
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStöber synthesis of salen-formaldehyde resin polymer- and carbon spheres with high nitrogen content and application of the corresponding Mn-containing carbon spheres as efficient electrocatalysts for the oxygen reduction reaction†
Tahereh Hosseinzadeh Sanatkara,
Alireza Khorshidi *a,
Rouhollah Yaghoubib,
Esmail Sohoulic and
Jamaladin Shakerid
*a,
Rouhollah Yaghoubib,
Esmail Sohoulic and
Jamaladin Shakerid
aDepartment of Chemistry, Faculty of Sciences, University of Guilan, Rasht, Guilan 41335-1914, Iran. E-mail: khorshidi@guilan.ac.ir
bDepartment of Chemistry, Faculty of Science, Tarbiat Modares University, Tehran, Iran
cYoung Researchers and Elites Club, Science and Research Branch, Islamic Azad University, Tehran, Iran
dDepartment of Chemistry, Isfahan University of Technology, Isfahan, 84156-83111, Iran
First published on 23rd July 2020
Abstract
Salen-formaldehyde (SF) resin polymer spheres were synthesized by the Stöber method from 4,4′-dihydroxysalen (N,N′-bis-(4-hydroxysalicylidene)-ethylenediamine; a tetradentate N2O2 Schiff base ligand) and formaldehyde. The salen precursor was prepared by condensation of ethylenediamine with 2,4-dihydroxybenzaldehyde in methanol. The SF resin colloidal spheres were also prepared by using Pluronic F127 and ammonia as a porogenic agent and catalyst, respectively (SF-P). In addition, corresponding Mn(II)-coordinated polymer spheres of the SF-P were synthesized (SF-P-Mn(II)). Corresponding monodispersed carbon spheres of all of the abovementioned samples were also obtained by pyrolysis technique. All of the products were characterized with conventional microscopic and spectroscopic techniques, as well as other physical methods such as BET analysis. It was found that carbonization of the SF resin spheres results in carbon spheres with specific surface areas in the range of 499–528 m2 g−1 and average pore sizes in the range of 2.58–3.08 nm. Nitrogen content of the SF-MWHT (obtained hydrothermally in a methanol/water mixture), and SF-P-C@Mn (obtained from carbonization of SF-P-Mn(II)) samples were as high as 27.5 wt% and 35.02 wt%, respectively. Finally, a glassy carbon electrode (GCE) modified with SF-P-C@Mn (SF-P-C@Mn/GCE) was prepared and its electrocatalytic activity was evaluated for oxygen reduction reaction (ORR) by linear sweep voltammetry (LSV). The LSV results showed that the SF-P-C@Mn/GCE has a higher current density and a lower negative potential in the ORR compared to GCE.
Introduction
Polymer and carbon spheres have attracted a myriad of research interest due to their interesting properties including high surface areas, electric conductivity, thermal stability, and biocompatibility. Their application has been extended to various fields such as catalyst preparation, separation, adsorption, energy storage/conversion, and biomedical engineering.1–6 In the past years, spherical carbon micro- and nanoparticles have been at the center of interest because of their unique features.7 Various strategies have been reported for the preparation of polymer colloids and their derived carbon-based nanomaterials.8–10Carbon nanospheres have been synthesized by several methods such as chemical vapor deposition (CVD), nanocasting with silica spheres as hard templates, hydrothermal carbonization of sugars, Stöber synthesis, soft-templating strategies via organic–organic self-assembly, etc.11 The well-known Stöber method was first reported in 1968 for the synthesis of colloidal silica spheres via controlled hydrolysis and condensation of Si-containing compounds such as tetraethyl orthosilicate (TEOS) with ammonia as a catalyst in aqueous alcoholic solution.12 Later, Liu et al. reported a modification to the Stöber synthesis for preparation of resorcinol formaldehyde (RF) polymer spheres with uniform and controllable particle size on submicrometer scale, and subsequent conversion of these spheres into carbon spheres via pyrolysis.13 Particle size of the resultant polymer- or carbon spheres can be controlled by varying the experimental parameters, including the alkyl chain of the alcohol, alcohol/water ratio, the amount of ammonia, formaldehyde, RF precursor, and so on.13 Resorcinol formaldehyde (RF) resins are interesting precursors which can be used to prepare highly porous carbon materials.14,15 On the other hand, various polymeric precursors, such as glucose,16 dopamine,17 tannic acid,18 phenol-formaldehyde resin19 and conducting polymers20 etc., have been successfully used to synthesize new uniform nanospheres, and provided new opportunities in the research area of colloidal and carbon spheres. The scope of applications of carbon materials have been made even more extensive by heteroatom-doping.21,22 Doping heteroatoms such as nitrogen, sulfur and phosphorus, even in small quantities in the carbon framework, can lead to significant enhancements in the performance of polymer colloids and their derived carbon-based nanomaterials.23,24 Nitrogen is the most widely used heteroatom that can be doped directly into the polymer colloids.
Schiff bases are a group of compounds that contain nitrogen atoms in their structure. They are characterized by an imine or azomethine group (–RC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), and are generally synthesized by condensation of primary amines with carbonyl compounds.25 They form an important class of ligands and have gained considerable attention due to their diverse applications such as catalysis,26 electrochemistry,27 bioscience,28 optics,29 host–guest chemistry,30 and molecular recognition.31 Moreover, Schiff bases have been successfully used to remove heavy metals selectively from aqueous media.32 Among Schiff bases, salen-type compounds are an interesting class of the tetradentate ligands with four donor sites (N2O2).33 As shown in Scheme 1, the 4,4′-dihydroxysalen ligand provides an ideal environment for equatorial coordination of various transition metals. While synthesis of polymer spheres with high nitrogen content is highly desirable for specific applications, there are few reports on preparation of such monodisperse polymer spheres.22,34
N–), and are generally synthesized by condensation of primary amines with carbonyl compounds.25 They form an important class of ligands and have gained considerable attention due to their diverse applications such as catalysis,26 electrochemistry,27 bioscience,28 optics,29 host–guest chemistry,30 and molecular recognition.31 Moreover, Schiff bases have been successfully used to remove heavy metals selectively from aqueous media.32 Among Schiff bases, salen-type compounds are an interesting class of the tetradentate ligands with four donor sites (N2O2).33 As shown in Scheme 1, the 4,4′-dihydroxysalen ligand provides an ideal environment for equatorial coordination of various transition metals. While synthesis of polymer spheres with high nitrogen content is highly desirable for specific applications, there are few reports on preparation of such monodisperse polymer spheres.22,34
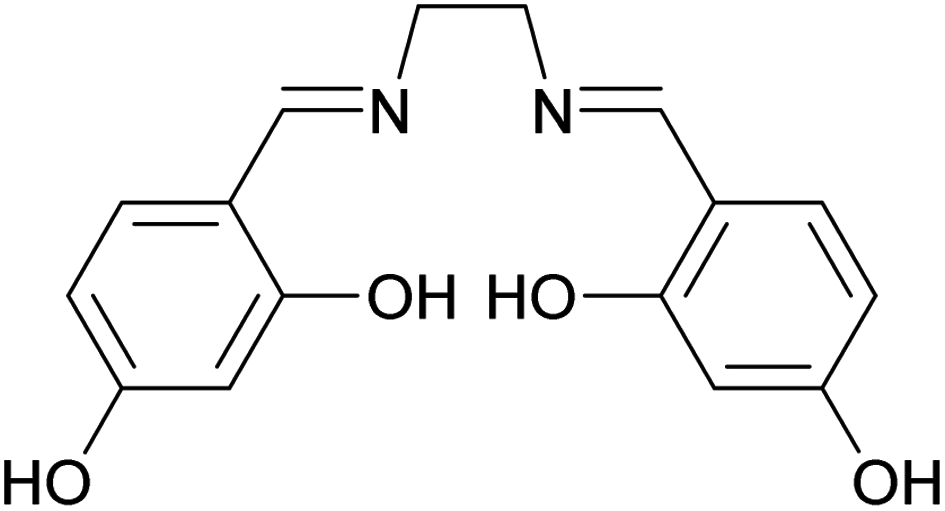 | ||
| Scheme 1 Molecular structure of 4,4′-dihydroxysalen. | ||
Carbon nanospheres have been recognized as promising electrocatalysts for the oxygen reduction reaction (ORR).13 N-doped mesoporous carbon nanospheres exhibit unique features, including enhanced dissociation of O2 within the pores, and improved wettability and charge/spin density on the carbon, to act as an electrode for the ORR.35 Moreover, decoration of the carbon nanospheres with electroactive metal nanoparticles can obviously enhance the ORR capacity.18
Herein, we report the synthesis of monodisperse salen-formaldehyde (SF) resin spheres by Stöber method using 4,4′-dihydroxysalen and formaldehyde as precursors, and Pluronic F127 as a template and porogenic agent. In addition, the corresponding polymeric metal complex spheres of the SF resin were obtained by deprotonation of the tetradentate (N2O2) salen moieties and coordination with Mn(II) by taking advantage of the chelating effect. Moreover, pyrolysis technique was used to prepare carbon spheres of the aforementioned resin polymers. Finally, Mn nanoparticles-loaded carbon spheres were successfully used as an electrocatalyst for the ORR process.
Experimental
Materials and methods
All of the chemicals including 2,4-dihydroxybenzaldehyde and ethylenediamine were purchased from Merck (http://www.merck.com) or Sigma-Aldrich (http://www.sigmaaldrich.com) Companies.All solvents and reagents were of analytical grade and used without further purification. A Heraeus CHN–O-Rapid elemental analyzer was used for elemental analyses. FT-IR spectra were recorded on a Bruker Alpha spectrophotometer using KBr pellets over the range of 4000–400 cm−1. UV-Vis spectra were recorded on a Rayleigh UV-1800 spectrophotometer. 1H NMR spectra were recorded in DMSO-d6 on a Bruker Avance III 300 MHz spectrometer at 298 K using tetramethylsilane as an internal standard. X-ray powder diffraction (XRD) data were acquired on a Philips PW1730 diffractometer with Cu Kα radiation (λ = 1.5406 Å). Field emission scanning electron microscopy (FESEM) images of the synthesized samples were obtained using a Tescan Mira III scanning electron microscope (Tescan, Czech Republic). A layer of Au was used for all samples before imaging. Energy dispersive X-ray technique (EDX) was used for surface elemental analysis. TEM imaging was performed on a Zeiss-EM10C microscope at an accelerating voltage of 100 kV. Brunauer–Emmett–Teller (BET) analyses were performed at 77 K on a Belsorp mini II gas adsorption analyzer, Japan. All of the samples were degassed at 200 °C for 2 h before BET analysis. X-ray photoelectron spectroscopy (XPS) analysis was performed by Gamma data-scienta ESCA 200 hemispherical analyzer equipped with an Al Kα (1486.6 eV) X-ray source.
Electrochemical measurements
All of the electrochemical measurements were carried out on a potentiostat/galvanostat of the type Autolab PGSTAT302N (Eco Chemie B. V., Switzerland) driven by the Nova 2.1.2 software. A conventional three-electrode setup including a modified or unmodified GCE working electrode, an Ag/AgCl (3.00 M KCl) reference electrode, and a Pt wire auxiliary electrode (Azar Electrode, Iran) was used. Some electrochemical techniques such as cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and amperometry were used in the experiments. The EIS measurements were performed at a frequency range of 10.0 kHz to 10 MHz, and 0.1 M KOH was utilized as a redox probe.Syntheses
Synthesis of the salen compound
The 4,4′-dihydroxysalen (N,N′-bis-(4-hydroxysalicylidene)-ethylenediamine) was synthesized by condensation of ethylenediamine with 2,4-dihydroxybenzaldehyde in methanol. Briefly, ethylenediamine (0.33 mL, 5.0 mmol) was added dropwise to a solution of 2,4-dihydroxybenzaldehyde (1.38 g, 10.0 mmol) in 100 mL of methanol at room temperature. The reaction mixture was then refluxed for 3 h with continuous stirring. After cooling down to room temperature, a yellow precipitate was filtered off and washed with methanol. Yield: 88%. Anal. calcd for C16H16N2O4 (MW = 300.31 g mol−1): C, 63.99; H, 5.37; N, 9.33. Found: C, 63.87; H, 5.45; N, 9.41%. IR (KBr pellet, cm−1): 3416 (s, νOH), 1639 (s, νCN), 1234 (m, νPh–O). UV-Vis: λmax (nm) (ε, M−1 cm−1) (DMSO): ∼280 (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750), 308 (21100), 390 (2100). 1H NMR (DMSO-d6, 300 MHz, ppm); δ: 13.58 (br, 2H7), 9.80 (br, 2H5), 8.37 (s, 2H2), 7.18 (d, 2H3, J = 8.4 Hz), 6.28 (dd, 2H4, J = 8.4 and 2.4 Hz), 6.27 (d, 2H6, J = 2.4 Hz), 3.79 (s, 4H1).
750), 308 (21100), 390 (2100). 1H NMR (DMSO-d6, 300 MHz, ppm); δ: 13.58 (br, 2H7), 9.80 (br, 2H5), 8.37 (s, 2H2), 7.18 (d, 2H3, J = 8.4 Hz), 6.28 (dd, 2H4, J = 8.4 and 2.4 Hz), 6.27 (d, 2H6, J = 2.4 Hz), 3.79 (s, 4H1).
Synthesis of the SF resin polymer spheres
Monodisperse SF resin spheres were synthesized by using 4,4′-dihydroxysalen and formaldehyde as precursors. In a typical synthesis, ammonia solution (25 wt%, 0.1 mL) was mixed with 16 mL of absolute methanol and 12 mL of deionized water and stirred for 1 h at 30 °C. Then, 4,4′-dihydroxysalen (0.55 g) was added and magnetically stirred for 30 min. Afterwards, 0.28 mL of 37% formaldehyde solution was added dropwise to the above solution. After few minutes, the solution became cloudy and stirring was continued for 24 h at the same temperature. A light-yellow solid product was obtained after centrifugation followed by air-drying at 100 °C, which was denoted as SF-MW (73% yield).The same procedure was repeated and right before separation of the product, a hydrothermal process was applied in a Teflon-lined autoclave at 100 °C for 24 h under a static condition. After cooling down to room temperature, the dark-yellow SF resin polymer spheres were separated by centrifugation and air-dried at 100 °C for 24 h. The product was denoted as SF-MWHT (70% yield).
In addition, hydrothermally processed SF resin polymer spheres were obtained in the same manner in 28 mL of absolute methanol (instead of a 16:12 mixture of MeOH:H2O mixture), which were denoted as SF-MHT (76% yield).
Pluronic F127-directed synthesis of the SF resin polymer spheres (SF-P)
The SF-P spheres were synthesized by the abovementioned precursors and triblock copolymer Pluronic F127. The overall process was similar to that described above section. First, ammonia solution (25 wt%, 0.1 mL) was mixed with 16 mL of absolute methanol and 12 mL of deionized water and stirred for 1 h at 30 °C. Then, 4,4′-dihydroxysalen (0.55 g) was added and magnetically stirred for 30 min. Afterwards, 0.27 g of triblock copolymer Pluronic F127 was added and stirred for 30 min. In the next step, 0.28 mL of 37% formaldehyde solution was added dropwise. After few minutes, the solution became cloudy and stirring was continued for 24 h at the same temperature. The light-yellow reaction mixture was then transferred into a Teflon-lined autoclave and heated at 100 °C for 24 h. After cooling down to room temperature, a dark-yellow solid product was obtained after centrifugation followed by air-drying at 100 °C, which was denoted as SF-P (68% yield).Synthesis of the Mn(II)-coordinated SF-P spheres (SF-P-Mn(II))
The overall process for the synthesis of SF-P-Mn(II) was similar to that described in above sections. First, ammonia solution (25 wt%, 0.1 mL) was mixed with 16 mL of absolute methanol and 12 mL of deionized water and stirred for 1 h at 30 °C. Then, 4,4′-dihydroxysalen (0.55 g) was added and magnetically stirred for 30 min. Afterwards, 0.27 g of triblock copolymer Pluronic F127 was added and stirred for 30 min. In the next step, 0.28 mL of 37% formaldehyde solution was added dropwise. After few minutes, the solution became cloudy and stirring was continued for 24 h at the same temperature. Then a methanolic solution of Mn(NO3)2·4H2O (2 mL, 0.1 M) was added dropwise to the reaction mixture and stirred for 8 h. It should be mentioned that after addition of the Mn(II) salt, color of the solution became dark red. Then the reaction mixture was transferred into a Teflon-lined autoclave and heated at 100 °C for 24 h. After cooling down to room temperature, the polymeric Mn(II) complex was collected by centrifugation and dried at 100 °C for 24 h (67% yield).Carbonization of the SF resin polymer spheres
For carbonization, the precursors were transferred in a quartz boat into a tube furnace and heated under N2 atmosphere. As shown in Fig. 1, first, the temperature was raised from room temperature to 350 °C with a heating rate of 5 °C min−1 and kept at 350 °C for 2 h. Next, the temperature was raised to 600 °C with a 5 °C min−1 heating rate and maintained at 600 °C for 4 h. Finally, the samples were cooled down to room temperature. The obtained samples from SF-MWHT, SF-P, and SF-P-Mn(II) were denoted as SF-C, SF-P-C, and SF-P-C@Mn, respectively.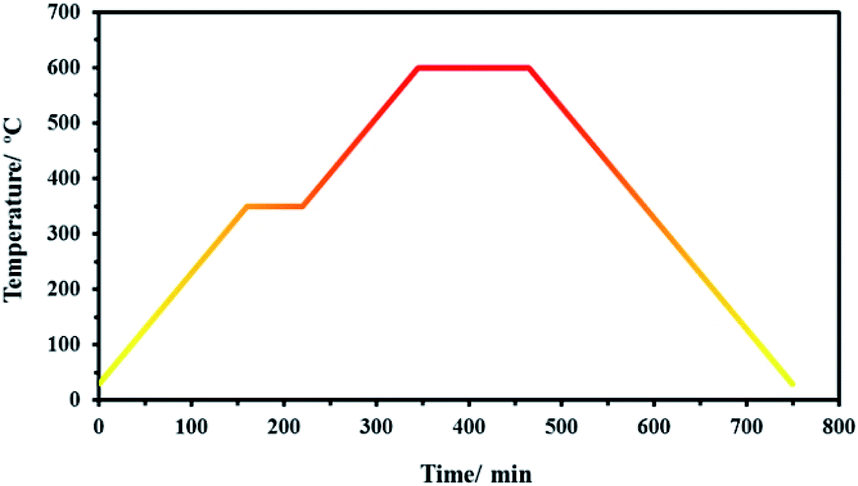 | ||
| Fig. 1 The heating program used for carbonization. | ||
Preparation of the modified carbon paste electrode
To prepare the SF-P-C@Mn-modified glassy carbon electrode, surface of the electrode was polished by alumina powder, washed with water, and sonicated in deionized water to eliminate any surface contamination. Then, 1.0 mg of the SF-P-C@Mn was mixed with 1.0 mL of water in a test tube and irradiated in an ultrasonic bath for 1 h. Then, 5 μL of this mixture was placed on the electrode in an oven at 50 °C to prepare the SF-P-C@Mn-modified glassy carbon electrode which was denoted as SF-P-C@Mn/GCE. The SF-C/GCE and SF-P-C/GCE were prepared in the same way.Results and discussion
Spectral characterization of the salen compound
In the FT-IR spectrum of 4,4′-dihydroxysalen (Fig. 2a), broad band of the phenolic O–H stretching (νO–H) was appeared at 3416 cm−1.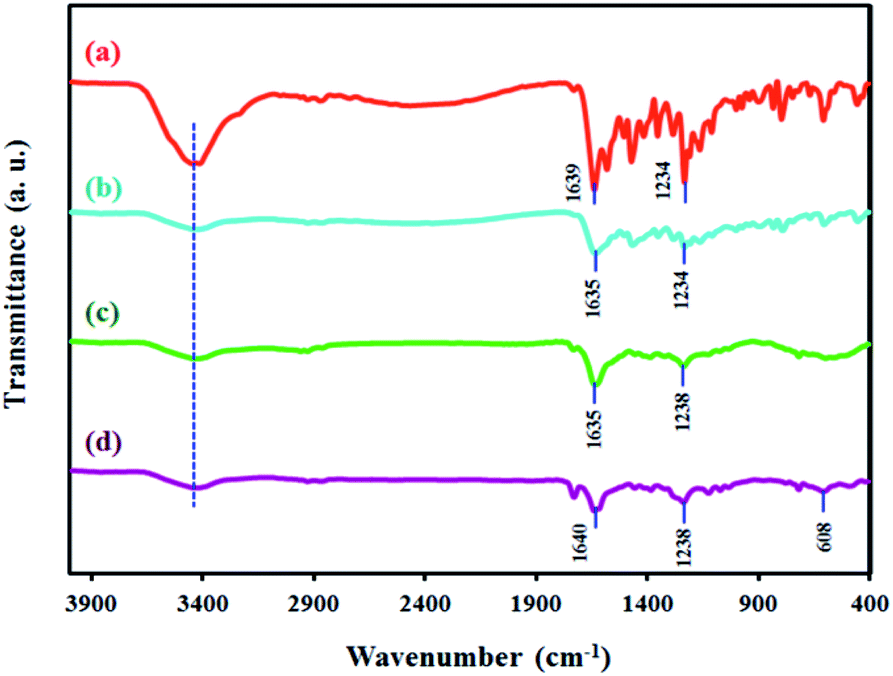 | ||
| Fig. 2 FT-IR spectra of (a) 4,4′-dihydroxysalen, (b) SF-MWHT, (c) SF-P-C, and (d) SF-P-C@Mn. | ||
Strong band of the azomethine moiety (νCN) was observed at 1639 cm−1, and that of the C–O stretching (νPh–O) was observed at 1234 cm−1.
Electronic absorption spectrum of the 4,4′-dihydroxysalen was recorded at room temperature in DMSO (2 × 10−5 M). As shown in Fig. 3, the UV-Vis spectrum of 4,4′-dihydroxysalen has three absorption bands at 280, 308 and 390 nm. The highly intense bands at 280 and 308 nm are assigned to π → π* and n → π* transitions of the salen, and the broad band at about 390 nm is attributed to intraligand charge-transfer.36
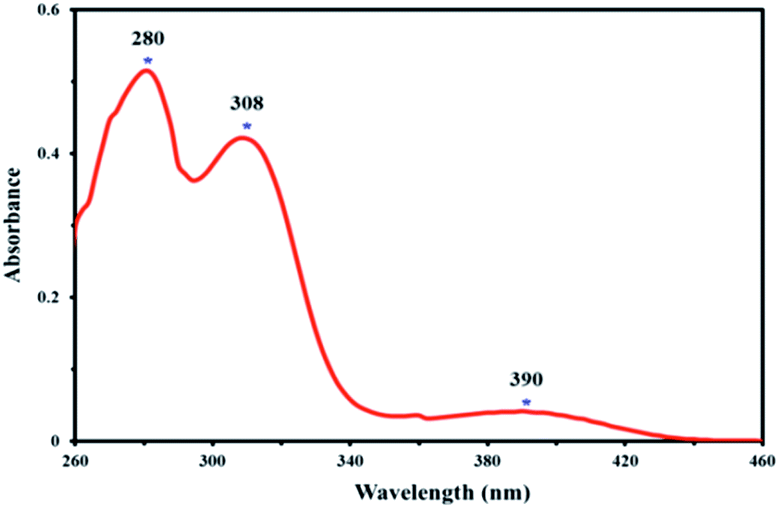 | ||
| Fig. 3 UV-Vis spectrum of 4,4′-dihydroxysalen in DMSO (2 × 10−5 M) at room temperature. | ||
1H NMR spectra of the salen Schiff base showed broad signals for phenolic OH7 and OH5 moieties at 13.58 and 9.80 ppm, respectively (Fig. 4). The aromatic protons resonated in the range of 6.18–7.18 ppm and unambiguously assigned according to their coupling constants (see Experimental). The expected singlet for the proton of the imine moiety (H2) was observed at 8.37 ppm, and the aliphatic protons (H1) were observed at 3.79 ppm as a singlet.
 | ||
| Fig. 4 1H NMR spectrum of 4,4′-dihydroxysalen in DMSO-d6. | ||
Characterization of the prepared resin polymer- and carbon spheres
A schematic illustration for preparation of the SF, SF-P and SF-P-Mn(II) resin polymer spheres and their corresponding carbon spheres is shown in Fig. 5. In a typical Stöber method, stable emulsion droplets are formed without any surfactant through hydrogen bonding interactions between water, methanol and 4,4′-dihydroxysalen Schiff base. Then, polymerization of 4,4′-dihydroxysalen and formaldehyde takes place inside the droplets by ammonia catalysis. During this process, 4,4′-dihydroxysalen, formaldehyde and ammonia react to form numerous condensation compounds such as hydroxymethyl substituted species that cross-link together and result in the formation of uniform colloidal SF polymer spheres. The ammonium cations not only accelerate the polymerization reaction between salen and formaldehyde in the emulsion, but also supply the positive charges distributing on the outer surface of polymer spheres that prevent the aggregation of the resin spheres.13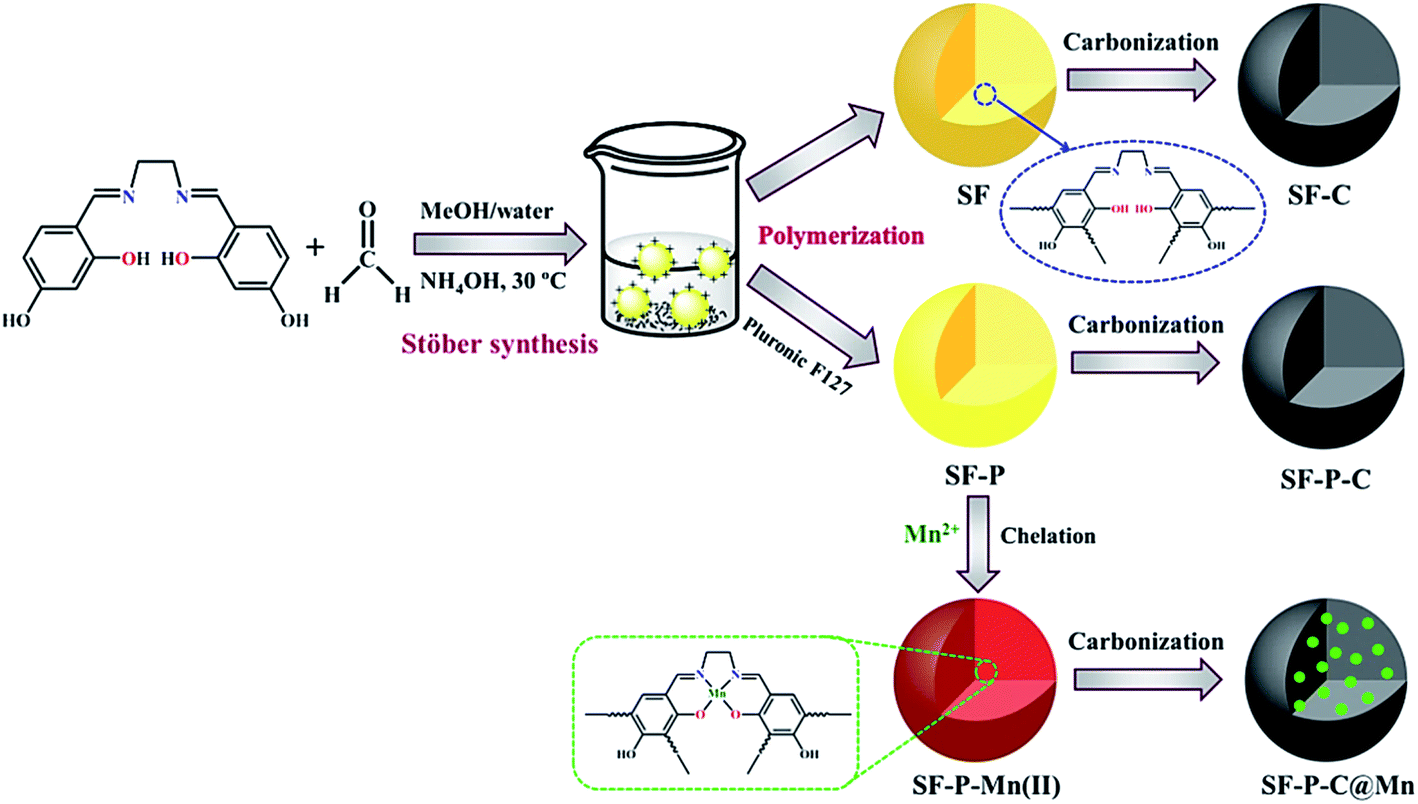 | ||
| Fig. 5 A schematic illustration for preparation of the SF, SF-P and SF-P-Mn(II) samples and their corresponding carbonization products. | ||
SEM images of the SF resin spheres before and after hydrothermal treatment (SF-MW and SF-MWHT, respectively) are shown in Fig. 6a and b.
 | ||
| Fig. 6 SEM images of (a) SF-MW, (b) SF-MWHT, and (c) SF-MHT resin polymer spheres. | ||
It is clear that the colloidal spheres had irregular shapes before hydrothermal treatment (Fig. 6a) and they became more spherical with smooth surfaces and reduced roughness after hydrothermal processing (Fig. 6b).37 It was also found that the solvent has an impact on the size of the obtained spheres. From Fig. 6c it is clear that in the case of the SF-MHT colloidal spheres, although absolute methanol as a solvent leads to the production of smoother spherical particles, instead it results in an increase in their size.
N2 adsorption–desorption isotherms of the SF samples are shown in Fig. 7. This study provided valuable information on the specific surface area and porosity of the prepared resin polymers. The SF resin spheres before and after hydrothermal treatment showed a type IV N2-sorption isotherm with distinct hysteresis loops in the range of p/p0 from 0.85 to 1.0, indicative of porous adsorbents with pores in the range of 1.5–100 nm. The SF-MHT sample, on the other hand, showed a type II N2-sorption isotherm, characteristic of powders with diameters exceeding micropores. The corresponding Barrett–Joyner–Halenda (BJH) curves (insets of Fig. 7), clearly indicate the presence of micro-, meso, and macropores in the SF-MW and SF-MWHT samples, and micro- and mesopores in the SF-MHT sample.
 | ||
| Fig. 7 N2 adsorption–desorption isotherms of (A) SF-MW, (B) SF-MWHT, and (C) SF-MHT samples. The insets show the corresponding BJH curves. | ||
Table 1 shows specific surface area (SBET), total pore volume (VTP) and mean pore diameter (DMP) values for each of the samples.
| Sample | SBET (m2 g−1) | VTP (cm3 g−1) | DMP (nm) |
|---|---|---|---|
| SF-MW | 10.88 | 0.11 | 41.21 |
| SF-MWHT | 15.78 | 0.18 | 46.72 |
| SF-MHT | 4.08 | 0.01 | 11.74 |
It is clear that the solvent system and hydrothermal treatment have significant positive effects on the morphology and physicochemical properties of the SF resin polymer spheres, which in turn may appear in their corresponding carbon spheres.
Based on the abovementioned results, the SF-MWHT sample was selected for further studies.
Nitrogen content of the SF-MWHT resin polymer spheres was determined by energy dispersive X-ray (EDX) analysis (Fig. 8). As the inset of Fig. 8a shows, nitrogen content of the SF-MWHT sample was about 27.5 wt%. EDX-mapping (Fig. 8b), on the other hand, revealed homogeneous distribution of the nitrogen atoms throughout the sample.
 | ||
| Fig. 8 (a) EDX analysis for the SF-MWHT resin polymer spheres, and (b) the corresponding elemental mapping. The inset shows the elemental analysis data. | ||
FT-IR spectrum of the SF-MWHT sample is shown in Fig. 2b and exhibits the anticipated bands for νO–H, νCN, and νPh–O at 3420, 1635, and 1234 cm−1, respectively.
Fig. 9 shows the SEM images of the carbonization products SF-C, SF-P-C and SF-P-C@Mn. It is clearly seen that the obtained carbon spheres have regular spherical shapes with smooth surfaces.
 | ||
| Fig. 9 SEM images of (a and d) SF-C, (b and e) SF-P-C, and (c and f) SF-P-C@Mn. The insets show the corresponding particle size distribution curves. | ||
After carbonization, however, some defects were observed on the surface of carbon particles as a result of shrinkage during the calcination process.18 It is noteworthy that the use of Pluronic F127 as a template resulted in an increase in particle size from about 140 nm for the SF-C to about 188 nm for the SF-P-C samples, respectively. The decrease in particle size to about 106 nm in the SF-P-C@Mn sample may be related to the presence of manganese species.
TEM imaging further demonstrated that the carbonization products were monodispersed and homogeneously spherical (Fig. 10).
 | ||
| Fig. 10 TEM images of (a, d) SF-C, (b, e) SF-P-C, and (c, f) SF-P-C@Mn. | ||
To further characterize the obtained carbon spheres, EDX analysis was performed. The results for the SF-C and SF-P-C samples are shown in Fig. S1 and S2,† and clearly indicate homogeneous distribution of the C, N, and O atoms in their structure. EDX spectrum of the SF-P-C@Mn sample is shown in Fig. 11a and confirms the presence of manganese in the carbonized spheres.
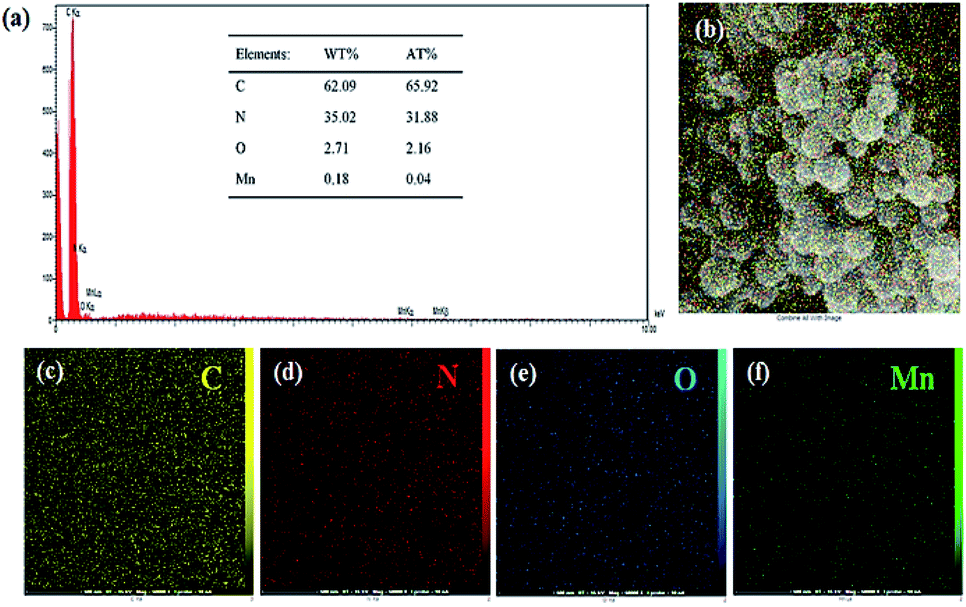 | ||
| Fig. 11 (a) EDX spectrum of the SF-P-C@Mn carbon spheres, and (b–f) the corresponding mapping images. The inset shows the elemental analysis data. | ||
Nitrogen and manganese content of the SF-P-C@Mn carbon spheres were found to be 35 and 0.18 wt%, respectively (table inset of Fig. 11a). Also, homogeneous distribution of the C, N, O and Mn constituents is evident from Fig. 11b–f.
Porosity of the carbonized samples was also studied. N2 adsorption–desorption isotherms of the carbonized samples are shown in Fig. 12. While the SF-C sample exhibited a type II isotherm, the other two showed a type IV isotherm with distinct hysteresis loops in the range of p/p0 from 0.3 to 1.0. Physico-chemical properties of the carbonized samples are listed in Table 2. Clearly, specific surface area (SBET), total pore volume (VTP), and mean pore diameter (DMP) of each sample was improved after calcination.
 | ||
| Fig. 12 N2 adsorption–desorption isotherms of (A) SF-C, (B) SF-P-C, and (C) SF-P-C@Mn carbon spheres. The insets show the corresponding BJH curves. | ||
| Sample | SBET (m2 g−1) | VTP (cm3 g−1) | DMP (nm) |
|---|---|---|---|
| SF-C | 499 | 0.33 | 2.54 |
| SF-P-C | 516.83 | 0.35 | 2.78 |
| SF-P-C@Mn | 528.49 | 0.41 | 3.08 |
In the case of the SF-C sample, a significant improvement in the BET surface area from 15.78 to 499 m2 g−1 was observed which can be attributed to the loss of water, NH4+ and other functional groups during the carbonization process.38
Total pore volume was also increased accordingly from 0.18 to 0.33 cm3 g−1. In the case of the SF-P-C and SF-P-C@Mn samples, on the other hand, removal of the template and complex formation effects may contribute to further enhance the above mentioned properties.11 The corresponding BJH curves (insets of Fig. 12), clearly indicate the presence of micro- and mesopores in the structure of SF, and micro-, meso-, and macropores in the SF-P and SF-P-C@Mn carbon spheres.
XRD patterns for the SF-C, SF-P-C and SF-P-C@Mn samples are shown in Fig. 13. As shown, the obtained carbon spheres are amorphous. It can be concluded that the use of Pluronic F127 or complexation with Mn(II) does not lead to crystallinity of the obtained carbon spheres. Broad diffraction peaks at 2 theta angles about 23°, can be ascribed to the (002) plane of amorphous carbon.22
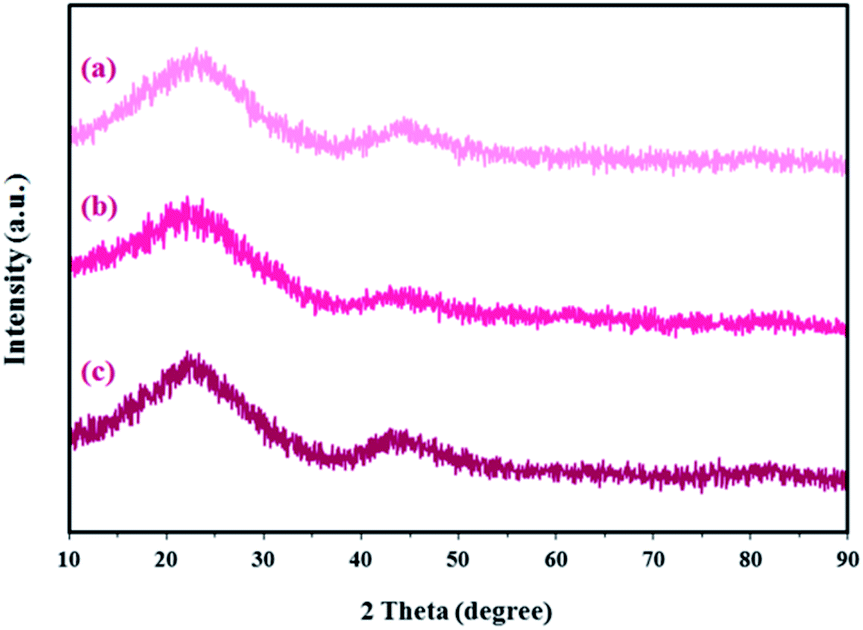 | ||
| Fig. 13 X-ray diffraction patterns for the (a) SF-C, (b) SF-P-C, and (c) SF-P-C@Mn samples. | ||
Fig. 2c and d show the FT-IR spectra of the SF-P-C and SF-P-C@Mn samples. Although most of the characteristic peaks of the parent scaffolds were present, band intensities were decreased to some extent. Also, in the FT-IR spectrum of SF-P-C@Mn (Fig. 2d), a new peak was appeared at 608 cm−1 which can be attributed to the Mn–O vibrations.39
XPS spectra in Fig. 14a–c show that C 1s, N 1s and O 1s characteristic peaks exist in SF-C, SF-P-C and SF-P-C@Mn, and Mn 2p is present in SF-P-C@Mn, which is in good agreement with EDX-mapping results.
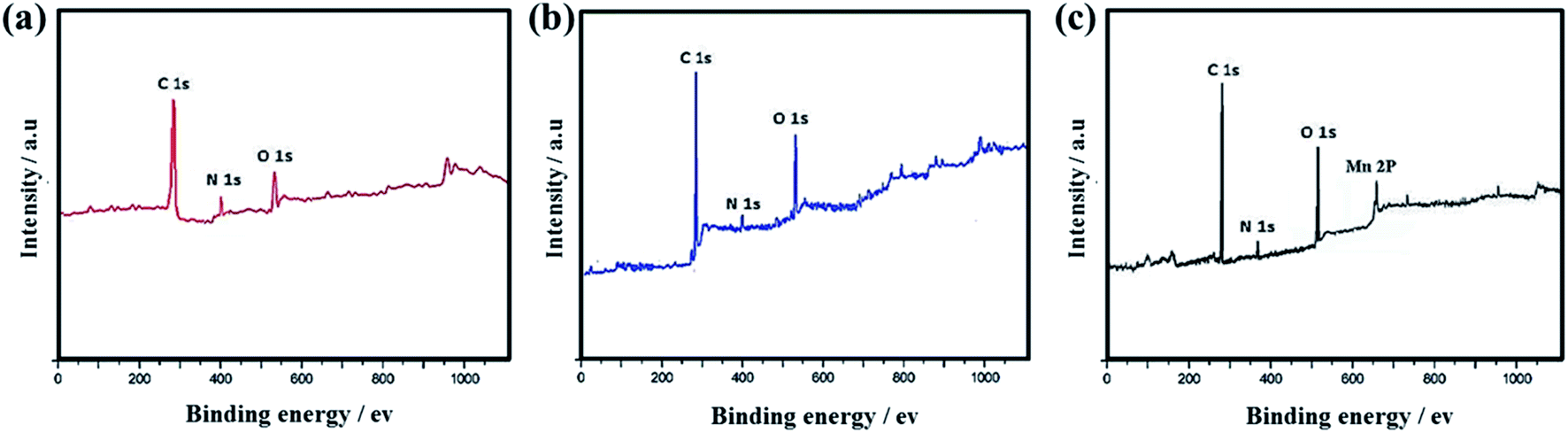 | ||
| Fig. 14 XPS survey spectra of (a) SF-C, (b) SF-P-C and (c) SF-P-C@Mn carbon spheres. | ||
Electrocatalytic performance
Eventually, catalytic activity of the SF-P-C@Mn sample was evaluated in the oxygen reduction reaction. LSVs of the GCE and MGCE in 0.1 M KOH are compared in Fig. 15. As shown, the ORR was carried out very late and very slowly at the surface of GCE, which indicates the lack of electrocatalytic performance.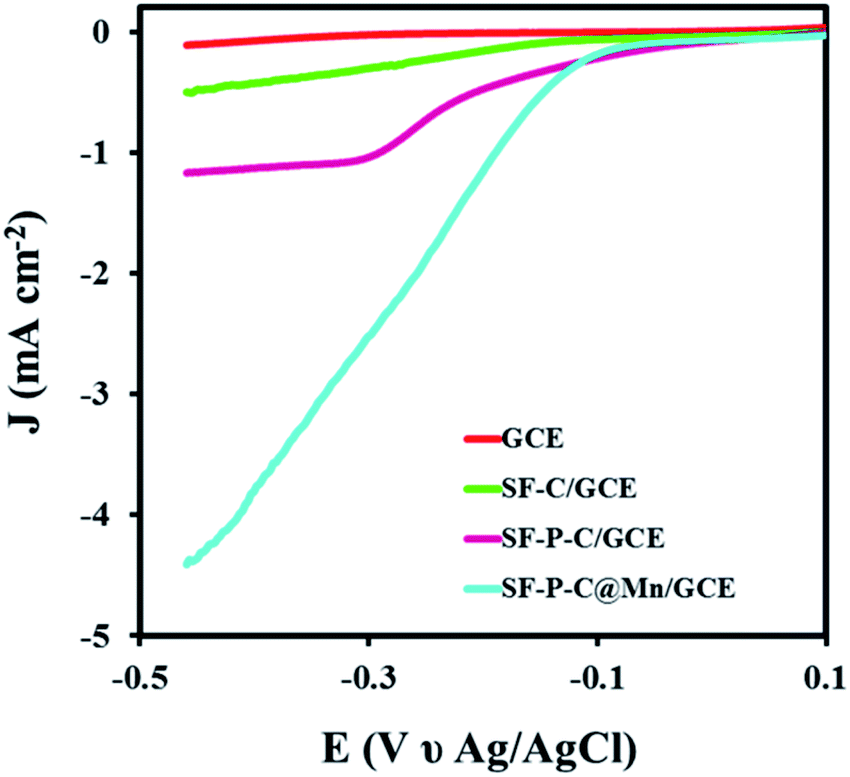 | ||
| Fig. 15 LSVs of GCE, SF-C/GCE, SF-P-C/GCE, and SF-P-C@Mn/GCE in 0.1 M KOH at a scan rate of 0.05 mV s−1. | ||
In contrast, the ORR for SF-C/GCE, SF-P-C/GCE, and SF-P-C@Mn/GCE was performed at −0.24, −0.18, and −0.11 V, respectively. The results show that SF-P-C@Mn/GCE has the higher potential for the reaction.
The Tafel slope and the exchange current density (J0) (Fig. 16) were calculated according to the following equation:
| E = RT/αnFlnJ0 − RT/αnFlnJ |
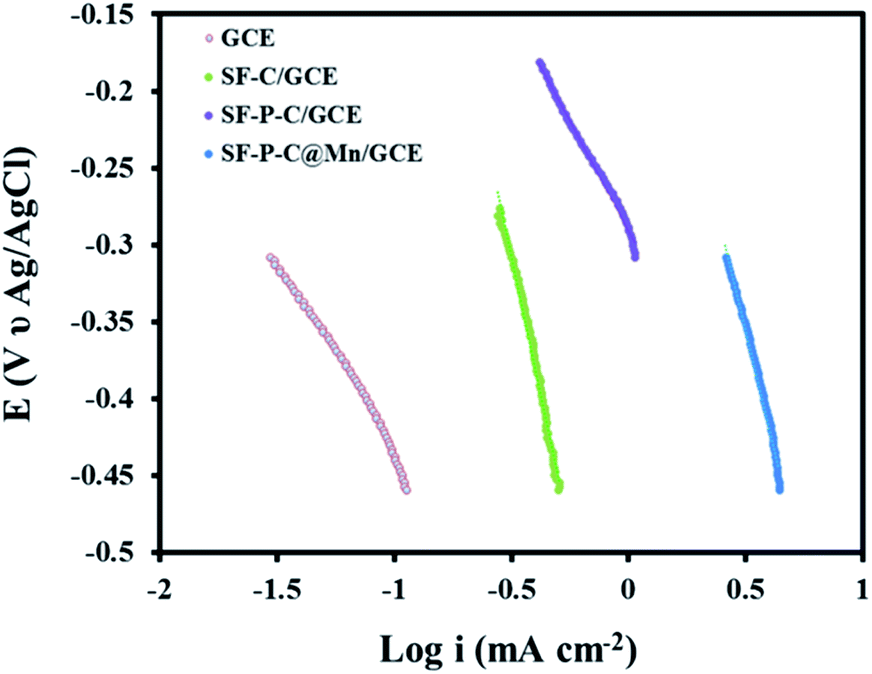 | ||
| Fig. 16 Tafel plot of GCE, SF-C/GCE, SF-P-C/GCE, and SF-P-C@Mn/GCE in 0.1 M KOH at a scan rate of 5 mV s−1. | ||
| Electrode | Tafel slope (mV dec−1) | Current density (mA cm−2) |
|---|---|---|
| GCE | 41.23 | 20.51 |
| SF-C/GCE | 73.41 | 33.29 |
| SF-P-C/GCE | 101.38 | 45.83 |
| SF-P-C@Mn/GCE | 138.12 | 70.32 |
In order to examine the resistance to charge transfer at the electrode surface during the ORR process, electrochemical impedance spectroscopy in 0.1 M KOH with a potential of −0.11 V within a frequency range of 10.0 kHz to 10 MHz with an amplitude of 10 mV AC was used. The corresponding Nyquist curves are shown in Fig. 17. As shown, resistance to charge transfer (RCT) of the SF-P-C@Mn/GCE is much lower than the SF-P-C/GCE, SF-C/GCE, and GCE which indicates high electrocatalytic performance of the modified electrode in the ORR process.
 | ||
| Fig. 17 Nyquist curves obtained from electrochemical impedance spectroscopy measurements in 0.1 M KOH. | ||
In order to determine the stability of the modified electrode in the ORR process, chronoamperometry technique was used. For this purpose, the SF-P-C@Mn/GCE was placed in 0.1 M KOH at a constant potential of −0.11 V for 3600 s, and the chronoamperogram was recorded (Fig. 18). Clearly, a constant current was obtained over the specified time period, which indicates durability and stability of the modified electrode.
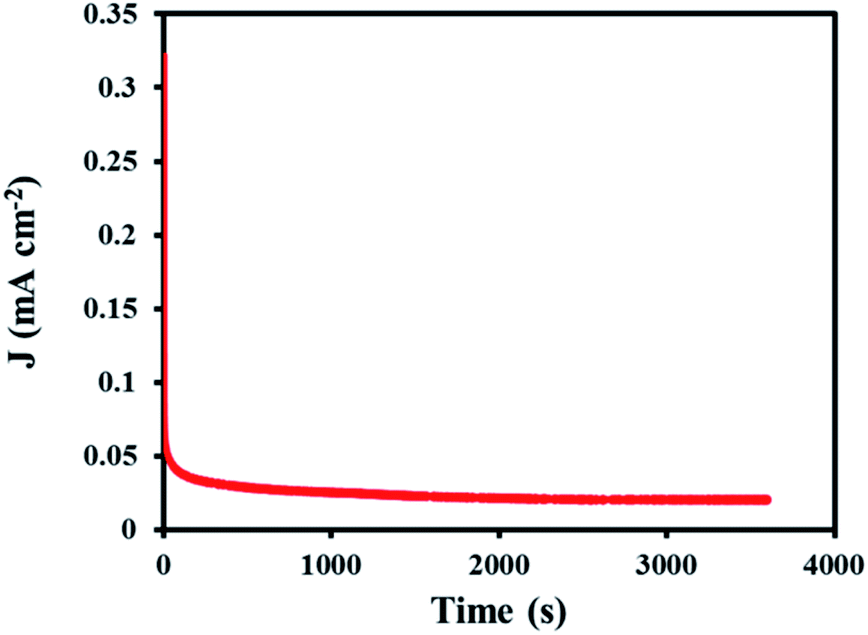 | ||
| Fig. 18 The recorded chronoamperogram for SF-P-C@Mn/GCE in 0.1 M KOH during 3600 s. | ||
Also, the stability of the SF-P-C@Mn/GCE was checked in the ORR process once a week consecutively for 4 weeks (with chronoamperometry technique). The results indicate that the desired SF-P-C@Mn/GCE surface has long-term stability and in the 4th week, only a 4.89% signal decrease was observed compared with the first week.
Conclusion
An extension of the Stöber method was used to prepare monodispersed salen-formaldehyde resin polymer- and carbon spheres with high nitrogen content. The results showed that the solvent system used has an impact on the properties of the obtained SF resin spheres. Pluronic F127 as a template and porogenic agent significantly improved the quality of the obtained resin spheres and their descendant carbon analogues. Mn(II)-coordinated polymer spheres of the SF resin were also synthesized.The SF-P-Mn(II) complex was then converted to SF-P-C@Mn spheres after carbonization under an inert atmosphere. Interesting physico-chemical properties of the obtained SF-P-C@Mn carbon particles, including a BET surface area of 528 m2 g−1 and a high nitrogen content of about 35 wt%, provided a suitable basis for modification of a glassy carbon electrode to be used in the oxygen reduction reaction. SF-P-C@Mn/GCE was successfully used as an electrocatalyst in the ORR process. The improved electrocatalytic activity may be attributed to the presence of Mn nanoparticles in the SF-P-C@Mn structure as was evident from the FT-IR and TEM analyses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of the Research Council of University of Guilan.References
- A. H. Lu, W. Schmidt, N. Matoussevitch, H. Bonnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer and F. Schuth, Angew. Chem., Int. Ed., 2004, 43, 4303–4306 CrossRef CAS PubMed.
- A. H. Lu, T. Sun, W. C. Li, Q. Sun, F. Han, D. H. Liu and Y. Guo, Angew. Chem., Int. Ed., 2011, 50, 11765–11768 CrossRef CAS PubMed.
- J. Wang, H. Liu, J. Diao, X. Gu, H. Wang, J. Rong, B. Zong and D. S. Su, J. Mater. Chem. A, 2015, 3, 2305–2313 RSC.
- T. Yang, R. Zhou, D. W. Wang, S. P. Jiang, Y. Yamauchi, S. Z. Qiao, M. J. Monteiro and J. Liu, Chem. Commun., 2015, 51, 2518–2521 RSC.
- Y. Fang, G. Zheng, J. Yang, H. Tang, Y. Zhang, B. Kong, Y. Lv, C. Xu, A. M. Asiri, J. Zi and F. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5366–5370 CrossRef CAS PubMed.
- L. Zhong, C. Anand, K. Lakhi, G. Lawrence and A. Vinu, Sci. Rep., 2015, 5, 12901–12908 CrossRef PubMed.
- W. Sun, M. Chen, S. Zhou and L. Wu, Langmuir, 2014, 30, 12011–12017 CrossRef CAS PubMed.
- J. Zhu, L. Liao, X. Bian, J. Kong, P. Yang and B. Liu, Small, 2012, 8, 2715–2720 CrossRef CAS PubMed.
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 1160–1165 CrossRef CAS.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- P. Zhang, Z. A. Qiao and S. Dai, Chem. Commun., 2015, 51, 9246–9256 RSC.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. Liu, S. Z. Qiao, H. Liu, J. Chen, A. Orpe, D. Zhao and G. Q. Lu, Angew. Chem., Int. Ed., 2011, 50, 5947–5951 CrossRef CAS PubMed.
- J. Xu, A. Wang and T. Zhang, Carbon, 2012, 50, 1807–1816 CrossRef CAS.
- A. H. Moreno, A. Arenillas, E. G. Calvo, J. M. Bermúdez and J. A. Menéndez, J. Anal. Appl. Pyrolysis, 2013, 100, 111–116 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- J. Zhao, C. L. Li and R. Liu, Nanoscale, 2018, 10, 5882–5887 RSC.
- M. Liu, C. Cai, J. Li, J. Zhao, W. Teng and R. Liu, J. Colloid Interface Sci., 2018, 528, 1–9 CrossRef CAS PubMed.
- R. Liu, F. Qu, Y. Guo, N. Yao and R. D. Priestley, Chem. Commun., 2013, 50, 478–480 RSC.
- G. Wu, Z. Chen, F. H. Garzon and P. Zelenay, ECS Trans., 2008, 16, 159–170 CrossRef CAS.
- X. Zhu, S. Wang, W. Huang, Y. Tian and X. Wang, Carbon, 2016, 105, 521–528 CrossRef CAS.
- J. C. Song, Z. Y. Lu and Z. Y. Sun, J. Colloid Interface Sci., 2014, 431, 132–138 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- X. Liu, C. Manzur, N. Novoa, S. Celedón, D. Carrillo and J. R. Hamon, Coord. Chem. Rev., 2018, 357, 144–172 CrossRef CAS.
- B. Huang, Y. Wang, S. Zhan and J. Ye, Appl. Surf. Sci., 2017, 396, 121–128 CrossRef CAS.
- Z. Wang, Y. Qin, C. Wang, L. Sun, X. Lu and X. Lu, Appl. Surf. Sci., 2012, 258, 2017–2021 CrossRef CAS.
- J. Ren, H. Xuan, W. Dai, Y. Zhu and L. Ge, Appl. Surf. Sci., 2018, 448, 609–617 CrossRef CAS.
- J. Tedim, S. Patrício, R. Bessada, R. Morais, C. Sousa, M. B. Marques and C. Freire, Eur. J. Inorg. Chem., 2006, 17, 3425–3433 CrossRef.
- L. Martínez-Rodríguez, N. A. Bandeira, C. Bo and A. W. Kleij, Chem.–Eur. J., 2015, 21, 7144–7150 CrossRef PubMed.
- Y. Xu, J. Meng, L. Meng, Y. Dong, Y. Cheng and C. Zhu, Chem.–Eur. J., 2010, 16, 12898–12903 CrossRef CAS PubMed.
- J. Wang, L. Huang, M. Xue, L. Liu, Y. Wang, L. Gao, J. Zhu and Z. Zou, Appl. Surf. Sci., 2008, 254, 5329–5335 CrossRef CAS.
- A. Erxleben, Inorg. Chim. Acta, 2018, 472, 40–57 CrossRef CAS.
- H. Tian, J. Liu, K. O'Donnell, T. Liu, X. Liu, Z. Yan, S. Liu and M. Jaroniec, J. Colloid Interface Sci., 2016, 476, 55–61 CrossRef CAS PubMed.
- T. Yang, J. Liu, R. Zhou, Z. Chen, H. Xu, S. Z. Qiao and M. J. Monteiro, J. Mater. Chem. A, 2014, 2, 18139–18146 RSC.
- D. Dey, G. Kaur, M. Patra, A. R. Choudhury, N. Kole and B. Biswas, Inorg. Chim. Acta, 2014, 421, 335–341 CrossRef CAS.
- C. Liu, J. Wang, J. Li, X. Hu, P. Lin, J. Shen, X. Sun, W. Han and L. Wang, J. Mater. Chem. A, 2016, 4, 11916–11923 RSC.
- S. Ren, M. Wang, C. Jia, C. Hao and X. Wang, Energy Technol., 2013, 1, 332–337 CrossRef CAS.
- A. Bayoudh, N. Etteyeb and F. Sediri, American Journal of Nanosciences, 2016, 2, 1–7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03815f |
| This journal is © The Royal Society of Chemistry 2020 |