
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ultrasonic-assisted synthesis of leather-derived luminescent graphene quantum dots: catalytic reduction and switch on–off probe for nitro-explosives†
Shamsa Kanwalabc,
Shanaz Jahanc and
Farukh Mansoor *a
*a
aDepartment of Chemistry, Khwaja Fareed University of Engineering and Information Technology, Rahim Yar Khan, Pakistan. E-mail: farukhmansoor.chemist@gmail.com
bCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS), Fuzhou, Fujian 350002, P. R. China
cICCBS, H.E.J Research Institute of Chemistry, University of Karachi, 75270, Karachi, Pakistan
First published on 17th June 2020
Abstract
The current research effort demonstrates the ultrasonic-assisted synthesis of highly fluorescent graphene quantum dots (GQDs) of ∼5 nm diameter. First, acid pyrolysis with ultrasonic hydrothermal co-cutting breaks down the coarse graphite into nanometric graphene sheets (GS) and graphene oxide sheets (GOS) with oxygen-rich functionalities. These functionalities were then used to break GOS into graphene oxide nanofibers (GONFs) and graphene oxide quantum dots (GOQDs). Finally, upon reduction, GOQDs lose oxygen linkages to produce fluorescent GQDs (quantum yield up to 27%). The as-developed GQDs were characterized with detailed optical and spectral studies through UV, PL, FTIR, TEM, AFM, XPS, XRD and other techniques. Notably, the synthesized GQDs were catalytically active to serve as a ratiometric fluorescence switch on–off probe for the reduction of toxic nitrophenols. Moreover, the GQDs detected nitrophenol derivatives at lower concentrations than previously reported analytical values. During the real sample analysis of spiked industrial water and exposed soil samples, a high selectivity and sensitivity of the applied method was achieved with a recovery of 99.7% to 101.3% at spiked concentrations of 400 nM to 100 nM, respectively. The detection limit of the photoluminescent probe for paranitrophenol was as low as 10 pM.
Introduction
GQDs are single atom thick, edge-bound nano-metric graphene fragments having electronic transport well-confined in all three spatial dimensions. Being parent material of GQDs, graphene is a semiconductor of zero bandgap, where the excitons present an infinite Bohr diameter.1 Therefore, a graphene fragment of any size (GQDs) will show quantum confinement effects, which are more prominent when their size ranges are 20 nm, along with a non-zero-bandgap and luminescence upon excitation. Tuning of this bandgap in graphene fragments or GQDs is possible through their size and surface chemistry modifications.2 DFT (density functional theory) calculations indicate that the bandgap of GQDs increases approximately 2 eV for a 20-aromatic-ring GQD and 7 eV for the benzene molecule.3 GQDs have tremendous properties that are well associated with their confinement and edge (like zigzag and armchair edges) effects. The engineering of such effects in nanomaterials4 and in-depth studies of the physical and chemical phenomena behind these required effects have opened avenues of materials science.Until now, little work has been performed on the optical properties directly associated with the edge effects and quantum confinement, where the focus led to the development of diverse chemical methods to attain the controllable synthesis of GQDs.5 On the other hand, GQDs fabrication through both top-down approaches (like hydrothermal graphene oxide reduction electron beam lithography6) and bottom-up approaches (based on chemical synthesis7,8) are mainly dominated by prolonged cutting/cracking strategies lacking the large-scale production of high quality GQDs with well-defined size and chemical functionalities. Currently, GQDs are desirable materials with exciting properties, leading to their potential applications in areas such as sensor devices,9 light emitting diodes,10 bio-imaging11 solar cells, quantum electronic devices, and environmentally benign devices.12 It is well known that the bandgap in graphene depends on its size, shape13 and the fraction of sp2 domains. Therefore, PL emission tuning can be performed by controlling the size and nature of the extended sp2 sites.14 Tuning these parameters eventually lead to graphene exhibiting the typical quantum yields of the carbon core-based nanomaterials.15 The exhibition of characteristic PL properties of dye-derived functional carbon nanodots16 having PL quantum yield as high as 90% and barely 30% for Se-doped GQDs17 has been reported.
We believe that it will be easier to extract and maintain size-controlled GQD synthesis when the starting material already has the small domain structure of the sp2 carbon. In short, the search of a facile material, fabrication strategy, spectral and optical tendencies is of prime importance to generate high-quality GQDs for their improved applications.18–20 Herein, we report a new and simple multistep-ultrasonic approach to synthesize GQDs with an extremely cheap, truly common and environment-friendly source. Moreover, the optimized parameters (including synthesis of GQDs, their size, shape effects, stability and solubility, surface functionalization, confinement and edge effects, bandgap, sp2 hybridization and electronic transitions along with luminescence effects and behaviours) are discussed in detail.
Nitrophenols, especially trinitrophenols (TNP), are heavy explosives,21,22 while the para and dinitrophenols (PNP, DNP) are major toxic/carcinogenic pollutants23 abundantly found in industrial and agricultural waste waters/effluents. Such phenol derivatives are stable, water-soluble and equally lethal to aquatic bodies.24 At the same time, nitrophenols are important in forensic research, land mine detection and the manufacturing of rocket fuels.21,22 The sensitive, selective and robust detection of such nitro-aromatic explosives/pollutants has attained wide attention and increased concerns regarding homeland security and public safety.25,26 On the other hand, the reduction of nitrophenols is of supreme importance for industrial products like paracetamol in pharma-industry, aniline and other dye-stuffs in chemical industry, anti-corrosion materials for paints and fuel lubricants and also for photographic materials.27,28 Several studies have shown the fabrication of environmentally safe/friendly and catalytically potent nano-tools (N-doped graphene, nano-ferrites and graphitic carbon nitride) to counter nitrophenols.29–31 Algarra and co-workers proposed carbon dots (CDs) as fluorescent sensors32 with a limit of detection just up to 2 μM. Similarly, efforts by Nian, Li and co-workers and Sun and co-workers lie in the same category for the detection limit up to 51 nM.33,34 Therefore, more selective and sensitive approaches are needed to detect lethal nitro-explosives up to the pico- and femto-molar levels.
Briefly, the objective of this study was to synthesize highly fluorescent GQDs from the burnt leather graphite as a carbon source, having excellent surface defects and edge structure for the catalytic performance, along with the low level detection of carcinogenic organics. Initially, the synthesis of GQDs was based on the oxidative carbonization of leather graphite named as GOS. Later, an ultrasonic cleavage accompanied with the hydrothermal route transformed the oxygenated graphite to the fibrous structure GONFs prior to the smaller sized GOQDs. The purposely incorporated oxidative groups facilitated the cracking of GOS and GONFs to GOQDs. In fact, GQDs were attained through continued ultrasonication-assisted hydrothermal co-cuttings of GONFs due to the frequent loss of newly added oxygen-bearing groups, along with effectively tuned optical properties. The overall synthetic approach comprised three steps, namely: (1) microwave-assisted pyrolysis, (2) ultrasonication treatment and (3) hydrothermal heating of burnt leather graphite in various solvents. In all above-mentioned procedures, carbonized leather graphite was added to the respective solvent system, and then treated with either ultrasonic, microwave pyrolysis or through hydrothermal heating to produce GQDs. The leather-derived GQDs served as ratiometric switch on–off fluorescent probes for the catalytic reduction of para-nitrophenol (PNP), 2,4-di-nitrophenol (DNP) and 2,4,6-tri-nitrophenol (TNP). For practical impact, the real sample analysis of industrial waste (water) and explosion-affected areas (soil) were performed. Results provided high selectivity and sensitivity with PL quenching up to pico-molar levels. Furthermore, interference species (such as certain polyaromatic hydrocarbon (PAHs), polychlorinated biphenyls (PCBs), and some heavy metals) did not alter the catalytic performance of the GQDs. Such attributes demonstrate the diverse potential of GQDs for further studies.
Materials and methods
Synthesis of GQDs
First, a raw leather piece was obtained from the international and famous leather goods brand Jafferjees, Karachi. 50 g of the burnt specimen was ground to make a fine black powder and then sieved with a mesh (10 μm size). Later, 0.5–12 g of the burnt leather graphite was mixed with varying concentrations of nitric acid (Table S1†) and ultrasonicated for different time intervals. The resulting yellowish-brown solution containing GQDs was centrifuged at 2000 rpm for 10 min to remove unreacted carbonized leather powder. The supernatant, which showed bright blue fluorescence under 365 nm (Fig. S1†) by UV lamp (Uvitec, UK), was collected and freeze-dried. Further purification steps were carried out by dissolving the GQDs in water/acetone (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) and centrifuging it at 12000 rpm for 15 min. These purification steps enabled the separation of excessive nitric acid from the GQDs. The resultant precipitates containing purified GQDs were collected, and finally dissolved in 10 mL of water for further use.
:3) and centrifuging it at 12000 rpm for 15 min. These purification steps enabled the separation of excessive nitric acid from the GQDs. The resultant precipitates containing purified GQDs were collected, and finally dissolved in 10 mL of water for further use.
Catalytic reduction of nitrophenols
For catalytic reduction, 50 μL PNP (14 mM) was mixed with 20 μL of NaBH4 (0.12 M) in the presence of 3 mL deionized water to form a homogenous reaction mixture. Then, 5 μL of the catalyst (5 nM or 5 × 1012 GQDs) was added to initiate the reduction process. The catalytic reduction of PNP to para-aminophenol (PAP) was monitored through UV-visible spectrophotometer, FTIR and NMR studies.Instrumentation
Transmission electron microscope (TEM) and atomic fluorescence microscopy (AFM) images were taken on a JEOL JEM 2100 TEM at an accelerating voltage of 200 kV and Agilent atomic force microscope, respectively. UV-visible absorption was carried out using an “Evolution” spectrophotometer. Fluorescence spectroscopy was performed on an LS 55 PerkinElmer spectrophotometer. The Fourier transform infrared spectral (FTIR) investigation was recorded on a 8900 Shimadzu HYPER. Experiments were performed by preparing KBr pellets in the frequency range of 400–4000 cm−1. X-ray photoelectron spectroscopy (XPS) analysis was performed by an ESCALAB 250 spectrometer (Thermo-VG Scientific Co., U.S.A.) with an ultrahigh vacuum generator. Powder X-ray diffraction (XRD) was analyzed by PANalytical-EMPYREAN.Results and discussion
Synthesis and characterization of GQDs
In this method, burnt leather graphite was treated via hydrothermal ultrasonication in the presence of nitric acid and other solvents without the involvement of any surface passivating agent or organic additives. Scheme 1 and Fig. S1† reveal that the ultrasonication time greatly affects the acid oxidative cracking and hydrothermal reduction of graphite by tuning their size. The size variation and surface structure defects were also found to tune the optical performance of synthesized GQDs (Fig. S2†).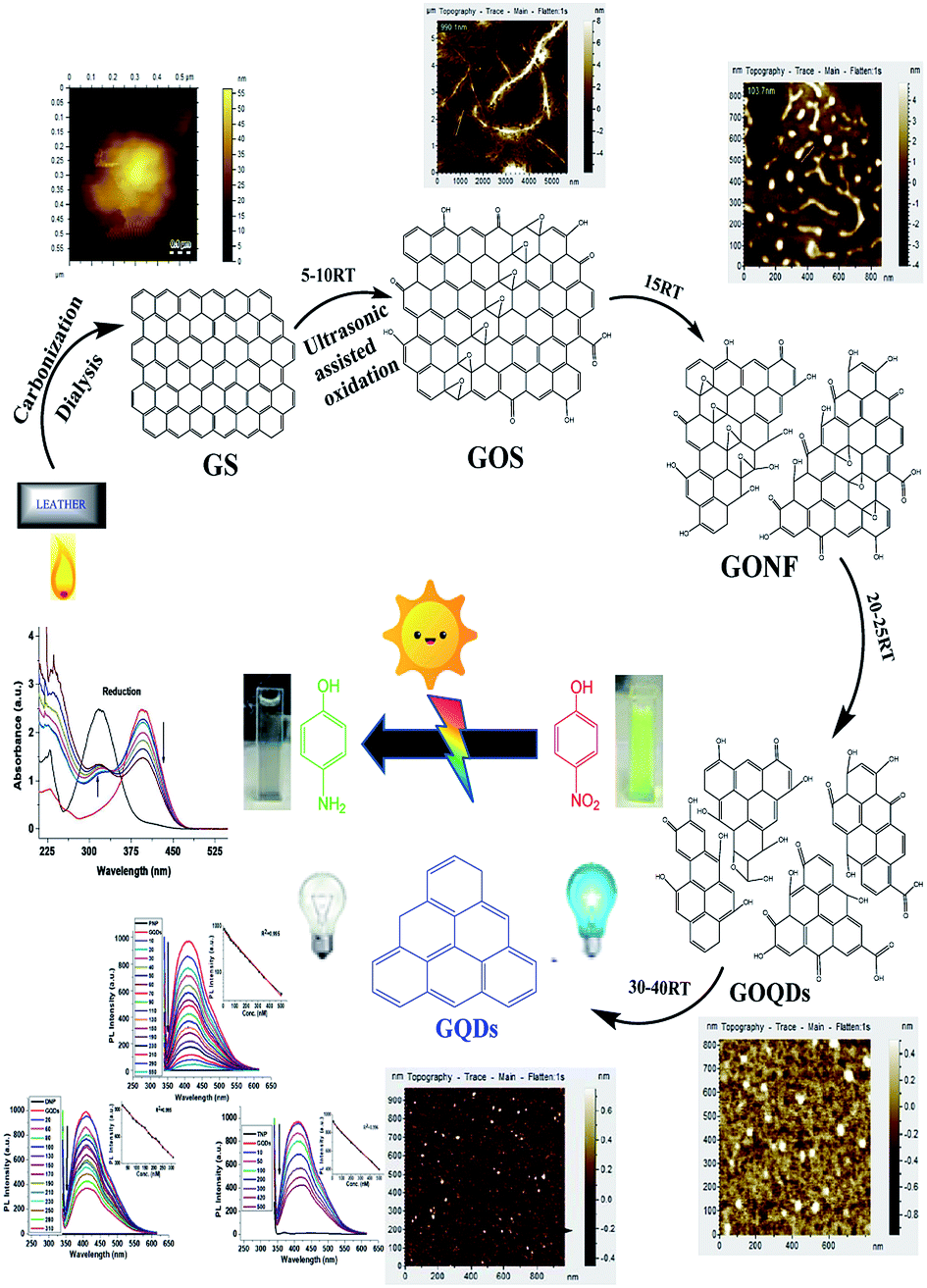 | ||
| Scheme 1 Schematic synthesis of GQDs with AFM images of different stages, along with their potential catalysis, as well as sensing ability. | ||
A similar phenomenon of acid oxidative hydrothermal cleavage in aqueous medium was prevalent in previous reports.20 However, in our case, the oxidative cleavage together with ultrasonication steered the process of cleavage, shortened GOS and incorporated the oxidative surface groups all under one platform. The step-by-step GOS cleavage and production of GONFs to GOQDs were evidently observed through AFM images (Scheme 1).
Similarly, TEM images (Fig. S2†) for the conversion of GOS to GONFs and GOQDs strongly support the AFM findings. Basically, acid oxidative-co-ultrasonic cutting confers the exfoliation of GOS, converting them to GOQDs all the way along the epoxy group channels.35
Undoubtedly, the prolonged ultrasonication-driven hydrothermal deoxidization aids in removing the bridging O atoms from the epoxy lines of GOQDs to finally produce GQDs.13 This is due to the fact that the ultrasonic waves generate alternating low and high-pressure waves in the mixture, leading to the formation and violent collapse of small vacuum bubbles. This cavitation causes high speed impinging liquid jets, deagglomeration and strong hydrodynamic shear-forces.35 In this way, the energy of ultrasonic waves leads to the pyrolysis of activated carbonized leather graphite through the generation of multiple oxygen groups containing functionalities and through the production of a zigzag surface structure during the cracking process of graphite. Similarly, upon prolonged ultrasonication, the graphene sheets ultimately break down into tiny GQDs by creating several physical and chemical defects on the graphene sheets and provide a myriad of active sites.35
The comparison of the UV-visible and PL spectra of GQDs synthesized with all three methods, i.e., on a hotplate, microwave-assisted and under ultrasonication, are shown in Fig. 1a. Notably, the current synthesis supports the effectiveness of an aqueous medium, which is similar to previous reports.36,37 However, after the detailed evaluation of different solvent effects on the synthesis and optical properties of GQDs, it was concluded that inorganic acids, especially nitric acid (pKa ∼ −1.5), exhibit strong oxidizing capacity due to their large positive reduction potential.38 Therefore, they incorporate various oxidative groups during the cracking of GOS. Various edge effects were produced during this cracking transition of GOS, which remarkably modulated the optical characteristics of GQDs, as compared to alcohols (Fig. 1b).
 | ||
| Fig. 1 (a) UV-visible spectra of GQDs (acid oxidation) through a hotplate, microwave and ultrasonication treatment, and their corresponding PL spectra. (b) UV-visible spectra of GQDs synthesized with different solvents, and their PL spectra are shown in the inset. (c) The representative UV-visible and PL spectra of GQDs (the inset shows the photograph of GQDs solution under 350 nm UV lamp). (d) The PLE spectrum of the as-synthesized GQDs with a detection wavelength of 420 nm and excited PL spectrum at 330 nm, suggesting the irradiation decay of activated electrons from the HOMO → LUMO transition (of free zigzag sites having carbene like triplet ground state energy level). | ||
The concentration of nitric acid was also optimized. It was found that as the concentration of nitric acid increased from 0.5 N to 2 N, the best result of oxidation was obtained at 1 N concentration in terms of the absorbance and PL performance (Table S1†). Moreover, it was observed that 1 N of nitric acid enhanced the PL intensity up to ∼7-folds, which simply contributed to the robustness. This made it into a more attractive candidate towards catalytic, as well as reductive, analytical studies (Fig. 1b). Contrarily, the non-acidic counterparts were different from the GQDs obtained under 1 N nitric acid oxidation (Table S1†). Acid-derived GQDs emit bright blue luminescence at 420 nm with a high quantum yield of 27% (taking quinine sulfate as a reference). Furthermore, the PL studies (Fig. S3a†) showed that GQDs developed under nitric acid oxidation exhibited excitation-dependent fluorescence behavior like the previously reported GQDs.5 Additionally, the developed GQDs were resistant to photobleaching, even after 24 hour exposure to UV light (Fig. S3b†).
Typical graphene shows a single π → π* transition of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond at 270 nm.39,40 In this study, GOS exhibited a π → π* blue-shifted transition of the –CC bond and n → π* transition of the –CO bond at around 250 and 298 nm, respectively (Fig. 1c). These are the corresponding peaks of the π → π* transition of the aromatic sp2 domains.41 As reported earlier, the PLE spectrum of GQDs showed two sharp peaks at 256 and 330 nm5, which confirm that like the 330 nm excitation, the 256 nm excitation can also induce the strongest PL emission at 420 nm. In Fig. 1d, the PLE peak at 330 nm corresponds to the 300 nm absorption band of GQDs, whereas the 256 nm PLE peak correlates well with the 250 nm absorption band originated from the π → π* transition. The PLE spectra of GQDs are directly related to both n → π* and π → π* transitions at 256 and 330 nm, rather than the commonly observed π → π* transition.42
C bond at 270 nm.39,40 In this study, GOS exhibited a π → π* blue-shifted transition of the –CC bond and n → π* transition of the –CO bond at around 250 and 298 nm, respectively (Fig. 1c). These are the corresponding peaks of the π → π* transition of the aromatic sp2 domains.41 As reported earlier, the PLE spectrum of GQDs showed two sharp peaks at 256 and 330 nm5, which confirm that like the 330 nm excitation, the 256 nm excitation can also induce the strongest PL emission at 420 nm. In Fig. 1d, the PLE peak at 330 nm corresponds to the 300 nm absorption band of GQDs, whereas the 256 nm PLE peak correlates well with the 250 nm absorption band originated from the π → π* transition. The PLE spectra of GQDs are directly related to both n → π* and π → π* transitions at 256 and 330 nm, rather than the commonly observed π → π* transition.42
These two electronic transitions of 256 nm (4.80 eV) and 330 nm (3.76 eV) can be regarded as the transition from the σ and π orbitals, i.e., from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO).5 In the UV-visible spectrum (Fig. 1c), newly observed bands at 250 nm and 300 nm arising from the triple carbenes at the free zigzag sites correspond to the HOMO-to-LUMO orbitals. The strong luminescence of GQDs may be attributed to the zig–zag edge structure of the triplet carbene.43 Moreover, we also propose that the observed luminescence is due to the irradiation decay of the activated electrons from HOMO to LUMO.42 Carbenes have two electronic configurations (triplet and singlet) at the ground state and their multiplicity is related to the energy difference (δE) between σ and π orbitals. The observed calculated energy difference between these two orbitals is 1.04 eV, which is within the required value of 1.5 eV for the triple carbenes, supporting the reasonable spectral assignment of both transitions.5
The Raman spectrum of GQDs in Fig. S4† displays two broad peaks at around 1385 cm−1 and 1595 cm−1, which are attributed to the disordered D band and the crystalline G band, respectively. The relative intensity of (ID/IG) is around 0.86, which indicates that they have structural similarity with graphite and are highly crystalline.44 The FTIR spectrum of graphitized leather after nitric acid oxidation-co-ultrasonication is shown in Fig. S5.† The band at 1707 cm−1 is assigned to the –CO vibration of the –COOH group located at the edges of GOS. The bands at 1638 cm−1 and 1358 cm−1 can be attributed to the skeletal vibration of –CC in GOS and the tertiary C–OH groups stretching, respectively.10 Another band appearing at 1046 cm−1 is due to the C–O–C stretching vibration. The epoxy groups gradually decreased, producing more –CO with sonication time as the GOS were cleaved to GONFs, as manifested by the appearance of an intense peak at 1707 cm−1 due to the –CO stretching mode. The previously reported oxidative cracking/shortening of graphite45 sheets5 and carbon nanotubes19 fibers42 truly support our findings regarding the ultrasonic/acid-assisted oxidative cracking of the carbonized leather source.
Evidently, from the FTIR characterization (Table S2†), the epoxy group signal was weakened and a strong –CO signal appeared as the sonication time increased. The epoxy groups simultaneously produced –CO, which was observed in the FTIR peak intensities of 10–25 RT, where the maximum production of the –CO groups was achieved within and at the surface of GONF and GOQDs. Further ultrasonication at 40 RT resulted in the reduction or deoxidization of the resulting GQDs (Scheme 1), which was confirmed by the disappearance of both C–O–C and –CO peaks, as shown in Table S2.†46
According to the previous findings based on hydrothermal reductions, the optical properties vary due to the variation in size, density and nature of the available sp2 sites in GQDs. Thus, the energy gap can be tuned by changing the size of GQDs. Therefore, by tailoring the energy gap of the GQDs, the size can be justified. This inverse relationship between the energy gap and size of the GQDs is linked to the ultrasonication time in our case, which can be seen from Table S2.† It is clear that as the ultrasonication time proceeds with the subsequent rise in temperature, the energy gap of the GQDs increases (0.82 eV to 1.04 eV) and their size decreases from ∼0.5 μm (in the case of GOS) to ∼5 nm (in the case of GQDs), as confirmed through AFM and TEM images (Scheme 1 and Fig. 2a and b), respectively.42
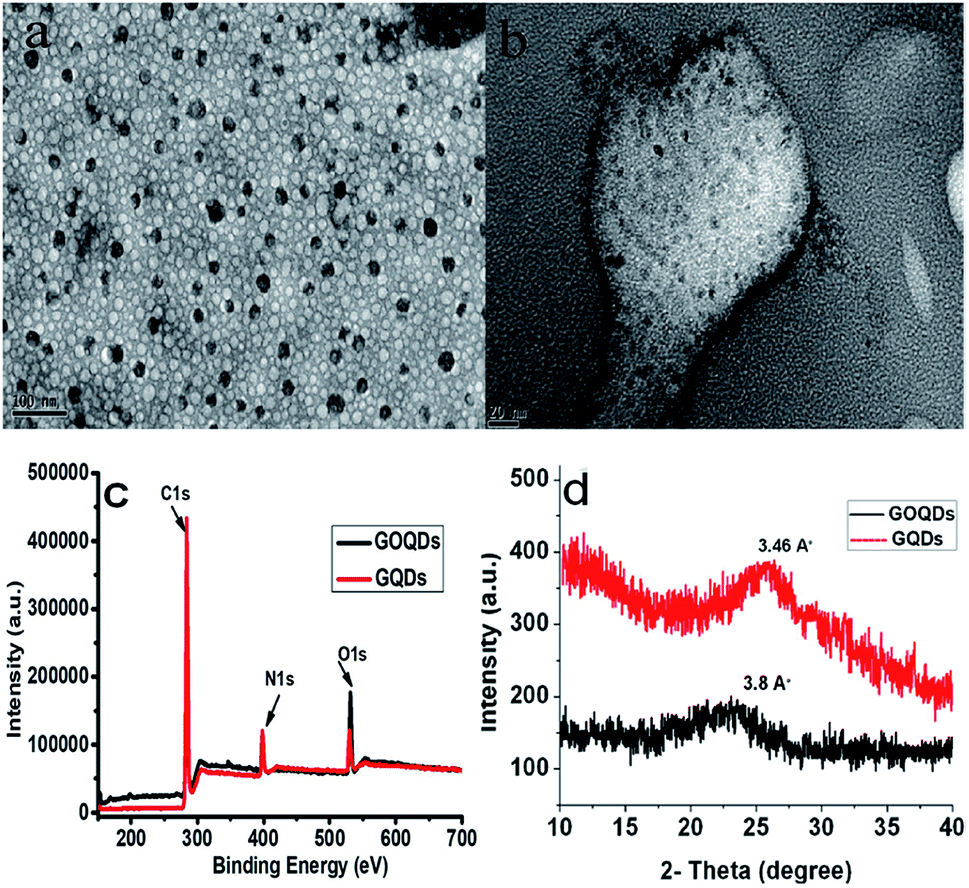 | ||
| Fig. 2 TEM images of GOQDs (a) and GQDs (b) synthesized at 25 min and 40 min ultrasonication, respectively. (c) XPS spectra of GOQDs and GQDs. (d) XRD patterns of GOQDs and GQDs. | ||
In order to further explore the chemical composition of GOQDs and GQDs, XPS analysis was performed. The full XPS spectra (Fig. 2c) show three prominent peaks: C 1s (284 eV), N 1s (398 eV), and O 1s (532 eV), indicating the presence of these elements.36,47,48 The findings of the XRD spectra (Fig. 2d), revealed that under the influence of ultrasonic-assisted acid oxidation at 25 min, the representative (002) spacing increased to 3.8 Å in the case of the GS fragments (GOQDs) when compared to bulk graphite, i.e., 3.34 Å5. However, upon further hydrothermal treatment of these GOQDs, the (002) spacing was reduced to 3.46 Å, which was very close to that of bulk graphite. In the case of GOQDs, the peak broadening effect at 2θ = 23.0° is due to the lattice distortion occurring in the stacking order of the graphite lattice as a result of the oxidative group incorporation.42 Furthermore, to investigate the stability of the as-synthesized GQDs towards ionic solutions, the effect of the NaCl concentration on the PL intensity of GQDs was analysed in a wide concentration range from 1 μM up to 10 mM. Fig. S6† validates that a negligible or no change in the PL intensity of GQDs was observed even with higher ionic concentrations after 24 h.
These results safely demonstrate that the as-synthesized GQDs exhibit unique surface properties, higher luminescence, and low photobleaching properties, as well as higher ionic stability, which present them as a potential candidate for potential applications in catalysis and in optoelectronic devices.
Catalytic reduction of nitrophenols
To evaluate the catalytic activity of the as-produced GQDs, we carried out the reduction of a series of nitrophenol compounds (PNP, DNP and TNP). It is well documented that under neutral or acidic conditions, nitrophenols exhibit a strong absorption peak at 317 nm, which on addition of GQDs in the presence of NaBH4, red shifts to 400 nm and the yellow solution turns colorless (Fig. 3a, c and e). The actual mechanism lies in the fact that in the presence of NaBH4, nitrophenols are converted into 4-nitrophenolate ions as the dominant species.49 As soon as the catalytically active GQDs were introduced into the reaction mixture, nitrophenolate ions were transformed into para-aminophenol (PAP), which appeared as a new peak at 315 nm with a concomitant decreased peak at 400 nm.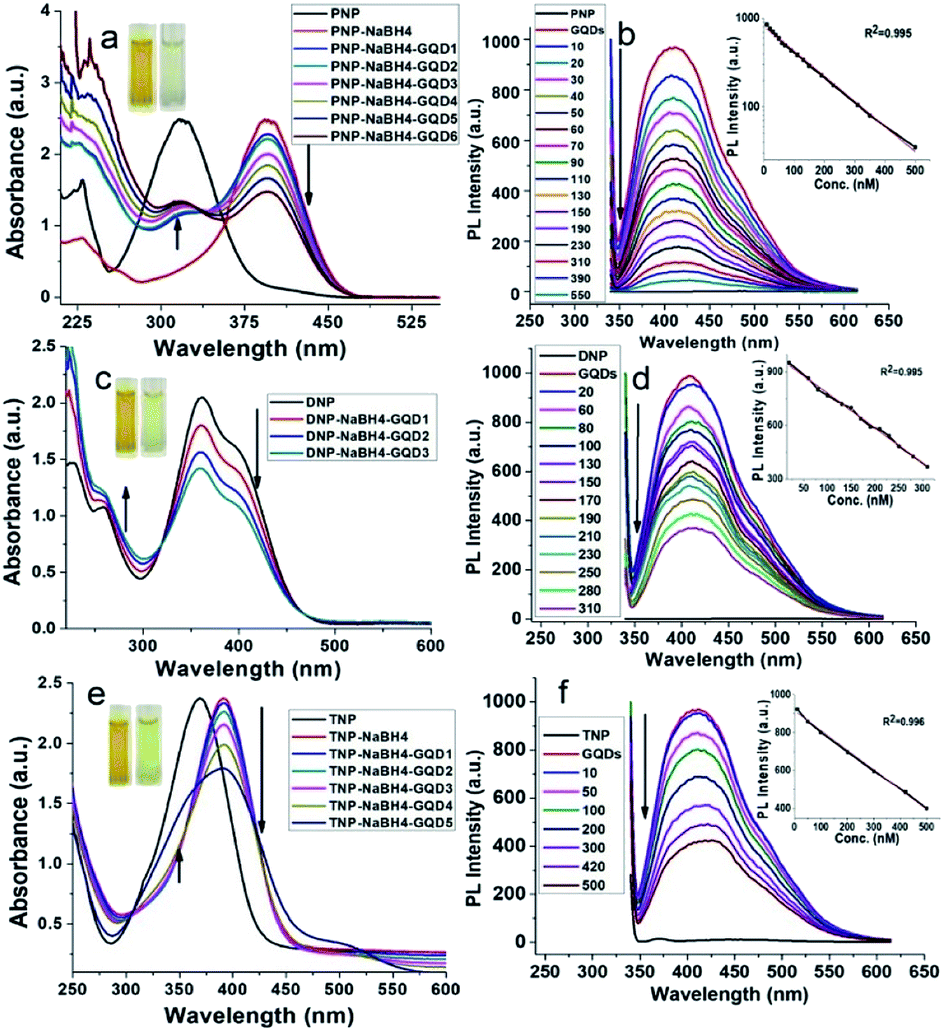 | ||
| Fig. 3 UV-visible (a, c and e) and PL spectra (b, d and f) of the catalytic performance of GQDs (concentrations = 10–60 μL GQDs for GQD1-GQD6) for 0.1 mM PNP (a and b) and 0.1 mM DNP (c and d) and 0.1 mM TNP (e and f) in the presence of NaBH4 (10 μL of 1 mM). The PL quenching spectra of GQDs are shown with 10–550 nM PNP (b), 20–310 nM DNP (d), and 10–500 nM TNP (f). The insets (b, d and f) represent the linear regression plots for the detection of PNP, DNP and TNP, respectively. | ||
Reduction of nitrophenols (0.1 mM) to PAP in the presence of NaBH4 (1 mM) by GQDs follow a pseudo-first-order reaction, which was monitored using an 8452A photodiode array spectrophotometer equipped with a stop-flow accessory. The reduction was rather fast and was completed within the mixing period. The peak at 400 nm (nitrophenolate ions) shifted to 315 nm, even with 20 μL GQDs. Therefore, under the same reaction conditions, it is difficult to achieve the reduction kinetics due to the fast reaction rate. We also found that no reaction was observed without the addition of GQDs, which is indicative of the inability of the self-reduction process under the same experimental conditions. Further kinetic studies under variable conditions are still under study in our group.
Fig. S7† represents the FTIR spectra for the catalytic reduction of PNP. As seen from the FTIR spectral data I of pure PNP, all corresponding vibrations of the –NO2 group (rocking, scissoring and wagging at 535, 629 and 755 cm−1, respectively)50 are present, which are predominantly absent in spectra II of the PNP-GQDs mixture. The sharp appearance of the N–H stretches of the primary amines at 3236 cm−1 and at 3410 cm−1 was observed. For the primary amines (RNH2), there are two bands in this region, i.e., asymmetrical (N–H), symmetrical (N–H) stretch, and N–H bending vibration at 1622 cm−1.29
The details of the corresponding groups and peak assignments are presented in Table S3.† The catalytic reduction of nitrophenols was also confirmed through an 1H NMR study (Fig. S8 and S9†). The 1H NMR data revealed that the appearance of an intense peak at 2.66 ppm in the presence and absence (under 2 h UV-irradiation) of NaBH4 gives an obvious clue for the in situ catalytic reduction of nitrophenols. It was observed that the signal for the NH2-group in the 1H NMR spectra after reduction was slightly down-field, which is possibly due to a side chain alkylation of the aminophenol while being adsorbed on the surface of the GQDs.51
Present method does not require the normally used dopant moieties involved in the graphene-based catalytic reduction of nitrophenols.30,52 The reduction of nitrophenols was also studied by PL studies (Fig. 3b, d and f). It was observed that the PL intensity was quenched upon the addition of different concentrations of nitrophenols into a fixed amount of GQDs. This PL quenching is probably due to the enhanced electron transfer from the carbene-like zigzag edges to nitrophenols, which is adsorbed over the surface of GQDs during its reduction.53
Similarly, the electron transfer has been also reported to adsorb hydrogen, subsequently favouring the hydrogenation reduction reaction.52 Typically, when GQDs are added to the reaction mixture, nitrophenols and borohydride ions (BH4−) are adsorbed on the surface of GQDs. Here, GQDs act as the medium to transfer electrons from the BH4− ions to the nitrophenols, resulting in the formation of phenolate ions. The aqueous medium supplies hydride ions (H−), leading to the resulting aminophenols54 (Scheme 1). Table S4† represents the ratiometric decrease in the fluorescence intensity of GQDs, with a limit of detection (LOD) of 10 pM, 65 pM and 80 pM for PNP, DNP and TNP, respectively.
Method sensitivity and selectivity
In order to study the sensing applications in real environmental samples, it is vital to investigate the effect of the interfering species, such as PAHs, PCBs, heavy metals and other pollutants.55,56 The selectivity of the reduction in the GQD-nitrophenol complex is shown in Fig. S10.† Our results indicate that the method selectivity was achieved for the catalytic reduction of nitrophenols in comparison to the blank sample (no GQDs was added), even in the presence of higher concentrations of interfering species. This indicates that GQDs (just like other fluorescent probes) exhibit good selectivity towards nitrophenols over other competing species.The applicability of the as-synthesized GQDs towards the catalytic reduction of nitrophenols was also monitored in real samples taken from industrial water and soil samples of bombing exposed area (Korangi industrial area and latest targeted area of suicide bombers in Karachi-Pakistan). For this purpose, the soil samples were taken in triplicates, filtered (30 mesh size), washed thoroughly with acetone and TNP were extracted in the form of picrate ions.57 The filtrate was diluted with deionized water and the concentration of TNP was detected (Table S5†). Similarly, for industrial water, the samples were spiked with varying concentrations of PNP. After the addition of GQDs, the PL intensity signals were monitored. The recoveries were found in the range from 97.3% to 101.7%, which indicated the reliability, efficacy and reproducibility of the present method.
Conclusions
The devised strategy explored the ultrasonic hydrothermal co-cutting of GS to deliver highly potent GQDs from a leather source for the first time. Notably, the ultrasonication time modification contributed towards size-controlled GQDs during the breakage and oxidative cleavage of GOS. Meanwhile, the roles of acids, solvents and types of ratiometric treatments were fully sorted and explained. The developed GQDs exhibited stable and strong visible emission and excellent luminescent properties without the incorporation of dopant moieties or surface functionalization. A series of GO products (like GOS, GONFs, GOQDs and GQDs) were synthesized in a sequence, which presented marvelous properties associated with their quantum confinement and edge effects. The significant quality-oriented GQDs possessed unique photoluminescence properties, water solubility, stability and lengthy PL lifetimes with no photobleaching. Finally, lethal and toxic nitrophenols were reduced to environment-friendly aminophenols through the catalytic effect of the prepared GQDs. Additionally, the sensing ability of GQDs was also tested for nitrophenols, and the LODs of such derivatives were found to be sharp at the picomolar level. Such efforts endorse GQDs to serve as excellent switch on–off probes for high-contrast bioimaging, biosensing, photovoltaics and drug delivery applications in future research.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We highly acknowledge the great guidance of Prof. Xueyuan Chen from FJIRSM, Fuzhou and Prof. Ju Huangxian from Nanjing University, Nanjing China. The work was conducted with funding from the Higher Education Commission of Pakistan (PM-IPFP/HRD/HEC/2011/0006).References
- A. K. Geim and K. S. Novoselov, in Nanoscience and Technology: A Collection of Reviews from Nature Journals, World Scientific, 2010, pp. 11–19 Search PubMed.
- S. Kim, S. W. Hwang, M.-K. Kim, D. Y. Shin, D. H. Shin, C. O. Kim, S. B. Yang, J. H. Park, E. Hwang and S.-H. Choi, ACS Nano, 2012, 6, 8203–8208 CrossRef CAS PubMed.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. S. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- S. Zhu, Y. Song, J. Wang, H. Wan, Y. Zhang, Y. Ning and B. Yang, Nano Today, 2017, 13, 10–14 CrossRef CAS.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- S. Schnez, F. Molitor, C. Stampfer, J. Güttinger, I. Shorubalko, T. Ihn and K. Ensslin, Appl. Phys. Lett., 2009, 94, 012107 CrossRef.
- X. Yan, X. Cui and L.-s. Li, J. Am. Chem. Soc., 2010, 132, 5944–5945 CrossRef CAS PubMed.
- X. Yan, X. Cui, B. Li and L.-s. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS PubMed.
- M. Bacon, S. J. Bradley and T. Nann, Part. Part. Syst. Char., 2014, 31, 415–428 CrossRef CAS.
- H. Zhao, Y. Chang, M. Liu, S. Gao, H. Yu and X. Quan, Chem. Commun., 2013, 49, 234–236 RSC.
- V. Gupta, N. Chaudhary, R. Srivastava, G. D. Sharma, R. Bhardwaj and S. Chand, J. Am. Chem. Soc., 2011, 133, 9960–9963 CrossRef CAS PubMed.
- H. Sun, L. Wu, N. Gao, J. Ren and X. Qu, ACS Appl. Mater. Interfaces, 2013, 5, 1174–1179 CrossRef CAS PubMed.
- D. Pan, L. Guo, J. Zhang, C. Xi, Q. Xue, H. Huang, J. Li, Z. Zhang, W. Yu, Z. Chen, Z. Li and M. Wu, J. Mater. Chem., 2012, 22, 3314–3318 RSC.
- A. Dato, V. Radmilovic, Z. Lee, J. Phillips and M. Frenklach, Nano Lett., 2008, 8, 2012–2016 CrossRef CAS PubMed.
- S. Sagbas and N. Sahiner, in Nanocarbon and its Composites, Elsevier, 2019, pp. 651–676 Search PubMed.
- L. Tong, X. Wang, Z. Chen, Y. Liang, Y. Yang, W. Gao, Z. Liu and B. Tang, Anal. Chem., 2020, 9, 6430–6436 CrossRef PubMed.
- S. Shen, J. Wang, Z. Wu, Z. Du, Z. Tang and J. Yang, Nanomaterials, 2020, 10, 375 CrossRef CAS PubMed.
- Y. Liu, Q. Zhou, Y. Yuan and Y. Wu, Carbon, 2017, 115, 550–560 CrossRef CAS.
- X. Hou, Y. Hu, P. Wang, L. Yang, M. M. Al Awak, Y. Tang, F. K. Twara, H. Qian and Y.-P. Sun, Carbon, 2017, 122, 389–394 CrossRef CAS PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan and G. Gao, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- Y. Dong, C. Chen, X. Zheng, L. Gao, Z. Cui, H. Yang, C. Guo, Y. Chi and C. M. Li, J. Mater. Chem., 2012, 22, 8764–8766 RSC.
- P. Hakey, W. Ouellette, J. Zubieta and T. Korter, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1428 CrossRef CAS PubMed.
- P. Brousseau and C. J. Anderson, Propellants, Explos. Pyrotech., 2002, 27, 300–306 CrossRef CAS.
- J. P. Agrawal, High energy materials: propellants, explosives and pyrotechnics, John Wiley & Sons, 2010 Search PubMed.
- J.-R. Chiou, B.-H. Lai, K.-C. Hsu and D.-H. Chen, J. Hazard Mater., 2013, 248, 394–400 CrossRef PubMed.
- M. Megharaj, H. Pearson and K. Venkateswarlu, Arch. Environ. Contam. Toxicol., 1991, 21, 578–584 CrossRef CAS.
- A. Rose, Z. Zhu, C. F. Madigan, T. M. Swager and V. Bulović, Nature, 2005, 434, 876 CrossRef CAS PubMed.
- M. B. Pushkarsky, I. G. Dunayevskiy, M. Prasanna, A. G. Tsekoun, R. Go and C. K. N. Patel, Proc. Natl. Acad. Sci., 2006, 103, 19630–19634 CrossRef CAS PubMed.
- Y. Du, H. Chen, R. Chen and N. Xu, Appl. Catal., A, 2004, 277, 259–264 CrossRef CAS.
- K.-I. Min, J.-S. Choi, Y.-M. Chung, W.-S. Ahn, R. Ryoo and P. Lim, Appl. Catal., A, 2008, 337, 97–104 CrossRef CAS.
- B. Qu, Z. Mu, Y. Liu, Y. Liu, R. Yan, J. Sun, Z. Zhang, P. Li and L. Jing, Environ. Sci.: Nano, 2020, 7, 262–271 RSC.
- B. B. Campos, R. Contreras-Cáceres, T. J. Bandosz, J. Jiménez-Jiménez, E. Rodríguez-Castellón, J. C. E. da Silva and M. Algarra, Carbon, 2016, 106, 171–178 CrossRef CAS.
- Y. Z. Fan, Y. Zhang, N. Li, S. G. Liu, T. Liu, N. B. Li and H. Q. Luo, Sens. Actuators, B, 2017, 240, 949–955 CrossRef CAS.
- X. Sun, J. He, Y. Meng, L. Zhang, S. Zhang, X. Ma, S. Dey, J. Zhao and Y. Lei, J. Mater. Chem. A, 2016, 4, 4161–4171 RSC.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- Q. Liang, W. Ma, Y. Shi, Z. Li and X. Yang, Carbon, 2013, 60, 421–428 CrossRef CAS.
- J. Ju and W. Chen, Biosens. Bioelectron., 2014, 58, 219–225 CrossRef CAS PubMed.
- A. Goyal, S. Bansal and S. Singhal, Int. J. Hydrogen Energy, 2014, 39, 4895–4908 CrossRef CAS.
- A. Cao, Z. Liu, S. Chu, M. Wu, Z. Ye, Z. Cai, Y. Chang, S. Wang, Q. Gong and Y. Liu, Adv. Mater., 2010, 22, 103–106 CrossRef CAS PubMed.
- Z. Luo, Y. Lu, L. A. Somers and A. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- J. Qian, D. Wang, F. H. Cai, W. Xi, L. Peng, Z. F. Zhu, H. He, M. L. Hu and S. He, Angew. Chem., Int. Ed., 2012, 51, 10570–10575 CrossRef CAS PubMed.
- L. Ren, J. Qiu and S. Wang, Compos. B Eng., 2013, 55, 548–557 CrossRef CAS.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272–12277 RSC.
- S. Zhuo, M. Shao and S.-T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- A. Incze, A. Pasturel and P. Peyla, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 212103 CrossRef.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2015, 10, 484–491 CrossRef PubMed.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS.
- M. X. Gao, C. F. Liu, Z. L. Wu, Q. L. Zeng, X. X. Yang, W. B. Wu, Y. F. Li and C. Z. Huang, Chem. Commun., 2013, 49, 8015–8017 RSC.
- X.-H. Li, X. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170–2174 RSC.
- R. J. Stedman and L. S. Miller, J. Org. Chem., 1967, 32, 3544–3547 CrossRef CAS.
- A. J. Abkowicz-Bieńko, Z. Latajka, D. C. Bieńko and D. Michalska, Chem. Phys., 1999, 250, 123–129 CrossRef.
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CrossRef CAS.
- Z. Naghshbandi, N. Arsalani, M. S. Zakerhamidi and K. E. Geckeler, Appl. Surf. Sci., 2018, 443, 484–491 CrossRef CAS.
- F. Yan, Z. Bai, F. Zu, Y. Zhang, X. Sun, T. Ma and L. Chen, Microchim. Acta, 2019, 186, 113 CrossRef PubMed.
- P. Ni, J. Xie, C. Chen, Y. Jiang, Y. Lu and X. Hu, Microchim. Acta, 2019, 186, 202 CrossRef PubMed.
- P. G. Thorne and T. F. Jenkins, Field Anal. Chem. Technol., 1997, 1, 165–170 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed characterization related to synthetic parameters, catalytic studies along with tables and graphs. See DOI: 10.1039/d0ra03715j |
| This journal is © The Royal Society of Chemistry 2020 |