
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fe3O4@SiO2@Au nanoparticles for MRI-guided chemo/NIR photothermal therapy of cancer cells†
Alexey Maximenko *ab,
Joanna Depciuch*a,
Natalia Łopuszyńskaa,
Malgorzata Stecc,
Żaneta Światkowska-Warkockaa,
Vadim Bayevb,
Piotr M. Zielińskia,
Jaroslaw Baranc,
Julia Fedotovab,
Władysław P. Węglarza and
Magdalena Parlinska-Wojtana
*ab,
Joanna Depciuch*a,
Natalia Łopuszyńskaa,
Malgorzata Stecc,
Żaneta Światkowska-Warkockaa,
Vadim Bayevb,
Piotr M. Zielińskia,
Jaroslaw Baranc,
Julia Fedotovab,
Władysław P. Węglarza and
Magdalena Parlinska-Wojtana
aInstitute of Nuclear Physics Polish Academy of Sciences, Radzikowskiego 152, 31-342 Kraków, Poland. E-mail: alexey.a.maximenko@gmail.com; joannadepciuch@gmail.com
bResearch Institute for Nuclear Problems of Belarusian State University, Bobruyskaya 11, 220030 Minsk, Belarus
cDepartment of Clinical Immunology, Institute of Pediatrics, Jagiellonian University Medical College, Św. Anny 12, Kraków, PL-30-663, Poland
First published on 15th July 2020
Abstract
Novel functionalized (biofunctionalization followed by cisplatin immobilization) Fe3O4@SiO2@Au nanoparticles (NPs) were designed. The encapsulation of Fe3O4 cores inside continuous SiO2 shells preserves their initial structure and strong magnetic properties, while the shell surface can be decorated by small Au NPs, and then cisplatin (cPt) can be successfully immobilized on their surface. The fabricated NPs exhibit very strong T2 contrasting properties for magnetic resonance imaging (MRI). The functionalized Fe3O4@SiO2@Au NPs are tested for a potential application in photothermal cancer therapy, which is simulated by irradiation of two colon cancer cell lines (SW480 and SW620) with a laser (λ = 808 nm, W = 100 mW cm−2). It is found that the functionalized NPs possess low toxicity towards cancer cells (∼10–15%), which however could be drastically increased by laser irradiation, leading to a mortality of the cells of ∼43–50%. This increase of the cytotoxic properties of the Fe3O4@SiO2@Au NPs, due to the synergic effect between the presence of cPt plus Au NPs and laser irradiation, makes these NPs perspective agents for potential (MRI)-guided stimulated chemo-photothermal treatment of cancer.
Introduction
Biocompatible Fe3O4@Au nanoparticles (NPs), where Fe3O4 are superparamagnetic cores coated with Au NPs, are building agents with multiple functions for early diagnostics and for new noninvasive therapies for previously incurable diseases. This makes them promising for theranostics, where imaging and therapy modalities are integrated within a single platform.1,2 Due to the superparamagnetic features of Fe3O4 cores, the NPs can be driven in biological objects by the external magnetic field gradient, and can thus be used for labeling of biomolecules and for drug delivery.3 Moreover, superparamagnetic Fe3O4 NPs exhibit strong magnetization in external magnetic field, which contribute to magnetic field perturbations. As a result the dephasing of protons is activated, leading to shortening of the T2 time (transverse or spin–spin) relaxation of the neighboring regions.4 Also superparamagnetic iron oxide NPs with ultrasmall size can influence the T1 time (longitudinal or spin-lattice) relaxation.5 This makes Fe3O4@Au NPs applicable as contrast agents in MR images. The presence of Au NPs, which have high X-ray absorption, make Fe3O4@Au NPs also promising for enhancing computed tomography imaging.6 On the other hand, superparamagnetic Fe3O4 NPs are attractive candidates for novel therapeutic approaches for cancer cell selective treatment. Thus, the superparamagnetic Fe3O4 NPs can be applied in magneto-mechanical cancer treatment, where NPs actuated through the use of super low frequency AC magnetic fields and contribute to cytoskeletal disruption and subsequent cell death, can be selectively enacted upon cancerous cells, while leaving healthy cells intact.7 Superparamagnetic Fe3O4 NPs also can be applied for magnetic particle hyperthermia, where the NPs exposed to alternating magnetic fields of relatively high frequencies (∼100 kHz), generate heat through Néel or Brownian relaxation, which leads to temperature increase, causing subsequent damage to the surrounding cells.8Au NPs demonstrate localized surface plasmon resonance,9 based on the collective oscillation of free electrons in conduction bands.10 These properties of gold NPs find an application in a minimally invasive technique, which is called photothermal therapy. In this therapy photothermal agents (gold NPs) convert the laser energy to heat, which kills the cancer cells. Moreover, Au NPs are widely used as a platform for drug delivery, for example cisplatin (cPt).11 In this case, gold NPs with immobilized cPt provide a synergetic effect in cancer treatment.12 And this synergetic effect could also be enhanced in combination with the photothermal therapy described above.13
Combination of magnetic iron oxide and gold NPs in the framework of one compact nanoparticle (below 40 nm), in which superparamagnetic properties of iron oxide NPs and surface plasmonic properties of gold NPs complement each other, can noticeably extend the theranostic potential of the system. However, attaching Au NPs to Fe3O4 is a challenging problem. One of the effective strategy, employs an intermediate adhesion layer of amino-modified silica, which coats the Fe3O4 NPs, and to which Au NPs are attached.14,15 The latter strategy allows to use hydrophobic iron oxide NPs prepared through thermal decomposition in non-aqueous media, which are nearly monodispersive.16,17 Silica shells also can effectively enhance biocompatibility, prevent corrosion and agglomeration of Fe3O4 NPs and improve their stability in aqueous solution. The presence of silanol functional groups on the surface of the core–shell Fe3O4@SiO2 NPs makes them dispersible in polar solvents such as water, and also provides easy further modification with other functional groups, thus enabling coupling with different drugs and biomolecules. The immobilization of cPt on the nanoparticles surface, can increase the therapeutic influence of the drug and make its impact more controllable. However, the combination all of the three components in one system is a challenging problem.
Here we design novel, targeted cPt-based Fe3O4@SiO2@Au NPs (Fig. 1), which will have an improved potential in magnetic resonance imaging (MRI)-guided chemo-photothermal stimulated treatment of cancer. For this purpose, our magnetic NPs were functionalized by cPt, a metallic platinum coordination compound with a square planar geometry, which shows high anticancer activity in a variety of tumors, and which is widely used in chemotherapy medication to treat a number of cancers.18 For the theranostic application, we tested the effectiveness of the NPs as contrast agents for MRI and investigated in vitro the chemo-phototherapy potential of the Fe3O4@SiO2@Au nanosystem on two colon cell lines: SW480 and SW620 in terms of cell viability (MTT assay) and morphological changes induced in tumor cells (optical microscopy). Because near-infrared (NIR) radiation can penetrate into biological tissues several centimeters deep, we chose a laser with an 808 nm wavelength, which is employed in photothermal therapy in the majority of cases.19 The morphology, structure and magnetic properties of the Fe3O4@SiO2 and Fe3O4@SiO2@Au nanosystems have been investigated using transmission electron microscopy (TEM), X-ray diffraction (XRD), Mössbauer spectroscopy and SQUID-magnetometry. The stability of the functionalized Fe3O4@SiO2@Au NPs and the verification of the effective process of biofunctionalization and cPt immobilization have been carried out using differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and Raman spectroscopy.
 | ||
| Fig. 1 Scheme of the Fe3O4@SiO2@Au NPs biofunctionalization process and process of the cPt immobilization. | ||
Experimental section
Materials
Tetraethyl ortho-silicate (TEOS, 99.0%), (3-aminopropyl)triethoxysilane (APTES, 99%), polyoxyethylene (5) nonylphenylether (Igepal CO-520), tetrakis(hydroxymethyl)phosphonium chloride (THPC, 80% solution in H2O), hydrogen tetrachloroaurate(III) hydrate (HAuCl4·3H2O, 99.9% metals basis), 16-mercaptohexadecanoic acid (16-MHDA), dimethylformamide (DMF), pentafluorophenyl (PFP), N,N-diisopropylethylamine (DIPEA) and N-cyclohexyl-N-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMC) were purchased from Sigma Aldrich (USA). Ferric chloride hexahydrate (FeCl3·6H2O, 99%), sodium oleate (NaOA, 95%) were purchased from Carl Roth. 1-Octadecene (ODE, tech. 90%), oleic acid (OA, tech. 90%), potassium carbonate (K2CO3, 99.0%) were purchased from Alfa-Aesar Chemicals. Cyclohexane (C6H12, 99.5%), hexane (C6H12, 97.0%), acetone (C3H6O, 99.5%), ammonia solution (NH3·H2O, 25%), chloroform (CHCl3), ethanol (C2H5OH, 96%) were bought from POCH. Ultrapure water was used throughout the experiments. All the chemicals were used as received without further purification.Synthesis of Fe3O4@SiO2@Au NPs
The process of synthesis involved three stages: (a) the synthesis of hydrophobic Fe3O4 NPs, (b) encapsulation of the Fe3O4 NPs in an SiO2 shell and (c) attaching the Au NPs to the Fe3O4@SiO2.Biofunctionalization of the Fe3O4@SiO2@Au NPs and immobilization of cPt on their surface
In order to attach the drugs to the stabilized Fe3O4@SiO2@Au NPs, 16-MHDA was used as a linker. This chemical compound has 16 carbon atoms, a thiol group (–SH) on one side and a carboxyl group on the other side. Thanks to the thiol group, the 16-MHDA is linked with the NPs surface and the COOH group is linked with cPt. Moreover, using this linker, the active surface of Au NPs was not coated, as it is in the case when e.g. polyethylene glycol (PEG) shell was used. First, the NPs were incubated with MHDA for overnight at 4 °C. After rinsing with DMF, the MHDA-covered NPs-structures were incubated in DMF solution of PFP, DIPEA and CMC, during 30 min at 25 °C. After repeated rinsing with DMF and centrifugation, a cPt solution was added and incubated for 30 min at 25 °C. The scheme of functionalization (biofunctionalization and immobilization of cPt on their surface) of Fe3O4@SiO2@Au NPs is shown in Fig. 1.TEM characterization
The morphology of the fabricated NPs was investigated by scanning transmission electron microscopy (STEM) using the high-angle annular dark-field detector (HAADF). Selected area electron diffraction (SAED) patterns were taken in the TEM mode. Energy dispersive X-ray spectroscopy (EDS) was used to analyze the chemical composition of the synthesized NPs. All these measurements were performed on an aberration-corrected FEI Titan electron microscope operating at 300 kV equipped with a FEG cathode. The particle size distribution was evaluated based on the HRSTEM images taken from different areas of the TEM grids.X-ray diffraction
The crystal microstructure of the fabricated NPs was analyzed using a two-circle laboratory diffractometer Panalytical X'Pert Pro. The measurements were performed in the standard θ–2θ geometry using Cu Kα radiation (λ = 0.15406 nm). The dispersion of the NPs was dried on a zero-background holder and was placed on a sample spinner with rotation time adjusted to 16 seconds. The data were collected in the range between 25–85° (2θ) with a step size of 0.08°.Mössbauer spectroscopy
Mössbauer spectra were recorded in transmission geometry at room temperature and at 16 K using the spectrometer MS4 (SeeCo, USA) with a 57Co/Rh source (12 mCi). Low temperature measurements were performed using a closed cycle refrigerator system CCS-850 (Janis Research Company, USA). The temperature was controlled using a Lakeshore temperature controller LS335 (Lake Shore Cryotronics, Inc., USA) with dual calibrated DT-670 sensors with an accuracy of ±0.005 K. The spectra were fitted using the MOSMOD software, assuming a Gaussian distribution of the hyperfine magnetic field (Hhf) and quadrupole splitting within the iron nuclei.22 Lorentzian line shape of source natural line width was determined from the Mössbauer spectrum of a pure 28 μm thick α-Fe foil. The spectrometer was calibrated to pure α-Fe by collecting the spectra of a standard α-Fe foil at room temperature and 16 K. All isomer shifts (IS) are presented in respect to an α-Fe standard.Magnetometry
A SQUID-magnetometer (MPMS XL, Quantum Design) was used to investigate the magnetic properties of iron-oxide NPs. The zero-field cooled (ZFC) temperature dependences of the magnetization were measured by cooling the sample from 300 K to 5 K and then applying a field of 100 Oe to record the magnetization, while the sample was heated from 5 K to 300 K. The field cooled (FC) measurements were made with a field of 100 Oe applied during the cooling process. The hysteresis curves were measured at 5 K and 300 K, respectively.UV-Vis measurements
The UV-Vis measurements were performed with a Lambda Bio20 instrument from PerkinElmer. The resolution was chosen to be 1 nm and the scan speed was 240 nm min−1. The spectral range was from 200 nm to 800 nm.MRI
To investigate the effectiveness of the NPs as contrast agents, MR imaging as well as relaxometry were performed using a 9.4T Bruker Biospec 94/20 MRI scanner. For the acquisition of the series of axial images of each sample, the RARE with variable repetition time TR (RAREVTR) and the MSME imaging sequences were used. Application of RAREVTR sequence allows to obtain simultaneously T1 and T2 relaxation times maps of samples with different concentrations of the NPs. MSME sequence was employed in order to increase the number of echoes and therefore to obtain better quality of T2 measured and T2-weighted images. For acquired T1 and T2 values, linear regression of relaxation rates: R1 = T1−1 and R2 = T2−1 vs. NPs concentration in samples was performed:| R1,2 = R1S,2S + r1,2C | (1) |
FTIR measurements
The Fourier transform infrared absorption (FT-IR) spectra in the wavelength range between 400–4000 cm−1 were acquired using an EXCALIBUR FTS-3000 spectrometer at room temperature and measured for the respective samples mixed with KBr. The sample was dried and sandwiched between two KRS-5 window disks. The 64 scans were averaged at a resolution of 4 cm−1. During the experiments, the spectrometer was purged with dry nitrogen. Baseline correction and normalization of FTIR spectra were applied.Calorimetric and TGA measurements
Calorimetric measurements were carried out under dry 5.0 pure nitrogen purge (25 mL min−1) on a TA Instruments' DSC 2500 differential scanning calorimeter equipped with a liquid nitrogen LN2P cooling pump. Thermal behaviour of samples placed in aluminium pans and crimped with hermetic lids, was studied in a temperature range from 5 °C to 250 °C with a maximum heating rate of 5 °C min−1. Calibrations of the temperature and enthalpy were performed using an indium standard. Additionally, modulated differential scanning calorimetry was employed to gain more thorough insight into thermal properties of the specimens. TGA experiments were carried out under flowing synthetic air (N5.0) on a TA Instruments' TGA 5500 high-resolution thermogravimetric analyser using platinum 100 μL pans with a maximum heating rate of 5 °C min−1. The specimens were either directly placed in open 100 μL platinum pans or previously enclosed inside aluminium pans to limit the evaporation rate and measured in a temperature range from the ambient temperature to 500 °C. The temperature calibration was carried out using nickel and alumel standards. Additionally, high resolution (HiRes TGA) mode was employed to gain more precise values of the experimental points.FT-Raman spectroscopy
FT-Raman spectra were recorded using a Nicolet NXR 9650 FT-Raman Spectrometer equipped with an Nd:YAG laser (1064 nm) and a germanium detector. The measurements were performed in the range from 150 cm−1 to 3.700 cm−1 with a laser power of 1 W. The unfocused laser beam was used with a diameter of approximately 100 μm and a spectral resolution of 8 cm−1. Raman spectra were processed by the Omnic/Thermo Scientific software based on 128 scans.Cell lines
As the in vitro model, two human colon cancer cell lines (SW480 and SW620) were used, which were obtained due to the courtesy of Prof. Caroline Dive, Paterson Institute for Cancer Research, University of Manchester. These cell lines were cultured in DMEM with high glucose (Corning, NY, USA) in a 37 °C humidified atmosphere with 5% CO2. All media were supplemented with 10% fetal bovine serum (FBS, Biowest, Nuaille, France) and gentamicin (50 μg mL−1), (PAN-Biotech, Aidenbach, Germany). The cells were cultured by bi-weekly passages and were regularly tested for Mycoplasma sp. contamination by PCR-ELISA kit (Roche, Mannheim, Germany) according to the manufacturers' instruction.Light microscopy images of cells
The images of cells at 100× magnification were taken using Microscope Olympus IX70 (Olympus Corporation, Tokyo, Japan).Light source and cells irradiation protocols
The irradiation of the cells was conducted by a low-intensity laser operating at 808 nm wavelength. An adjustable power supply was connected to the setup to enable control of the power output of the laser with intensity of 100 mW cm−2 and irradiation time of 5 minutes. The description of the all samples, which were studied in this experiment, is presented in Table 1.| Sample | Name of the samples in the manuscript |
|---|---|
| Control samples of SW480 and SW620 cell lines (cells without addition of other substances and without laser irradiation) | Ctrl |
| Cells cultured with Fe3O4@SiO2@Au NPs | C@NPs |
| Cells cultured with 5 μM of cisplatin | C@cPt |
| Cells cultured with functionalized Fe3O4@SiO2@Au NPs | C@cPtNPs |
| Control samples irradiated by the 808 nm laser | C@808 |
| Cells cultured with Fe3O4@SiO2@Au NPs and irradiated by the 808 nm laser | C@808NPs |
| Cells cultured with 5 μM cisplatin and irradiated by the 808 nm laser | C@808cPt |
| Cells cultured with functionalized Fe3O4@SiO2@Au NPs and irradiated by the 808 nm laser | C@808cPtNPs |
MTS assay
Cytotoxic activity of NPs, cPt, laser irradiation and combination of these three factors against human colon cancer cells (SW480 and SW620) was determined using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, Madison, WI). Briefly, the cells were cultured in flat-bottom 96-well plates (Sarstedt, Numbrecht, Germany) at a density 1 × 104 per well in DMEM medium containing 10% FBS. After 24 h, 20 μL of 50 μg mL−1 functionalized Fe3O4@SiO2@Au NPs as well as non-functionalized solutions were added to the cells. After additional 24 hours of culture, 20 μL of MTS (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega) dye solution was added per well and incubated for 1.5 h. The quantity of formazan product, directly proportional to the number of living cells in culture, was detected by absorbance measurements at 490 nm with a 96-well plate reader (Spark® Tecan, Mannedorf, Switzerland).Analysis of cell viability data
The obtained MTS assay results are represented as the means ± SEM (the standard error of the mean). The quantitative results were finally compared with the T test. P value < 0.05 was considered to be statistically significant. Moreover, in the MTS test, data for each treatment point shows an average from three parallel wells. The data were analyzed and presented graphically using the Origin Lab2019b software.Results and discussion
Characterization of nanoparticles
The three stages of Fe3O4@SiO2@Au NPs synthesis are schematically represented in the Fig. 2a. In the first stage, synthesis of hydrophobic monodisperse spherical Fe3O4 NPs was made by thermal decomposition of iron precursor using oleic acid as surfactant.16,17 The purpose of the second stage was encapsulating of the Fe3O4 NPs by a full SiO2 shell by reverse microemulsion method.20 At this stage two subsequent ligand exchange reactions occurred on the surface of the Fe3O4 NPs. At first, the ligand exchange was between chemically adsorbed oleic acid and surfactant Igepal CO-520, and as a result Igepal CO-520 was chemically adsorbed on the Fe3O4 NPs surface. Then, TEOS was added to the mixture where it hydrolyzed, performed ligand exchange with Igepal CO-520 and chemically adsorbed on the Fe3O4 NPs surface. The adsorbed TEOS forms an SiO2 shell after its condensation process. At the end of the second stage, the surface of the core–shell NPs was functionalized with amino groups using APTES, where the zeta potential value of the NPs surface was increased to 40 mV allowing thus for sequential adsorption of Au NPs on the surface of Fe3O4@SiO2 NPs was allowed.15 At the third stage Au NPs were fabricated by direct reduction method, where HAuCl4·3H2O was used as gold precursor and tetrakis (hydroxymethyl)phosphonium chloride (THPC) as reducing agent.21,23 The fabricated Au NPs were mixed with the solution of functionalized Fe3O4@SiO2 and adsorbed on their surface. As a result, Fe3O4@SiO2@Au NPs were obtained. The morphology investigation and chemical characterization of the NPs at every stage of the synthesis were made by transmission electron microscopy (Fig. 2). In Fig. 2b–d we present STEM HAADF images of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@Au NPs, respectively. In Fig. 2e, the SAED pattern for the Fe3O4 NPs after synthesis is shown. The EDX maps of Fe3O4@SiO2 NPs and Fe3O4@SiO2@Au NPs are presented in Fig. 2f and g. Moreover, the distributions of Fe3O4 NPs size, SiO2 shell thickness and Au NPs size obtained from the analysis of STEM HAADF images can be found in Fig. 2h–j. As it can be seen from Fig. 2b and h in the first stage of synthesis, monodispersed Fe3O4 NPs with the average diameter of 11 nm were fabricated. The diffraction rings on the SAED pattern, Fig. 2e, were indexed with the planes corresponding to inverse spinel crystal structure typical for Fe3O4.23 The successful and uniform encapsulation of Fe3O4 NPs by the SiO2 shell in the second stage of the synthesis is presented in Fig. 2c and f, where a well-defined core–shell structure with one iron-oxide core surrounded by a homogeneous SiO2 shell is observed. The average thickness of the shell is around 10 nm (Fig. 2i). In the third stage the Fe3O4@SiO2 core–shell NPs were decorated with Au NPs with an average size of 4 nm, Fig. 2d, g and j. From Fig. 2d and g, it can be seen that every Fe3O4@SiO2 core–shell nanoparticle has a few Au NPs on its surface.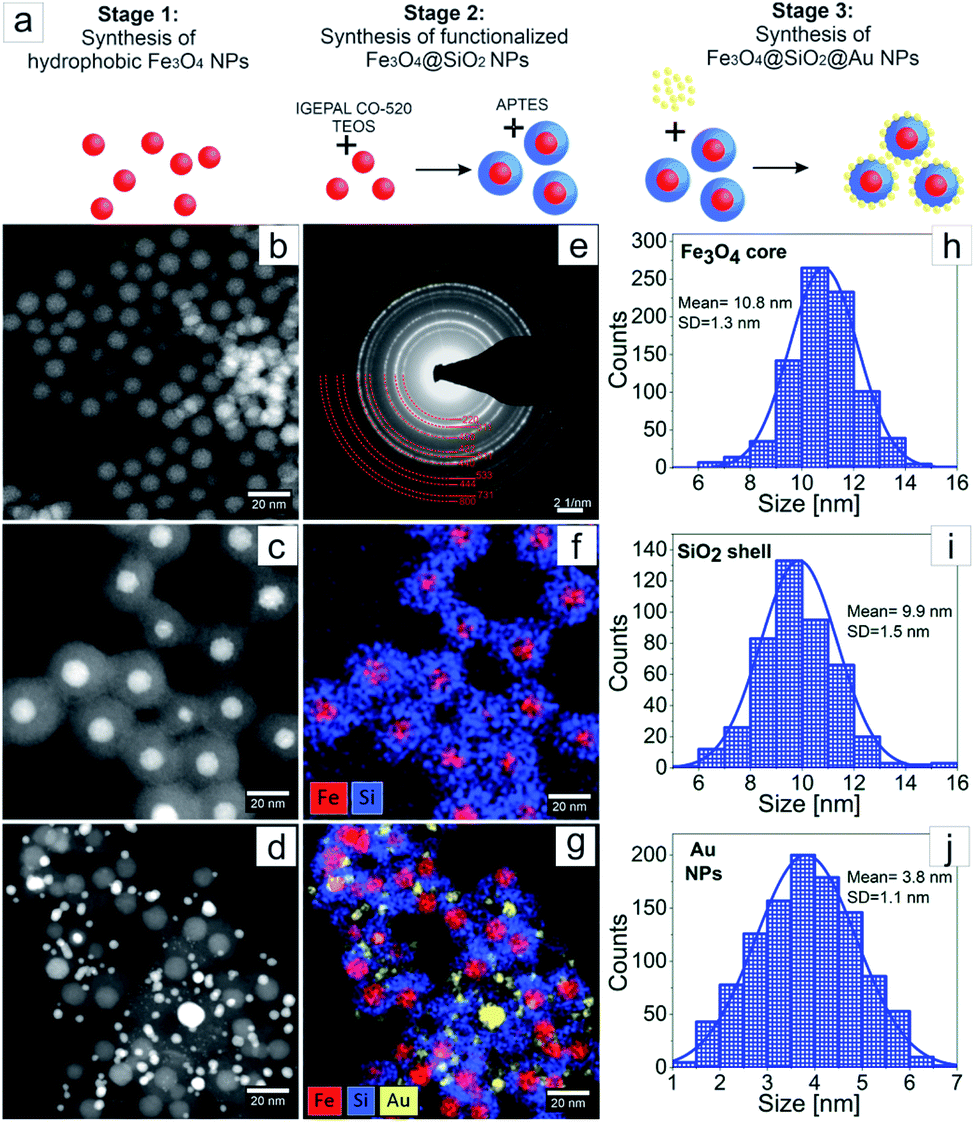 | ||
| Fig. 2 Schematic diagram of the three stages of Fe3O4@SiO2@Au NPs synthesis (a); HAADF STEM overview of Fe3O4 (b), Fe3O4@SiO2 (c) and Fe3O4@SiO2@Au (d) NPs; SAED pattern of Fe3O4 NPs (e); EDX elemental maps of Fe, Si, and Au distribution in Fe3O4@SiO2 (f) and Fe3O4@SiO2@Au (g) NPs; the distributions of: Fe3O4 NPs size (h), SiO2 shell thickness (i) and Au NPs size (j). | ||
The UV-Vis absorption spectra of different Fe3O4 NPs with and without a SiO2 shell as well as Fe3O4@SiO2 decorated by Au NPs are presented in Fig. 3. In the UV-Vis spectrum of pure Fe3O4 NPs, no obvious peaks were observed. However, encapsulation of the magnetic NPs in a 10 nm SiO2 shell, caused the appearance of a peak at 250 nm in the spectra of the Fe3O4@SiO2 NPs. This peak corresponds to the created core–shell structure of the NPs, and may originate from the changes of the band gap caused by the quantum size effect and surface effect of nanostructures,24 as well as from the Fe–O–Si bonds of the core–shell NPs.25 Moreover, decorated Fe3O4@SiO2 NPs with 4 nm Au NPs caused the appearance of a broad peak around ∼540 nm, which originates from the gold NPs.26 Interestingly, the peak at 250 nm, which was visible in the UV-Vis spectrum of Fe3O4@SiO2, was shifted towards lower wavelengths. This can suggest, that the synthesis of Au NPs on the Fe3O4@SiO2 NPs surface caused changes in the Fe–O–Si bonds, which are, as most number of chemical bonds, sensitive for the local environmental changes.25
 | ||
| Fig. 3 UV-Vis spectra of: Fe3O4 NPs (black colour), Fe3O4@SiO2 NPs (red colour) and Fe3O4@SiO2@Au (blue colour). | ||
In the FTIR spectra of pure Fe3O4 NPs, Fe3O4@SiO2 NPs and Fe3O4@SiO2@Au (Fig. 4) a peak at 590 cm−1 corresponding to Fe3O4 is observed. Moreover, peaks at 1110 cm−1, 920 cm−1, 790 cm−1, 670 cm−1 originating from FeOH are noticed. Furthermore, peaks characteristic for γ-FeO(OH) and α-FeO(OH) are visible in FTIR spectra of Fe3O4 NPs at 1026 cm−1, 1161 cm−1, 753 cm−1 and 890 cm−1, 797 cm−1 wavenumbers, respectively.27 In the FTIR spectra of Fe3O4@SiO2 NPs and Fe3O4@SiO2@Au a peak at 1560 cm−1 corresponding to C–N vibration is visible. The presence of these peaks indicates that the Fe3O4 NPs were successfully modified by the SiO2 groups, thus forming Fe3O4@SiO2 nanoparticles.28
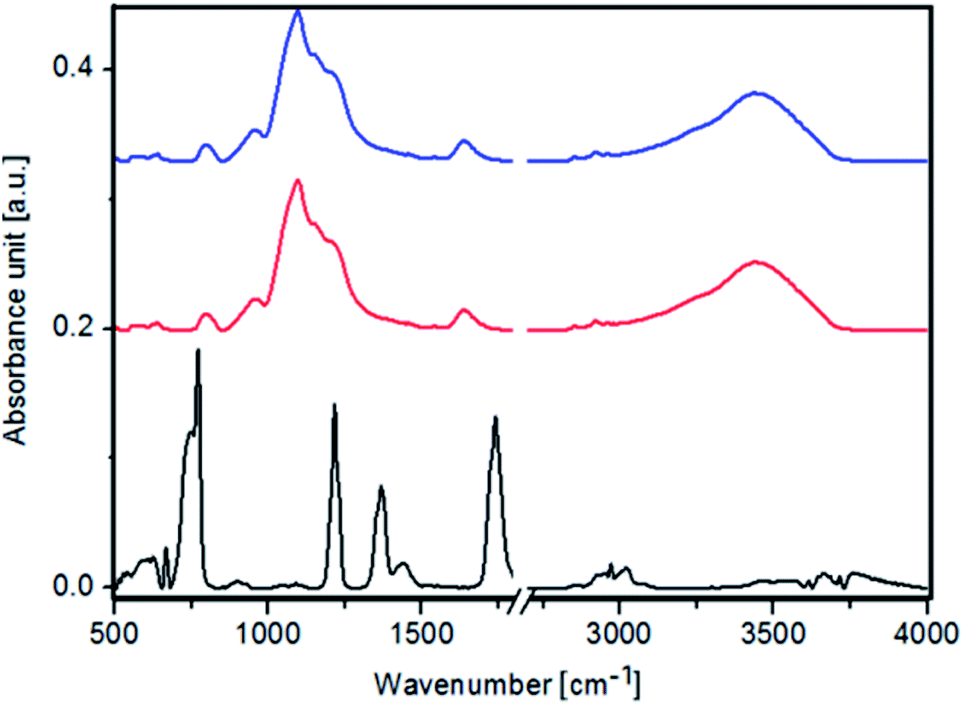 | ||
| Fig. 4 FTIR spectra of: Fe3O4 NPs (black color), Fe3O4@SiO2 NPs (red color) and Fe3O4@SiO2@Au (blue color). | ||
The crystalline structure of the NPs fabricated at every stage of the synthesis was investigated using XRD (Fig. 5). The XRD pattern of Fe3O4 was refined with the Rietveld method using the Fullprof software,29 where for peaks fitting a Voigt function in the Thompson-Cox-Hastings approximation was applied.30 The prominent peaks at 30.17° (220), 35.54° (311), 43.20° (400), 53.59° (422), 57.13° (511), 62.74° (440) and 74.23° (533) matched the magnetite phase. The calculated lattice constant in the Fullprof program was a = 8.361(1) Å. The obtained value is the average value of the lattice constant for bulk magnetite (a = 8.39 Å)31 and for bulk maghemite (a = 8.33 Å),32 what is typical for Fe3O4 NPs.33 The XRD pattern of the Fe3O4@SiO2 core–shell NPs looks almost the same as the pattern for pure Fe3O4 NPs, except the broad peak, which originates from the amorphous SiO2. The XRD pattern of the Fe3O4@SiO2@Au NPs contains extra wide peaks, which can be assigned to the (111), (200), (220), (311) and (222) diffraction peaks of small Au NPs with fcc structure.34 This allows to suggest that the encapsulation process of Fe3O4 in the SiO2 shell has no influence on the initial structure of the Fe3O4 NPs, which does also not change during the Au NPs deposition, due to good protection function of the SiO2 shell.35
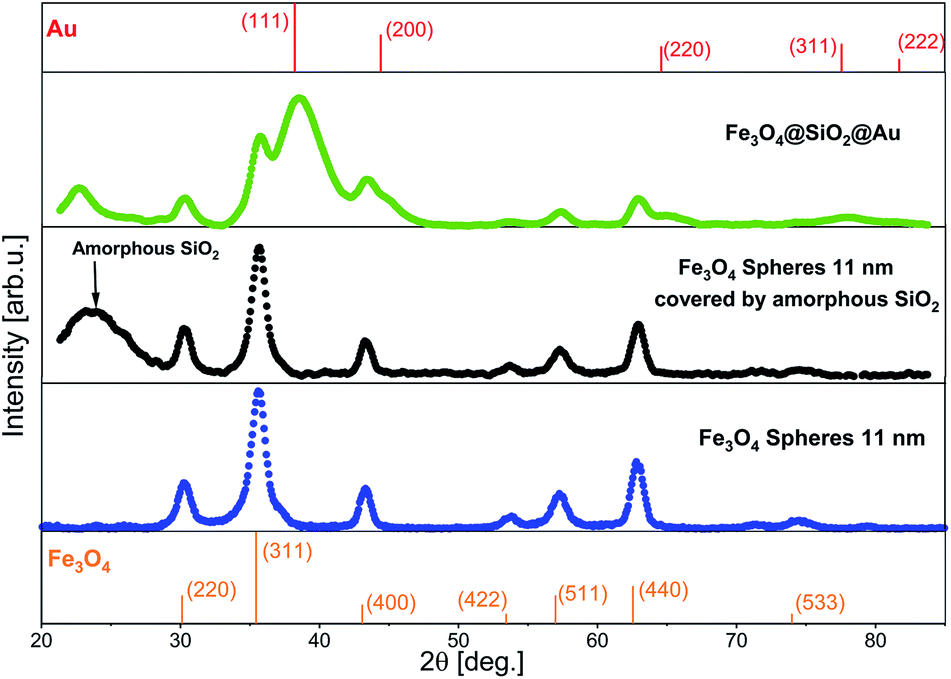 | ||
| Fig. 5 XRD patterns of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@Au NPs. On the top and on the bottom of the image standard references of Au and of Fe3O4 are added.36,37 | ||
In order to ensure that the encapsulation into the SiO2 shell has no significant influence on the initial magnetic properties of the Fe3O4 cores, and to evaluate the application potential of the investigated nanosystems for magnetic resonance imaging, we made magnetometry of the pure Fe3O4 NPs and the Fe3O4@SiO2 core–shell NPs at room temperature (300 K) and at 5 K (Fig. 6a and b). As seen from the TEM images (Fig. 2), the SiO2 shell entirely covers the nanoparticle and protects the iron-oxide core from the environment, preventing its further oxidation. This means that possible changes of magnetic properties of the NPs may occur only at the stage of Fe3O4 NPs encapsulation into SiO2 shell, when the iron-oxide core can be oxidized. Also, as can be seen from Fig. 2b and c, the SiO2 shell is the main reason for the relative decrease of iron oxide content in the particles, which will influence the total value of saturation magnetization of the NPs. It was found that at 300 K, pure and core–shell NPs exhibited superparamagnetic behavior with no evident coercivity or remanence.38 For Fe3O4@SiO2 NPs saturation magnetization is lower (MS(300 K) = 15 emu g−1) compared to pure NPs (MS(300 K) = 46 emu g−1). The reduced saturation magnetization of the Fe3O4@SiO2 NPs, resulting in decreased magnetic anisotropy and attributed to the relative decrease of iron oxide content in the particles, is mainly explained by the presence of the silica shell coating.39 It also worth mentioning that in the case of pure Fe3O4 NPs, the measured values of MS can be reduced by the presence of the protective shell of oleic acid, which prevents the interaction of iron-based NPs with the atmosphere.33,40 The temperature dependence of the magnetic moment M(T) of the Fe3O4 and Fe3O4@SiO2 NPs under an applied magnetic field (Hext = 100 Oe) after zero-field cooling (ZFC) and after field cooling (FC) was measured from 300 K to 5 K (Fig. 6c). This dependence allows to determine the blocking temperature (TB) of the NPs, being the temperature above which the majority of the particles becomes superparamagnetic, and above which the FC and ZFC magnetization curves coincide. For Fe3O4@SiO2 NPs TB = 88 K, while for the pure Fe3O4 NPs TB is higher (TB = 132 K). The smaller value of TB for the encapsulated Fe3O4 NPs can be explained by the reduced dipole–dipole interaction between the iron-based cores of the Fe3O4@SiO2 NPs in comparison to pure Fe3O4 NPs. Likewise, very interesting results were obtained from FC (Hext = 50 kOe) magnetization hysteresis loops recorded at 5 K for pure Fe3O4 and Fe3O4@SiO2 NPs. As expected at this temperature, the hysteresis loops are opened and the NPs are in ferromagnetic state. Also the hysteresis loops have a clear negative shift from the origin along the field axis and evidence the presence of an exchange bias field (HEB), which originates from the interface exchange coupling between ferro(ferri) (FM)/antiferromagnetic (AFM).41 The presence of the exchange bias field may result in higher TB values and coercivity fields (HC), in comparison to particles, where such fields are absent. For pure Fe3O4 NPs the values HEB = 46 Oe, HC(5 K) = 287 Oe are close to the values HEB = 43 Oe, and HC(5 K) = 268 Oe for Fe3O4@SiO2 NPs (see the insets in Fig. 6a and b). Chalasani et al. showed that in Fe3O4 NPs produced by the thermal decomposition method, the exchange bias fields originate from the presence of trace amounts of wustite (FeO) and thus this higher content of FeO is responsible for the larger exchange bias fields.42 The matching of the values HEB and HC allows to conclude that encapsulation of Fe3O4 cores by the SiO2 shells does not influence the structure of the Fe3O4 cores. As it can be expected, the attachment of small Au gold NPs does not influence the magnetic properties of the Fe3O4 NPs and Fe3O4@SiO2@Au NPs can be easily attracted by a magnet (Fig. 6d).
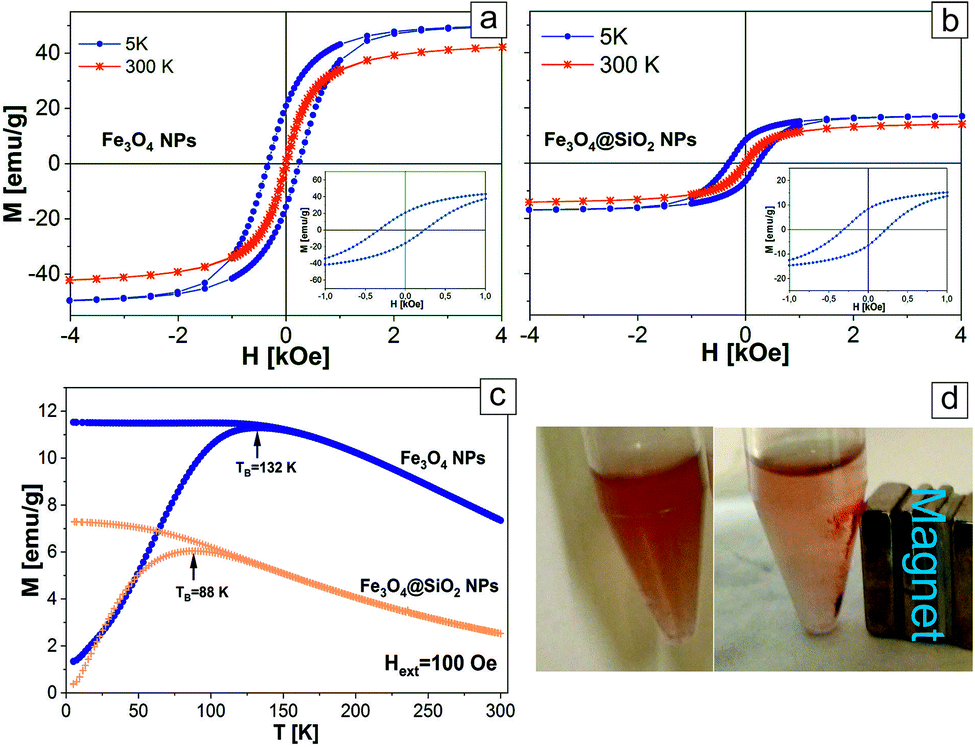 | ||
| Fig. 6 The hysteresis loops of pure Fe3O4 NPs (a) and Fe3O4@SiO2 NPs (b) measured at 5 K and 300 K. The insets are an expanded view of the low-field region; (c) ZFC-FC curves of pure Fe3O4 NPs and Fe3O4@SiO2 NPs; (d) Fe3O4@SiO2@Au NPs without magnetic field (left) and attracted by the magnet (right). | ||
For local structure characterization of the as fabricated Fe3O4 cores and after their encapsulation in the SiO2 shell, Mössbauer spectroscopy measurements were performed. Fitted 57Fe Mössbauer spectra are shown in Fig. 7, while hyperfine parameters of sub-spectra and their relative contribution (area ratio) are summarized in Table 2. It is established by Chlan et al.43 and Senn et al.,44 that above the Verwey transition temperature (125 K), stoichiometric magnetite has a cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry with inverse spinel cubic structure. The cell unit (Fe3+)A (Fe3+Fe2+)B consists of Fe3+ ions at the A-site (tetrahedral) and Fe2+ ions at the B-site (octahedral). At tetrahedral sites, Fe atoms are in a +3 state, while in B positions the mean valence is +2.5. Mössbauer spectra of the as fabricated Fe3O4 core samples and after their encapsulation in the SiO2 shell at room temperature (Fig. 7a and c), are characterized with singlet IS = 0.32 mm s−1, without resolved quadrupole splitting corresponding to Fe3+ ions. The observed broadened baseline in spectrum of NPs, is most probably caused by the presence of an unresolved magnetic sextet with weighted average isomer shift ∼0.6 mm s−1 corresponding to Fe2.5+ ions, which indicates a superparamagnetic state. At low temperatures, the structure of magnetite is monoclinic with a Cc space group symmetry.44 After Verwey transition, the unit cell becomes four times bigger than cubic, doubled in the c direction, and with Fe ions residing in 16 B inequivalent positions (8 Fe3+ and 8 Fe2+) and 8 A inequivalent positions.43 The spectrum of Fe3O4 NPs (Fig. 7b) obtained at 16 K, is deconvoluted into three sextets with parameters corresponding to Fe3+ ions at A site (IS = 0.21 mm s−1 and Hhf = 51.1 T), Fe3+ ions at B site (IS = 0.58 mm s−1 and Hhf = 51.7 T) and Fe2+ ions at B site (IS = 0.96 mm s−1 and Hhf = 48.8 T), which is in good agreement with the values presented in ref. 43. It is worth mentioning, that according to the results of spectra approximation (Table 2), the ratio of the contribution of sextet of Fe3+ ions at A site to the contribution of sextets of Fe ions at B site is 0.56, which is very close to the reference value of stoichiometric magnetite.45,46 At the same time the relative contribution of subspectrum attributed to Fe2+ ions at B site is less than expected (only 17% against ∼30% expected). The lower contribution of Fe2+ ions in Mössbauer spectrum could be due to the partial oxidation of Fe3O4 NPs leading to the formation of some amount of maghemite or wustite at the surface of NPs. The Mössbauer spectra of the Fe3O4 cores after their encapsulation in the SiO2 shell (Fig. 7d) are very similar to those of the Fe3O4 NPs, but with slightly smaller values of IS and Hhf. As it was mentioned by Lyubutin et al.,47 the shell material can induce pressure on the NPs and this effect can change the Mössbauer parameters. From the other side, the shift of IS and Hhf values may be attributed to the formation of interfacial structure.48 The above-mentioned results confirm that the as-fabricated Fe3O4 NPs and those encapsulated by the SiO2 shell possess a very similar crystal structure.
m symmetry with inverse spinel cubic structure. The cell unit (Fe3+)A (Fe3+Fe2+)B consists of Fe3+ ions at the A-site (tetrahedral) and Fe2+ ions at the B-site (octahedral). At tetrahedral sites, Fe atoms are in a +3 state, while in B positions the mean valence is +2.5. Mössbauer spectra of the as fabricated Fe3O4 core samples and after their encapsulation in the SiO2 shell at room temperature (Fig. 7a and c), are characterized with singlet IS = 0.32 mm s−1, without resolved quadrupole splitting corresponding to Fe3+ ions. The observed broadened baseline in spectrum of NPs, is most probably caused by the presence of an unresolved magnetic sextet with weighted average isomer shift ∼0.6 mm s−1 corresponding to Fe2.5+ ions, which indicates a superparamagnetic state. At low temperatures, the structure of magnetite is monoclinic with a Cc space group symmetry.44 After Verwey transition, the unit cell becomes four times bigger than cubic, doubled in the c direction, and with Fe ions residing in 16 B inequivalent positions (8 Fe3+ and 8 Fe2+) and 8 A inequivalent positions.43 The spectrum of Fe3O4 NPs (Fig. 7b) obtained at 16 K, is deconvoluted into three sextets with parameters corresponding to Fe3+ ions at A site (IS = 0.21 mm s−1 and Hhf = 51.1 T), Fe3+ ions at B site (IS = 0.58 mm s−1 and Hhf = 51.7 T) and Fe2+ ions at B site (IS = 0.96 mm s−1 and Hhf = 48.8 T), which is in good agreement with the values presented in ref. 43. It is worth mentioning, that according to the results of spectra approximation (Table 2), the ratio of the contribution of sextet of Fe3+ ions at A site to the contribution of sextets of Fe ions at B site is 0.56, which is very close to the reference value of stoichiometric magnetite.45,46 At the same time the relative contribution of subspectrum attributed to Fe2+ ions at B site is less than expected (only 17% against ∼30% expected). The lower contribution of Fe2+ ions in Mössbauer spectrum could be due to the partial oxidation of Fe3O4 NPs leading to the formation of some amount of maghemite or wustite at the surface of NPs. The Mössbauer spectra of the Fe3O4 cores after their encapsulation in the SiO2 shell (Fig. 7d) are very similar to those of the Fe3O4 NPs, but with slightly smaller values of IS and Hhf. As it was mentioned by Lyubutin et al.,47 the shell material can induce pressure on the NPs and this effect can change the Mössbauer parameters. From the other side, the shift of IS and Hhf values may be attributed to the formation of interfacial structure.48 The above-mentioned results confirm that the as-fabricated Fe3O4 NPs and those encapsulated by the SiO2 shell possess a very similar crystal structure.
 | ||
| Fig. 7 Mössbauer spectra of the Fe3O4 NPs acquired at 290 K (a) and 16 K (b) and of the Fe3O4@SiO2 NPs acquired at 290 K (c) and 16 K (d). | ||
| Sample (temperature) | Sub-spectrum | IS, mm s−1 | Hhf, T | Hhf-sigma, T | E, mm s−1 | Area ratio, % |
|---|---|---|---|---|---|---|
| Fe3O4 NPs (290 K) | Fe3+/Fe2.5+ | 0.32 | — | — | — | 26 |
| Fe3O4 NPs (16 K) | Fe3+ at A site | 0.21 | 51.1 | 0.14 | 0.04 | 36 |
| Fe3+ at B site | 0.58 | 51.7 | 0.12 | −0.02 | 47 | |
| Fe2+ at B site | 0.96 | 48.8 | 5.5 | −0.24 | 17 | |
| Fe3O4@SiO2 NPs (290 K) | Fe3+/Fe2.5+ | 0.32 | — | — | — | 26 |
| Fe3O4@SiO2 NPs (16 K) | Fe3+ at A site | 0.20 | 49.4 | 0.11 | 0.04 | 35 |
| Fe3+ at B site | 0.38 | 50.8 | 0.07 | −0.04 | 53 | |
| Fe2+ at B site | 0.90 | 47.2 | 6.1 | −0.47 | 12 |
MR imaging of Fe3O4@SiO2
In order to evaluate the theranostic potential of the studied nanoparticles, we investigated the MRI enhancing function of the active element of the nanoparticle (i.e. Fe3O4@SiO2), which can influence the MRI contrast. In this experiment we investigate only the active element of the nanoparticle because it was the only component exhibiting strong magnetic properties, causing local perturbations of the magnetic field, which resulted in signal loss due to significant shortening of the transversal relaxation time T2. In addition, neither Au nanoparticles (see Fig. 2g) nor MHDA linker (see Fig. 1) form a continuous shell around the Fe3O4@SiO2 NPs. As a result, the Au NPs and attached drugs did not interfere to prevent water molecules from possible direct contact with the surface of the active element. Alternative behavior was observed for the case of continuous shell observed for NPs studied, for example, by Hou et al.14 where Fe3O4@SiO2 NPs were covered by 15 nm gold continuous shell. In our case the water molecules were very close to the surface of Fe3O4@SiO2 NPs and the dephasing of protons was activated by magnetic field perturbations in the direct vicinity of the Fe3O4@SiO2 NPs. Thus, for MRI contrasting properties evaluation, we calculated the values of the relaxation rates: R1 and R2 from the measured relaxation times for Fe3O4@SiO2 active element of the NPs. The dependences of the relaxation rates R1 and R2 on the concentration of Fe3O4@SiO2 nanoparticles are presented in Fig. 8a and b respectively. The effect of increasing NPs concentration on the relaxation rate is significantly higher for R2, especially in low concentration range, most interesting for potential clinical application of the theranostic agents. In a concentration range up to 0.1828 mmol L−1, there was a very good linear dependence of nanoparticles concentration on the R2 relaxation rate (R2 above 0.99). For higher concentrations, the effect of T2 shortening was less pronounced, which can be attributed to clustering of NPs, as their concentration in the solution increases.49 This effect of NPs clustering was observed visually in samples with the highest concentration (Fe3O4 concentrations: 0.7311 and 0.3655 mmol L−1), therefore the specific relaxivity r2 was determined only for unclustered NPs. Linear regression results for R2 relaxation rates were: R2S = 1.894 ± 0.033 s−1, r2 = 579.5 ± 4.3 s−1 mmol−1 L. Conversely, the effect of T1 shortening was practically not observed for low NPs concentrations up to 0.0457 mmol L−1, with T1 values characteristic for distilled water, and in higher concentration range, it was much weaker than observed for T2. This effect was confirmed by other papers5,14 and can be attributed to limited access of water molecules to superparamagnetic center (caused by SiO2 coating), which is crucial for r1 enhancement. Only significantly higher concentrations of Fe3O4@SiO2 NPs, provide enough superparamagnetic centers to effectively change the T1 relaxation time of the whole sample.50 Linear regression results for R1 relaxation rate were: R1S = 0.3113 ± 0.0020 s−1, r1 = 0.5665 ± 0.0070 s−1 mmol−1 L, illustrating three orders of magnitude difference between r2 and r1 relaxivities.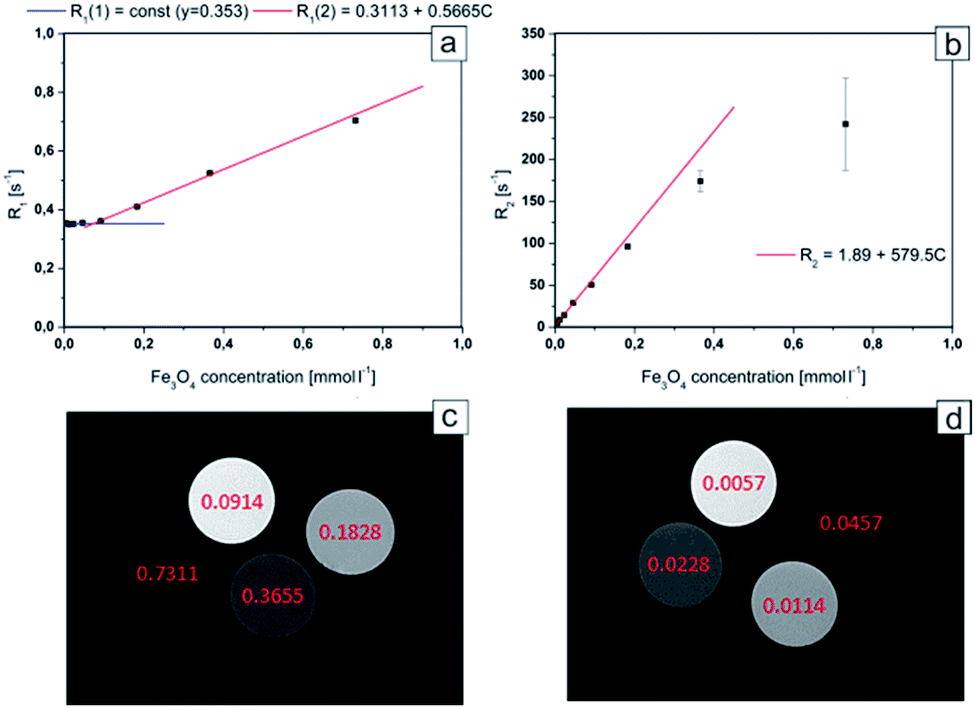 | ||
| Fig. 8 R1 relaxation rate dependence on Fe3O4 concentration for Fe3O4@SiO2 nanoparticles (a); R2 relaxation rate dependence on Fe3O4 concentration for Fe3O4@SiO2 nanoparticles (b); axial images of samples containing different concentration of Fe3O4@SiO2 nanoparticles. MSME sequence: TR: 6000 ms, TE: 8 ms, FA: 180°, FOV: 40 × 40 mm, slice thickness: 2 mm; for TE = 8 ms (c) and 105 ms (d) respectively. Concentrations of Fe3O4 are marked in pictures (in mmol L−1). | ||
The obtained results showed that the examined nanocapsules exhibit a strong effect of R2 relaxation rates. In lower concentration range, which is more interesting in view of possible clinical applications, the effect of T1 relaxation time is very small, and will have negligible effect on the MR image contrast change. Therefore, the investigated Fe3O4@SiO2 nanoparticles, exhibit very strong T2 contrasting properties (“negative” contrast), which is greatly visible in T2-weighted images (see Fig. 8c and d). Here, the MR images for high and low NPs concentration ranges are compared separately, to illustrate the possibility of effective contrasting in both cases. For high concentrations however, much shorter echo time (TE) in MRI pulse sequences have to be applied to get reasonable images.
Stability of the biofunctionalized Fe3O4@SiO2@Au NPs with immobilized cPt on their surface
In order to evaluate the stability of the functionalized Fe3O4@SiO2@Au NPs and to verify the effectiveness of the biofunctionalization and immobilization process, Raman spectroscopy, as well as TGA and DSC analysis, have been performed.In this study, we show a successful biofunctionalization of Fe3O4@SiO2@Au, which is very important for an effective delivery of the drug to cancer cells. For this purpose, Raman spectra of four samples i.e. cPt (Fig. 9, black spectrum), pure MHDA (Fig. 9, red spectrum), the MHDA capped Fe3O4@SiO2@Au NPs (Fig. 9, blue spectrum) and Fe3O4@SiO2@Au NPS + MHDA + cPt (Fig. 9, green spectrum) were acquired. In the Raman spectrum of the MHDA sample a peak of the thiol group (–SH) at 2900 cm−1 is visible.51 The disappearance of this peak in the biofunctionalized and cPt containing samples confirms a successful attachment of the MHDA to the surface of gold NPs. Moreover, in the Raman spectrum of Fe3O4@SiO2@Au + MHDA + cPt (Fig. 9, green spectrum), a peak corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations (1680 cm−1) is observed.52 This group is responsible for linking cPt with biofunctional surfactants on the Fe3O4@SiO2@Au surface.51 Both observations provide an evidence on the success of Fe3O4@SiO2@Au biofunctionalization and cPt immobilization.
O vibrations (1680 cm−1) is observed.52 This group is responsible for linking cPt with biofunctional surfactants on the Fe3O4@SiO2@Au surface.51 Both observations provide an evidence on the success of Fe3O4@SiO2@Au biofunctionalization and cPt immobilization.
 | ||
| Fig. 9 Raman spectra of: cPt (black spectrum), the pure MHDA (red spectrum), the Fe3O4@SiO2@Au + MHDA (blue spectrum), Fe3O4@SiO2@Au + MHDA + cPt (green spectrum). | ||
The results of TGA experiments presented in Fig. 10 a, were measured for liquid functionalized Fe3O4@SiO2@Au NPs and were acquired either directly in an open pan or using a hermetic aluminium container with a hermetic lid, automatically punctured right before loading the sample into the TGA furnace. While the curve registered in the conventional TGA mode shows only strong evaporation of the solvent, the data recorded during the HiRes mode experiment (solid line), indicate two major weight loss regions. First, a small, but still pronounced decrease of mass starts slightly above 93 °C (7.14% weight loss), and the second, much larger one, follows from ca. 103 °C (92.8% weight loss). Most probably both observed effects are due to the conversion of the solvent itself, rather than of the active pharmaceutical ingredient.
 | ||
| Fig. 10 Conventional (dashed line) and HiRes mode (solid line) TGA (a) and DSC (b) experiments performed for functionalized Fe3O4@SiO2@Au NPs. The inset presents components of the heat flow signal registered in MT DSC experiment. | ||
The DSC thermogram registered upon heating is dominated by a broad peak starting around 147 °C, originating from an endothermic process reflecting a decomposition of the sample. Nevertheless, a group of small thermal anomalies is also visible between 95 °C and 122 °C (Fig. 10b). A closer inspection of the heat flow curve measured during the MTDSC experiment (inset in Fig. 10b), revealed one exothermic thermal effect followed by two endothermic ones. Further analysis of the heat flow signal allowed to conclude that these anomalies are strictly kinetic, since they are contributing to the non-reversing (kinetic) component of the measured heat flow.
The results of both TGA and DSC measurements, indicate that the specimen is thermally stable at least up to 90 °C. However, further study of the anomalies revealed during DSC and TGA experiments above 90 °C are necessary and might shed new light on decomposition kinetics of the functionalized Fe3O4@SiO2@Au NPs.
Simulation of combined chemo-phototherapy of colon cancer cells
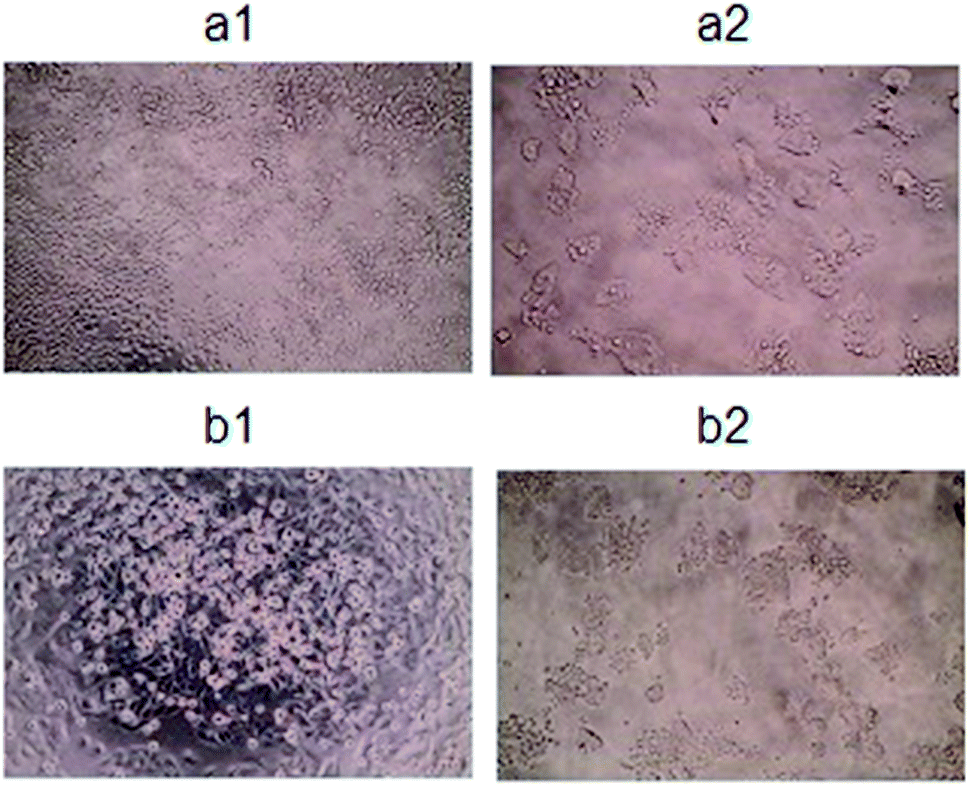 | ||
| Fig. 11 Microscopy images of colon cancer cells (SW480, SW620) morphology: Ctrl (a1 and b1); C@808cPtNPs (a2 and b2, respectively). | ||
 | ||
| Fig. 12 Viability of colon cancer cells: SW480 (a); SW620 (b) after addition of cPt, Fe3O4@SiO2@Au, functionalized Fe3O4@SiO2@Au and laser irradiation in the presence of these three systems. Data was considered as significant when *p < 0.05 vs. control. | ||
Laser irradiation of the cells cultured with cPt caused 12% and 8% mortality of the SW480 and SW620 cell lines, respectively, which is around three and four times higher, than the mortality effect of cells cultured with cPt, but not subjected to irradiation.
Cells cultured with non-functionalized Fe3O4@SiO2@Au NPs and irradiated by laser, exhibited a very high mortality in comparison with other samples. This effect was caused by the Au NPs decorating the Fe3O4@SiO2 NPs. Gold NPs show photothermal effects of plasmon-resonance, which depend, among others, on the NPs size. Therefore, it can be assumed, that Au NPs were used as photosensitizers in this study. The temperature effects, correlated with the nanoparticle sizes were theoretically explained by Jain et al.63 They showed the relationship existing between the size of the Au NPs diameter and the optical absorption or scattering properties of these nanoparticles using the Mie theory. Indeed, for the gold nanoparticles with ∼20 nm diameter, the main contribution to the total extinction of light was absorption. Interestingly, with the increase of the nanoparticle size up to ∼40 nm, the values of light extinction were dependent on the scattering effects. When the particle size was ∼80 nm, the extinction of gold nanoparticles was caused by both, absorption and scattering.64 Moreover, Jiang et al. calculated, using experimental data as well as the Mie theory, the dependence between the particle size and photothermal conversion efficiency of spherical gold nanoparticles and they showed, that smaller nanoparticles can more efficiently transduce the energy from light to heat than larger Au NPs.65 The highest, ∼50% and ∼43% mortality of the SW480 and SW620 cells were observed in the case of C@808cPtNPs samples. This could be caused by synergistic effect of both components cPt and Au NPs, as well as the enhancement by laser irradiation.66
Conclusions
In conclusion, a new type of compact Fe3O4@SiO2@Au nanoparticles (below 40 nm in size) for theranostic applications was designed, synthesized and investigated. The fabricated nanoparticles were uniform, well dispersed in water and had low distribution in sizes of the spherical Fe3O4 core (≈11 nm), the thickness of SiO2 shell (≈10 nm), and Au nanoparticles (≈4 nm) deposited on SiO2 shell. The investigated nanoparticles showed pronounced superparamagnetic properties at room temperature with high saturation magnetization, which is caused by Fe3O4 core. The superparamagnetic state was confirmed by Mössbauer spectroscopy, which showed a short relaxation time of the NPs magnetic cores. It was found that the structure and magnetic properties of the Fe3O4 core were stable and were very similar before and after their encapsulation in the SiO2 shell, which stabilized them and made dispersible in water. The studied nanoparticles exhibited very strong T2 contrasting properties, which is nicely visible in T2-weighted images, and which made the nanoparticles applicable in diagnostics, as contrast agents in MRI. The investigated nanoparticles also demonstrate low toxicity, because of the biocompatible SiO2 continuous shell, which reliably prevents the contact of the Fe3O4 magnetic core with the environment. Moreover, functionalized nanoparticles showed a synergetic effect of investigated nanoparticles and anticancer drugs in chemo-photothermal stimulated treatment of cancer. All of the as mentioned properties of the nanoparticles showed their potential in magnetic resonance imaging (MRI)-guided chemo-photothermal stimulated treatment of cancer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Monika Kula PhD is acknowledged for the use of the Raman and UV-Vis instrument located at The Franciszek Górski Institute of Plant Physiology, Polish Academy of Sciences. Partial financial support by Pik-Instruments is greatly acknowledged. Also, the authors acknowledge prof. Dr hab. Jerzy W. Mietelski and Dr Barbara Petelenz from the Institute of Nuclear Physics Polish Academy of Sciences for making NPs fabrication process possible. Natalia Łopuszyńska acknowledges the fellowship with the project InterDokMed no. POWR.03.02.00-00-I013/16.References
- Y. Ma, J. Huang, S. Song, H. Chen and Z. Zhang, Small, 2016, 12, 4936–4954 CrossRef CAS PubMed.
- M. V. Efremova, Y. A. Nalench, E. Myrovali, A. S. Garanina, I. S. Grebennikov, P. K. Gifer, M. A. Abakumov, M. Spasova, M. Angelakeris, A. G. Savchenko, M. Farle, N. L. Klyachko, A. G. Majouga and U. Wiedwald, Beilstein J. Nanotechnol., 2018, 9, 2684–2699 CrossRef CAS PubMed.
- B. K. Sodipo and A. A. Aziz, J. Magn. Magn. Mater., 2016, 416, 275–291 CrossRef CAS.
- H. Bin Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef.
- A. Alipour, Z. Soran-Erdem, M. Utkur, V. K. Sharma, O. Algin, E. U. Saritas and H. V. Demir, Magn. Reson. Imaging, 2018, 49, 16–24 CrossRef PubMed.
- D. Kim, S. Park, H. L. Jae, Y. J. Yong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS PubMed.
- A. M. Master, P. N. Williams, N. Pothayee, N. Pothayee, R. Zhang, H. M. Vishwasrao, Y. I. Golovin, J. S. Riffle, M. Sokolsky and A. V. Kabanov, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- G. Salas, J. Camarero, D. Cabrera, H. Takacs, M. Varela, R. Ludwig, H. Dähring, I. Hilger, R. Miranda, M. D. P. Morales and F. J. Teran, J. Phys. Chem. C, 2014, 118, 19985–19994 CrossRef CAS.
- H. Jo, D. Yoon, A. Sohn, D. W. Kim, Y. Choi, T. Kang, D. Choi, S. W. Kim and L. P. Lee, J. Phys. Chem. C, 2014, 118, 18659–18667 CrossRef CAS.
- Z. Zhang, J. Wang and C. Chen, Adv. Mater., 2013, 25, 3869–3880 CrossRef CAS PubMed.
- J. Tan, T. J. Cho, D. H. Tsai, J. Liu, J. M. Pettibone, R. You, V. A. Hackley and M. R. Zachariah, Langmuir, 2018, 34, 154–163 CrossRef CAS PubMed.
- D. H. Tsai, T. J. Cho, S. R. Elzey, J. C. Gigault and V. A. Hackley, Nanoscale, 2013, 5, 5390–5395 RSC.
- M. Mirrahimi, Z. Abed, J. Beik, I. Shiri, A. Shiralizadeh Dezfuli, V. P. Mahabadi, S. Kamran Kamrava, H. Ghaznavi and A. Shakeri-Zadeh, Pharmacol. Res., 2019, 143, 178–185 CrossRef CAS PubMed.
- X. Hou, X. Wang, R. Liu, H. Zhang, X. Liu and Y. Zhang, RSC Adv., 2017, 7, 18844–18850 RSC.
- A. Gao, H. Liu, L. Hu, H. Zhang, A. Hou and K. Xie, Chin. Chem. Lett., 2018, 29, 1301–1304 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J. E. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- M. V. Kovalenko, M. I. Bodnarchuk, R. T. Lechner, G. Hesser, F. Schäffler and W. Heiss, J. Am. Chem. Soc., 2007, 129, 6352–6353 CrossRef CAS PubMed.
- S. Dasari and P. Bernard Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- M. Norouzi, B. Nazari and D. W. Miller, Electrospun-based systems in cancer therapy, Elsevier Ltd., 2017 Search PubMed.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- D. G. Duff, A. Baiker, I. Gameson and P. P. Edwards, Langmuir, 1993, 9, 2310–2317 CrossRef CAS.
- D. G. Rancourt and J. Y. Ping, Nucl. Instrum. Methods Phys. Res., Sect. B, 1991, 58, 85–97 CrossRef.
- F. J. Douglas, D. A. MacLaren, N. Maclean, I. Andreu, F. J. Kettles, F. Tuna, C. C. Berry, M. Castro and M. Murrie, RSC Adv., 2016, 6, 74500–74505 RSC.
- P. Ball and L. Garwin, Nature, 1992, 355, 761–764 CrossRef.
- A. Ribera, I. W. C. E. Arends, S. De Vries, J. Pérez-Ramírez and R. A. Sheldon, J. Catal., 2000, 195, 287–297 CrossRef CAS.
- S. Petersen, A. Barchanski, U. Taylor, S. Klein, D. Rath and S. Barcikowski, J. Phys. Chem. C, 2011, 115, 5152–5159 CrossRef CAS.
- L. Yang, J. Tian, J. Meng, R. Zhao, C. Li, J. Ma and T. Jin, Molecules, 2018, 23, 562 CrossRef PubMed.
- W. Jiang, J. Wu, Y. Shen, R. Tian, S. Zhou and W. Jiang, Nano, 2016, 11, 1–13 Search PubMed.
- J. Rodríguez-Carvajal, Tech. Report, CEA-CNRS, Saclay, Fr Search PubMed.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- L. Qiao, Z. Fu, J. Li, J. Ghosen, M. Zeng, J. Stebbins, P. N. Prasad and M. T. Swihart, ACS Nano, 2017, 11, 6370–6381 CrossRef CAS PubMed.
- C. Pecharromán, J. E. Gonzalez-Carreno and T. Iglesias, Phys. Chem. Miner., 1995, 22, 21–29 CrossRef.
- U. Klekotka, D. Satuła, S. Spassov and B. Kalska-Szostko, Colloids Surf., A, 2018, 537, 452–459 CrossRef CAS.
- S. Devi, B. Singh, A. K. Paul and S. Tyagi, Anal. Methods, 2016, 8, 4398–4405 RSC.
- R. Roto, Y. Yusran and A. Kuncaka, Appl. Surf. Sci., 2016, 377, 30–36 CrossRef CAS.
- H. Swanson and E. Tatge, Circular of the Bureau of Standards no. 539: Standard X-ray Diffraction Powder Patterns, United States Department of Commerce, Washington, 1953, vol. I Search PubMed.
- H. E. Swanson, H. F. McMurdie, M. C. Morris and E. H. Evans, Standard X-ray Diffraction Powder, United States Department of Commerce, Washington, 1967, vol. 25 Search PubMed.
- L. Mohammed, H. G. Gomaa, D. Ragab and J. Zhu, Particuology, 2017, 30, 1–14 CrossRef CAS.
- M. Yang, L. Gao, K. Liu, C. Luo, Y. Wang, L. Yu, H. Peng and W. Zhang, Talanta, 2015, 131, 661–665 CrossRef CAS PubMed.
- K. Petcharoen and A. Sirivat, Mater. Sci. Eng., B, 2012, 177, 421–427 CrossRef CAS.
- Z. Swiatkowska-Warkocka, K. Kawaguchi, H. Wang, Y. Katou and N. Koshizaki, Nanoscale Res. Lett., 2011, 6, 1–7 CrossRef PubMed.
- R. Chalasani and S. Vasudevan, J. Phys. Chem. C, 2011, 115, 18088–18093 CrossRef CAS.
- V. Chlan, J. Zukrowski, A. Bosak, Z. Tarnawski and J. M. Honig, Phys. Rev. B, 2018, 98, 125138 CrossRef CAS.
- M. S. Senn, I. Loa, J. P. Wright and J. P. Attfield, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 125119 CrossRef.
- A. Yurenya, A. Nikitin, A. Garanina, R. Gabbasov, M. Polikarpov, V. Cherepanov, M. Chuev, A. Majouga and V. Panchenko, J. Magn. Magn. Mater., 2019, 474, 337–342 CrossRef CAS.
- I. S. Lyubutin, C. R. Lin, Y. V. Korzhetskiy, T. V. Dmitrieva and R. K. Chiang, J. Appl. Phys., 2009, 106, 034311 CrossRef.
- I. S. Lyubutin, N. E. Gervits, S. S. Starchikov, C. R. Lin, Y. T. Tseng, K. Y. Shih, C. C. Wang, I. H. Chen, Y. L. Ogarkova and N. Y. Korotkov, Smart Mater. Struct., 2016, 25, 015022 CrossRef.
- M. Abbas, B. P. Rao, N. Islam and S. M. Naga, Ceram. Int., 2014, 40, 1379–1385 CrossRef CAS.
- Y. V. Bogachev, A. V. Nikitina, A. A. Kostina, V. A. Sabitova, V. V. Pankov, T. G. Shutava, E. G. Petrova, D. A. Kotsikau, V. O. Natarov and K. S. Livanovich, Appl. Magn. Reson., 2017, 48, 715–722 CrossRef CAS.
- W. Zhang, L. Liu, H. Chen, K. Hu, I. Delahunty, S. Gao and J. Xie, Theranostics, 2018, 8, 2521–2548 CrossRef CAS PubMed.
- A. Deręgowska, J. Depciuch, R. Wojnarowska, J. Polit, D. Broda, H. Nechai, M. Gonchar and E. Sheregii, Int. J. Biol. Biomol. Agric. Food Biotechnol. Eng., 2013, 7, 1–4 Search PubMed.
- P. Bazylewski, R. Divigalpitiya and G. Fanchini, RSC Adv., 2017, 7, 2964–2970 RSC.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed.
- A. M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351–1371 CrossRef CAS PubMed.
- S. Choi, B. I. Sang, J. Hong, K. J. Yoon, J. W. Son, J. H. Lee, B. K. Kim and H. Kim, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef CAS PubMed.
- X. Huang and M. A. El-Sayed, Alexandria Med. J., 2011, 47, 1–9 CrossRef.
- C. F. Bohren and D. R. Huffman, Absorption and scattering of light by small particles, Wiley-VCH Verlag GmbH & Co. KGaA, 1998 Search PubMed.
- J. Fatisson, I. R. Quevedo, K. J. Wilkinson and N. Tufenkji, Colloids Surf., B, 2012, 91, 198–204 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- G. Sonavane, K. Tomoda and K. Makino, Colloids Surf., B, 2008, 66, 274–280 CrossRef CAS PubMed.
- A. Leibovitz, J. C. Stinson, W. B. McCombs, C. E. McCoy, K. C. Mazur and N. D. Mabry, Cancer Res., 1976, 36, 4562–4569 CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- K. S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 20331–20338 CrossRef CAS.
- K. Jiang, D. A. Smith and A. Pinchuk, J. Phys. Chem. C, 2013, 117, 27073–27080 CrossRef CAS.
- J. G. Mehtala, S. Torregrosa-Allen, B. D. Elzey, M. Jeon, C. Kim and A. Wei, Nanomedicine, 2014, 9, 1939–1955 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03699d |
| This journal is © The Royal Society of Chemistry 2020 |