
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient Ce–Co composite oxide decorated Au nanoparticles for catalytic oxidation of CO in the simulated atmosphere of a CO2 laser†
Qiang Fang‡
a,
Hailian Li‡a,
Qingquan Lin *a,
Kuo Liu*b,
Yang Suc,
Guodong Huoa,
Xuhua Zoua,
Xiufeng Xua,
Haisheng Weia and
Shixue Qi*a
*a,
Kuo Liu*b,
Yang Suc,
Guodong Huoa,
Xuhua Zoua,
Xiufeng Xua,
Haisheng Weia and
Shixue Qi*a
aInstitute of Applied Catalysis, College of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, China. E-mail: Tsqlin@ytu.edu.cn; Qishixue@ytu.edu.cn
bResearch Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: kuoliu@rcees.ac.cn
cDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 16th June 2020
Abstract
Gold nanoparticles have a high activity for CO oxidation, making them suitable to be used in a CO2 laser which maintains its efficiency and stability via the recombination of CO and O2 produced by the CO2 decomposition. However, the high concentration of CO2 in the working environment greatly reduces the activity of the catalyst and makes the already unstable gold nanoparticles even more so. A novel Au/Ce-Co-Ox/Al2O3 gold catalyst, prepared by a deposition precipitation method in this study, displays high activity and good stability for CO oxidation in a simulated atmosphere of a CO2 laser with the feed gases containing a high concentration of CO2 up to 60 vol% but a low concentration of O2 for the stoichiometric reaction with CO. An excellent performance for CO oxidation under CO2-rich conditions could be achieved by decorating the surface of the Al2O3 support with Ce–Co composite oxides. The strong interaction between gold and the composite support, accompanied by the increase of labile lattice oxygen species and the decrease of surface basicity, led to a high CO oxidation rate and resistance towards CO2 poisoning.
1. Introduction
The catalytic oxidation of carbon monoxide (CO), one of the simplest and most widely investigated reactions in the research field of heterogeneous catalysis, has played a significant role in different fields, such as CO oxidation in O2-rich gases for gas masks and automotive applications,1–4 CO oxidation in H2-rich atmospheres (CO-PROX) for fuel cells,2,5 CO oxidation in CO2-rich gases for carbon dioxide lasers (CO2 laser).6,7 The CO2 laser, which is considered to be the most efficient and powerful laser, emitting a laser beam by the decomposition of CO2 molecules in the discharge, needs to prolong the working life and ensure the quality of laser output via recombining CO and O2,7,8 especially in a sealed high-frequency CO2 laser with a normal working temperature of about 200 °C. However, scientists have been facing an extremely troublesome question for three or more decades, which is that the catalysts deactivated rapidly during CO oxidation, and one of the main reasons for this is the accumulation of carbonate species and the coverage of the active sites on the surface of the catalysts, caused by feed gases or reaction intermediates.9–11 The atmosphere in the CO2 laser, containing a high concentration of CO2, as high as 60 vol%, and stoichiometric or even lower O2/CO ratio for O2 formed by CO2 dissociation, is very harsh for catalysts.12 Thus, it is urgent that new stable catalysts are designed with high efficiency and resistance to CO2 poisoning under CO2-rich conditions.Over the past decades, various platinum group metal (PGM) catalysts for CO oxidation have been widely developed.5,13 In particular, because gold (Au) nanoparticles (NPs) were discovered, showing excellent catalytic performance towards CO oxidation even at very low temperatures, Au-based catalysts have been extensively and systematically investigated.14,15 When compared with PGM catalysts, Au catalysts have the advantages of a higher catalytic activity for CO oxidation, a wider working temperature range, abundant reserves and less price fluctuation, and thus, Au catalysts became promising catalysts for CO oxidation.9,16 Many researchers investigated the effect of a small amount of CO2 on the performance of the used Au catalysts and found the prepared catalysts had some resistance to CO2 poisoning in the CO-PROX catalysts.17–20 However, little attention has been paid to the effect of high concentration of CO2 and few Au catalysts can be utilized well in atmospheres with high concentrations of CO2.21 Therefore, it is necessary to study the characteristics and behaviour of Au catalysts in the feed gases containing a high concentration of CO2.7,21 Also, investigation of the behaviour and characteristics of the Au-based catalysts under CO2-rich conditions can help with the fundamental understanding of the Au catalysts.
In order to design a stable Au catalyst under a high concentration of CO2, acidic supports were firstly considered because less carbonate species may be formed on the surface of them22,23 and alumina is a suitable material for this. However, alumina supported Au NPs (Au/Al2O3) have long been considered to have a relatively low catalytic activity, because Al2O3 is an inert carrier, unlike TiO2 and other reducible carriers which are active, despite its large specific surface area and high thermal stability.24 Fortunately, a series of active Au catalysts has been prepared by doping metal oxides on the surface of the alumina to increase the surface oxygen species on the catalyst surface and thus significantly improving the catalytic activity for the CO oxidation in an O2-rich atmosphere.7,22,23 Hedge and co-workers found that the oxygen storage materials (OSM) could be good dopants which they applied for Pd catalysts and highly improved the reaction rates of CO oxidation.25,26
Based on the reasons discussed previously, in order to improve catalytic activities and stability of the alumina supported Au catalysts for CO oxidation in the simulated CO2-rich atmosphere of the CO2 laser, firstly the surface of the alumina was decorated with the Ce–Co composite oxygen storage oxides, and a novel Au/Ce-Co-Ox/Al2O3 catalyst was designed using a deposition precipitation method, which improved the strong interaction between the Au and the support, and meanwhile the labile oxygen species increased and the basicity on the surface of the support decreased. Finally, after being decorated, the prepared Au/Ce-Co-Ox/Al2O3 catalyst showed a much superior performance for the reaction of CO oxidation in the simulated atmospheres of the CO2 laser containing a low concentration of O2 (0.5 vol%) and a high concentration of CO2 (as high as 60 vol%). Experimental and characterization results showed that the added lattice oxygen species and less strongly alkaline site of the decorated Au/Ce-Co-Ox/Al2O3 catalyst both played a crucial role in achieving high activity and good stability.
2. Experimental
2.1 Preparation of CeOx and CoOx modified Al2O3 composites
The preparation process of Ce-Co-Ox/Al2O3 was as follows: firstly, 0.34 mL of Ce(NO3)2 solution (0.70 mol L−1) and 0.03 mL Co(NO3)2 solution (2.72 mol L−1) were mixed together and diluted to 1.5 mL with deionized water. Then, 0.94 g of the commercially available spherical support: γ-Al2O3 (Alumina Corporation of China, 20–40 mesh, ∼157 m2 g−1) was mixed with the solution. Finally, after mixing and aging for 2 h, the sample was dried at 120 °C for 4 h, and then calcined at 850 °C for 4 h. The CeOx and CoOx modified Al2O3 composite obtained was denoted as Ce-Co-Ox/Al2O3.2.2 Preparation of supported Au catalysts
The Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 catalysts were both synthesized by a deposition–precipitation method. Firstly, an aqueous solution of HAuCl4 (0.516 g, 19.12 gAu L−1) was diluted to 2.0 mL with deionized water. Then, the solution was placed into a clean beaker and diluted with ultra-pure water, and the pH value was adjusted to 8 by adding dropwise an aqueous solution of NaOH (1 mol L−1). Afterwards, 1 g Ce-Co-Ox/Al2O support was added into the solution. The sample was washed in ammonia solution at a pH of 9–10 to remove residual chloride ions, and then dried in air at 70 °C for 12 h. The sample obtained was denoted as Au/Ce-Co-Ox/Al2O3.An Au/Al2O3 catalyst was also prepared using the same procedure, but the pH value was adjusted to 10, in order to obtain slightly larger Au NPs.
2.3 Activity test
Before the activity tests, all the catalysts were pretreated with H2 at 300 °C for 1 h. The CO conversion test was conducted in a CO2-rich atmosphere. The composition of raw gas was: 60 vol% CO2 + 1 vol% CO + 0.5 vol% O2, balanced with N2. The gas mixture passed through the U-shaped quartz reactor at a speed of 100 mL min−1. The reaction temperature was 220 °C. An Agilent 7820A gas chromatograph was used to determine the CO content. For the CO conversion test in CO2 free atmosphere, the composition of raw gas contained 1 vol% CO + 0.5 vol% O2, balanced with N2. The gas mixture passed through the U-shaped quartz reactor at a speed of 100 mL min−1. The reaction temperature was 220 °C.The reaction rate was tested with a gas flow of 200 mL min−1, and the gas compositions and reaction temperature were exactly the same as those shown previously. A portion (0.01 g) of catalyst and 0.09 g of diluent (Al2O3) were loaded into the reaction tube. The CO content was determined by a gas chromatography.
2.4 Catalyst characterization
The crystal phase structures of the carriers and catalysts were determined using a PANalytical PW3040/60 X'Pert PRO X-ray diffraction (XRD) instrument. The Cu Kα monochromatized radiation (λ = 0.1541 nm) was used with a scan speed of 20° min−1 and a scan range of 10–80°.A Thermo Scientific IRIS Intrepid II ICP was used to determine the content of Au in the catalyst. A portion (0.1 g) of sample was put into a beaker, and the Au on the surface of the catalyst was soaked with aqua regia solution. Then the Au was washed with deionized water, and the eluent was transferred into a volumetric flask.
Transmission electron microscopy (TEM) images were recorded using a Hitachi 7700 microscope operated at 120 kV. Prior to making the observations, the catalysts were pretreated with H2 or air at 300 °C for 1 h and then suspended in ethanol with ultrasonic dispersion for >10 min. One or two droplets of the dispersed solution were dropped on a microgrid carbon polymer supported on a copper grid. By measuring 300 particles from randomly selected areas, the particle size distribution was acquired.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was be used to determine the particle size of the Au. A FEI Tecnai G2 F30 S-Twin field emission transmission electron microscope with an acceleration voltage of 300 kV was also used. Prior to the observations, the powder catalysts were collected after peeling off the outside of the samples (a physical method that does not change the nanoparticle size of Au but could increase the content of it in order to easily observe more Au particles). Then, the samples were finely ground in an agate mortar and subsequently dispersed ultrasonically in ethanol for 5 min. After a period of time, 1–2 drops of the supernatant were dropped on to the micro grid, followed by drying.
Oxygen temperature programming desorption (O2-TPD) was evaluated using an automatic PCA-1200 chemisorption analyzer. A portion (100 mg) of the dried sample was reduced with H2 (30 mL min−1) at 300 °C for 1 h. After cooling to room temperature, the catalyst was purged with He (20 mL min−1) for 20 min, and then with O2 (20 mL min−1) for 30 min to absorb the O2. The heating rate was 10 °C min−1.
Hydrogen temperature-programmed reduction (H2-TPR) was conducted using an automatic PCA-1200 Chemisorption analyzer. The catalysts were firstly purged with N2 (30 mL min−1) at 120 °C for 30 min. After cooling to room temperature, 10 vol% H2/Ar (20 mL min−1) was introduced onto the surface of the catalysts, and the temperature was increased to 900 °C at 10 °C min−1. The H2 consumption was monitored by a TCD detector.
Carbon dioxide temperature programming desorption (CO2-TPD) was evaluated using an automatic PCA-1200 chemisorption analyzer. At first, 100 mg of dried sample was reduced with H2 (30 mL min−1) at 300 °C for 1 h. After cooling to room temperature, the catalyst was purged with He (20 mL min−1) for 20 min, and then with CO2 (20 mL min−1) for 30 min to absorb the CO2. The heating rate was 10 °C min−1.
3. Results and discussion
3.1 Activity
The Au targeted at 1 wt% was deposited on γ-Al2O3, CoOx/Al2O3, CeOx/Al2O3 and Ce-Co-Ox/Al2O3, and the resultant catalysts had actual Au loadings of 0.71 wt%, 0.46 wt%, 0.79 wt% and 0.89 wt%, respectively, as measured by ICP, and their BET surface areas were similar (see Table S1, ESI†). The actual Au loadings obtained varied with the different supports, and the Ce–Co composite oxide had more adsorption sites for Au, which was possibly because of the higher isoelectric point originating from the co-decoration.27Fig. 1a shows the profiles of CO conversions as a function of reaction time over the Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 catalysts in a simulated CO2-rich atmosphere of a high-frequency CO2 laser at the usual working temperature of 220 °C and a high space velocity of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL gcat−1 h−1 in order to accelerate the deactivation of catalyst and compare their performances. Considering the big changes of the two curves, a significant effect of the type of support used is easily observed. As shown in Fig. 1a, the Au/Al2O3 catalyst had a very low activity for this reaction which was reflected by the following phenomenon. The Au/Al2O3 catalyst only gave a CO conversion of approximately 35% at the start of reaction and the CO conversion decreased slowly as the reaction time was extended, and the remaining CO conversion was maintained at only 15%, about half of the original activity after nearly 1500 min of testing. However, on the Au/Ce-Co-Ox/Al2O3 catalyst, at the beginning of CO oxidation, the CO conversion reached as high as 78%, 2.2 times that of the unmodified Au/Al2O3 catalyst. After a long time of activity test, a conversion of approximately 55% over Au/Ce-Co-Ox/Al2O3 was maintained, corresponding to a retention of 70% of the original activity and 3.7 times higher than that of the Au/Al2O3 catalyst. In addition, a well-known active and stable reducible oxide supported Au catalyst (1 wt% Au/Fe2O3–HgI) synthesized by Haruta's laboratory and obtained from Haruta Gold Inc., was compared with our Au catalyst. As shown in Fig. 1a (inset), the Au/Fe2O3–HgI catalyst had a very high initial activity, but suffered from a faster deactivation. Under the same conditions, although the initial activity of our Au/Ce-Co-Ox/Al2O3 catalyst was slightly lower than that of Au/Fe2O3–HgI, the Au/Ce-Co-Ox/Al2O3 catalyst obviously lost its activity much more slowly. After 50 min, the CO conversion over Au/Ce-Co-Ox/Al2O3 became higher than that over 1 wt% Au/Fe2O3–HgI, demonstrating that a much better stability was found on Au/Ce-Co-Ox/Al2O3.
000 mL gcat−1 h−1 in order to accelerate the deactivation of catalyst and compare their performances. Considering the big changes of the two curves, a significant effect of the type of support used is easily observed. As shown in Fig. 1a, the Au/Al2O3 catalyst had a very low activity for this reaction which was reflected by the following phenomenon. The Au/Al2O3 catalyst only gave a CO conversion of approximately 35% at the start of reaction and the CO conversion decreased slowly as the reaction time was extended, and the remaining CO conversion was maintained at only 15%, about half of the original activity after nearly 1500 min of testing. However, on the Au/Ce-Co-Ox/Al2O3 catalyst, at the beginning of CO oxidation, the CO conversion reached as high as 78%, 2.2 times that of the unmodified Au/Al2O3 catalyst. After a long time of activity test, a conversion of approximately 55% over Au/Ce-Co-Ox/Al2O3 was maintained, corresponding to a retention of 70% of the original activity and 3.7 times higher than that of the Au/Al2O3 catalyst. In addition, a well-known active and stable reducible oxide supported Au catalyst (1 wt% Au/Fe2O3–HgI) synthesized by Haruta's laboratory and obtained from Haruta Gold Inc., was compared with our Au catalyst. As shown in Fig. 1a (inset), the Au/Fe2O3–HgI catalyst had a very high initial activity, but suffered from a faster deactivation. Under the same conditions, although the initial activity of our Au/Ce-Co-Ox/Al2O3 catalyst was slightly lower than that of Au/Fe2O3–HgI, the Au/Ce-Co-Ox/Al2O3 catalyst obviously lost its activity much more slowly. After 50 min, the CO conversion over Au/Ce-Co-Ox/Al2O3 became higher than that over 1 wt% Au/Fe2O3–HgI, demonstrating that a much better stability was found on Au/Ce-Co-Ox/Al2O3.
 | ||
| Fig. 1 The CO conversions (a) and their specific rates (b) over Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 for CO oxidation in CO2-rich atmospheres. Reaction conditions: 60 vol% CO2 + 1 vol% CO + 0.5 vol% O2, balanced with N2. Weight hourly space velocity (WHSV) = 120000 (a) and 1200000 mL g−1 h−1 (b), temperature: 220 °C. The inset in (a) shows CO conversions over the Au/Ce-Co-Ox/Al2O3 and 1%Au/Fe2O3–HgI catalysts. | ||
Further investigation of the intrinsic catalytic activity was made by means of the specific reaction rates of the two catalysts (Fig. 1b). The specific reaction rate over Au/Ce-Co-Ox/Al2O3 was more superior to that on the Au/Al2O3 catalyst and this was consistent with the results of the CO conversion method mentioned previously. The reaction rate over Au/Ce-Co-Ox/Al2O3 catalyst was calculated to be 0.22 molCO molAu−1 s−1 when the test was started, and it was 3.8 times that of Au/Al2O3 catalyst (0.057 molCO molAu−1 s−1). When the reaction had proceeded for 150 min, a more striking observation was that the reaction rate of the Au/Ce-Co-Ox/Al2O3 catalyst had reached 0.16 molCO molAu−1 s−1, which was nearly 6 times that of the Au/Al2O3 catalyst (0.025 molCO molAu−1 s−1). Thus, it was further confirmed that co-decoration of Al2O3 by CeOx and CoOx had a significant promotional effect on the catalytic performance of the alumina-based Au catalysts.
The activity of CO oxidation on the Au/Ce-Co-Ox/Al2O3 and Au/Al2O3 catalysts in the feed gases with or without CO2 was studied (Fig. S1, ESI†). At a very high space velocity of 120000 mL gcat−1 h−1, the CO conversion over the Au/Al2O3 catalyst decreased from 48% (no CO2 in the feed gases) to 36% (60 vol% CO2 in the feed gases) at the start of reaction, and then the CO conversion quickly decreased by nearly half to 20% as the reaction time progressed. However, on the Au/Ce-Co-Ox/Al2O3 catalyst, it is interesting that the CO conversions were both very high, reaching as high as 82% (no CO2 in the feed gases) and 78% (60 vol% CO2 in the gas feed) at the beginning of the CO oxidation. In order to show the promotional effect of CeCoOx on the stability in a CO2-rich atmosphere, ratios of CO conversion in the CO2 rich atmosphere (60 vol% CO2) to that in the atmosphere without CO2 (0 vol% CO2) over Au/Ce-Co-Ox/Al2O3 and Au/Al2O3 were calculated (Fig. 2). Compared with Au/Al2O3, Au/Ce-Co-Ox/Al2O3 had a higher ratio, indicating that Au/Ce-Co-Ox/Al2O3 exhibited a better tolerance to high concentrations of CO2. Similarly, the reaction rates over the Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 catalysts almost showed the same trend, as shown in Fig. S2 (ESI).† Meanwhile, compared with the reaction rate obtained in the atmosphere without CO2, the reaction rate of CO oxidation over Au/Ce-Co-Ox/Al2O3 maintained one-half or more (0.22 vs. 0.42 molCO molAu−1 s−1) when reacted in a CO2-rich atmosphere, which further indicated that the decoration of CeOx and CoOx had indeed improved the catalytic activity of CO oxidation.
 | ||
| Fig. 2 Ratios of CO conversion in the CO2 rich atmosphere to that in the atmosphere without CO2 on the Au/Ce-Co-Ox/Al2O3 and Au/Al2O3 catalysts. | ||
In addition, the CO conversions on Au/Ce-Co-Ox/Al2O3, Au/CeOx/Al2O3 and Au/CoOx/Al2O3 were also obtained at a space velocity of 120000 mL gcat−1 h−1, and the results are shown in Fig. 3. The Au/CeOx/Al2O3 catalysts had a higher original activity, which was about twice that on the Au/CoOx/Al2O3 catalyst, which may be due to the lower Au loading of Au/CoOx/Al2O3. Furthermore, the Au/Ce-Co-Ox/Al2O3 catalyst not only had a high original activity, but also possessed better reaction stability than that on Au/CeOx/Al2O3 during CO oxidation. Therefore, co-decoration with CeOx and CoOx could promote the stability of CO oxidation in a CO2-rich atmosphere and it is important to clarify the promotional principle.
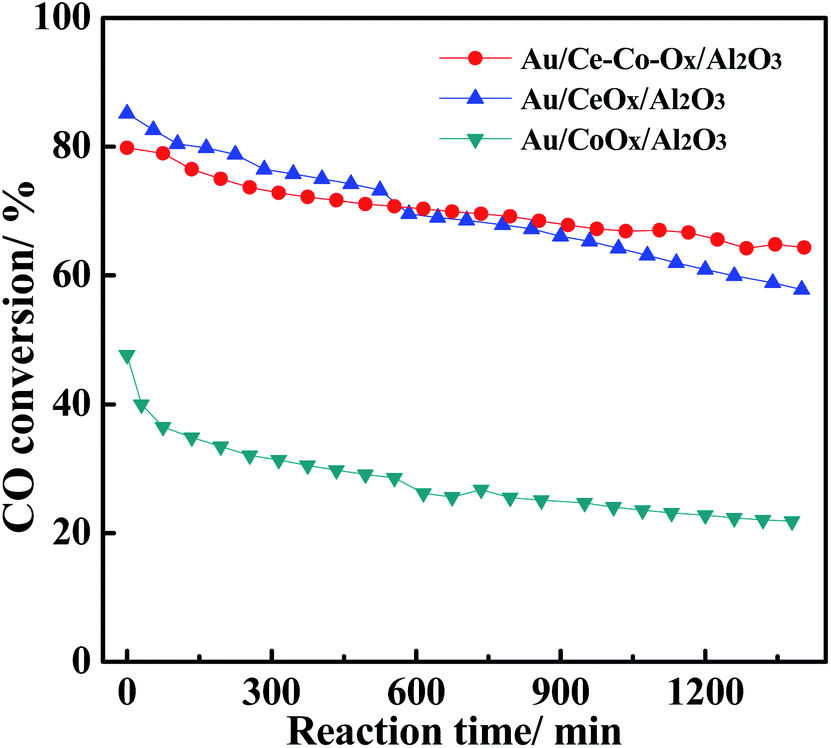 | ||
| Fig. 3 The CO conversion on the Au/CeOx/Al2O3, Au/CoOx/Al2O3 and Au/Ce-Co-Ox/Al2O3 catalysts. Reaction conditions: 1 vol% CO + 0.5 vol% O2, balanced N2, WHSV = 120000 mL g−1 h−1, temperature = 220 °C. | ||
3.2 Au particle sizes and strong metal-support interaction (SMSI)
To reveal the unique effect of co-decorating oxides on the catalytic performance of CO oxidation on the Au/Ce-Co-Ox/Al2O3 catalyst in O2-poor and CO2-rich feed gases, further characterizations were carried out to explore the essential influence of the modification of CeOx and CoOx on the structure of the Au active sites. Firstly, the structure of the supports and the distribution of Au NPs of the two catalysts were studied using XRD and TEM techniques. As shown in Fig. 4, three peaks located at 37°, 45° and 66° could be assigned to the diffraction peaks of Al2O3 (PDF 87-0597), which were found in all the samples, and the diffraction peaks (28°, 33°, 56.3°) were also observed and attributed to CeO2. However, the diffraction peak of Co3O4 or other CoOx was absent, because CoOx might be highly dispersed on the surface of Al2O3. In addition, no diffraction peaks of Au species, including Au0 or Au2O3, were observed on the surface of the Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 catalysts, indicating that the Au species might also be highly dispersed. | ||
| Fig. 4 XRD diffraction patterns of Al2O3 (a), Au/Al2O3 (b), Ce-Co-Ox/Al2O3 (c) and Au/Ce-Co-Ox/Al2O3 (d). | ||
In order to confirm the conclusions given previously, TEM was conducted. As shown in Fig. 5, the Au NPs were both well dispersed on the surface of Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 catalysts with a mean particle size of (3.0 ± 0.5) nm and (3.3 ± 0.5) nm, respectively. It has been reported by many researchers that the diameter of the Au particle could affect the CO oxidation activity, and the optimum particle size of Au might be in the range of 0.5–5 nm.28 The Au particle size in this study was within this range, guaranteeing a high CO oxidation rate. In order to further verify the dispersion of Au particles in the Au/Ce-Co-Ox/Al2O3 catalyst, HADDF-STEM was also conducted. As shown in Fig. 5e, the bright spot represents Au NPs, whereas the large pale white patch is Ce-Co-Ox, indicating that most of the Au NPs were attached to the surface of Ce-Co-Ox. A uniform special distribution of the Au, Ce and Co elements can also be observed from the results of the energy dispersive spectrometry (EDS) mapping of the HAADF-STEM image (Fig. 6). The spectra of energy dispersive X-ray spectroscopy (EDX) line scan demonstrated a similar distribution of the Au, Ce and Co elements along one particle (Fig. S3, ESI†), indicative of their close contact. The close contact between Au NPs and Ce-Co-Ox can lead to a strong interaction between Au and Ce-Co-Ox,29,30 thus benefiting the CO oxidation.
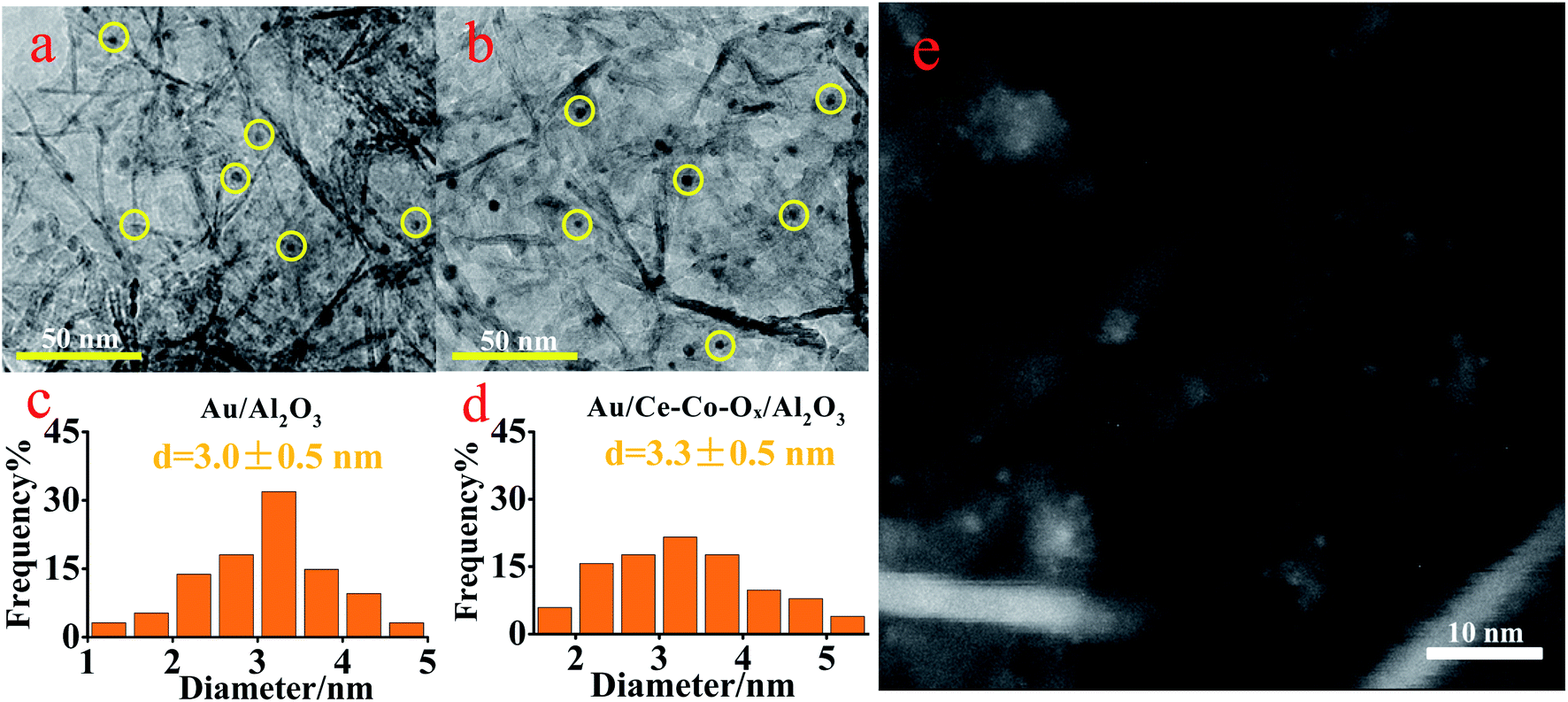 | ||
| Fig. 5 The TEM images of the Au/Al2O3 (a) and Au/Ce-Co-Ox/Al2O3 catalysts (b) with their Au particle size distributions (c and d) and HADDF-STEM image of the Au/Ce-Co-Ox/Al2O3 catalyst (e). | ||
 | ||
| Fig. 6 HADDF-STEM image (a) and EDS mapping (b) of the Au/Ce-Co-Ox/Al2O3 catalyst. | ||
The difference in the mean particle sizes of the two catalysts was insignificant, which might not be the main reason for the different CO oxidation activity and stability. Thus, it can be proposed that the Au/Ce-Co-Ox/Al2O3 had a much higher reactivity for the SMSI between Au and the Ce-Co-Ox/Al2O3 composite support, and the chemical properties of Ce-Co-Ox composite oxides were likely to benefit the CO oxidation reaction.
3.3 Oxygen species on the surface
In order to investigate the chemical properties of the supports, the adsorption/desorption capacity of the oxygen species on the surface of the supports were measured using the O2-TPD and H2-TPR experiments. Prior to the O2-TPD tests, the catalyst samples were reduced in situ with H2 at 300 °C for 60 min and then purged with He for 30 min. As shown in Fig. 7, the desorption peaks of the two catalysts at 140 °C were similar at low temperatures, which can be mainly attributed to adsorbed oxygen species on the Au NPs28 and the desorption peak of Au/Ce-Co-Ox/Al2O3 was a little larger than that of Au/Al2O3. Also, a broad peak at ∼260–550 °C was observed over Au/Al2O3, which could be ascribed to the desorption of the lattice oxygen. The lower the desorption temperature, the easier the labile lattice oxygen species could react with CO on the support surface. After the co-decoration of CeOx and CoOx, the desorption temperature of the lattice oxygen shifted to a lower temperature (210–600 °C) over the Au/Ce-Co-Ox/Al2O3 catalyst and its peak area increased by nearly 200% (the peak areas of Au/Al2O3 and Au/Ce-Co-Ox/Al2O3 after integration were 68 and 120, respectively). This means that more labile lattice oxygen species were generated on the surface of the support and reacted easily with the adsorbed CO.30–32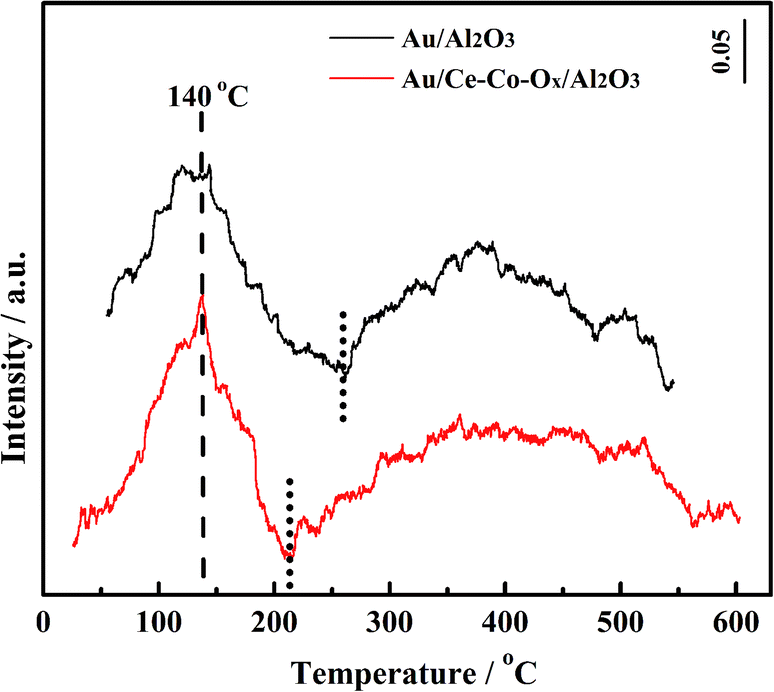 | ||
| Fig. 7 The O2-TPD profiles of the Au/Ce-Co-Ox/Al2O3 and Au/Al2O3 catalysts. | ||
It has been reported previously by many researchers that the adsorbed oxygen can directly combine with CO on the catalyst to produce CO2 over Au/Al2O3, and its lattice oxygen cannot participate in this reaction.33,34 However, Widmann and Behm35,36 reported that the lattice oxygen on the reducible support surface could participate in CO oxidation and oxygen vacancies could be formed. Then, the adsorbed oxygen will replace the oxygen vacancy and form a new cycle.35–38 The conclusion that the lattice oxygen species of CeOx and CoOx can participate in CO oxidation was also confirmed by the 18O isotope labelling experiment conducted by Madier et al.39 and Amin et al.40 Therefore, it could be inferred that both the adsorbed and lattice oxygen species contributed to CO oxidation in this study.
The previous results were verified by the H2-TPR. As shown in Fig. 8, two H2 consumption peaks at 196 °C and 792 °C were observed over Au/Al2O3, and ascribed to the reduction of Au species and the lattice oxygen of Al2O3, respectively. When Al2O3 was decorated with CeOx and CoOx, three peaks were observed, which can be assigned to the reduction of CoOx (283–483 °C), CeOx (483–572 °C),1,2 and lattice oxygen of Al2O3 (675 °C) possibly due to the H2 spill over from CoOx or CeOx, respectively. After Au was deposited on the Co-Ox/Al2O3 composite support, the reduction peaks of the Au species, CoOx, CeOx and Al2O3 all shifted to lower temperatures (98 °C, or 98–165 °C for the overlap of the reduction peak of partial Au species), demonstrating that the SMSI between Au and the composite support was increased. It was clearly observed that after co-decoration and Au loading, more labile oxygen species were generated. The labile oxygen species were important for improving the reaction rate on catalysts for CO oxidation, for example, on Ce-Co-O.35–40 A synergistic effect in the CeO2 and Co3O4 compound was found by Luo et al.37 in the Ce-Co-O catalyst, resulting in a better activity of the CO oxidation. Thus, it can be inferred that the synergistic effect was also present in our catalyst, increasing the amount of labile oxygen species. In general, the more oxygen species adsorbed by the Au NPs, the more labile lattice oxygen species there were on the surface of the support, jointly promoting the catalytic activity on the Au/Ce-Co-Ox/Al2O3 catalyst. Therefore, the Au/Ce-Co-Ox/Al2O3 catalyst was greatly improved due to the increase of the number of labile oxygen species.
 | ||
| Fig. 8 The H2-TPR profiles of the Au/Ce-Co-Ox/Al2O3 and Au/Al2O3 catalysts. | ||
3.4 Stability of the carbonate species
Generally, the acidity and basicity of the support have a great influence on the catalyst's performance for accumulation of carbonate species, especially in the simulated CO2-rich atmosphere of the CO2 laser, and thus, this was explored with CO2-TPD. The higher temperature of the CO2 desorption peak, the higher the strength of the alkaline sites. As shown in Fig. 9, an obvious peak of CO2 desorption at the lower temperature of ∼100 °C was observed for both catalysts, which was assigned as the desorption of CO2 from weaker surface alkaline sites. This reaction was carried out at 220 °C, and at this temperature the CO2 adsorbed on these weaker alkaline sites would be easily desorbed, and there would be little or no accumulation of carbonate species. Therefore, the activity of these two catalysts was not affected by these weak alkaline sites. Meanwhile, large broad peaks of CO2 desorption, assigned as medium (220–550 °C) and strong alkali sites (>550 °C),37 were also found on the surface of Au/Al2O3, and were significantly larger than those found on the Au/Ce-Co-Ox/Al2O3 catalyst, therefore the Au/Ce-Co-Ox/Al2O3 catalyst had less strong alkali sites and could accumulate less carbonate species to maintain its high activity during the reaction. The CO2 was reported to have a negative role on CO oxidation, because the carbonate species formed on the catalyst surface from the reaction between CO2 and the lattice oxygen could inhibit the recovery of vacancies by O2.41 However, the presence of Ce-Co-Ox led to less carbonate species on the catalyst surface in the CO2-rich atmosphere (Fig. 10), alleviating the negative effect of CO2. As summarized in Fig. 10, the more labile lattice oxygen increased the probability of the recovery of the vacancies by O2. The reasons given previously may be why the Au/Ce-Co-Ox/Al2O3 catalyst was much more active and stable but the Au/Al2O3 catalyst performance was much lower and it lost its activity rapidly in a CO2-rich atmosphere. | ||
| Fig. 9 The CO2-TPD profiles of Au/Ce-Co-Ox/Al2O3 and Au/Al2O3. | ||
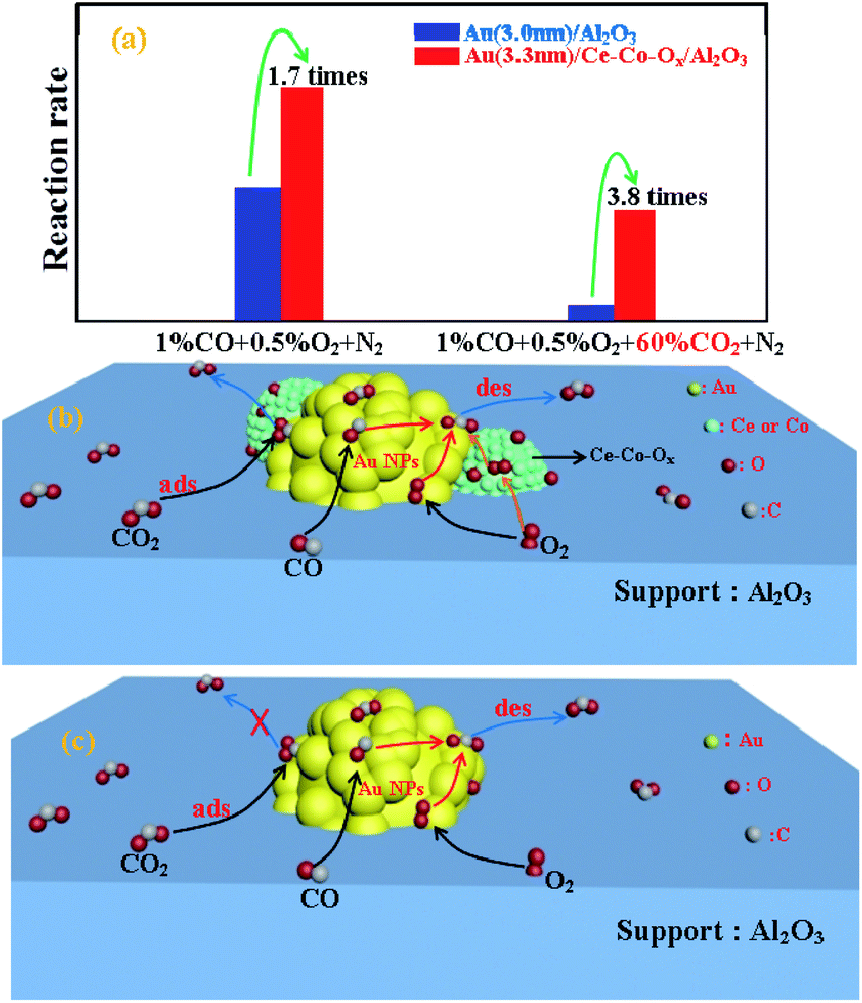 | ||
| Fig. 10 The CO oxidation specific rates (a) and the mechanism on Au/Ce-Co-Ox/Al2O3 (b) and Au/Al2O3 (c). | ||
4. Conclusions
In summary, a novel Au/Ce-Co-Ox/Al2O3 catalyst, possessing nearly the same mean particle size as Au/Al2O3, was prepared by co-decoration of CeOx and CoOx on the Au/Al2O3 catalyst. The co-decoration of the Ce–Co composite oxides dramatically improved the strong interaction between the gold and the support, increased the labile lattice oxygen species, and meanwhile decreased the strength of the alkaline sites on the surface of the support, and thus greatly improved its performance for CO oxidation in the simulated atmosphere of the CO2 laser where the feed gases contained a low concentration of O2 and a high concentration of CO2. This work clarified the catalyst design for CO oxidation in CO2-rich atmospheres of the sealed CO2 laser.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21606189, 21808193) and Natural Science Foundation of Shandong Province (ZR2015BQ011).Notes and references
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590 CrossRef.
- X. Du, W. Han, Z. Tang and J. Zhang, New J. Chem., 2019, 43, 14872 RSC.
- X. Zhai, C. Liu, Q. Chang, C. Zhao, R. Tan, H. Peng, D. Liu, P. Zhang and J. Gui, New J. Chem., 2018, 42, 18066 RSC.
- J. Saavedra, T. Whittaker, Z. Chen, C. J. Pursell, R. M. Rioux and B. D. Chandler, Nat. Chem., 2016, 8, 584 CrossRef CAS PubMed.
- J. A. Macken, S. K. Yagnik and M. A. Samis, IEEE J. Quantum Electron., 1989, 25, 1695 CrossRef CAS.
- Q. Lin, C. Han, H. Su, L. Sun, T. Ishida, T. Honma, X. Sun, Y. Zheng and C. Qi, RSC Adv., 2017, 7, 38780 RSC.
- C. Willis and J. G. Purdon, J. Appl. Phys., 1979, 50, 2539 CrossRef CAS.
- S. Xie, H. Dai, J. Deng, H. Yang, W. Han, H. Arandiyan and G. Guo, J. Hazard. Mater., 2014, 279, 392 CrossRef CAS PubMed.
- A. Luengnaruemitchai, S. Osuwan and E. Gulari, Int. J. Hydrogen Energy, 2004, 29, 429 CrossRef CAS.
- A. Luengnaruemitchai, D. T. K. Thoa, S. Osuwan and E. Gulari, Int. J. Hydrogen Energy, 2005, 30, 981 CrossRef CAS.
- A. Leba, T. Davran-Candan, Z. I. Önsan and R. Yıldırım, Catal. Commun., 2012, 29, 6 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 2, 405 CrossRef.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- J. Saavedra, H. A. Doan, C. J. Pursell, L. C. Grabow and B. D. Chandler, Science, 2014, 345, 1599 CrossRef CAS PubMed.
- Z. Wu, H. Zhu, Z. Qin, H. Wang, J. Ding, L. Huang and J. Wang, Fuel, 2013, 104, 41 CrossRef CAS.
- T. Shodiya, O. Schmidt and N. Hotz, J. Catal., 2013, 300, 63 CrossRef CAS.
- S. Zhang, K. An, C. Fang, Z. Zhang, Q. Liu, S. Lu and Y. Liu, Catal. Today DOI:10.1016/j.cattod.2019.04.041.
- E. de O. Jardim, S. Rico-Francés, F. Coloma, E. V. Ramos-Fernández, J. Silvestre-Albero and A. Sepúlveda-Escribano, Appl. Catal., A, 2014, 487, 119 CrossRef CAS.
- S. D. Gardner, G. B. Hoflund, B. T. Upchurch, D. R. Schryer, E. J. Kielin and J. Schryer, J. Catal., 1991, 129, 114 CrossRef CAS.
- Q. Lin, B. Qiao, Y. Huang, L. Li, J. Lin, X. Y. Liu, A. Wang, W.-C. Li and T. Zhang, Chem. Commun., 2014, 50, 2721 RSC.
- W.-Z. Li, L. Kovarik, D. Mei, M. H. Engelhard, F. Gao, J. Liu, Y. Wang and C. H. F. Peden, Chem. Mater., 2014, 26, 5475 CrossRef CAS.
- J. Saavedra, C. J. Pursell and B. D. Chandler, J. Am. Chem. Soc., 2018, 140, 3712–3723 CrossRef CAS PubMed.
- P. Bera, K. C. Patil, V. Jayaram, G. N. Subbanna and M. S. Hegde, J. Catal., 2009, 9, 293 Search PubMed.
- T. Baidya, A. Gupta, P. A. Deshpandey, G. Madras and M. S. Hegde, J. Phys. Chem. C, 2009, 113, 4059 CrossRef CAS.
- T. Ishida, H. Koga, M. Okumura and M. Haruta, Chem. Rec., 2016, 16, 2278 CrossRef CAS PubMed.
- R. Si, J. Liu, K. Yang, X. Chen, W. Dai and X. Fu, J. Catal., 2014, 311, 71 CrossRef CAS.
- Z. Gao, C. Li, G. Fan, L. Yang and F. Li, Appl. Catal., B, 2018, 265, 523 CrossRef.
- Y. Zhang, X. Yang, Y. Zhou, G. Li, Z. Li, C. Liu, M. Bao and W. Shen, Nanoscale, 2016, 8, 18626 RSC.
- H. H. Kung, M. C. Kung and C. K. Costello, J. Catal., 2003, 216, 425 CrossRef CAS.
- L. Xue, C. Zhang, H. He and Y. Teraoka, Appl. Catal., B, 2007, 75, 167 CrossRef CAS.
- C. K. Costello, J. H. Yang, H. Y. Law, Y. Wang, J.-N. Lin, L. D. Marks, M. C. Kung and H. H. Kung, Appl. Catal., A, 2003, 243, 15 CrossRef CAS.
- C. K. Costello, M. C. Kung, H. S. Oh, Y. Wang and H. H. Kung, Appl. Catal., A, 2002, 232, 159 CrossRef CAS.
- D. Widmann, A. Krautsieder, P. Walter, A. Bruckner and R. J. Behm, ACS Catal., 2016, 6, 5005 CrossRef CAS.
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740 CrossRef CAS PubMed.
- J.-Y. Luo, M. Meng, X. Li, X.-G. Li, Y.-Q. Zha, T.-D. Hu, Y.-N. Xie and J. Zhang, J. Catal., 2008, 254, 310 CrossRef CAS.
- M. Okumura, J. M. Coronado, J. Soria, M. Haruta and J. C. Conesa, J. Catal., 2001, 203, 168 CrossRef CAS.
- Y. Madier, C. Descorme, A. M. Le Govic and D. Duprez, J. Phys. Chem. B, 1999, 103, 10999 CrossRef CAS.
- H. M. A. Amin and H. Baltruschat, Phys. Chem. Chem. Phys., 2017, 19, 25527 RSC.
- S. Wu, W. Sun, J. Chen, J. Zhao, Q. Cao, W. Fang and Q. Zhao, J. Catal., 2019, 377, 110 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The equation for calculating the reaction rate, and the characterization data of physical properties, HADDF-STEM & EDX. See DOI: 10.1039/d0ra03558k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |