
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly green approach towards aromatic nitro group substitutions: catalyst free reactions of nitroimidazoles with carbon nucleophiles in water†
Syed Raza Shah,
Muhammad Ali,
Muhammad U. Anwar and
Ahmed Al-Harrasi*
and
Ahmed Al-Harrasi*
Natural and Medical Sciences Research Center, University of Nizwa, Nizwa 611, Sultanate of Oman. E-mail: aharrasi@unizwa.edu.om; Fax: +96825446612; Tel: +96825446328
First published on 8th June 2020
Abstract
We have successfully developed a flexible green aqueous approach for the formation of a carbon–carbon bond by the reaction of highly-enolizable carbanions (mostly derived from 1,3-dicarbonyl compounds) with an aromatic carbon bearing a nitro group. The key step involves a nucleophilic displacement reaction. All newly synthesized compounds were unambiguously characterized via various spectroscopic techniques including NMR, mass spectrometry and single-crystal X-ray diffraction as applicable. We believe that our study will be useful in providing new insights into catalyst-free water-mediated nucleophilic substitution reactions.
Introduction
In the context of green chemistry, the best solvent is no solvent at all, conversely, on the likelihood that if a solvent is ever essential, water is a sizable substitute to propose. Water is one of the lesser-studied solvents for everyday syntheses either at the laboratory or at the industrial scale. In contemporary times, several reactions were revealed in water even though water-insoluble substrates were utilized in suspensions. There is a growing awareness across the chemical community to streamline the synthetic methodologies employing sustainable technologies.1 In general, organic solvents are preferred by chemists but are worrisome for their threatening environmental impacts. However, water holds an undisputable position among all solvents as an ideal solvent.2 Water has been less extensively utilized as a solvent for practical synthetic procedures because of its inherent incapability to solubilize organic materials and the decomposition of most of the moisture-sensitive chemicals. However, several important reactions pertaining to C–H bond conversions into C–hetero atom bonds were studied in water.3 Furthermore, rhodium-catalyzed 1,4-addition of organoboron reagents represents a valuable approach to transition metal-catalyzed asymmetric C–C bond formation.4 The regio- and stereoselective hydroxylation of steroids in water, by employing manganese porphyrins and β-cyclodextrins as catalysts and iodosobenzene as an oxidant, has led to the formation of C–O bonds.5 The reactivity of numerous natural products in aqueous suspensions has been sophisticatedly corroborated by Sharpless.6 Recently, numerous reactions including Wittig reaction,7 Mannich-type reactions,8 and intramolecular Diels–Alder reaction9 were studied in water.There is a large band of nucleophilic substitution and addition reactions that were studied in non-aqueous media.10–31 Herein, we successfully report that the SNAr reaction can be effectively performed in aqueous conditions while using carbon-based ambident nucleophiles at somewhat elevated temperatures.
Results and discussion
Hefty amounts of organic solvents compared to particular reagents in a particular reaction are customarily crucial for any chemical reaction to take place. The majority of the post-reaction procedures are heavily dependent upon solvents. Approximately 20 million metric tons per annum of organic solvents are produced industrially,32 of which excessive quantities are consumed as nonrenewable solvents that make it a highly unfeasible and unsustainable practice. Replacing toxic solvents with either water or using “No Solvent” is a highly demanding approach for chemical syntheses because of its many green implications. One of the most fundamental chemical processes is the aromatic nucleophilic substitution reaction, of which, replacement of aromatic nitro group(s) by carbon nucleophiles is of utmost interest.The leaving ability of the nitro group (nucleofugicity) is much higher than that of I, Br, Cl and F, but this mostly occurs when an aromatic nucleus possesses two or more nitro groups.33 However, it is a well-conceived fact that a direct displacement of a nitro group requires somehow inductive activation by electron-withdrawing group(s).34 We herein report a green approach involving aromatic nitro group substitution reactions of nitroimidazoles with carbon nucleophiles in water.
Initially, we screened a range of solvents (THF, CH3CN, C2H5OH, DCM, and DMSO) to replace the aromatic nitro group of imidazole with carbon nucleophiles with and without organic or inorganic bases (Scheme 1).
 | ||
| Scheme 1 Optimization of the reaction conditions (where R1 = –CH2CH2–OH, R2 = –CH3 entry 2, Table 2). | ||
Interestingly, no product formation was attained for entries 4, 7 and 8 (Table 1), as depicted in Scheme 1. We assumed that the byproduct nitrous acid (HNO2) was captured and neutralized by an equimolar amount of base and therefore proved to be an inhibitor to initiate the reaction. This finding is suggestive of the fact that the presence of an acid is paramount to success in this reaction. This was further confirmed when the same reaction was conducted in ethanol (entry 3, Table 1) and water (entry 6, Table 1) but without any additive and yielded the desired adduct in appreciable amounts. However, we did not encounter any solubility issues while using water as the solvent.
| Entry | Nucleophiles | Electrophilic substrate | T (°C) | t (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 |  |
 |
Reflux | 0.5 | 72 |
| 2 |  |
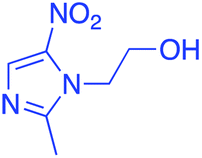 |
Reflux | 3 | 70 |
| 3 |  |
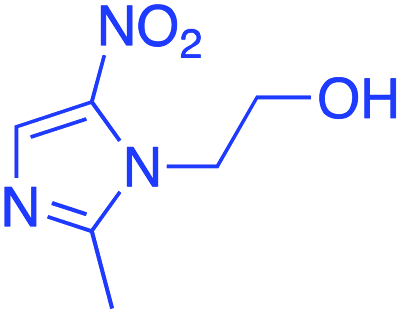 |
Reflux | 6 | 66 |
| 4 |  |
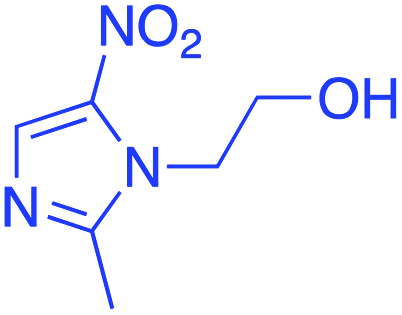 |
Reflux | 50 | 30 |
| 5 |  |
 |
Reflux | 48 | 65 |
| 6 | 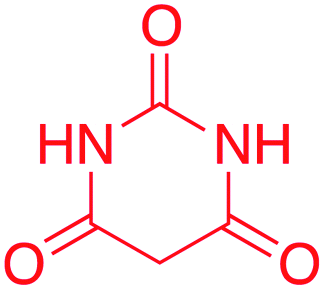 |
 |
Reflux | 48 | 69 |
| 7 | 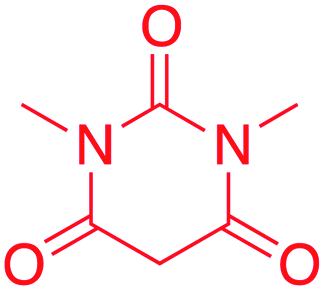 |
 |
Reflux | 48 | 72 |
| 8 |  |
 |
Reflux | 80 | 20 |
| 9 |  |
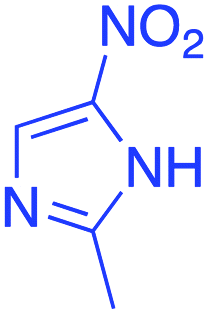 |
Reflux | 44 | 73 |
| 10 | 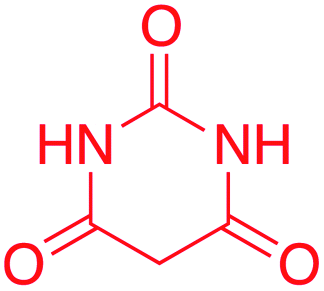 |
 |
Reflux | 48 | 77 |
| 11 |  |
 |
Reflux | 48 | 78 |
In order to check the scope of the reaction, a range of carbon nucleophiles and nitroimidazoles as effective electrophilic reagents were investigated (Scheme 2, entries 1–11). We believe that carbon nucleophiles were generated in situ in their enolized forms in water as the solvent. These enolized species are perfect ambident nucleophiles35 that can readily be captured by electrophiles. To the best of our knowledge, this is the first example of mono-nitro-substituted aromatic nuclei in which the nitro groups are being replaced by some ambident carbon nucleophiles.
 | ||
| Scheme 2 Reaction of active methylenes with nitroimidazoles. | ||
Water as a green solvent has been employed and has made the overall process extremely attractive for synthetic chemists. In the nitroimidazole nucleus, the sp2-hybridized carbon atom that bears the nitro group is directly attached to an sp3-nitrogen atom. Further, another sp2-nitrogen atom is also present. These two nitrogen atoms sufficiently withdraw electron density and polarize the ring. Then, the presence of the nitro group further activates the ring so a nucleophilic substitution reaction is not unusual stuff to occur. In such typical nucleophilic aromatic substitution reactions, the addition–elimination mechanism is suggested to occur most likely.36 The whole reaction is completed in aqueous media without using any catalyst and any other hazardous solvents/reagents. Initially, we realized that the enol forms of the active methylenes 1, 2, 3 and 4 sufficiently acidify the reaction mixtures (pH drops to 3). We observed that active methylenes are solely soluble in water, while the pH of the solution was found to be around 5. However, when the nitroimidazole was added to the same solution, the pH of the reaction mixture suddenly dropped to 3. In our opinion, this fact could possibly be explained by hypothesizing that one of the oxygen atoms of the nitro group is protonated and therefore the resulting conjugate acid is responsible for this drop in pH. During the process, the nitro group adopts Nef's37 like resonating structures, which are depicted in Scheme 3. The activation of the nitro group through protonation rendered it a highly labile leaving group. Meanwhile sp2 carbon atom bonded to the nitro group becomes more viable for nucleophilic displacement. This was to our surprise that a nucleophilic substitution reaction occurred without the presence of any other electron-withdrawing group on the imidazole ring. We also noticed that nitrous acid (HNO2) that is produced during the course of the reaction, produced oximes as a result of reaction with active methylenes. Upon refluxing the reaction mixture, there was always a 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 7:3 ratios of the addition product versus oxime formation. Similar observations were observed in literature involving reactions between active methylenes and HNO2.38
:2 to 7:3 ratios of the addition product versus oxime formation. Similar observations were observed in literature involving reactions between active methylenes and HNO2.38
 | ||
| Scheme 3 Plausible mechanism/s for the aromatic nucleophilic displacement reaction. | ||
However, in order to enhance the yield of the addition product, an extra equivalent of active methylene was employed. Most of the adducts showed high melting points (155 to 300 °C). The newly synthesized compounds were structurally characterized by different analytical techniques including IR, mass spectrometry (ESI+), and 1H and 13C NMR spectroscopy. The single-crystal X-ray diffraction was used for the characterization of the products (1, 3 and 5) and unambiguously confirmed the structures (Fig. S23–S25 of ESI†). A plausible mechanism for the reaction is illustrated in Scheme 3.
We opine that the reaction may proceed in two distinct ways, as depicted in Scheme 3. In the first instance, enolized active methylene protonates the oxygen atom of nitroimidazole, followed by the formation of an ion pair in which only carbanion attacks the electrophilic center of the substrate. We did not observe any O-alkylated product. We believe that the naked oxygen anion is masked through hydrogen bonding with water molecules. In an alternative pathway, after the activation of the electrophilic carbon via the protonation of the oxygen atom of nitroimidazole, water replaces the nitro group. In the next step, carbanion replaces a protonated hydroxyl group to form a carbon–carbon bond. A DFT study is underway to pin down the most plausible way as to how this reaction really proceeds.
Experimental
IR spectra (KBr) were recorded on a Bruker FT-IR IFS48 spectrophotometer. EI mass spectra were obtained using HR-ESI-MS: Agilent Technologies, 6530. Accurate mass and data were tabulated as m/z. 1H-NMR and 13C-NMR spectra were recorded in DMSO-d6 using Bruker 600 MHz and 150 MHz spectrophotometers, respectively. Splitting patterns are as follows; s, singlet; d, doublet; dd, double doublets; t, triplet; m, multiplet. Chemical shifts are reported in δ (ppm) and coupling constants are measured in Hz. The progress of all the reactions was monitored via TLC, which was performed on 2.0 × 5.0 cm aluminum sheets pre-coated with silica gel 60F254 to a thickness of 0.25 mm (Merck). The chromatograms were visualized under ultraviolet light (254–366 nm) or iodine vapours. The title compounds were synthesized and characterized satisfactorily.General procedure for the synthesis of compounds 1–11
A 50 mL round bottom flask was charged with 1 mmol of active methylene(s) and 1 mmol of corresponding nitroimidazole(s), followed by the addition of 10 mL of distilled water. The reaction mixture was refluxed from several minutes to several hours until a colored precipitate(s) appeared. Variation in the color of various reaction mixtures during the course of the reaction is shown in Fig. S27–S37.† Crude solids so obtained were filtered and washed with water and then methanol. After washing the solids, they were dissolved in hot methanol and then kept at room temperature for crystallization.6-Hydroxy-5-(1-(2-hydroxyethyl)-2-methyl-1H-imidazol-5-yl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (1)
Yield: 72% (193 mg) as pale yellow powder; purification: filtration and crystallization in water; mp = 251–252 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.08 (s, O121H), 11.10 (s, N1&52H), 7.55 (s, C81H), 5.08 (t, J = 5.5 Hz, O181H), 4.08 (t, J = 5.1 Hz, C172H), 3.68 (t, J = 4.7 Hz, C162H), 2.59 (s, C193H); 13C NMR (150 MHz, DMSO-d6) δ = 173.47 (C6), 161.02 (C2&4), 139.39 (C10), 129.33 (C7), 112.91 (C8), 81.21 (C3), 59.89 (C17), 49.27 (C16), 10.29 (C19). FTIR (cm−1): 1645, 1584, 1520, 1205; HRMS (ESI+): calculated for C10H12N4O3S (M + H+) 269.29, found 269.09.6-Hydroxy-5-(1-(2-hydroxyethyl)-2-methyl-1H-imidazol-5-yl)pyrimidine-2,4(1H,3H)-dione (2)
Yield: 70% (176 mg) as pale yellow powder; purification: filtration and recrystallization in water; mp = 211–212 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.37 (s, N1&52H), 7.04 (s, C81H), 5.06 (s, O181H), 3.96 (t, J = 5.5 Hz, C172H), 3.55 (t, J = 5.4 Hz, C162H), 2.56 (s, C193H); 13C NMR (150 MHz, DMSO-d6) δ 164.11 (C2 & 4), 151.98 (C6), 142.44 (C10), 131.02 (C7), 117.31 (C8), 74.07 (C3), 59.64 (C17), 47.23 (C16), 11.44 (C19); FTIR (cm−1): 1684, 1626, 1547.; HRMS (ESI+): calculated for C10H12N4O4 (M + H+) 253.23, found 253.09.6-Hydroxy-5-(1-(2-hydroxyethyl)-2-methyl-1H-imidazol-5-yl)-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (3)
Yield: 66% (185 mg) as light pink crystalline solid; purification: filtration and recrystallization in water; mp = 163–164 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.49 (s, O121H), 7.12 (s, C81H), 4.99 (t, J = 5.2 Hz, O181H), 3.94 (t, J = 5.4 Hz, C172H), 3.51 (t, J = 5.3 Hz, C162H), 3.07 (s, C19&206H), 2.60 (s, C213H); 13C NMR (150 MHz, DMSO-d6) δ 161.95(C2&4), 152.89(C6), 142.54(C10), 131.74(C7), 116.87(C8), 74.15(C3), 59.69(C17), 47.27(C16), 27.04(C19&20), 11.38(C21); FTIR (cm−1): 1683, 1602, 1568, 1530; HRMS (ESI+): calculated for C12H16N4O4 (M + H+) 281.28, found 281.10.3-Hydroxy-2-(1-(2-hydroxyethyl)-2-methyl-1H-imidazol-5-yl)cyclohex-2-en-1-one (4)
Yield: 30% (70 mg) as light brown solid; purification: chromatography: DCM:MeOH (9.5:0.5 to 3:1) over silica; mp = 186–187 °C; 1H NMR (600 MHz, DMSO-d6) δ 14.89 (s, O121H), 7.56 (s, C81H), 4.97 (t, J = 5.3 Hz, O181H), 4.02 (t, J = 5.1 Hz, C172H), 3.65 (t, J = 5.0 Hz, C162H), 2.48 (s, C193H), 2.29 (t, J = 6.3 Hz, C1&54H), 1.82 (p, J = 6.3 Hz, C62H); 13C NMR (150 MHz, DMSO-d6) δ 188.43(C2&4), 138.37(C10), 132.90(C7), 114.12(C8), 102.04(C3), 60.29(C17), 48.68(C16), 35.39(C1&5), 20.87(C6), 11.16(C19); FTIR (cm−1): 2921, 1600, 1515; HRMS (ESI+): calculated for C12H16N2O3 (M + H+) 237.27, found 237.12.
2-(5-(6-Hydroxy-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-2-methyl-1H-imidazol-1-yl)ethyl benzoate (5)
Yield: 65% (240 mg) dark brown product; purification: filtration; mp = 225–226 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.77 (s, O121H), 10.85 (s, N1&52H), 7.89 (d, J = 7.7 Hz, C23&272H), 7.65 (d, J = 7.4 Hz, C251H), 7.53 (t, J = 7.4 Hz, C24&262H), 7.20 (s, C81H), 4.44 (t, J = 4.5 Hz, C172H), 4.43 (t, J = 4.6 Hz, C162H), 2.64 (s, C193H); 13C NMR (150 MHz, DMSO-d6) δ 174.80 (C6), 165.51(C4), 162.10 (C20), 159.54 (C2), 142.62 (C10), 133.54 (C25), 130.37(C22), 129.22 (C23&27), 129.16 (C7), 128.90 (C24&26), 117.47 (C8), 78.57(C3), 63.37(C17), 43.92 (C16), 11.20(C19); HRMS (ESI+): calculated for C17H16N4O4S (M + H+) 373.41, found 373.09; FTIR (cm−1): 1706, 1665, 1635, 1577, 1526.2-(5-(6-Hydroxy-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-2-methyl-1H-imidazol-1-yl)ethyl benzoate (6)
Yield: 69% (249 mg) as white powder; purification: DCM: MeOH (9.5:0.5 to 3:1) over silica; mp = 197–198 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.64 (s, O121H), 9.45 (s, N1&52H), 7.90 (d, J = 7.7 Hz, C23&272H), 7.66 (t, J = 7.4 Hz,, C251H), 7.52 (t, J = 7.6 Hz, C24&262H), 7.15 (s, C81H), 4.45 (t, J = 5.1 Hz, C172H), 4.42 (t, J = 5.2 Hz, C162H), 2.64 (s, C193H); 13C NMR (150 MHz, DMSO-d6) δ 165.51(C4&20), 164.01(C2), 151.94(C6), 142.10(C10), 133.53(C25), 131.65(C22), 129.22(C7,23&27), 128.87(C24&26), 116.96(C8), 74.02(C3), 63.33(C17), 43.79(C16), 11.13(C19); HRMS (ESI+): calculated for C17H16N4O5 (M + H+) 357.34, found 357.11; FTIR (cm−1): 2963, 1714, 1566.
2-(5-(6-Hydroxy-1,3-dimethyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-2-methyl-1H-imidazol-1-yl)ethyl benzoate (7)
Yield: 72% (273 mg) as light pink crystalline solid; purification: filtration and recrystallization from ethanol; mp = 181–182 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.66 (s, O121H), 7.86 (d, J = 7.7 Hz, C23&272H), 7.64 (t, J = 7.4 Hz, C251H), 7.49 (t, J = 7.6 Hz, C24&262H), 7.16 (s, C81H), 4.42 (t, J = 4.0 Hz, C172H), 4.41 (t, J = 4.1 Hz, C162H), 3.03 (s, C28&296H), 2.65 (s, C193H); 13C NMR (150 MHz, DMSO-d6) δ 165.52(C4&20), 161.86(C2), 152.79(C6), 142.07(C10), 133.47(C25), 132.24(C22), 129.19(7,23&27), 128.76(C24&26), 116.95(C8), 74.46(C3), 63.44(C17), 48.63(methanol), 43.84(C16), 27.03(C28&29), 11.14(C19).; FTIR (cm−1): 1724, 1679, 1575; HRMS (ESI+): calculated for C19H20N4O5 (M + H+) 385.39, found 385.15.2-(5-(2-Hydroxy-6-oxocyclohex-1-en-1-yl)-2-methyl-1H-imidazol-1-yl)ethyl benzoate (8)
Yield: 20% (65 mg) as greenish yellow solid; purification: filtration and recrystallization in ethanol; mp = 151–152 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.89 (d, J = 7.7 Hz, C23&272H), 7.64 (t, J = 7.4 Hz, C251H), 7.50 (t, J = 7.6 Hz, C24&262H), 6.61 (s, C81H), 4.33 (t, J = 5.4 Hz, C172H), 4.11 (t, J = 5.4 Hz, C162H), 2.44 (s, C193H), 2.15 (t, J = 6.3 Hz, C1&54H), 1.76 (p, J = 6.3 Hz, C62H).; 13C NMR (150 MHz, DMSO-d6) δ 190.04(C2&4), 165.58(C20), 141.70(C10), 133.48(C25), 131.29(C22), 129.35(C23&27), 129.20(C7), 128.80(C24&26), 120.32(C8), 100.33(C3), 63.74(C17), 42.96(C16), 36.28(C1&5), 21.11(C6), 11.98(C19); HRMS (ESI+): calculated for C19H20N2O4 (M + H+) 341.38, found 341.14; FTIR (cm−1): 2937, 1713, 1588.6-Hydroxy-5-(2-methyl-1H-imidazol-5-yl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (9)
Yield: total 73% (166 mg) as grey solid; purification: filtration washed with ethanol; mp = 186–187 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.33 (s, O121H), 13.10 (s, N161H), 11.09 (s, N1&52H), 7.43 (s, C81H), 2.53 (s, C153H); 13C NMR (150 MHz, DMSO-d6) δ 173.41 (C6), 161.02 (C2&4), 138.96 (C10), 130.29 (C7), 108.99 (C8), 81.46 (C3), 11.17 (C15); FTIR (cm−1) 1644, 1590, 1522; HRMS (ESI+) calculated for C8H8N4O2S (M + H+) 225.25, found 225.02.6-Hydroxy-5-(2-methyl-1H-imidazol-5-yl)pyrimidine-2,4(1H,3H)-dione (10)
Yield: total 77% (161 mg) as pink solid; purification techniques: filtration washed with ethanol; mp = 214–215 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.17 (s O121H), 13.17 (s, N161H), 9.67 (s, N1&52H), 7.33 (s, C81H), 2.52 (s, C153H); 13C NMR (150 MHz, DMSO-d6) δ 163.13(C2&4), 151.09 (C6), 137.89 (C10), 131.60 (C7), 107.46 (C8), 77.32 (C3), 11.13 (C15); FTIR (cm−1) 1687, 1637, 1580; HRMS (ESI+) calculated for C8H8N4O3 (M + H+) 209.18, found 209.04.6-Hydroxy-1,3-dimethyl-5-(2-methyl-1H-imidazol-5-yl)pyrimidine-2,4(1H,3H)-dione (11)
Yield: total 78% (185 mg) pink solid product; purification: DCM:MeOH (9.5:0.5 to 3:1) over silica; mp = 206–207 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.24 (s, O121H), 7.45 (s, C81H), 3.15 (s, C17&186H), 2.53 (s, C153H); 13C NMR (150 MHz, DMSO-d6) δ 161.21 (C2&4), 152.24 (C6), 138.49 (C10), 131.95 (C7), 108.53(C8), 78.28 (C3), 27.26 (C17&18), 11.49 (C15); FTIR (cm−1): 1633, 1592, 1555; HRMS (ESI+) calculated for C10H12N4O3 (M + H+) 237.24, found 237.04.
Conclusion
In conclusion, we have developed a flexible green aqueous approach for the formation of a carbon–carbon bond formation by the reaction of highly enolizable carbanions with an aromatic carbon bearing a nitro group. The key step involves a nucleophilic displacement reaction. This facile and green protocol will be practical for catalyst-free water-mediated nucleophilic substitution reactions. The current protocol can possibly be extended to the synthesis of active pharmaceutical ingredients or active intermediates. More studies are underway in our laboratories.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the University of Nizwa for the financial support of this project through the scholarship of SRS.References
- T. Welton, Proc. R. Soc. A, 2015, 471, 20150502 Search PubMed.
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed.
- C. I. Herrerías, X. Yao, Z. Li and C.-J. Li, Chem. Rev., 2007, 107, 2546–2562 CrossRef PubMed.
- C.-G. Feng, Z.-Q. Wang, C. Shao, M.-H. Xu and G.-Q. Lin, Org. Lett., 2008, 10, 4101–4104 CrossRef CAS PubMed.
- J. Yang and R. Breslow, Angew. Chem., Int. Ed., 2000, 39, 2692–2695 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- J. Dambacher, W. Zhao, A. El-Batta, R. Anness, C. Jiang and M. Bergdahl, Tetrahedron Lett., 2005, 46, 4473–4477 CrossRef CAS.
- T. Hamada, K. Manabe and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 7768–7769 CrossRef CAS PubMed.
- H. Yanai, A. Saito and T. Taguchi, Tetrahedron, 2005, 61, 7087–7093 CrossRef CAS.
- H. W. Moore, Chem. Soc. Rev., 1973, 2, 415–455 RSC.
- M. F. Sartori, Chem. Rev., 1963, 63, 279–296 CrossRef CAS.
- R. P. Shishkina and V. N. Berezhnaya, Russ. Chem. Rev., 1994, 63, 139–146 CrossRef.
- M. Aguilar-Martinez, N. A. Macias-Ruvalcaba, J. A. Bautista-Martinez, M. Gomez, F. J. Gonzalez and I. Gonzalez, Curr. Org. Chem., 2004, 8, 1721–1738 CrossRef CAS.
- S. Giorgi-Renault, J. Renault, P. Gebel-Servolles, M. Baron, C. Paoletti, S. Cros, M. C. Bissery, F. Lavelle and G. Atassi, J. Med. Chem., 1991, 34, 38–46 CrossRef CAS PubMed.
- T. S. Lin, S. Xu, L. Zhu, L. A. Cosby and A. C. Sartorelli, J. Med. Chem., 1989, 32, 1467–1471 CrossRef CAS PubMed.
- L. Salmon-Chemin, E. Buisine, V. Yardley, S. Kohler, M.-A. Debreu, V. Landry, C. Sergheraert, S. L. Croft, R. L. Krauth-Siegel and E. Davioud-Charvet, J. Med. Chem., 2001, 44, 548–565 CrossRef CAS PubMed.
- E. V. M. dos Santos, J. W. d. M. Carneiro and V. F. Ferreira, Bioorg. Med. Chem., 2004, 12, 87–93 CrossRef CAS PubMed.
- J.-C. Lien, L.-J. Huang, J.-P. Wang, C.-M. Teng, K.-H. Lee and S.-C. Kuo, Bioorg. Med. Chem., 1997, 5, 2111–2120 CrossRef CAS PubMed.
- M. G. Johnson, H. Kiyokawa, S. Tani, J. Koyama, S. L. Morris-Natschke, A. Mauger, M. M. Bowers-Daines, B. C. Lange and K.-H. Lee, Bioorg. Med. Chem., 1997, 5, 1469–1479 CrossRef CAS PubMed.
- J. M. Miguel del Corral, M. A. Castro, M. Gordaliza, M. L. Martín, A. M. Gamito, C. Cuevas and A. S. Feliciano, Bioorg. Med. Chem., 2006, 14, 2816–2827 CrossRef CAS PubMed.
- K. Takagi, M. Matsuoka, K. Hamano and T. Kitao, Dyes Pigm., 1984, 5, 241–251 CrossRef CAS.
- M. Matsuoka, K. Takagi, H. Obayashi, K. Wakasugi and T. Kitao, J. Soc. Dyers Colour., 1983, 99, 257–260 CrossRef CAS.
- J. Catalan, F. Fabero, M. Soledad Guijarro, R. M. Claramunt, M. D. Santa Maria, M. de la C. Foces-Foces, F. Hernandez Cano, J. Elguero and R. Sastre, J. Am. Chem. Soc., 1990, 112, 747–759 CrossRef CAS.
- V. K. Tandon, R. B. Chhor, R. V Singh, S. Rai and D. B. Yadav, Bioorg. Med. Chem. Lett., 2004, 14, 1079–1083 CrossRef CAS PubMed.
- V. K. Tandon, R. V Singh and D. B. Yadav, Bioorg. Med. Chem. Lett., 2004, 14, 2901–2904 CrossRef CAS PubMed.
- V. K. Tandon, D. B. Yadav, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 3288–3291 CrossRef CAS PubMed.
- V. K. Tandon, D. B. Yadav, R. V Singh, M. Vaish, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 3463–3466 CrossRef CAS PubMed.
- V. K. Tandon, D. B. Yadav, R. V Singh, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 5324–5328 CrossRef CAS PubMed.
- V. K. Tandon, H. K. Maurya, D. B. Yadav, A. Tripathi, M. Kumar and P. K. Shukla, Bioorg. Med. Chem. Lett., 2006, 16, 5883–5887 CrossRef CAS PubMed.
- V. K. Tandon, D. B. Yadav, H. K. Maurya, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem., 2006, 14, 6120–6126 CrossRef CAS PubMed.
- V. K. Tandon, H. K. Maurya, A. Tripathi, G. B. ShivaKeshava, P. K. Shukla, P. Srivastava and D. Panda, Eur. J. Med. Chem., 2009, 44, 1086–1092 CrossRef CAS PubMed.
- J. H. Clark, T. J. Farmer, A. J. Hunt and J. Sherwood, Int. J. Mol. Sci., 2015, 16, 17101–17159 CrossRef CAS PubMed.
- J.-I. Hayami, M. Asahi, R. Tamura and N. Ono, Bull. Inst. Chem. Res., Kyoto Univ., 1980, 58, 222–227 CAS.
- J. Cornelisse and E. Havinga, Chem. Rev., 1975, 75, 353–388 CrossRef CAS.
- F. Carey and R. Sundberg, Advanced Organic Chemistry. Part B: Reactions and Synthesis. Fourth Edition, Springer, 3rd edn, 2001, vol. 6 Search PubMed.
- E. J. Fendler and J. H. Fendler, J. Chem. Soc., Perkin Trans. 2, 1972, 1403–1407 RSC.
- J. U. Nef, Justus Liebigs Ann. Chem., 1894, 280, 263–291 CrossRef CAS.
- E. Iglesias and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1989, 343–346 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1995006 and 1995009. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03640d |
| This journal is © The Royal Society of Chemistry 2020 |