DOI:
10.1039/D0RA03674A
(Paper)
RSC Adv., 2020,
10, 27794-27804
Novel drug isolated from mistletoe (1E,4E)-1,7-bis(4-hydroxyphenyl)hepta-1,4-dien-3-one for potential treatment of various cancers: synthesis, pharmacokinetics and pharmacodynamics†
Received
24th April 2020
, Accepted 9th July 2020
First published on 24th July 2020
Abstract
(1E,4E)-1,7-Bis(4-hydroxyphenyl)hepta-1,4-dien-3-one (DHDK) is a novel curcuminoid analogue isolated from mistletoe. DHDK exhibits better anti-tumour activity, higher bioavailability and superior stability than curcumin. DHDK is difficult to isolate from Viscum coloratum, but it can be synthesised. MTT (methylthiazolyldiphenyl tetrazolium bromide) assay was used to evaluate the in vitro cytotoxic activity of synthesised DHDK on 12 cancer cell lines. Results showed that DHDK exhibited excellent potential as an anticancer agent, especially for breast and lung cancer. Efficacy was further evaluated in vivo by using MCF-7 breast cancer models. DHDK showed a dose-dependent relationship without weight reduction, mortality growth inhibition or tissue toxicity. Pharmacokinetics and tissue distribution statistics were determined by LC-ESI-MS/MS. This work provided preliminary data on this natural compound and could open up new prospects for changing related parameters to improve drug efficacy.
1. Introduction
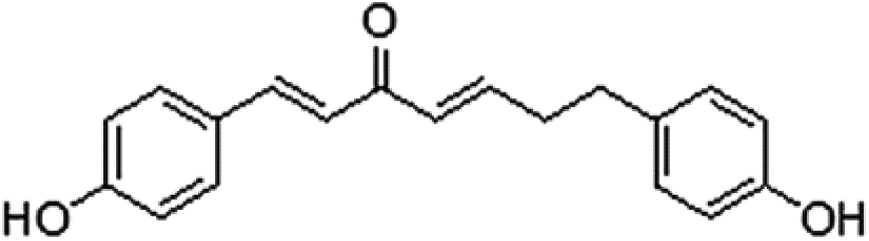
Cancer is a life-threatening problem, and 1.35 billion new cases are reported annually worldwide. It is the second leading cause of death globally, and was responsible for an estimated 9.6 million deaths in 2018.1 Breast cancer is the most common malignancy in females in numerous countries.2 Many types of cancer treatments, such as surgery, chemotherapy and radiation therapy, are available. Some people with cancer undergo only one treatment, but most patients are subjected to a combination of treatments. However, severe side effects and increasing multidrug resistance have limited the clinical application of such treatments.3,4 In this regard, new drugs must be developed. Natural products have been the focus of research because of their advantages of unique structure, abundant biological activity and multidrug resistance elimination. Curcumin is a polyphenol naturally generated from the rhizome of Curcuma longa L, which has long been used in traditional Chinese herbal medicine for treatment of various diseases. Curcumin has attracted much attention for its pharmacological activities, especially its anti-tumour ability with various mechanisms.5–8 Although curcumin has anti-tumour efficacy and few side effects, its low water solubility, fast degradation and low bioavailability hinder its potential medical applications.9 Extracts of Viscum coloratum (Kom.) Nakai (mistletoe) exhibit numerous biological activities. Extracts of this whole plant are used for treatment of various tumours in Europe,10 such as breast,11 bladder12 and lung cancers.13 Mistletoe extract is administered as a supportive treatment in cancer therapy in numerous patients.14 Mistletoe (Viscum album) preparations consist of active ingredients, such as mistletoe lectins and viscotoxins, which show anti-tumour properties by causing cell cycle delay or arrest and inducing apoptosis,15 influencing tumour angiogenesis16 and exerting immune-potentiating activities that may strengthen the host defence system against cancer.17 The components of mistletoe demonstrate anti-multidrug resistance potential in vitro.18 We obtained (1E,4E)-1,7-bis(4-hydroyphenyl)hepta-1,4-dien-3-one (DHDK, Fig. 1), a new diaryl heptanone from V. coloratum.19 DHDK is a new curcuminoid analogue that has better anti-tumour activity, higher bioavailability and better stability than curcumin.20
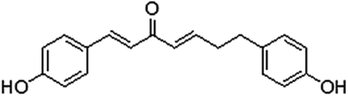 |
| | Fig. 1 (1E,4E)-1,7-Bis(4-hydroyphenyl)hepta-1,4-dien-3-one. | |
The isolation of natural DHDK from V. coloratum is difficult and complicated. To our knowledge, methods for synthesis of this compound have not been reported. Thus, we developed a total synthesis route in eight steps by using 3-(4-hydroxyphenyl)propanoic acid and hydroxybenzaldehyde to provide raw materials for further amplifying the industrial and clinical production of DHDK. The pharmacokinetics and pharmacodynamics of DHDK have also not been investigated. The current study involved the preliminary evaluation of DHDK for drug efficacy in vivo and in vitro and the determination of its pharmacokinetics and tissue distribution. Curcumin has been widely studied in different fields. We infer that DHDK would obtain greater attention than curcumin given it's great advantages in terms of it's chemical and physical properties. Results can provide important basic laboratory database support for further clinical or other studies.
2. Results and discussion
2.1. Synthesis of DHDK
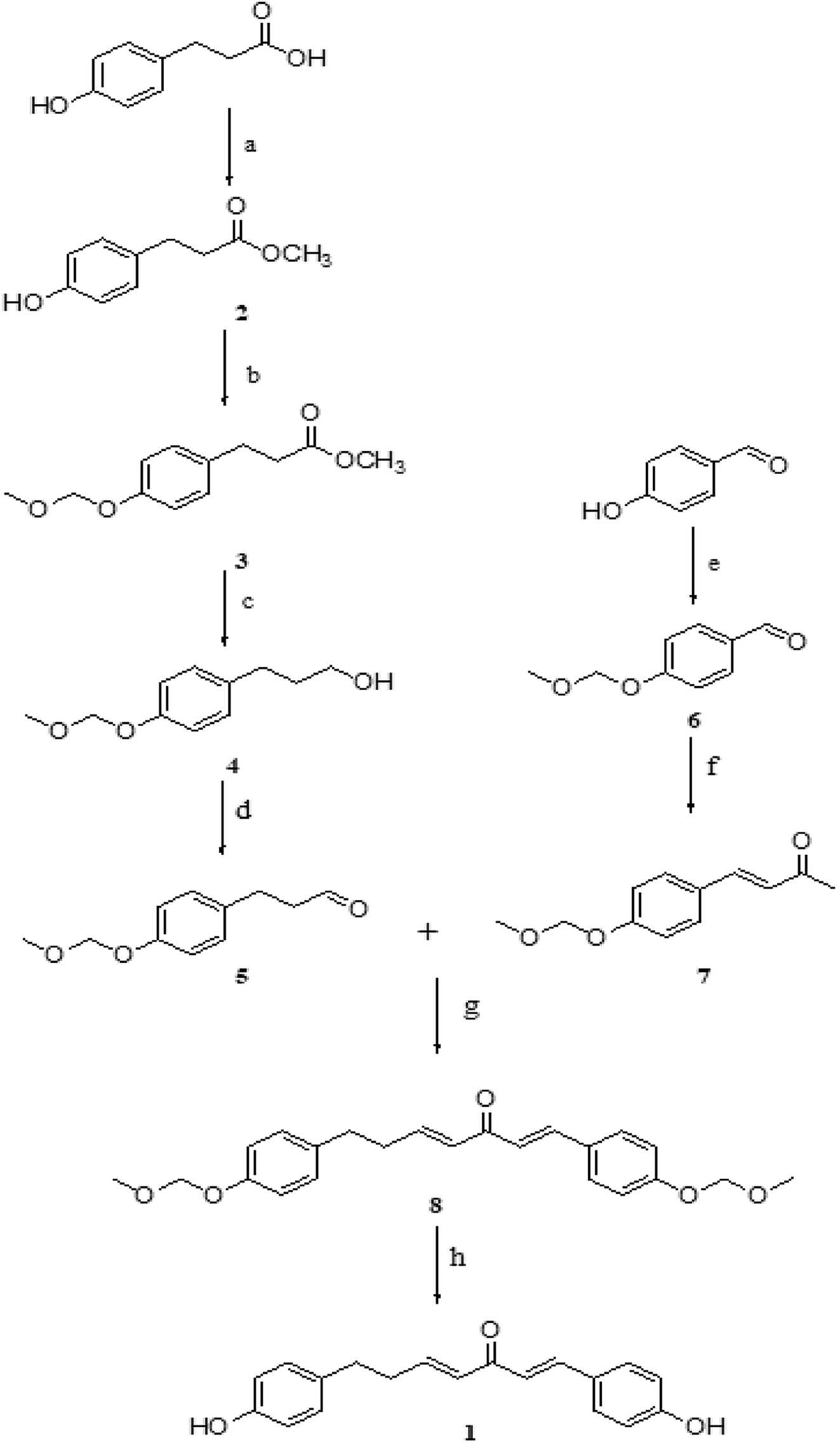
Inspired by the synthesis of curcumin, our strategy involved the synthesis of DHDK by aldol reaction. Our route began with commercially available 3-(4-hydroxyphenyl)propanoic acid and hydroxybenzaldehyde. The processes were shown in Scheme 1. 3-(4-Hydroxyphenyl)propanoic acid and methanol were used to generate compound 2 (3-(4-hydroxyphenyl)-propanoic acid methyl ester) at 35 °C. Compound 2 and hydroxybenzaldehyde were protected with bromomethyl methyl ether to produce compound 3 (4-(methoxymethoxy)-benzene propanoic acid methyl ester) (1H NMR spectra shown in Fig. S1†) and compound 6 (4-(methoxymethoxy)benzaldehyde), respectively. Compound 5 (4-(methoxymethoxy)-benzenepropanal) was afforded by the oxidation of compound 4 (4-(methoxymethoxy)-benzene propanol) (1H NMR spectra shown in Fig. S2†). Compound 4 was produced by the reduction of compound 3 with lithium aluminium hydride. The condensation of compound 6 and acetone afforded compound 7 (3-[4-(methoxymethoxy)-phenyl]-but-3-en-2-one). Compound 8 (1,7-bis(4-(methoxymethoxy)phenyl)hepta-1,4-dien-3-one) was generated by the condensation of compound 5 and 7. Finally, the treatment of compound 8 with HCl furnished the desired product 1 (1H NMR, 13C NMR, MS are shown in Fig. S3–S5†).
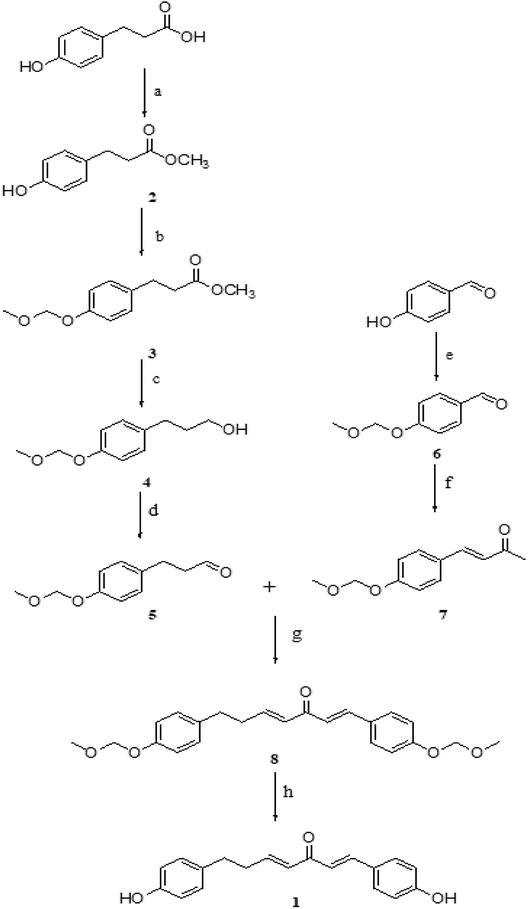 |
| | Scheme 1 Reagents and conditions: (a) CH3OH, H2SO4, 4 h, 35 °C (b) DIPEA, DCM, acetone, MOMBr, 2 h, reflux (c) LiAlH4, dry THF, 3 h, rt (d) DMP, DCM, NaHCO3, 4 h, 0 °C (e) MOMBr, K2CO3, acetone, 5 h, reflux (f) H2O, 10% NaOH, acetone, rt (g) LDA, THF, 5 h, −78 °C (h) HCl (6 mol L−1), methanol, 15 min, 60 °C, reflux. | |
2.2. Cytotoxicity in vitro
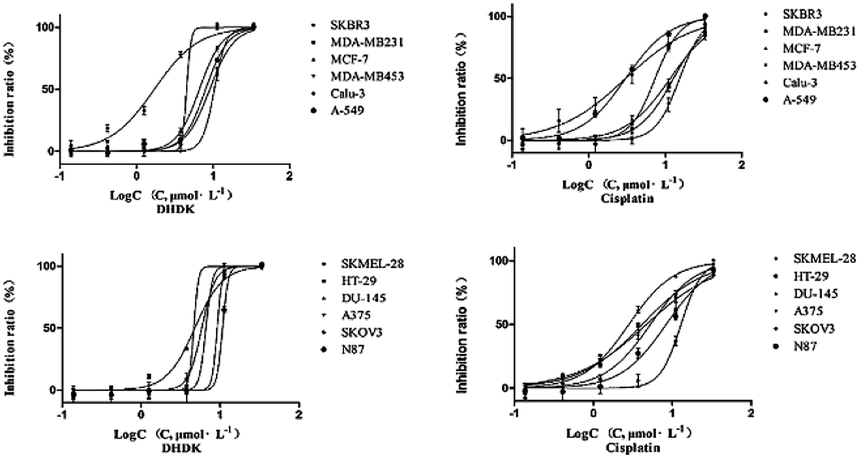
2.2.1 Cytotoxicity against cancer cell lines. DHDK showed cytotoxic activity in the micromolar range on 12 cancer cell lines. In particular, DHDK showed higher potency than CDDP (cis-platinum) against four different human breast cancer cell lines (SKBR3, MDA-MB231, MCF-7 and MDA-MB453) and human skin malignant melanoma cell line SKMEL-28. The IC50 value of the five cancer cell lines treated with DHDK were significantly lower than that of cells treated with CDDP. There was no significant difference in IC50 value between the DHDK group and CDDP group on colon cancer cell (HT-29) and lung cancer cell (Calu-3). The IC50 of other tumour cells treated with DHDK was significantly higher than that of cells treated with CDDP but was less than 11 μmol L−1 (3.25 μg mL−1). The activity of DHDK against different cancer cell lines indicated its potential as an anticancer agent. In addition, DHDK showed stronger potency against the four human breast cancer cell lines than the positive control CDDP. The IC50 value of the SKBR3 cell line was as low as 1.71 μmol L−1 (Table 1). The curves of the inhibitory effect of DHDK and cisplatin on the growth of different tumour cell lines are shown in Fig. 2.
Table 1 The IC50 values (μmol L−1) of DHDK and CDDP against cancer cell linesa
| Cell line |
Cancer type |
IC50, μmol L−1 |
| DHDK |
Cisplatin |
| IC50: half maximal inhibitory concentration; * significant difference compared to corresponding cisplatin group p < 0.05. |
| SKBR3* |
Breast |
1.71 ± 0.10 |
3.04 ± 0.17 |
| MDA-MB231* |
Breast |
4.49 ± 046 |
12.81 ± 0.58 |
| MCF-7* |
Breast |
9.09 ± 0.46 |
11.16 ± 0.60 |
| MDA-MB453* |
Breast |
10.42 ± 0.92 |
15.43 ± 0.83 |
| Calu-3 |
Lung |
6.65 ± 0.42 |
7.39 ± 0.25 |
| A-549* |
Lung |
8.01 ± 0.44 |
3.03 ± 0.24 |
| SKMEL-28* |
Skin |
9.26 ± 0.57 |
12.94 ± 0.96 |
| HT-29 |
Colon |
4.79 ± 0.54 |
4.71 ± 0.17 |
| DU-145* |
Prostate |
4.62 ± 0.22 |
2.82 ± 0.14 |
| A375* |
Melanoma |
6.64 ± 0.40 |
5.28 ± 0.18 |
| SKOV3* |
Ovary |
9.59 ± 0.44 |
4.15 ± 0.30 |
| N87* |
Stomach |
10.81 ± 0.50 |
8.35 ± 0.30 |
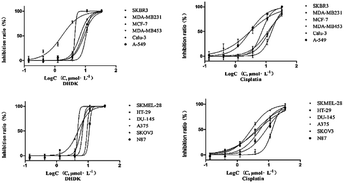 |
| | Fig. 2 Curves of the inhibitory effects of DHDK and cisplatin on the growth of different tumour cell lines. | |
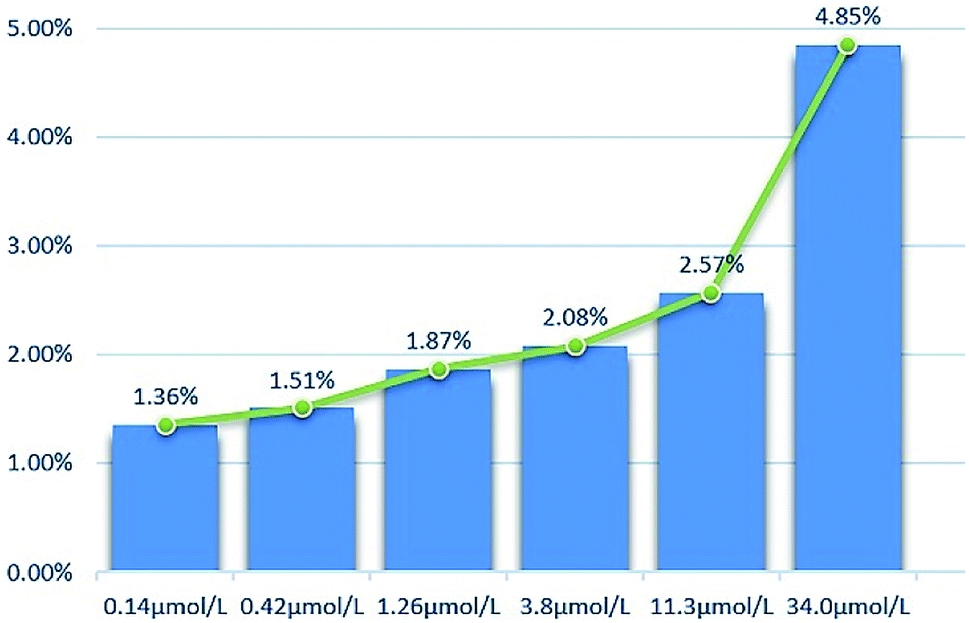
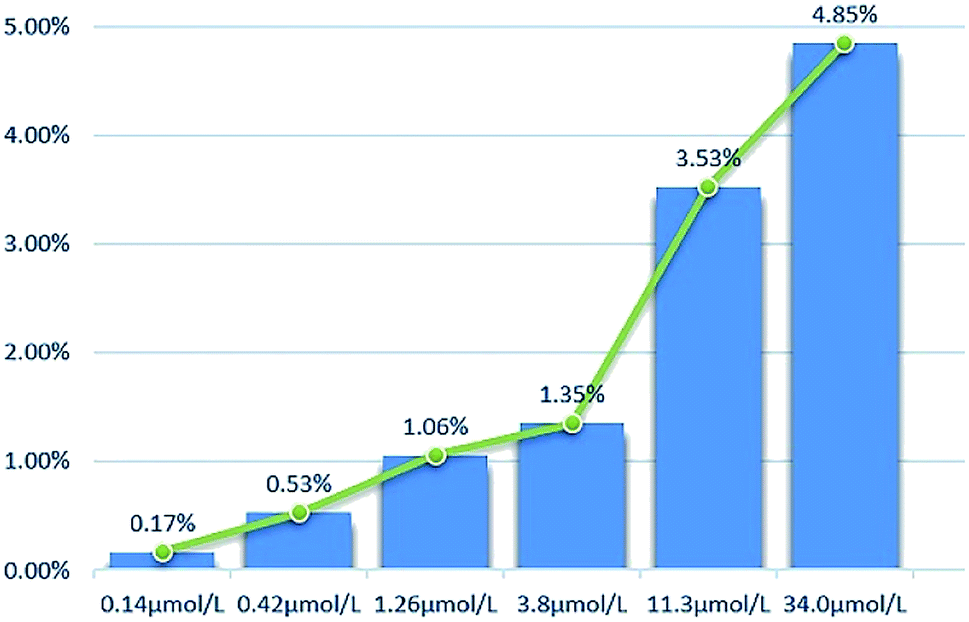
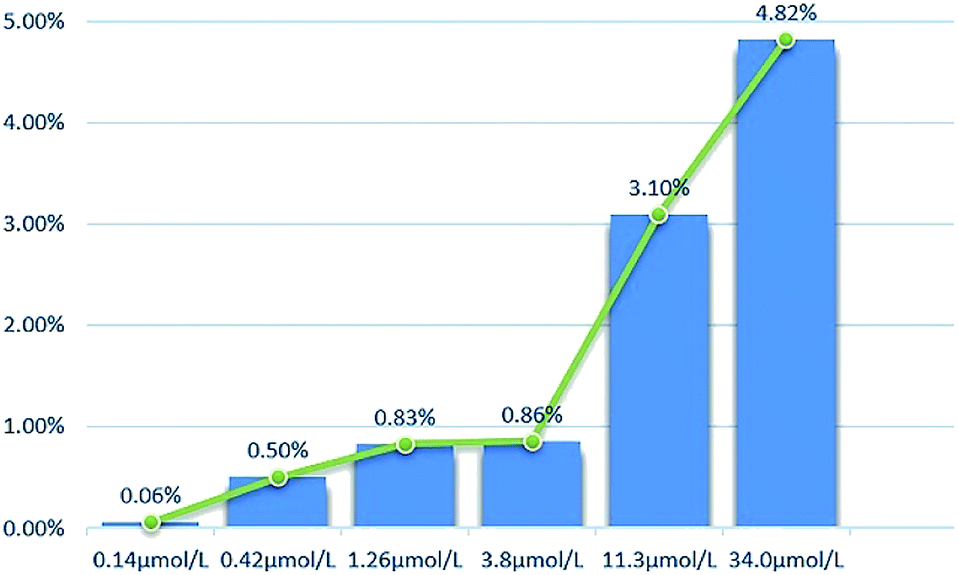
2.2.2 Cytotoxicity against normal cell lines. The toxicity of different concentrations of DHDK on normal cell lines (LO2 human normal liver cells, GES-1 human normal gastric epithelial cells, BEAS-2B human normal lung epithelial cell) was investigated (Table 2). The results showed that DHDK has little inhibitory effect on normal cells, and the inhibition rate of each group is less than 5%, which is much smaller than the inhibition rate of DHDK on cancer cells at the same concentration. This indicates that DHDK has certain selectivity for the toxic effects of cancer cells and normal cells. Inhibition rate of different DHDK concentrations to different normal cell lines are shown in Table 3 (Fig. 3–5).
Table 2 OD values of different DHDK concentrations to different normal cell lines
| Group |
OD value (mean ± SD) |
| LO2 |
GES-1 |
BEAS-2B |
| Control group |
0.69 ± 0.0091 |
0.71 ± 0.022 |
0.66 ± 0.0086 |
| 0.14 μmol·L−1 |
0.69 ± 0.013 |
0.71 ± 0.0031 |
0.66 ± 0.0017 |
| 0.42 μmol·L−1 |
0.68 ± 0.039 |
0.71 ± 0.0098 |
0.66 ± 0.0028 |
| 1.26 μmol·L−1 |
0.68 ± 0.022 |
0.71 ± 0.0051 |
0.66 ± 0.0027 |
| 3.8 μmol·L−1 |
0.68 ± 0.031 |
0.70 ± 0.0035 |
0.66 ± 0.0010 |
| 11.3 μmol·L−1 |
0.68 ± 0.0066 |
0.69 ± 0.0097 |
0.64 ± 0.0028 |
| 34.0 μmol·L−1 |
0.67 ± 0.025 |
0.68 ± 0.0075 |
0.63 ± 0.0030 |
| Blank group |
0.42 ± 0.0014 |
0.042 ± 0.0020 |
0.041 ± 0.0005 |
Table 3 Inhibition rate of different DHDK concentrations to different normal cell lines
| Group |
Inhibition rate (%) |
| LO2 |
GES-1 |
BEAS-2B |
| Control group |
0 |
0 |
0 |
| 0.14 μmol·L−1 |
1.36 |
0.17 |
0.059 |
| 0.42 μmol·L−1 |
1.51 |
0.53 |
0.50 |
| 1.26 μmol·L−1 |
1.87 |
1.06 |
0.83 |
| 3.8 μmol·L−1 |
2.08 |
1.35 |
0.86 |
| 11.3 μmol·L−1 |
2.57 |
3.53 |
3.10 |
| 34.0 μmol·L−1 |
4.17 |
4.85 |
4.82 |
| Blank group |
— |
— |
— |
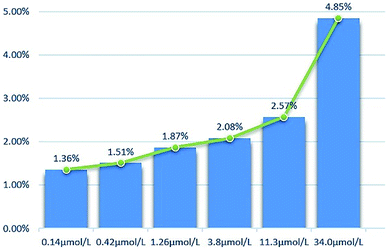 |
| | Fig. 3 Inhibition rate of different DHDK concentrations to LO2 human normal liver cell. | |
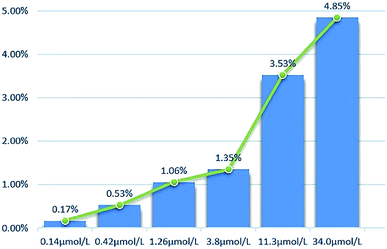 |
| | Fig. 4 Inhibition rate of different DHDK concentrations to GES-1 human normal gastric mucosal cell. | |
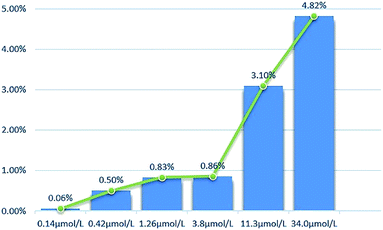 |
| | Fig. 5 Inhibition rate of different DHDK concentrations to BEAS-2B human normal lung epithelial cell. | |
2.3. Drug efficacy in vivo
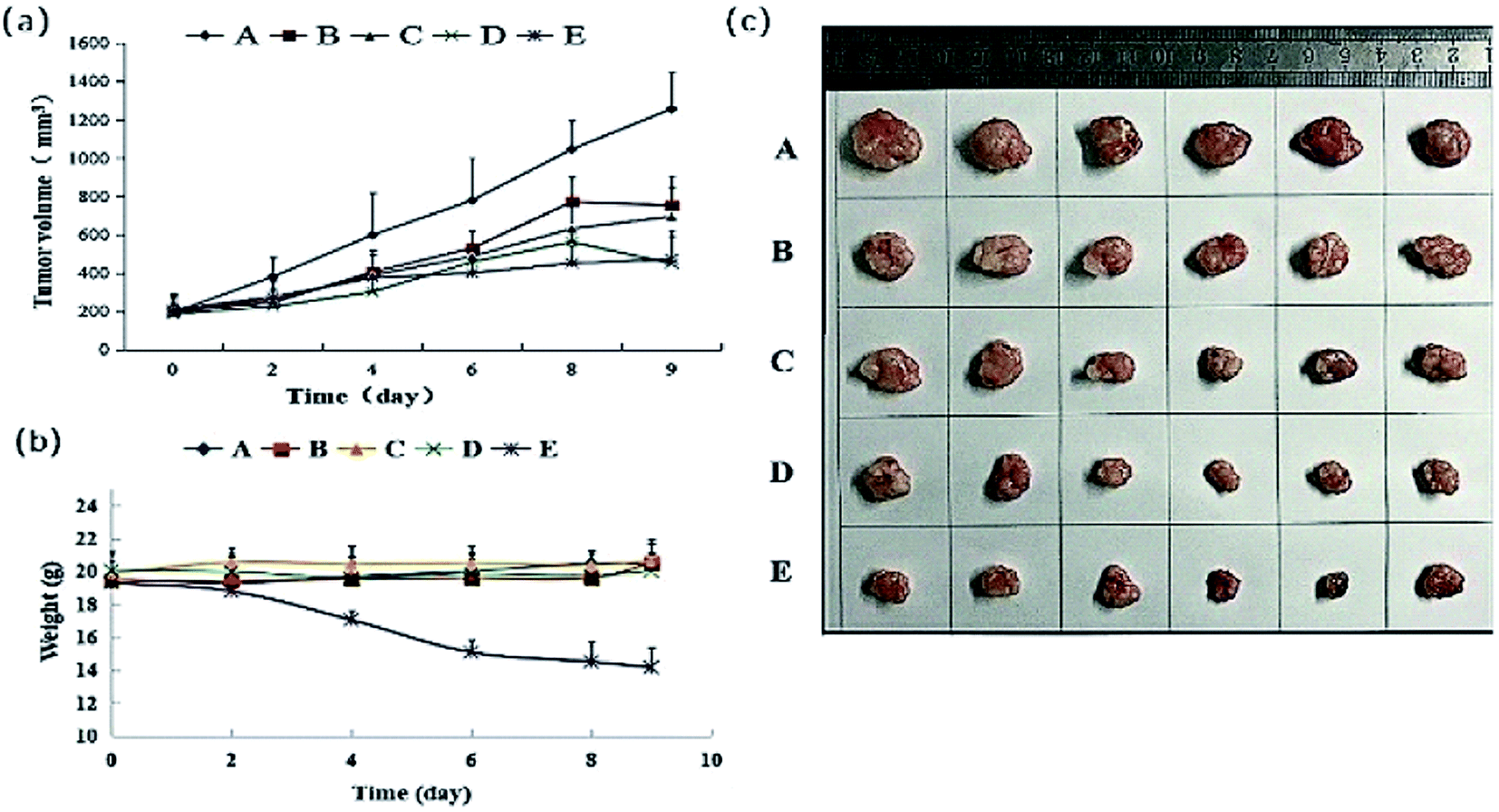
All of the test groups showed inhibition of tumour proliferation with the descending order of 40 mg kg−1 (high-dose group), 20 mg kg−1 (medium-dose group) and 10 mg kg−1 (low-dose group), the results were shown in Fig. 6. A high dosage was more effective than a low dosage. The inhibition rates determined as tumour weight in 40, 20 and 10 mg kg−1 groups were 47.85%, 35.22% and 28.44% respectively, which were lower than that positive control group (64.10%) (Table 4). The tumour weight and tumour inhibition rate of each administration group and cisplatin group were significantly lower than that of the blank group (p < 0.01). The tumour weight and tumour inhibition rate of each administration group were significantly lower than that of the cisplatin group (p < 0.01). From the Fig. 6(b) and Table S2,† we can see that DHDK groups have stable weight during the nine days, while the positive control group has an obvious weight loss. From this phenomenon we can infer that DHDK might have better drug safety than cisplatin. Overall, 40 mg kg−1 DHDK exhibited the highest tumour inhibition rate compared with the other two groups.
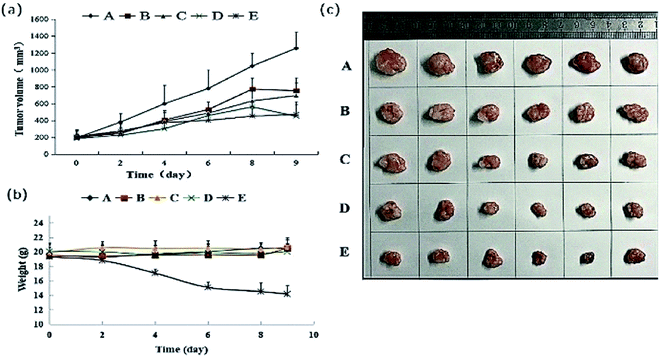 |
| | Fig. 6 (a) Mean tumour volume–time curve of various groups after tail vein injection of DHDK solution. (b) Mean weight–time curve of various groups after tail vein injection of DHDK solution. (c) Tumour picture of various groups obtained after the mice were sacrificed ((A) negative control group, (B) low-dose group, (C) medium-dose group, (D) high-dose group, (E) positive control group, n = 6). | |
Table 4 Tumour weight and inhibition rate of various groupsa
| Group |
Tumour weight (mean) |
Inhibition rate (IR, %) |
| (A) Negative control group, (B) low-dose group, (C) medium-dose group, (D) high-dose group, (E) positive control. ** Significant difference compared to A, p < 0.01; ## significant difference compared to (E), p < 0.01. |
| A |
834.36 ± 41.00 |
— |
| B |
597.04 ± 38.19** |
28.44**## |
| C |
540.53 ± 63.76** |
35.22**## |
| D |
435.13 ± 140.10**# |
47.85**## |
| E |
299.53 ± 109.60**# |
64.10** |
2.4. Determination of biochemical indexes in serum and plasma
The results of the measured biochemical indicators are shown in the Table 5. The results showed that the level of ALT in each administration group was slightly lower than those in the blank group, but there was no significant difference between the administration groups and the blank group. There was no significant difference between the levels of BUN, CRE and AST in each administration group and that in the blank group. This result initially indicated that DHDK has no potential liver and kidney toxicity.
Table 5 The values of ALT, ALT, BUN, CRE in various groups
| Groups |
BUN (mmol L−1) mean ± SD |
CRE (μmol L−1) mean ± SD |
AST (U L−1) mean ± SD |
ALT (U L−1) mean ± SD |
| Blank |
9.14 ± 0.084 |
86.37 ± 5.97 |
51.11 ± 2.88 |
37.82 ± 6.80 |
| Low dose group |
10.16 ± 1.35 |
78.85 ± 3.90 |
50.90 ± 3.04 |
39.18 ± 0.81 |
| Medium dose group |
9.74 ± 0.47 |
87.97 ± 7.63 |
50.93 ± 0.81 |
39.70 ± 2.75 |
| High dose group |
9.86 ± 0.33 |
82.97 ± 2.97 |
52.98 ± 3.07 |
38.41 ± 0.67 |
2.5. Pathological tissue sections
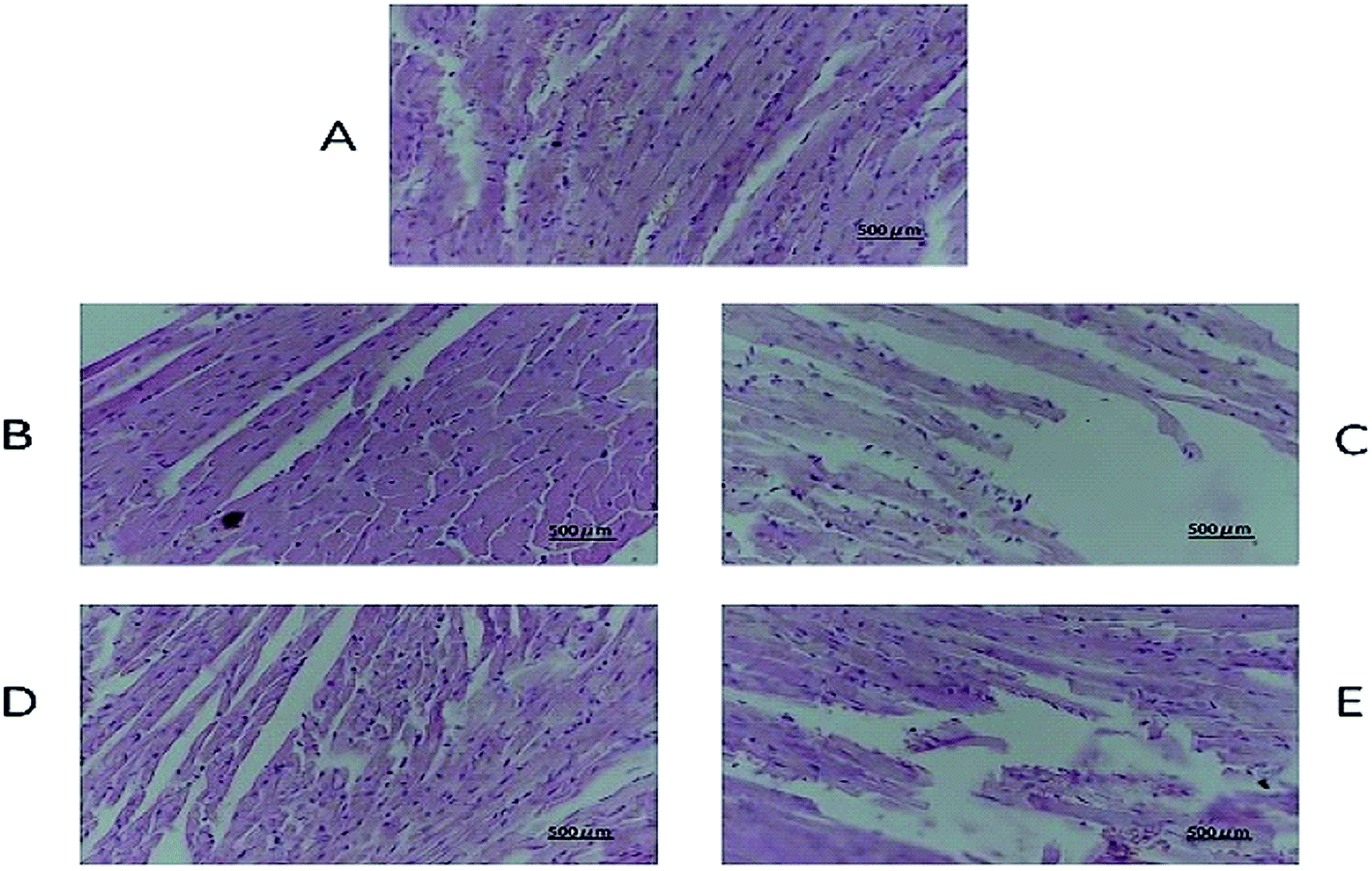
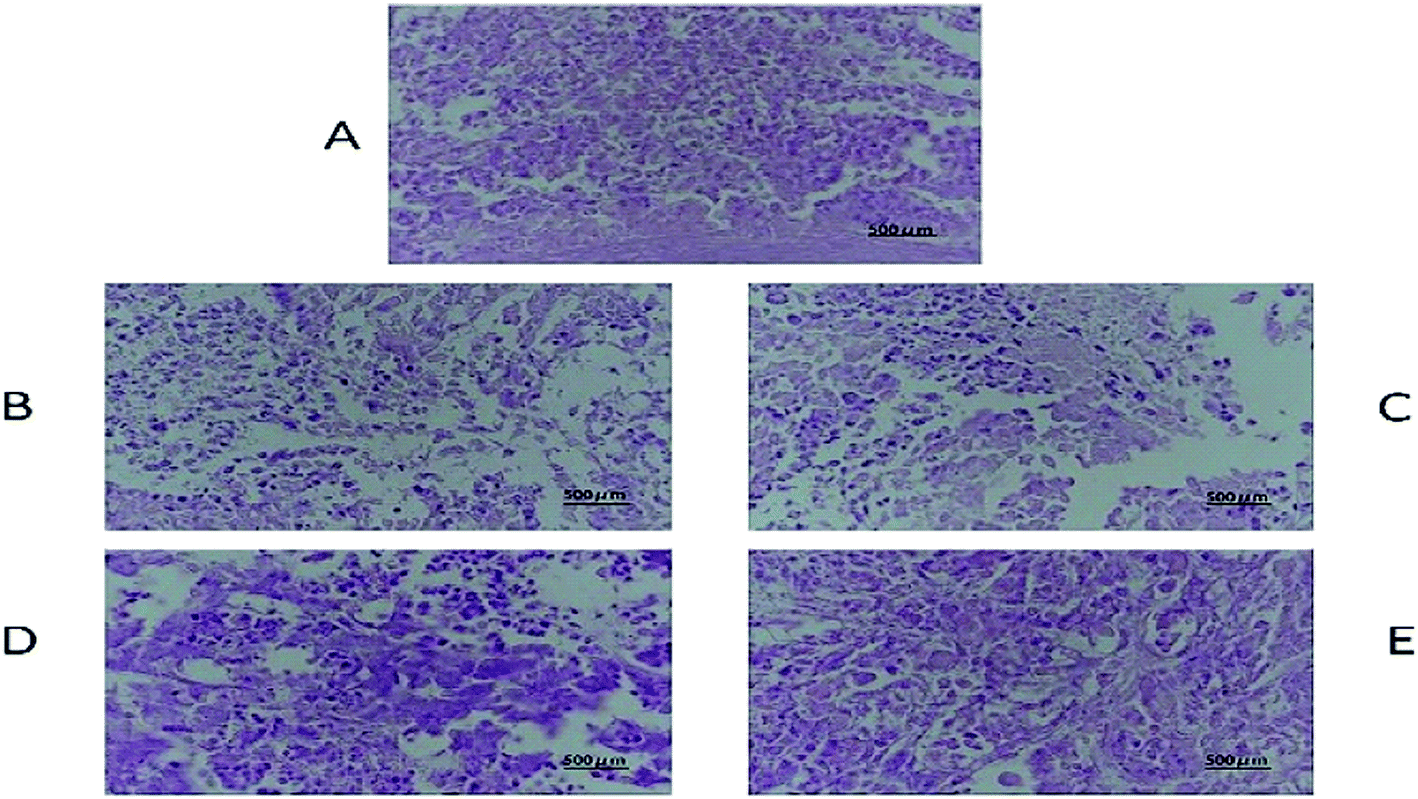
The tissue slices of female nude mice are shown in Fig. 7, 8 and S6–S10.† No toxicity was observed in tissues exposed to different dosages of DHDK. This finding could be due to the fact that DHDK does not accumulate in tissues. Myocardial fibre fracture was observed in the nude mice of the positive control group (Fig. 7). This phenomenon has also been reported in other related studies.21–23 The tumour slices are shown in Fig. 8. A small amount of punctate nucleolysis and a low degree of necrosis were detected in the low-dose group. Analysis of the pathological sections showed point necrosis and dissolved nucleus in the medium-dose group. A large amount of tumour necrosis with nuclear envelope breakdown and dissolved nucleus was observed in the high-dose group. Moreover, a large amount of tumour cell necrosis with nucleolysis was observed in the positive control group. The results provide further evidence that DHDK shows efficacy against anti-MCF-7 breast cancer cells. Overall, based on the analyses of tissue slices and efficacy in vivo, DHDK exhibits anti-tumour effects, and potential advantages in drug safety over cisplatin.
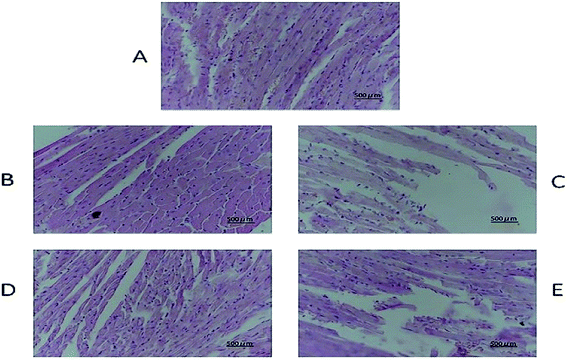 |
| | Fig. 7 Heart histopathology of various groups obtained after the mice were sacrificed ((A) negative control group, (B) low-dose group, (C) medium-dose group, (D) high-dose group, (E) positive control group). | |
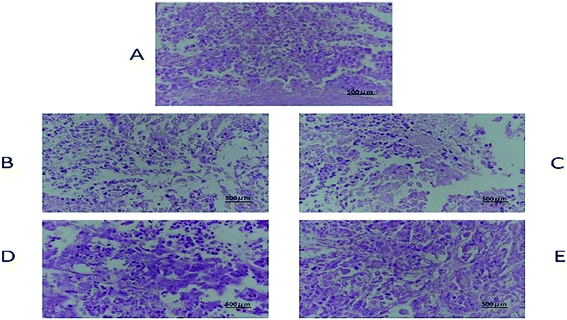 |
| | Fig. 8 Tumour histopathology of various groups obtained after the mice were sacrificed ((A) negative control group, (B) low-dose group, (C) medium-dose group, (D) high-dose group, (E) positive control group). | |
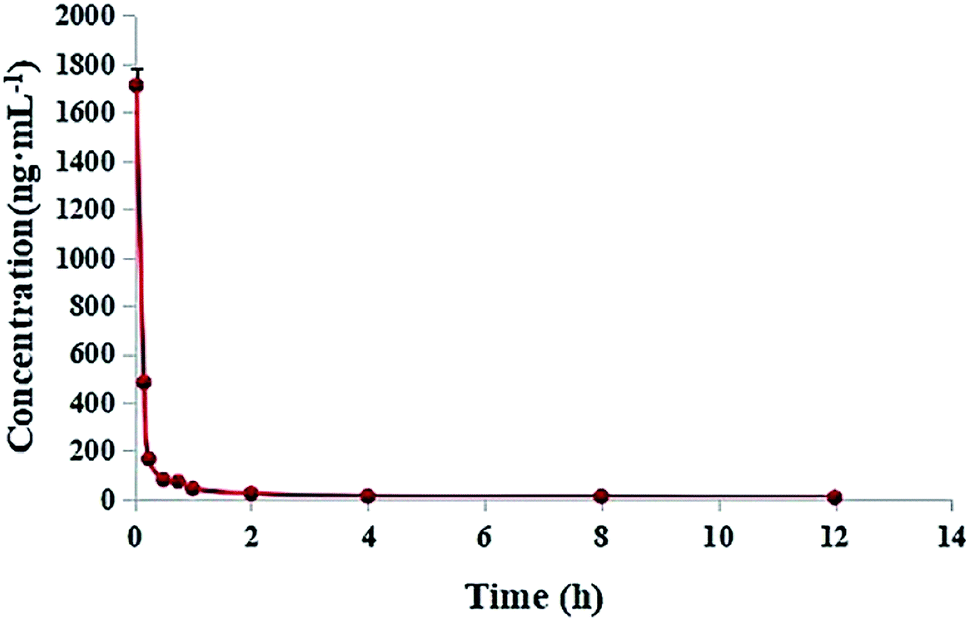
2.6. Pharmacokinetic study
Pharmacokinetic parameters were determined by the two-compartmental analysis of plasma concentration versus time by using DAS2.0. The results indicated that DHDK belongs to the group of fast metabolites. The main pharmacokinetic parameters are listed in Tables 6 and S3.† At the beginning, the plasma concentration of DHDK rapidly decreased, the curve was shown in Fig. 9. Compared with cisplatin, DHDK has poor pharmacokinetic parameters. We obtained the pharmacokinetic parameters of cisplatin from ref. 24, the main pharmacokinetic parameters were shown in Table 7. The dosage of DHDK is 1.4 times higher than cisplatin, but the ‘relative bioavailability’ of DHDK was only 1.28% of cisplatin. Despite the low AUC(0–t) of DHDK, the anti-tumour effect was proved to be good according to the efficacy evaluation in vivo (tumour inhibition rate of dose 40 mg kg−1 was 47.85% on mice). Therefore, we can optimize the pharmacokinetic behavior to gain a more satisfying AUC. DHDK has a slower clearance rate than cisplatin, which means less excretion through the prototype, so DHDK is less toxic in the kidney than cisplatin. DHDK has a much shorter half-life than cisplatin, indicating a higher metabolic rate and/or higher affinity with red blood cells than cisplatin. The distribution volume of DHDK is extraordinarily less than the average blood volume of a rat. This may be because the initial plasma concentration drops so fast that the C0 value and its error are too large, which leads to the calculated V1 being too small. This also shows that DHDK might have a strong affinity with blood cells and has a tendency to quickly distribute in blood cells. Above all, DHDK might have stronger anti-tumor effect and lower renal toxicity than cisplatin. We need to further study the changing process of DHDK in the body in detail and optimize pharmacokinetic parameters based on this process. For example, we could use structural modification or drug delivery systems to optimize pharmacokinetic parameters, such as ABI-007 paclitaxel nanoparticles of American Biological Company,25 which can significantly improve the pharmacokinetic parameters to obtain better pharmacodynamics. PD showed that the tumour inhibition rates of DHDK in 40 mg kg−1, 20 mg kg−1, and 10 mg kg−1 were 47.85%, 35.22%, and 28.44%, respectively. The tumour inhibition rate of cisplatin group (5 mg kg−1) was 64.10%. The dosage of DHDK high-dose group was 8 times higher than cisplatin group and after dose conversion, the AUC(0–t) of DHDK high-dose group was 7.3% of cisplatin group, but the tumour inhibition rate of DHDK in the high-dose group was 74.6% of the cisplatin group. This data further suggested that DHDK might have a better anti-cancer effect than cisplatin by improving pharmacokinetic parameters in vivo.
Table 6 Main pharmacokinetic parameters of DHDK after tail vein injection of 14 mg kg−1 DHDK solution (n = 6)a
| Parameters |
Unit |
Mean |
SD |
| SD: standard deviation. |
| t1/2α |
h |
0.064 |
0.021 |
| t1/2β |
h |
1.79 |
0.047 |
| AUC0→t |
μg L−1 h |
459.47 |
193.20 |
| AUC0→∞ |
μg L−1 h |
534.32 |
202.71 |
| V1 |
L kg−1 |
0.037 |
0.009 |
| CL |
L kg h−1 |
0.008 |
0.002 |
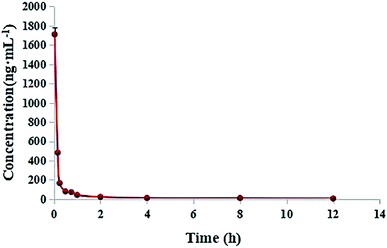 |
| | Fig. 9 Mean plasma concentration–time curves of DHDK after the tail vein injection of 14 mg kg−1 DHDK solution (n = 6). | |
Table 7 Main pharmacokinetic parameters of DHDK and cisplatin
| Parameters |
Unit |
DHDK |
Cisplatin |
| t1/2α |
h |
0.064 |
0.3 |
| t1/2β |
h |
1.79 |
23 |
| AUC0→t |
μg L−1 h |
459.47 |
36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| V1 |
L kg |
0.008 |
9.2 |
| CL |
L kg h−1 |
0.037 |
0.23 |
2.7. Tissue distribution
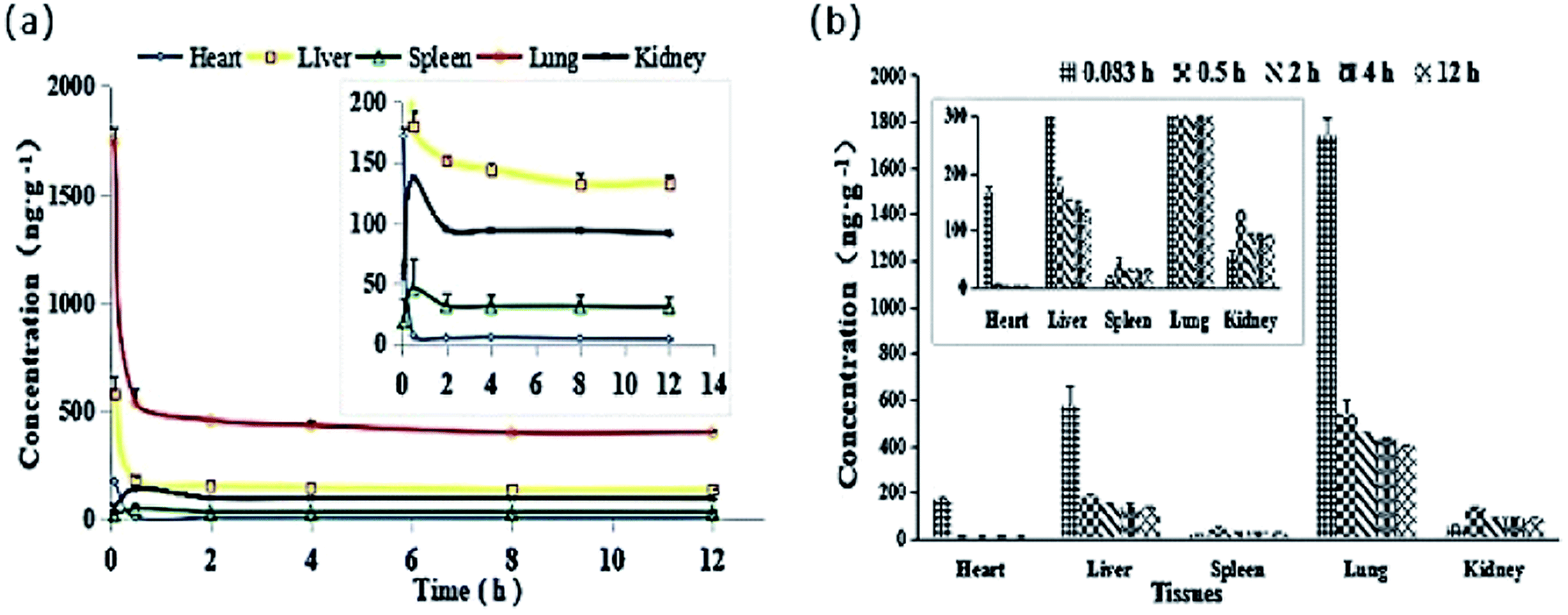
The tissue distribution of DHDK at different time intervals is shown in Fig. 10 and Table S4.† The DHDK concentration in different tissues in rats followed the order of lung > liver > heart > kidney > spleen. However, the concentration in the tissues was low and rapidly declined except for in the kidney and spleen, consistent with the results of the pharmacokinetic study. The concentration of DHDK in the lung tissue was higher than that in the other tissues. This phenomenon is consistent with the in vitro cytotoxicity result in Calu-3 lung cancer cell line. Therefore, the application of DHDK in lung cancer treatment can be further developed. For example, DHDK spray could be developed to increase its concentration in the lung and reduce the content in other tissues. The clearance rates followed the order of kidney > spleen > heart > lung > liver (shown in Table 8).
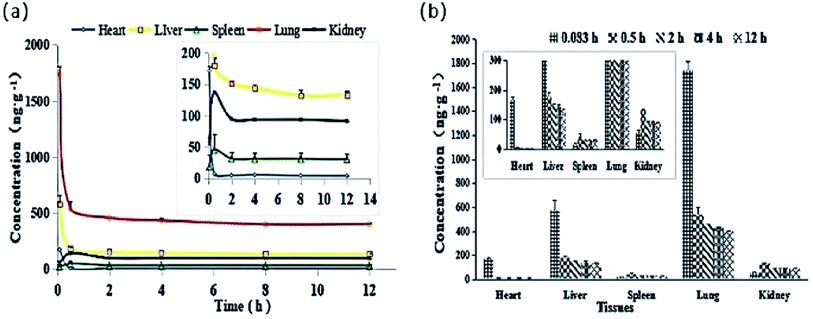 |
| | Fig. 10 (a) Mean tissue concentration–time curves of DHDK after tail vein injection of 14 mg kg−1 DHDK solution (n = 6). (b) Mean DHDK concentrations in various tissues at indicated time points after tail vein injection of 14 mg kg−1 DHDK solution (n = 6). | |
Table 8 Main pharmacokinetic parameters of DHDK after tail vein injection of DHDK solution (mean ± SD, n = 5)a
| Parameters |
Unit |
Heart |
Liver |
Spleen |
Lung |
Kidney |
| SD: standard deviation. |
| AUC0→∞ |
μg L−1 h |
413.54 ± 364.13 |
2453.46 ± 424.52 |
206.81 ± 84.27 |
1627.09 ± 298.71 |
57.23 ± 32.28 |
| CL |
L kg h−1 |
50.93 ± 26.52 |
5.846 ± 1.02 |
81.66 ± 45.73 |
8.89 ± 1.97 |
244.93 ± 9.95 |
3. Materials and methods
3.1. Chemicals and reagents
Methanol, tetrahydrofuran, hydrochloride, triethylamine, formic acid, acetonitrile, anhydrous ethanol and dimethyl sulphoxide of HPLC grade were purchased from Concord Technology (Tianjin, China). Sulfuric acid, anhydrous ether, potassium bicarbonate and bromomethyl methyl ether were obtained from Tianjin Kaixin Chemical Industry (Tianjin, China). Lithium aluminum hydride, anhydrous sodium sulphate, sodium hydroxide, sodium chloride, N,N-diisopropylehylamine and N,N-lithium diisopropylamine were acquired from Tianjin Fuyu Fine Chemical (Tianjin, China). Anhydrous potassium carbonate, phosphoric acid and isopropanol were supplied by Comio Chemical Reagent (Tianjin, China). Petroleum ether, ethyl acetate, dichlorimethane, methyl tertiary butyl ether (MTBE), methanol, anhydrous alcohol and acetone were supplied by Shandong Yuwang Industry (Shandong, China). Normal saline, Tween 80 and 95% ethanol were bought from Shenyang Weilong Trade Limited (Shenyang, China). Purified water was purchased from Shenyang Pharmaceutical University and further purified by passing through a 0.22 μm filter. 3-(4-Hydroxyphenyl)propanoic acid and hydroxybenzaldehyde were obtained from Liaoning Benyuan Pharmaceutical (Benxi, China). DHDK was synthesised by Shenyang Pharmaceutical University, Pharmaceutical Analysis Teaching and Research Section. Curcumin was purchased from Chengdu Chroma Biotechnology (Chengdu, China). All other chemical reagents used were of analytical grade. Methylthiazolyldiphenyl tetrazolium bromide (MTT) and cis-platinum were acquired from Sigma-Aldrich (St. Louis, MO, USA).
3.2. Synthesis of DHDK
The total synthesis route is shown in Scheme 1.
3-(4-Hydroxyphenyl)-propanoic acid methyl ester 2. 3-(4-Hydroxyphenyl) propanoic acid (2.8 g, 16.67 mmol) was dissolved in MeOH (40 mL), concentrated sulphuric acid (0.4 mL) was added dropwise and allowed to react at 35 °C for 4 h. A portion of the methanol was removed by concentration under reduced pressure. 10 mL of water was added to the solution and adjusted to pH 6 by adding saturated sodium bicarbonate. The mixture was extracted with EtOAc three times, washed with brine, dried with anhydrous Na2SO4 and concentrated in a rotary evaporator, the resulting product (2.8 g, white powder) was pure enough for the next step (95% yield).
4-(Methoxymethoxy)-benzene propanoic acid methyl ester 3. Compound 2 (2.8 g, 15.54 mmol) and N,N-diisopropylethylamine (3.5 mL, 21.65 mmol) were dissolved in anhydrous dichloromethane, stirred for 30 min and 50 mL of acetone and 1.7 mL of bromomethyl methyl ether (21.65 mmol) were added. The mixture was heated to reflux, monitored by TLC and cooled to room temperature. The mixture was washed with dilute hydrochloric acid (20 mL, 1.0 mol L−1) and water, extracted with EtOAc three times, dried with anhydrous Na2SO4 and concentrated in a rotary evaporator. The resulting crude product was purified by silica gel column chromatography (5% ethyl acetate in hexane) to give 2.93 g of compound 3 as white crystals (84% yield).
4-(Methoxymethoxy)-benzene propanol 4. Lithium aluminium hydride (1.5 g, 40.18 mmol) was dispersed in tetrahydrofuran (50 mL) at 0 °C. Lithium aluminium hydride in tetrahydrofuran (10 mL) was added dropwise to compound 3 (2.93 g, 13.05 mmol) tetrahydrofuran solution. After stirring at room temperature for 3 h, 10 mL of ethyl acetate was slowly added dropwise and quenched by adding 10 mL of water. The pH was adjusted to 6 by adding diluted hydrochloric acid. The mixture was extracted with EtOAc three times. The organic layers were combined, washed with brine, dried with anhydrous Na2SO4 and concentrated in a rotary evaporator. The resulting product (2.41 g, colorless oily liquid) was pure enough for the next step (94% yield).
4-(Methoxymethoxy)-benzenepropanal 5. DMP (1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one) (10.4 g, 24.56 mmol), compound 4 (2.41 g, 12.28 mmol) and sodium bicarbonate (2.09 g, 24.56 mmol) were sequentially added to DCM (30 mL). The mixture was stirred at 0 °C for 4 h and monitored via TLC. Sodium thiosulphate was used to quench the reaction. The layers were washed with saturated ammonium chloride, combined the organic layers, dried with anhydrous sodium sulphate and concentrated in a rotatory evaporator. The resulting crude residual was purified using a silica gel column (3% ethyl acetate in hexane) to give 2.22 g of compound 5 as colorless transparent oily liquid (93% yield).
4-(Methoxymethoxy)-benzaldehyde 6. Hydroxybenzaldehyde (2.5 g, 20.47 mmol) and anhydrous potassium carbonate (14 g, 81.89 mmol) were added to acetone (100 mL) and stirred for 30 min. Bromomethyl methyl ether (2 mL, 26.61 mmol) was then added. After heating to reflux for 5 h, the mixture was cooled to room temperature and filtered. The filtrate was extracted with EtOAc three times, washed with diluted hydrochloric acid (20 mL, 1.0 mol L−1), washed with water twice, dried with anhydrous Na2SO4 and concentrated in vacuo. After confirmation by TLC, the resulting product (3.06 g, light yellow liquid) was pure enough for next step without further purification (90% yield).
(E)-4-(4-(Methoxymethoxy)phenyl)but-3-en-2-one 7. Compound 6 (3.06 g, 18.41 mmol) was dissolved in acetone (30 mL), water (5 mL) and aqueous 10% NaOH (15 mL) were sequentially added at 0 °C with nitrogen protection and allowed to react at 25 °C. The reaction was monitored by TLC. The mixture was extracted by EtOAc three times. The organic layers were combined, washed with brine, dried with anhydrous Na2SO4 and concentrated in a rotary evaporator. The resulting product was purified by silica gel column chromatography to give 3.19 g of compound 7 as white crystals (84% yield).
1,7-Bis(4-(methoxymethoxy)-phenyl)hepta-1,4-dien-3-one 8. Compound 7 (3.19 g, 18.41 mmol) was dissolved in THF (50 mL) and cooled to −78 °C. The system was dried and protected with nitrogen, slowly added with lithium diisopropylamide (19.33 mmol) and allowed to react for 1 h. Compound 5 (2.22 g, 11.43 mmol) in tetrahydrofuran (10 mL) was then slowly added to the mixture, allowed to react for 5 h, quenched with water and extracted with EtOAc three times. The organic layers were combined, washed with brine, dried with anhydrous Na2SO4 and concentrated in a rotary evaporator. The crude residue (reddish brown oily liquid) was used in the next step without further purification (36% yield).
(1E,4E)-1,7-Bis(4-hydroxyphenyl)hepta-1,4-dien-3-one 1. Compound 8 was dissolved in methanol (50 mL), hydrochloric acid (10 mL, 6 mol L−1) was quickly added at 60 °C and allowed to react for 15 min. The mixture was extracted with EtOAc three times, washed with brine, dried with anhydrous Na2SO4 and concentrated in a rotary evaporator. The resulting product was purified by open silica gel column chromatography and octadecyl silyl chromatography to give 0.67 g of compound 1 as yellow powder (55% yield, 96% purity).
3.3. Drug efficacy in vitro
3.3.1 Cell culture information. Calu-3, A-549 (human lung cancer), A375 (human skin melanoma), SKMEL-28 (human skin melanoma), DU-145 (human prostate carcinoma), N87 (human gastric epithelial), SKBR3, MDA-MB231, MCF-7, MDA-MB453 (human breast adenocarcinoma), SKOV3 (human ovarian cancer) and HT-29 (human colorectal carcinoma) cell lines were obtained from the American Type Culture Collection (ATCC). The cells were cultured in DMEM or RPMI-1640 media at 37 °C in an atmosphere of 5% CO2 and 100% relative humidity under sterile conditions to maintain continuous logarithmic growth. The media were supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin.
3.3.2 MTT cell proliferation assay. With CDDP as positive control, MTT assay was used to determine the inhibition rate of DHDK on the proliferation of the 12 tumour cell lines and 3 normal cell lines. The anti-tumour spectrum of DHDK was then initially screened. The cells in the logarithmic growth phase (5 × 104 cells per mL, 100 μL) were separately seeded in 96-well flat-bottomed microculture plates (approximately 4000–5000 cells per well). The edges of the wells were filled with sterile PBS. After culture for 24 h, the cells were completely adhered to the wall and the original culture solution was removed. The final concentrations of DHDK in the wells were as follows: 0.14, 0.41, 1.2, 3.7, 11.3 and 33.3 μmol L−1 in 200 μL of media. The positive control was CDDP with concentrations of 0.14, 0.42, 1.26, 3.8, 11.3 and 34.0 μmol L−1. Each concentration in the treatment groups as well as the blank and control groups were tested in sextuplicate (n = 6). After incubation for 72 h, the cells in each well were added with 100 μL MTT (5 mg mL−1). The plates were incubated for 4 h, the supernatant was aspirated and 150 μL of DMSO was added to each well. The plates were shaken for 10 min until the crystals were completely dissolved. Absorbance (A value) was determined and processed using the GraphPad Prism v5.01 software. Results were used to draw the dose–response inhibition curves of the different tumour cell lines. IC50 values were then calculated.
3.4. Preparation of DHDK micelles
DHDK micelles were prepared by thin-film hydration. Specific amounts of DHDK and Tween 80 were dissolved in 3 mL of ethanol. The solution was evaporated by rotary evaporation at 50 °C to obtain a thin film. The film was dispersed with 0.9% normal saline solution at 50 °C and vortexed for 5 min to obtain DHDK micelles.
3.5. Drug efficacy in vivo
3.5.1 Establishment of MCF-7 breast cancer model. The human breast cancer cell MCF-7 was trypsinised and centrifuged at 1000 rpm for 5 min. The supernatant was removed, and the residue was washed twice with PBS and centrifuged again. The supernatant was removed, and the residue was resuspended in the culture medium. Finally, the residue was adjusted to a concentration of 5 × 107 cells per mL with physiological saline and injected (0.2 mL, 1 × 107) into the antebrachium right axillary of a nude mouse. Tumour formation time, growth, infiltration and metastasis were observed and recorded in detail.
3.5.2 Dosage regimen. Thirty female nude mice were randomly divided into five groups (n = 6 per group). DHDK micelles were injected into nude mouse models of well-established MCF-7 xenograft tumour every day for 9 days. Dosages of 10, 20 and 40 mg kg−1 were administered to mice in low-, medium- and high-dosage groups, respectively. The positive group (cisplatin) was established by administering 5 mg kg−1 cisplatin to the same conditioned female nude mice via the tail vein for 9 days. The blank control group consisted of mice that were not given any drug. Tumour volume and mouse weight were calculated and measured during the experiments.
3.6. Determination of biochemical indexes in serum and plasma
Twenty four female mice were randomly divided into four groups (n = 6 per group). DHDK micelles were injected into mice every day for 9 days. Consistent with the dosing regimen in vivo efficacy evaluation, the mice were killed on the last day after administration, and plasma and serum were taken. Take the appropriate amount of mice serum and follow the instructions in the kit to determine the AST, ALT and CRE in the serum. Take the appropriate amount of mice plasma and follow the instructions in the kit to determine the BUN in the plasma.
3.7. Pathological tissue sections
Histopathological technique is used for comprehensive observation of specific tissue samples and the state of structure. In more than 100 years of application and development, this technique has become an important technology in life sciences and is the gold standard for disease diagnosis in clinical practice. In the current study, we evaluated the conditions of the pathological sections of different tissues. At the end of the 9 day treatment, the tumour-bearing nude mice were anesthetised and killed to collect fresh tissues (heart, liver, spleen, lung, kidney, brain and tumour). The tissues were soaked and fixed with 10% formalin solution, embedded in paraffin and cut into 4 μm paraffin sections. The sections were subjected to HE staining to detect the toxicity of DHDK.
3.8. Pharmacokinetics in vivo
3.8.1 Dosage regimen and sample collection. Based on in vivo and in vitro activities, DHDK micelle injection was conducted for PK studies at the dosage of 14 mg kg−1 by tail vein. Prior to treatment, the rats were fasted for 12 h and allowed to drink freely. Blood samples (0.3 mL) were collected from the orbital venous plexus into a heparinised centrifuge tube at 0, 0.033, 0.167, 0.25, 0.5, 0.75, 1, 2, 4, 8 and 12 h. The blood samples were centrifuged. Plasma was obtained and stored at −80 °C until analysis.
3.8.2 LC-ESI-MS/MS condition. LC-ESI-MS/MS (Waters I-Class UPLC, Waters Xevo TQ-S) was used to determine plasma concentration by using a Thermo Hypersil GOLD C18 column (2.1 mm × 100 mm, 1.9 μm). The mobile phase consisted of 0.1% formic acid in water (A) and acetonitrile (B). The gradient elution conditions were 0–2.5 min (A:B = 55:45 to 15:85), 2.5–2.6 min (A:B = 15:85 to 55:45) and 2.6–3.5 min (A:B = 55:45 to 55:45); injection volume of 2 μL; and flow rate of 0.3 mL min−1. The scanning mode of MS was MRM (multi-reaction monitoring). DHDK was determined with precursor ion at m/z 294.98 and product ion at m/z 106.67. Internal standard curcumin was analysed with precursor ion at m/z 369.1 and product ion at m/z 284.8.
3.8.3 Sample pretreatment. The rat plasma was thawed and vortexed. The plasma (100 μL) was added with 10 μL of the internal standard and 10 μL of methanol, vortexed for 1 min and added with 2 mL of MTBE. The mixture was vortexed for 5 min and centrifuged at 12000 rpm at 4 °C for 5 min. The supernatant was blow dried at 37 °C, and the residue was redissolved with 100 μL of the initial mobile phase (water: acetonitrile = 55:45) and vortexed for 3 min to dissolve. The mixture was centrifuged at 12000 rpm for 3 min at 4 °C, and 2 μL of the supernatant was injected into the system.
3.9. Tissue distribution
3.9.1 Dosage regimen and sample collection. Twenty-four SD rats (12 males and 12 females) were randomly divided into four groups (n = 6 per group) and intravenously injected with 14 mg kg−1 DHDK micelles. The rats were fasted 12 h before drug administration but were allowed to drink freely. The rats were killed by cervical dislocation at 0.25, 1, 4 and 24 h (n = 6 per time-point) after intravenous administration. Heart, liver, spleen, lung and kidney were excised and thoroughly rinsed of residual blood in ice-cold saline before gently blotting on absorbent paper and weighing. The tissues were homogenised, and the homogenates were stored at −80 °C until analysis. The analytical method performed was the same as that in the pharmacokinetics study.
3.10. Ethical statement
All in vivo experimental studies were performed in accordance with the Guideline for Animal Experimentation of Shenyang Pharmaceutical University and with the approval from the Animal Ethics Committee of Shenyang Pharmaceutical University (Approval number: 211002000310215).
4. Conclusion
This study involves synthesis of DHDK, determination of its in vitro and in vivo drug efficacy with respect to dosage and analyses of tissue slices, pharmacokinetics and tissue distribution. DHDK is a novel drug that can be completely synthesised by asymmetric aldol condensation. DHDK has anti-tumour effect in vitro, especially against breast and lung cancers. This study optimized the dosage and the in vivo effect of DHDK was positively related to the dosage applied. The high-dose group showed better antitumor effect than the other two dose groups and was without toxicity. DHDK has little inhibitory effect on normal cells, which indicates that DHDK has certain selectivity for the toxic effects of cancer cells and normal cells. The results of the measured biochemical indicators of AST, ALT, BUN and CRE indicated that DHDK has no potential liver and kidney toxicity. The result of tissue distribution showed that the drug is easy to distribute in lung tissue, so the effect in treating lung cancer may be need further research.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors are grateful for the Natural Science Foundation of Liaoning Province for funding toward financial support (No. 20180551065)
References
- World Health Organization, Cancer. Fact Sheet No. 297, http://www.who.int/mediacentre/factsheets/fs297/en/, accessed 01 August 2015 Search PubMed.
- L. Scotti, F. J. Mendonça Júnior and F. F. Ribeiro, et al., Natural product inhibitors of topoisomerases: review and docking study, Curr. Protein Pept. Sci., 2018, 19, 275–291 CAS.
- F. Ries, M. Dicato and M. Pauly, et al., The Genetic Basis of Multidrug Resistance, Pathol., Res. Pract., 1992, 188, 804–807 CrossRef.
- R. Buckingham, J. Fitt and J. Sitziaz, et al., Patients' experiences of chemotherapy: side-effects of carboplatin in the treatment of carcinoma of the ovary, Eur. J. Cancer Care, 1997, 6, 59–71 CrossRef CAS PubMed.
- D. Thaloor, A. K. Singh and G. S. Sidhu, et al., Inhibition of angiogenic differentiation of human umbilical vein endothelial cells by curcumin, Cell Growth Differ., 1998, 9, 305–312 CAS.
- J. van E. Marjan, E. Teuling and C. M. S. Yvonne, et al., Research time- and dose-dependent effects of curcumin on gene expression in human colon cancer cells, J. Carcinog., 2004, 3, 17 Search PubMed.
- J. M. Holy, Curcumin disrupts mitotic spindle structure and induces micronucleation in MCF-7 breast cancer cells, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2002, 518, 71–84 CrossRef CAS.
-
(a) F. Yang, G. P. Lim and A. N. Begum, et al., Curcumin Inhibits Formation of Amyloid B Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid In Vivo, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS PubMed;
(b) R. W. McDonald, W. Bunjobpon and T. Liu, et al., Synthesis and anticancer activity of nordihydroguaiaretic acid (NDGA) and analogues, Anti-Cancer Drug Des., 2001, 16, 261–270 CAS;
(c) T. Choshi, S. Horimoto and C. Y. Wang, et al., Synthesis of dibenzoylmethane derivatives and inhibition of mutagenicity in Salmonella typhimurium, Chem. Pharm. Bull., 1992, 40, 1047 CrossRef CAS PubMed;
(d) J. Ishida, M. Kozuka and H. K. Wang, et al., Antitumor-promoting effects of cyclic diarylheptanoids on Epstein-Barr virus activation and two-stage mouse skin carcinogenesis, Cancer Lett., 2000, 159, 135–140 CrossRef CAS PubMed.
- P. Anand, et al., Bioavailability of Curcumin: Problems and Promises, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- R. Mishra, M. K. Das and S. Singh, et al., Articulatin-D induces apoptosis via activation of caspase-8 in acute T-cell leukemia cell line, Mol. Cell. Biochem., 2017, 426, 87–99 CrossRef CAS PubMed.
- R. Grossarth-Maticek and R. Ziegler, Randomised and non-randomised prospective controlled cohort studies in matched-pair design for the long-term therapy of breast cancer patients with a mistletoe preparation (Iscador): a re-analysis, Eur. J. Med.Res., 2006, 11, 485–495 CAS.
- P. J. Goebell, T. Otto and J. Suhr, et al., Evaluation of an Unconventional Treatment Modality with Mistletoe Lectin to Prevent Recurrence of Superficial Bladder Cancer: A Randomized Phase ii Trial, J. Urol., 2002, 168, 72–75 CrossRef PubMed.
- G. B. Sela, M. Wollner and L. Hammer, et al., Mistletoe as complementary treatment in patients with advanced non-small-cell lung cancer treated with carboplatin-based combinations: a randomised phase II study, Eur. J. Cancer, 2013, 49, 1058–1064 CrossRef PubMed.
- C. Maletzki, M. Linnebacher and R. Savai, et al., Mistletoe lectin has a shiga toxin-like structure and should be combined with other Toll-like receptor ligands in cancer therapy, Cancer Immunol. Immunother., 2013, 62, 283–1292 CrossRef PubMed.
-
(a) M. Harmsma, M. Ummelen and W. Dignef, et al., Effects of mistletoe (Viscum album L.) extracts Iscador on cell cycle and survival of tumor cells, Arzneim. Forsch., 2006, 56, 474–482 CAS;
(b) E. Kovacs, S. Link and U. Toffol-Schmidt, Cytostatic and cytocidal effects of mistletoe (Viscum album L.) quercus extract Iscador, Arzneim.-Forsch., 2006, 56, 467–473 CAS;
(c) G. Seifert, P. Jesse and A. Laengler, et al., Molecular mechanisms of mistletoe plant extract-induced apoptosis in acute lymphoblastic leukemia in vivo and in vitro, Cancer Lett., 2008, 264, 218–228 CrossRef CAS PubMed.
- W. B. Park, S. Y. Lyu and J. H. Kim, et al., Inhibition of tumor growth and metastasis by Korean mistletoe lectin is associated with apoptosis and antiangiogenesis, Cancer Biother.Radiopharm., 2001, 16, 439–447 CrossRef CAS PubMed.
-
(a) M. Schink, W. Tröger and A. Dabidian, et al., Mistletoe Extract Reduces the Surgical Suppression of Natural Killer Cell Activity in Cancer Patients. A Randomized Phase III Trial, Forsch. Komplementarmed, 2007, 14, 9–17 Search PubMed;
(b) L. Heinzerling, B. V. Von and C. Liebenthal, et al., Immunologic effector mechanisms of a standardized mistletoe extract on the function of human monocytes and lymphocytes in vitro, ex vivo, and in vivo, J. Clin. Immunol., 2006, 26, 347–359 CrossRef PubMed;
(c) B. Liu, H. J. Bian and J. K. Bao, Plant lectins: potential antineoplastic drugs from bench to clinic, Cancer Lett., 2010, 287, 1–12 CrossRef CAS PubMed.
- U. Weissenstein, M. Kunz and K. Urech, et al., Interaction of standardized mistletoe (Viscum album) extracts with chemotherapeutic drugs regarding cytostatic and cytotoxic effects in vitro, BMC Complementary Altern. Med., 2014, 14, 6 CrossRef PubMed.
- Y. L. Zhao, X. Y. Wang and L. X. Sun, et al., Cytotoxic constituents of Viscum coloratum, Z. Naturforsch. C, 2012, 67, 129–134 CAS.
- D. Lou, L. Zhang and W. Sun, et al., Determination of Curcumin in Rat Plasma by HPLC and Study of its Pharmacokinetics, Chin. J. Mod. Appl. Pharm., 2011, 28, 870–873 CAS.
- E. S. E. El-Awady, Y. M. Moustafa and D. M. Abo-Elmatty, et al., Cisplatin-induced cardiotoxicity: mechanisms and cardioprotective strategies, Eur. J. Pharmacol., 2011, 650, 335–341 CrossRef CAS PubMed.
- G. Chvetzoff, B. Bonnotte and B. Chauffert, Anticancer chemotherapy. prevention of toxicity, La Presse Médicale, 1999, 27, 2106–2112 Search PubMed.
- V. B. Pai and D. M. C. Nahata, Cardiotoxicity of Chemotherapeutic Agents, Drug Saf., 2000, 22, 263–302 CrossRef CAS PubMed.
- L. I. wen Jun, Z. H. E. N. Yong-Xing and L. I. Yan-Fu, Pharmacokinetics of carboplatin and cisplatin in rats, Chin. J. Pharm., 1991, 22, 5 Search PubMed.
- N. K. Ibrahim, N. Desai and S. Legha, et al., Phase I and pharmacokinetic study of ABI-007, a Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel, Clin. Cancer Res., 2002, 8, 1038–1044 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03674a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*