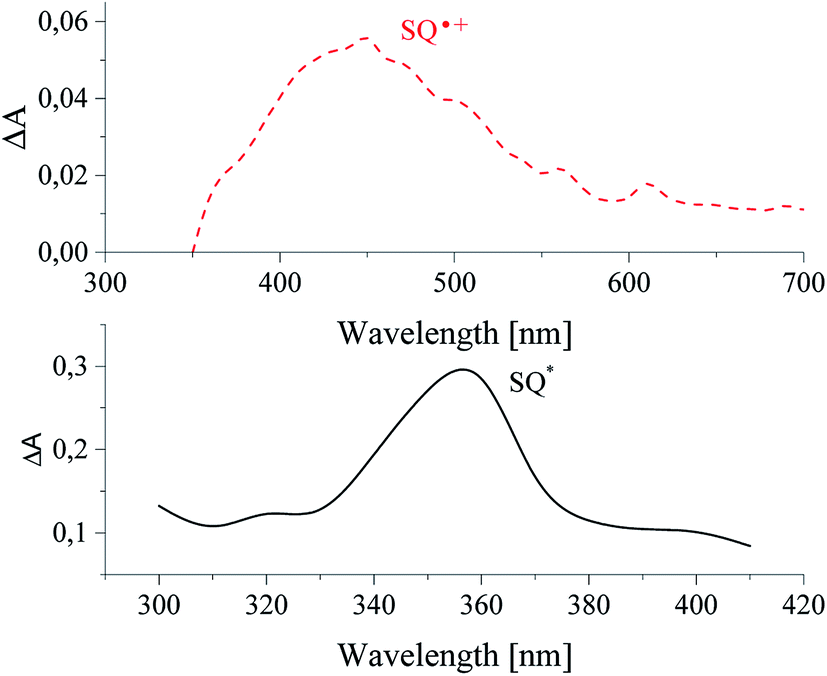 | ||
| Fig. 8 Transient absorption spectra recorded for sensitizer alone (down) and in presence of diphenyliodonium hexafluorophosphate (up) in acetonitrile solution. | ||
An addition of diphenyliodonium salt results in disappearance of the excited state absorption band, and new absorption band with maximum at 460 nm is formed. That is assigned to the product of oxidation of sensitizer (radical cation of squaraine) (see Fig. 8 upper curve).
The curves in Fig. 9 shown the disappearance of the excited singlet state of sensitizer alone (lower curve) and in a presence of diphenyliodonium salt (upper curve). It was found, that an addition of diphenyliodonium salt results in a decrease of lifetime of excited state of photosensitizer by about 1 μs, and increases the rate constant of decay of sensitizer from 4.29 × 105 s−1 to 8.37 × 105 s−1.30 The lifetime was changed with an increasing concentration of coinitiator. Basing on Stern–Volmer relationship, the rate constant of quenching of excited state of sensitizer was calculated. The values kq depend on the chemical structure of diphenyliodonium salt and are equal 0.16 × 106 M−1 s−1 and 2.41 × 106 M−1 s−1 for salts I1 and I81, respectively.30
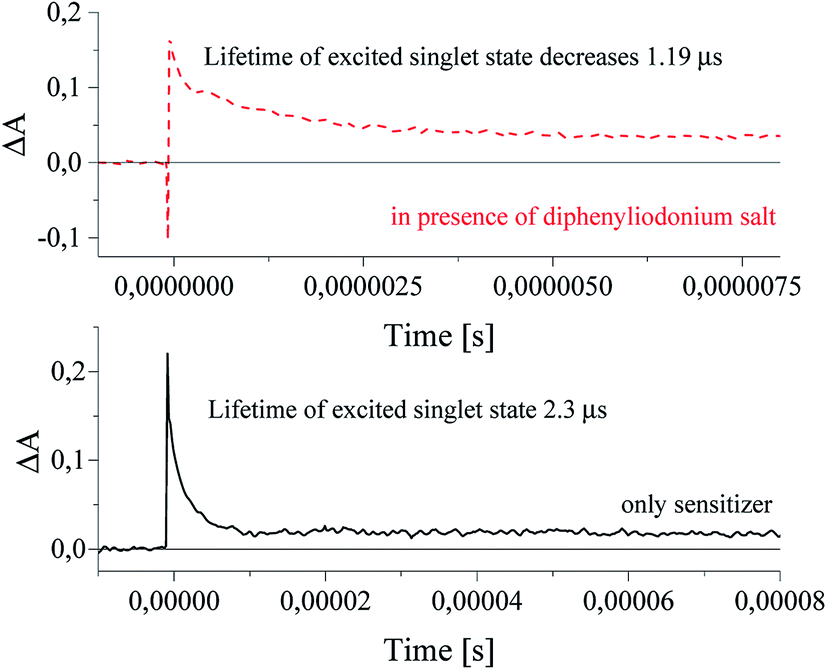 | ||
| Fig. 9 The kinetic curves recorded 1 μs after laser pulse at 380 nm. The concentration of diphenyliodonium salt was 2 × 10−3 M. | ||
In presence of an alkyltriphenylborate salt, photoexcited squaraine undergoes immediate disappearance, that was observed at 360 nm wavelength. The time of decay of excited sensitizer is about 2.33 μs, but the rate constant of the decay is equal 3.31 × 107 s−1. A new absorption band at 480–500 nm assigned to the radical anion of sensitizer was appear (Fig. 10).
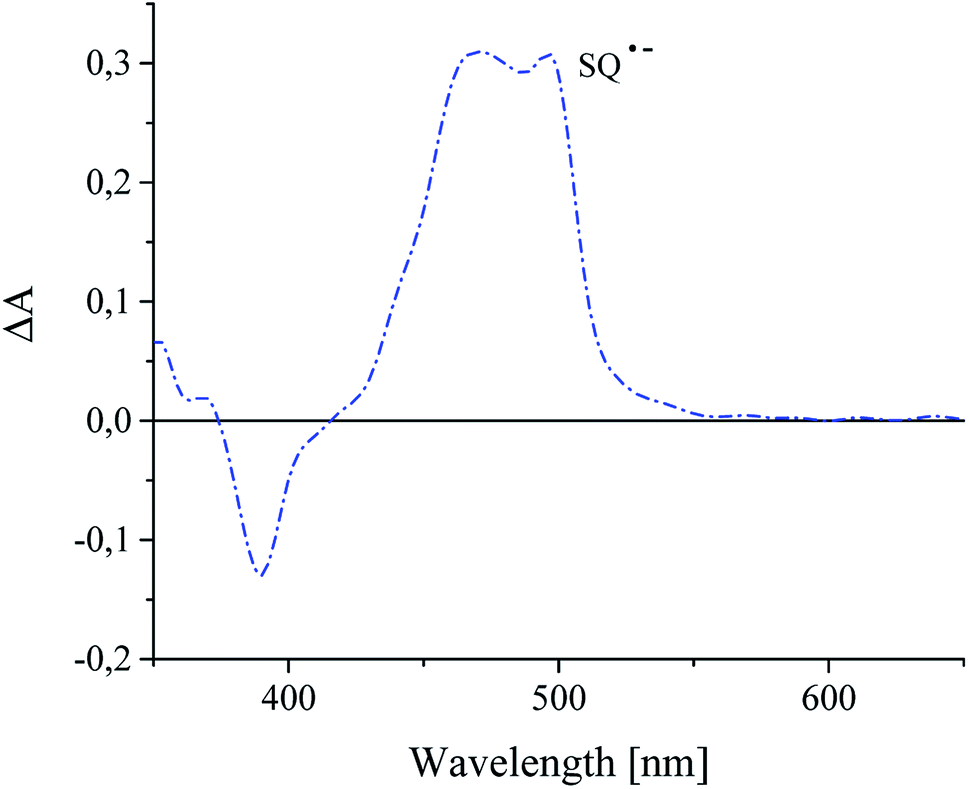 | ||
| Fig. 10 Transient absorption spectra of recorded for squaraine dye in presence of tetramethylammonium n-butyltriphenylborate recorded 100 ns after laser pulse in acetonitrile as a solvent. | ||
It was found, that the times of formation and disappearance of product of oxidization and reduction of squaraine dyes are different.8 Their values for dye-based radical cation are 14 ns and 260 ns, but for dye-based radical anion are 10 ns and 1.14 μs, respectively.32
On the basis of nanosecond laser flash photolysis results, it was found that n-butyltriphenylborate anion is oxidized, but diphenyliodonium cation is reduced by photoexcited sensitizer. The initiating radicals are formed in the secondary reactions. These results are in good accordance with thermodynamic parameters calculated for both electron transfer processes and given in Table 1.
Conclusions
UV-Vis irradiation from the range 300 nm to 500 nm initiates radical photopolymerization of multifunctional acrylic monomer comprising a squaraine dye and iodonium salts with distinct substitution pattern or/and alkyltriphenylborate salt. One of reasons of addition of onium salt to a bimolecular photoinitiator is the significant increase the polymerization rate which is very important in systems that need a fast cure. The negative values of free energy change for the electron transfer process suggest that in such photoinitiating systems the intermolecular electron transfer occurs. The free radicals are formed as a results of electron transfer processes between sensitizer and both coinitiators. In first, an electron transfer from photoexcited sensitizer to diphenyliodonium salt (ET1), and second an electron transfer from borate salt to sensitizer in excited state (ET2) occur. Both processes take place simultaneously but with different rates. The rate of electron transfer ET1 depends on the an electron acceptor structure and changes from 0.16 × 106 M−1 s−1 and 2.41 × 106 M−1 s−1. The photoinitiating systems composed of sensitizer and an electron donor and an electron acceptor are more efficient initiators for triacrylate polymerization under visible light than the selected commercially available photoinitiators acting at the same conditions.From our results, the studied systems: squaraine dye/borate salt/iodonium salt should be considered as remarkable photoinitiators, which is valuable for practical and industrial applications. The kinetic results also shown, that the systems composed of squaraine/diphenyliodonium salt/alkyltriphenylborate salt may be considered as an attractive alternative for UV-near visible light commercially available photoinitiators. New dye mediated photoinitiators are important for new and advanced applications, especially in the field of visible light curing.
Conflicts of interest
There are no conflicts to declare.References
- F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevée, Acc. Chem. Res., 2016, 49, 1980–1989 CrossRef CAS PubMed.
- J. Ortyl, M. Topa, I. Kamińska-Borek and R. Popielarz, Eur. Polym. J., 2019, 116, 45–55 CrossRef CAS.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- E. Hola, M. Pilch, M. Galek and J. Ortyl, Polym. Chem., 2020, 11, 480–495 RSC.
- E. Hola, J. Ortyl, M. Jankowska, M. Pilch, M. Galek, F. Morlet-Savary, B. Graff, C. Dietlin and J. Lalevée, Polym. Chem., 2020, 11, 922–935 RSC.
- F. A. Ogliari, C. Ely, C. L. Petzhold, F. F. Demarco and E. Piva, J. Dent., 2007, 35, 583–587 CrossRef CAS PubMed.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307–1315 CrossRef CAS.
- J. Kabatc, K. Kostrzewska, K. Jurek, M. Kozak, A. Balcerak and Ł. Orzeł, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 471–484 CrossRef CAS.
- Y. Lin and J. W. Stransbury, Polymer, 2003, 44, 4781–4789 CrossRef CAS.
- H. J. Timpe, S. Ulrich, C. Decker and J. P. Fouassier, Macromolecules, 1993, 26, 4560–4566 CrossRef CAS.
- J. Kirschner, J. Paillard, M. Bouzrati-Zeerelli, J. M. Becht, J. E. Klee, S. Chelli, S. Lakhdar and J. Lalevée, Molecules, 2019, 24, 2913 CrossRef CAS PubMed.
- P. Xiao, F. Dumur, B. Graff, J. Zhang, F. Morlet-Savary, D. Gigmes, J. P. Fouassier and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 567–575 CrossRef CAS.
- M. Bouzrati-Zerelli, M. Maier, C. Dietlin, F. Morlet-Savary, J. P. Fouassier, J. E. Klee and J. Lalevée, Dent. Mater., 2016, 32, 1226–1234 CrossRef CAS PubMed.
- D. Wang, P. Garra, J. P. Fouassier, B. Graff, Y. Yagci and J. Lalevée, Polym. Chem., 2019, 10, 4991–5000 RSC.
- M. Abdallah, A. Hijazi, B. Graff, J. P. Fouassier, G. Rodghiero, A. Gualandii, F. Dumur, P. G. Cozzi and J. Lalevée, Polym. Chem., 2019, 10, 872–884 RSC.
- H. Salmi, H. Tar, A. Ibrahim, C. Ley and X. Allonas, Eur. Polym. J., 2013, 49, 2275–2279 CrossRef CAS.
- H. Tar, D. S. Esen, M. Aydin, Ch. Ley, N. Arsu and X. Allonas, Macromolecules, 2013, 46, 3266–3272 CrossRef CAS.
- J. Qiu and J. Wei, J. Polym. Res., 2014, 21, 1–7 Search PubMed.
- T. Brömme, D. Oprych, J. Horst, P. S. Pinto and B. Strehmel, RSC Adv., 2015, 5, 69915–69924 RSC.
- A. Balcerak and J. Kabatc, RSC Adv., 2019, 9, 28490–28499 RSC.
- K. Kawamura, J. Schmitt, M. Barnet, H. Salmi, Ch. Ley and X. Allonas, Chem.–Eur. J., 2013, 19, 12853–12858 CrossRef CAS PubMed.
- Y. He, W. Zhou, F. Wu, M. Li and E. Wang, J. Photochem. Photobiol., A, 2004, 162, 463–471 CrossRef CAS.
- H. Yong, W. Zhou, G. Liu, L. M. Zhen and E. Wang, J. Photopolym. Sci. Technol., 2000, 13, 253–254 CrossRef CAS.
- O. I. Tarzi, X. Allonas, C. Ley and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2594–2603 CrossRef CAS.
- E. Andrzejewska, Polym. Int., 2017, 66, 366–381 CrossRef CAS.
- M. Sangermano, N. Razza and J. V. Crivello, Macromol. Mater. Eng., 2014, 299, 775–793 CrossRef CAS.
- P. Xiao, F. Dumur, J. Zhang, M. Bouzrati-Zerelli, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1806–1815 CrossRef CAS.
- J. V. Crivello and U. Bulut, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5217–5231 CrossRef CAS.
- F. Morlet-Savary, J. E. Klee, F. Pfefferkorn, J. P. Fouassier and J. Lalevée, Macromol. Chem. Phys., 2015, 216, 2161–2170 CrossRef CAS.
- K. Kostrzewska, J. Ortyl, R. Dobosz and J. Kabatc, Polym. Chem., 2017, 8, 3464–3474 RSC.
- J. Kabatc, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1575–1589 CrossRef CAS.
- J. Kabatc, K. Kostrzewska, M. Kozak and A. Balcerak, RSC Adv., 2016, 6, 103851–103863 RSC.
- P. Xiao, J. Zhanga, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- P. Xiao, F. Dumur, T. T. Bui, F. Goubard, B. Graff, F. Morlet-Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, ACS Macro Lett., 2013, 2, 736–740 CrossRef CAS.
- S.-Y. Park, K. Jun and S.-W. Oh, Bull. Korean Chem. Soc., 2005, 26, 428–432 CrossRef CAS.
- R. J. Damico, Org. Chem., 1964, 29, 1971 CrossRef CAS.
- J. Kabatc, J. Ortyl and K. Kostrzewska, RSC Adv., 2017, 7, 41619–41629 RSC.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- M. D. Goodner and C. N. Bowman, Macromolecules, 1999, 32, 6552–6559 CrossRef CAS.
- E. Frick, H. A. Ernst, D. Voll, T. J. A. Wolf, A.-N. Unterreiner and C. Barner-Kowollik, Polym. Chem., 2014, 5, 5053–5068 RSC.
- F. G. Bordwell and T.-Y. Lynch, J. Am. Chem. Soc., 1989, 111, 7558–7562 CrossRef CAS.
- M. L. Coote, C. Y. Lin and H. Zipse, The stability of carbon-centered radicals, in Carbon-Centered Free Radicals and Radical Cations: Structure, Reactivity, and Dynamics, ed. M. D. E. Forbes, John Wiley & Sons, Inc., Publication, 2010, pp. 83–104 Search PubMed.
- F. Sun, N. Zhang, J. Nie and H. Du, J. Mater. Chem., 2011, 21, 17290–17296 RSC.
- B. R. Nayak and L. J. Mathias, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5661–5670 CrossRef CAS.
- J. Kabatc, K. Kostrzewska and K. Jurek, RSC Adv., 2016, 6, 74715–74725 RSC.
| This journal is © The Royal Society of Chemistry 2020 |