
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluoxetine scaffold to design tandem molecular antioxidants and green catalysts†
Giovanni Ribaudo a,
Marco Bortolib,
Alberto Ongaroa,
Erika Oselladorec,
Alessandra Gianoncellia,
Giuseppe Zagottoc and
Laura Orian*b
a,
Marco Bortolib,
Alberto Ongaroa,
Erika Oselladorec,
Alessandra Gianoncellia,
Giuseppe Zagottoc and
Laura Orian*b
aDipartimento di Medicina Molecolare e Traslazionale, Università degli Studi di Brescia, Viale Europa 11, 25123 Brescia, Italy
bDipartimento di Scienze Chimiche, Università degli Studi di Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: laura.orian@unipd.it
cDipartimento di Scienze del Farmaco, Università degli Studi di Padova, Via Marzolo 5, 35131 Padova, Italy
First published on 15th May 2020
Abstract
Fluoxetine finds application in the treatment of depression and mood disorders. This selective serotonin-reuptake inhibitor (SSRI) also contrasts oxidative stress by direct ROS scavenging, modulation of the endogenous antioxidant defense system, and/or enhancement of the serotonin antioxidant capacity. We synthesised some fluoxetine analogues incorporating a selenium nucleus, thus expanding its antioxidant potential by enabling a hydroperoxides-inactivating, glutathione peroxidase (GPx)-like activity. Radical scavenging and peroxidatic activity were combined in a water-soluble, drug-like, tandem antioxidant molecule. Selenofluoxetine derivatives were reacted with H2O2 in water, and the mechanistic details of the reaction were unravelled combining nuclear magnetic resonance (NMR), electrospray ionisation-mass spectrometry (ESI-MS) and quantum chemistry calculations. The observed oxidation–elimination process led to the formation of seleninic acid and cinnamylamine in a trans-selective manner. This mechanism is likely to be extended to other substrates for the preparation of unsaturated cinnamylamines.
Introduction
Fluoxetine hydrochloride (N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine HCl) is a selective serotonin-reuptake inhibitor (SSRI), which was approved by the FDA in 1987 for the treatment of depression.1,2 This drug, marketed as Prozac by Eli Lilly, enhances the serotoninergic tone by increasing the concentration of the neurotransmitter in the synaptic cleft by inhibiting the serotonin transporter.3 From a structural point of view, it is constituted by a racemic mixture of R(−)-fluoxetine and S(+)-fluoxetine, which show a moderate difference in terms of inhibitory activity that becomes more evident in the corresponding metabolites.4 Fluoxetine has been approved worldwide for the treatment of major depression, but its activity on a wide spectrum of mood disorders has been reported.5Further studies unveiled that fluoxetine may protect against the adverse effects of different types of immune system stressors and contrast, through a combination of mechanisms, oxidative damage. This feature is thought to play a primary role in neuroprotection, since brain is very susceptible to oxidative stress due to its high energetic requirement. Moreover, growing evidence suggests that oxidative stress and an abnormally increased generation of reactive oxygen species (ROS) may be implicated in the pathogenesis of many psychiatric and degenerative disorders.6–10 Considering the mechanistic aspects, the insurgence of such diseases may be the result of ROS-related damage, i.e. lipid peroxidation, DNA or protein oxidation, and mitochondrial damage. From a biochemical and clinical point of view, the antioxidant effects of antidepressant agents were observed in animal models by measuring variations of GSH, malondialdehyde, nitric oxide and isoprostanes concentrations.11–13
At molecular level, it has been highlighted that fluoxetine exerts its antioxidant effects through a combination of mechanisms, involving direct ROS scavenging, modulation of the expression and functioning of enzymatic and non-enzymatic components of the endogenous antioxidant defence system, and/or enhancement of the serotonin antioxidant capacity.14,15 Concerning the first putative mechanism, the activity of fluoxetine against ROS has been thoroughly studied from a biochemical and computational point of view. Although the compound demonstrated an antioxidant role, its direct contribution to ROS scavenging is of a minor entity with respect to that of its metabolites.16,17 In addition, fluoxetine contrasts oxidative stress also by increasing extracellular concentration of serotonin, which is a known strong antioxidant.17–19 This neurotransmitter, together with its N-acetyl metabolite, exerts neuroprotection by modulating oxidative burst mechanism and/or the production of superoxide anion radical.19 Fluoxetine is also thought to act indirectly on oxidative stress by modulating the expression of enzymes such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx).14 Finally, recent findings suggest that fluoxetine may enhance cellular antioxidant capacity by tuning mitochondrial redox parameters and through the upregulation of thioredoxin (Trx), a dithiol-disulfide oxidoreductase that can facilitate H2O2 scavenging.20 Nevertheless, the indirect antioxidant role that fluoxetine plays in neuroprotection is still debated. Particularly, Dalmizrak et al. reported that long-term use of fluoxetine may be connected with glutathione reductase (GR) deficiency,21 while Byeon et al. pointed out that SSRIs might mediate oxidative stress in aquatic invertebrates.22
In the above described scenario, we have synthesised chimeric derivatives of fluoxetine that have been modified to incorporate a selenium nucleus, thus providing an additional mechanism to enhance its molecular antioxidant performance (Fig. 1).
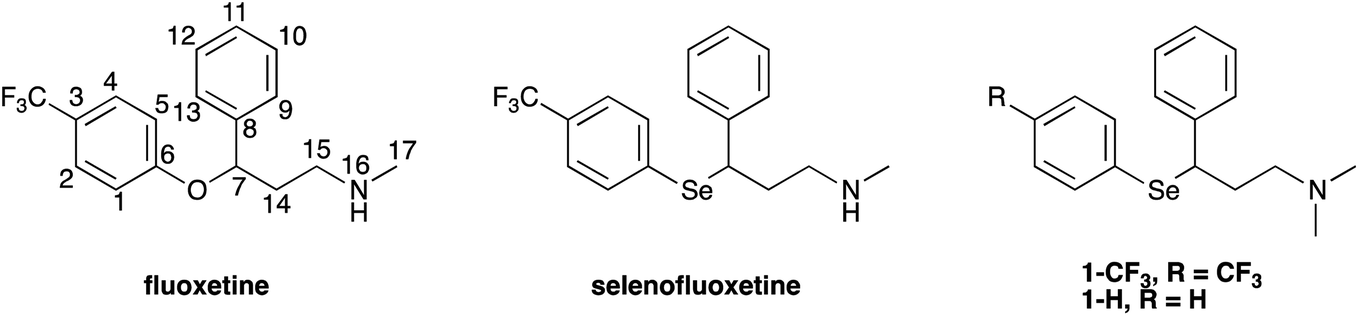 | ||
| Fig. 1 Fluoxetine and its derivatives studied in this work. | ||
Besides the radical scavenging activity via HAT (Hydrogen Atom Transfer) mechanism, recently reported for fluoxetine,17 selenofluoxetine and its derivatives have the capacity of reducing hydroperoxides and H2O2 as GPx mimics.23–25 The biological role of selenium is a long-debated issue since its identification in GPx, an enzyme able to protect cells from oxidative stress by inactivating hydroperoxides.26–30 As often happens, nature inspired medicinal chemists and synthetic selenium-based compounds were developed through the years to mimic GPx activity as well as to exert antioxidant activity through other mechanisms, such as metal binding.23–25,31 Among GPx mimics, ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) is the most popular since it was employed in clinical trials.32 Aryl and alkyl selenides,33,34 together with aryl diselenides,35–38 have been prepared and tested in vitro and in vivo. In addition, unsubstituted alkylphenyl selenides and diphenyldiselenide have been adopted to study the reactivity towards peroxides as models for understanding the behaviour of organoselenides in biological environments.39 Since selenides are largely used in organic synthesis to catalyze oxidation in presence of H2O2, combined experimental and theoretical mechanistic studies on model organoselenides are important also for designing selenium based organocatalysts.40–45
The rationale that guided the design of the selenium-containing analogues of fluoxetine aims to combine the antioxidant radical scavenging capacity, within the CNS, of the parent compound with the GPx-like activity of organic selenides in a water-soluble drug-like small molecule (tandem antioxidant). Thus, selenofluoxetine and its derivatives here reported represent models for the experimental and theoretical investigation of their elementary, but biologically as well as chemically significant, reaction with H2O2. As will be discussed in the following, the oxidation–elimination process observed for the studied compounds leads to the production of a cinnamylamine. Such scaffold is found in many drugs like flunarizine (calcium antagonist) and naftifine (antifungal). It must be noted that other strategies for the synthesis of allylamines were previously reported in the literature, such as the preparation from alcohols and amines through alkoxyphosphonium salts,46 the reduction of secondary amides followed by N-methylation47 or the reduction of propargylamines.48 Nevertheless, the cited methods require hazardous and expensive reactants, harsh experimental conditions (temperature and pressure) and do not guarantee stereoselectivity. By contrast, the approach we propose efficiently proceeds in aqueous medium, it is trans-selective and requires only H2O2 as oxidant agent. Nuclear magnetic resonance (NMR) and electrospray ionisation-mass spectrometry (ESI-MS) combined with quantum chemistry calculations are used to characterise intermediates and products and to draw energy profiles based on which the mechanistic details are unravelled.
Experimental
Chemistry
Commercially available chemicals were purchased from Sigma-Aldrich and used without any further purification if not specified elsewhere. NMR experiments were performed on a Bruker Avance III 400 spectrometer (frequencies: 400.13, 100.62, and 76.37 MHz for 1H, 13C, and 77Se nuclei, respectively) equipped with a multinuclear inverse z-field gradient probe head (5 mm). For data processing, TopSpin 4.0.8 software was used and the spectra were calibrated using solvent signal (1H-NMR, δH = 7.26 ppm for CDCl3, δH = 2.50 ppm for DMSO-d6, δH = 4.79 ppm for D2O; 13C-NMR, δC = 77.16 ppm for CDCl3, δC = 39.52 ppm for DMSO). Multiplicities are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad; dd, doublet of doublets. Mass spectra were recorded by direct infusion ESI on a Thermo Fisher Scientific LCQ Fleet ion trap mass spectrometer and on a Waters Xevo G2 QTof high-resolution mass spectrometer (HRMS). The purity profile of the compounds was assayed by HPLC using a Varian Pro-Star system equipped with a Biorad 1706 UV-VIS detector and an Agilent C-18 column (5 μm, 4.6 × 150 mm). An appropriate ratio of water (A) and acetonitrile (B) was used as mobile phase with an overall flow rate of 1 mL min−1. The general method for the analyses is reported here: 0 min (90% A–10% B), 5 min (90% A–10% B), 25 min (10% A–90% B), 30 min (90% A–10% B), and 32 min (90% A–10% B). The purity of all compounds was ≥96%, unless otherwise stated (254 nm).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:0.5). After 2 hours, the solution was left to cool to room temperature. A solid precipitate of N,N-dimethyl-3-oxo-3-phenylpropan-1-aminium chloride salt formed and the solid was filtered with a Buchner funnel, washed with cold acetone (3 × 10 mL) and with hexane (1 × 10 mL). Yield 3.48 g (98%); white solid; 1H-NMR (400 MHz, DMSO): δH (ppm) 10.57 (br, 1H, NH), 8.02 (d, 2H, J = 7.2 Hz, Ph-H), 7.69 (t, 1H, J = 7.4 Hz, Ph-H), 7.57 (t, 2H, J = 7.6 Hz, Ph-H), 3.63 (t, 2H, J = 7.2 Hz, C(O)CH2), 3.40 (t, 2H, J = 7.2 Hz, CH2N), 2.80 (s, 6H, N(CH3)2); 13C-NMR (101 MHz, DMSO): δC (ppm) 196.8 (s, C(O)), 135.9 (s, Ph-C), 133.7 (s, 2C, Ph-C), 128.8 (s, Ph-C), 128.0 (s, 2C, Ph-C), 51.8 (s, CH2N), 42.2 (s, 2C, N(CH3)2), 33.1 (s, C(O)CH2); (HRMS ESI+) m/z calcd for C11H16NO+ [M + H]+: 178.1232; found: 178.1308.:2.5:0.5) and was stopped after 2 hours when the starting material spot was no longer detected. The solvent was evaporated under reduced pressure obtaining the compound as hydrochloride salt. Yield 1.88 g (96%), white solid; 1H NMR (400 MHz, DMSO): δH (ppm) 10.90 (br, 1H, NH), 7.52–7.49 (m, 2H, Ph-H), 7.45–7.35 (m, 3H, Ph-H), 5.31 (dd, J = 9.1, 5.2 Hz, 1H, CH(Cl)), 3.23–3.18 (m, 1H, CH(Cl)CHAHB), 3.10–3.04 (m, 1H, CH(Cl)CHAHB), 2.76 (s, 6H, N(CH3)2), 2.63–2.44 (m, 2H, CH2N); 13C-NMR (101 MHz, DMSO): δC (ppm) 141.0 (s, Ph-C), 129.3 (s, 2C, Ph-C), 129.2 (s, 2C, Ph-C), 127.5 (s, Ph-C), 61.1 (s, CH(Cl)), 42.7 (s, CH2N), 42.4 (s, 2C, N(CH3)2), 33.6 (s, CH(Cl)CH2); (HRMS ESI+) m/z calcd for C11H17ClN+ [M + H]+: 198.1044, found: 198.1043.:7.5:0.5). Yield 112 mg (34%); yellow oil; 1H-NMR (400 MHz, CDCl3): δH (ppm) 7.49 (d, J = 8.0 Hz, 2H, Ph-H), 7.43 (d, J = 8.4 Hz, 2H, Ph-H), 7.29–7.19 (m, 5H, Ph-H), 4.49–4.46 (m, 1H, CH(Se)), 2.38–2.22 (m, 4H, CHCH2CH2N), 2.22 (s, 6H, N(CH3)2); 13C-NMR (101 MHz, CDCl3): δC (ppm) 141.72 (s, Ph-C), 134.9 (s, 2C, Ph-C), 134.7 (s, Ph-C), 129.7 (q, J = 32.6 Hz, Ph-C), 128.6 (s, 2C, Ph-C), 127.9 (s, 2C, Ph-C), 127.4 (s, Ph-C), 125.6 (q, J = 3.7 Hz, 2C, Ph-C), 124.2 (q, J = 272.19 Hz, CF3), 57.8 (s, CH(Se)), 46.3 (s, CH2N), 45.4 (s, 2C, N(CH3)2), 34.0 (s, CH(Se)CH2); (HRMS ESI+) m/z calcd for C18H21F3NSe+ [M + H]+: 388.0786, found: 388.0866.:7.5:0.5). Yield 155 mg (38%); yellow oil; 1H-NMR (400 MHz, CDCl3): δH (ppm) 7.43–7.40 (m, 2H, Ph-H), 7.28–7.18 (m, 8H, Ph-H), 4.38–4.33 (m, 1H, CH(Se)), 2.31–2.19 (m, 4H, CH(Se)CH2CH2N), 2.17 (s, 6H, N(CH3)2); 13C-NMR (101 MHz, MeOD): δC (ppm) 140.6 (s, Ph-C), 135.5 (s, Ph-C), 128.8 (s, 2C, Ph-C), 128.4 (s, Ph-C), 128.3 (s, 2C, Ph-C), 128.0 (s, 2C, Ph-C), 127.3 (s, Ph-C), 127.2 (s, 2C, Ph-C), 56.4 (s, CH(Se)), 43.7 (s. CH2N), 42.1 (c, 2C, N(CH3)2), 30.5 (s, CH(Se)CH2); (HRMS ESI+) m/z calcd for C17H22NSe+ [M + H]+: 320.0912, found: 320.1072.
:2.5:0.5). After 2 hours, the solution was left to cool to room temperature. A solid precipitate of N,N-dimethyl-3-oxo-3-phenylpropan-1-aminium chloride salt formed and the solid was filtered with a Buchner funnel, washed with cold acetone (3 × 10 mL) and with hexane (1 × 10 mL). Yield 3.48 g (98%); white solid; 1H-NMR (400 MHz, DMSO): δH (ppm) 10.57 (br, 1H, NH), 8.02 (d, 2H, J = 7.2 Hz, Ph-H), 7.69 (t, 1H, J = 7.4 Hz, Ph-H), 7.57 (t, 2H, J = 7.6 Hz, Ph-H), 3.63 (t, 2H, J = 7.2 Hz, C(O)CH2), 3.40 (t, 2H, J = 7.2 Hz, CH2N), 2.80 (s, 6H, N(CH3)2); 13C-NMR (101 MHz, DMSO): δC (ppm) 196.8 (s, C(O)), 135.9 (s, Ph-C), 133.7 (s, 2C, Ph-C), 128.8 (s, Ph-C), 128.0 (s, 2C, Ph-C), 51.8 (s, CH2N), 42.2 (s, 2C, N(CH3)2), 33.1 (s, C(O)CH2); (HRMS ESI+) m/z calcd for C11H16NO+ [M + H]+: 178.1232; found: 178.1308.:2.5:0.5) and was stopped after 2 hours when the starting material spot was no longer detected. The solvent was evaporated under reduced pressure obtaining the compound as hydrochloride salt. Yield 1.88 g (96%), white solid; 1H NMR (400 MHz, DMSO): δH (ppm) 10.90 (br, 1H, NH), 7.52–7.49 (m, 2H, Ph-H), 7.45–7.35 (m, 3H, Ph-H), 5.31 (dd, J = 9.1, 5.2 Hz, 1H, CH(Cl)), 3.23–3.18 (m, 1H, CH(Cl)CHAHB), 3.10–3.04 (m, 1H, CH(Cl)CHAHB), 2.76 (s, 6H, N(CH3)2), 2.63–2.44 (m, 2H, CH2N); 13C-NMR (101 MHz, DMSO): δC (ppm) 141.0 (s, Ph-C), 129.3 (s, 2C, Ph-C), 129.2 (s, 2C, Ph-C), 127.5 (s, Ph-C), 61.1 (s, CH(Cl)), 42.7 (s, CH2N), 42.4 (s, 2C, N(CH3)2), 33.6 (s, CH(Cl)CH2); (HRMS ESI+) m/z calcd for C11H17ClN+ [M + H]+: 198.1044, found: 198.1043.:7.5:0.5). Yield 112 mg (34%); yellow oil; 1H-NMR (400 MHz, CDCl3): δH (ppm) 7.49 (d, J = 8.0 Hz, 2H, Ph-H), 7.43 (d, J = 8.4 Hz, 2H, Ph-H), 7.29–7.19 (m, 5H, Ph-H), 4.49–4.46 (m, 1H, CH(Se)), 2.38–2.22 (m, 4H, CHCH2CH2N), 2.22 (s, 6H, N(CH3)2); 13C-NMR (101 MHz, CDCl3): δC (ppm) 141.72 (s, Ph-C), 134.9 (s, 2C, Ph-C), 134.7 (s, Ph-C), 129.7 (q, J = 32.6 Hz, Ph-C), 128.6 (s, 2C, Ph-C), 127.9 (s, 2C, Ph-C), 127.4 (s, Ph-C), 125.6 (q, J = 3.7 Hz, 2C, Ph-C), 124.2 (q, J = 272.19 Hz, CF3), 57.8 (s, CH(Se)), 46.3 (s, CH2N), 45.4 (s, 2C, N(CH3)2), 34.0 (s, CH(Se)CH2); (HRMS ESI+) m/z calcd for C18H21F3NSe+ [M + H]+: 388.0786, found: 388.0866.:7.5:0.5). Yield 155 mg (38%); yellow oil; 1H-NMR (400 MHz, CDCl3): δH (ppm) 7.43–7.40 (m, 2H, Ph-H), 7.28–7.18 (m, 8H, Ph-H), 4.38–4.33 (m, 1H, CH(Se)), 2.31–2.19 (m, 4H, CH(Se)CH2CH2N), 2.17 (s, 6H, N(CH3)2); 13C-NMR (101 MHz, MeOD): δC (ppm) 140.6 (s, Ph-C), 135.5 (s, Ph-C), 128.8 (s, 2C, Ph-C), 128.4 (s, Ph-C), 128.3 (s, 2C, Ph-C), 128.0 (s, 2C, Ph-C), 127.3 (s, Ph-C), 127.2 (s, 2C, Ph-C), 56.4 (s, CH(Se)), 43.7 (s. CH2N), 42.1 (c, 2C, N(CH3)2), 30.5 (s, CH(Se)CH2); (HRMS ESI+) m/z calcd for C17H22NSe+ [M + H]+: 320.0912, found: 320.1072.NMR study of the reaction with H2O2
In order to study the oxidation reaction of compounds 1-CF3 and 1-H, their reaction with H2O2 was investigated by 1H-NMR in an aqueous environment at room temperature (22 °C). The compounds were used as hydrochloride salts (28 mM: 1-CF3 = 4.9 mg, 11.6 μmol and 1-H = 5.6 mg, 17.6 μmol) and dissolved in D2O, t0 spectra were registered with 8 scans, 1 dummy scan and a delay time d1 of 2 s. Afterwards, H2O2 (1.1 eq., H2O2 3% w/w solution: 1-CF3 = 15 μL, 1-H = 18 μL) was added and the spectra were registered every 2 or 5 minutes until the reaction was complete (47 min for 1-CF3 and 104 min for 1-H) (8 scans, 1 dummy scan, d1 of 2 s). The reaction was studied plotting the results of the integration of the signals corresponding to the protons bound to the carbon adjacent to the selenium atom for the starting material, and the two diastereoisomers and the allylic hydrogens for the cinnamylamine. 1H–77Se HMBC NMR spectra were acquired using a repetition delay of 1 s; a total of 300 experiments of 16–40 scans were accumulated and processed with a magnitude calculation; an evolution delay of 33.3 ms was used for 1H–77Se long-range coupling constants; the spectral width was 13 ppm in F2 and 1000 ppm in F1. Zero-filling in the F1 and F2 dimensions, multiplication with a Gaussian function (in F2) and a squared sine function (in F1) were performed prior to 2D Fourier transformation.ESI-MS study of the reaction with H2O2
Mechanistic ESI-MS studies were performed under the same experimental conditions (concentration and temperature) used in the NMR analysis, except for the fact that Milli-Q water was used instead of D2O. Solutions were diluted 1:1000 in methanol before the analysis. Spectra were recorded by direct infusion ESI on a Thermo Fisher Scientific (Waltham, MA) LCQ Fleet ion trap mass spectrometer. ESI parameter for positive ionisation mode are here described: 4.0 kV spray voltage, 225 °C capillary temperature, 5 μL min−1 flow rate. For negative ionisation mode: 5.0 kV spray voltage, 180 °C capillary temperature, 5 μL min−1 flow rate.
Computational methodology
For hydrogen atom transfer (HAT) reactions, geometry optimisations of the reactants and products were performed in the gas phase without any constraint, using the M06-2X functional49 combined with the 6-31G(d) basis set, as implemented in Gaussian 16.50 Spin contamination was checked for the doublet ground state species to assess the reliability of the wavefunction. To confirm the nature of the stationary points and to obtain the thermodynamic corrections at 1 atm and 298 K, frequency calculations at the M06-2X/6-31G(d) level of theory were run to ascertain that only positive frequencies were present. In order to obtain more accurate energy values, single-point energy calculations were performed at M06-2X/6-311+G(d,p) in the gas phase, and subsequently, at the same level of theory, in benzene and water using the continuum Solvation Model based on Density (SMD).51 This level of theory is denoted in the text (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d,p). The choice fell on benzene and water because the model an apolar and a polar environment, respectively.52 Energy barriers were calculated for the most reactive sites (identified on the basis of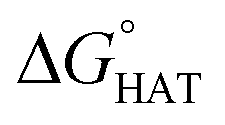 ), at the (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d,p) level of theory. Analysis of the single imaginary frequency of transition states confirmed that the normal mode involved was correct for the HAT process. Energy barriers were calculated, referring to the free reactants in the gas phase as well as in the solvent.
), at the (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d,p) level of theory. Analysis of the single imaginary frequency of transition states confirmed that the normal mode involved was correct for the HAT process. Energy barriers were calculated, referring to the free reactants in the gas phase as well as in the solvent.
The quantum chemistry calculations for the Se oxidation mechanism were performed using the Amsterdam Density Functional (ADF).53–55 The energy profiles were obtained from geometries and energies computed by using the OLYP functional,56,57 which is known to perform well for reactivity studies on organic compounds, and it has been recently benchmarked58 and applied59 to organic dichalcogenides. OLYP was combined with the TZ2P basis set for all the atoms.60 The TZ2P basis set is of triple-ζ quality and has been augmented with two sets of polarisation functions. Core shells of the atoms (1s for C, F, N and O and up to 3p for Se) were treated by using the frozen-core approximation. Scalar relativistic effects were treated using the Zeroth Order Regular Approximation (ZORA).61–63 The numerical integration was performed by using the fuzzy-cell integration scheme developed by Becke.64,65 Energy minima and transition states have been verified through vibrational analysis. All minima were found to have zero imaginary frequencies and all transition states have one that correspond to the mode of the reaction under consideration. For single point calculations in water the conductor-like screening Model was employed (COSMO), as implemented in ADF.66–68 Water was parameterised using a dielectric constant of 78.39 and a solvent radius of 1.93 Å.
Results and discussion
Synthesis of the selenium-based fluoxetine analogues
Selenofluoxetine closely resembles fluoxetine, in which oxygen was substituted with a selenium atom. N,N-dimethyl derivatives 1-CF3 and 1-H (Fig. 1) were designed to simplify synthetic procedures and were used for the experimental reactivity studies. Compounds 1-CF3 and 1-H, were obtained through a multi-step synthesis. Acetophenone was initially subjected to a Mannich reaction with dimethylamine hydrochloride and formaldehyde giving compound 2.69 The Mannich base was then reduced with sodium borohydride providing compound 3 as two enantiomers,70 and subsequently chlorinated with thionyl chloride obtaining the intermediate synthon 4 (Scheme 1).71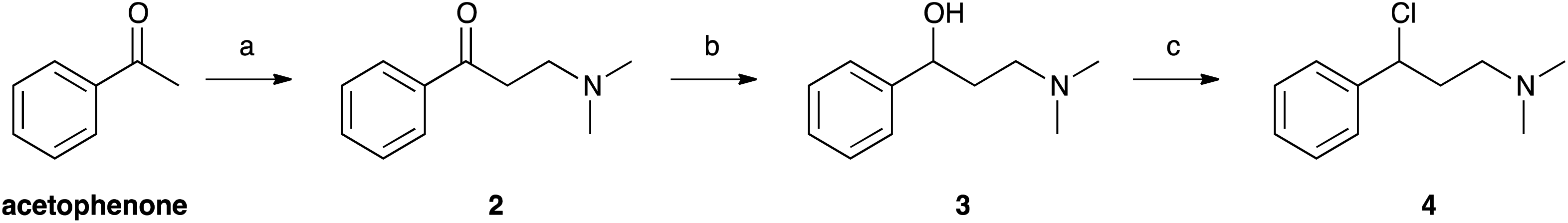 | ||
| Scheme 1 (a) dimethylamine HCl/HCOH/EtOH; (b) NaBH4/KOH/MeOH; (c) SOCl2. | ||
The chloride was then displaced by the appropriate selenide nucleophile, formed in situ by the reduction of the corresponding diphenyl diselenide with sodium borohydride, providing 1-CF3 and 1-H (Scheme 2; please refer to ESI† for detailed experimental procedures and analytical data). Particularly, 1,2-bis(4-(trifluoromethyl)phenyl)diselane (compound 5) was obtained through the reaction with elemental selenium of the Grignard reagent formed from p-trifluoromethyl bromobenzene (see Fig. S1–S22† for 1H, 13C and mass spectra).72,73 The unsubstituted diphenyl diselenide is commercially available.
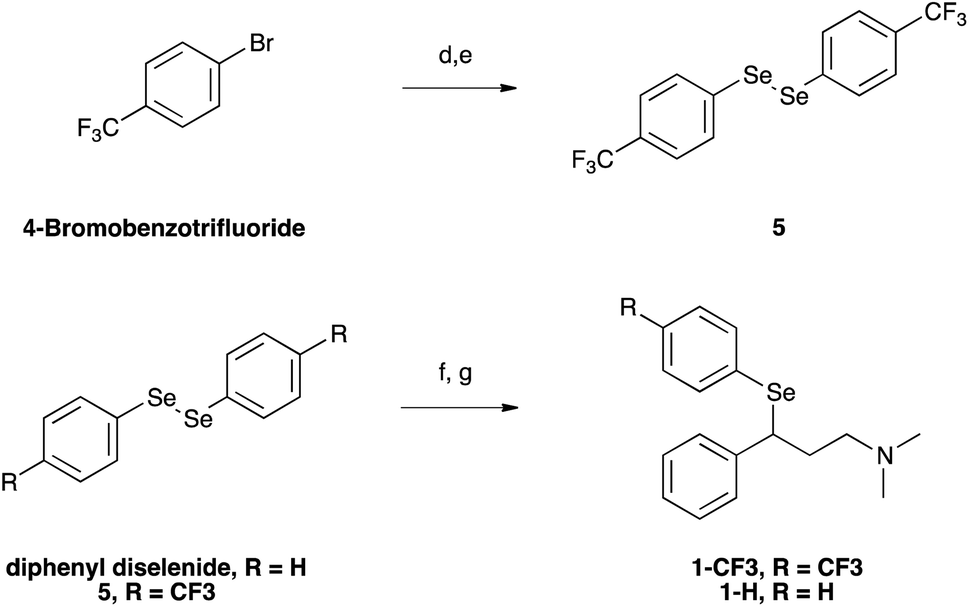 | ||
| Scheme 2 (d) Mg/Et2O; (e), Se; (f) NaBH4/KOH/EtOH; (g) compound 3/EtOH. | ||
Oxidation by H2O2: NMR results
In our experimental model reaction, the process initiates upon addition of H2O2 and evolves to an oxidation-triggered elimination, which is peculiar of selenides having protons in the β-position with respect to the chalcogen nucleus (Scheme 3). It is a highly trans-selective process affording olefins, and it was demonstrated that it occurs through a syn mechanism.74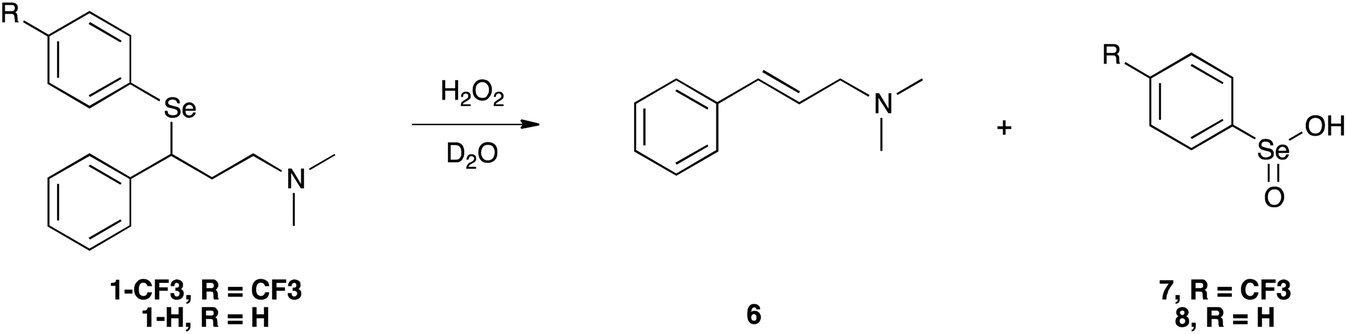 | ||
| Scheme 3 Oxidation of 1-CF3 and 1-H by H2O2. This scheme represents the overall reaction leading to the formation of cinnamylamine and seleninic acid, which were experimentally observed as final products. | ||
The first step of the process consists in the oxidation of the starting selenide to the corresponding selenoxide. It has been reported that this reaction can occur using hydrogen peroxide (H2O2), meta-chloroperoxybenzoic acid (mCPBA) and ozone.39,75,76 This process promotes the formation of a new chiral centre, giving rise to two enantiomers or to two diastereoisomers, depending on the initial compound. Then, an intramolecular syn elimination takes place: a proton is transferred from the β-position to the oxygen of the selenoxide, and the selenium–carbon bond breaks leading to the formation of a carbon–carbon double bond (Scheme 4).77
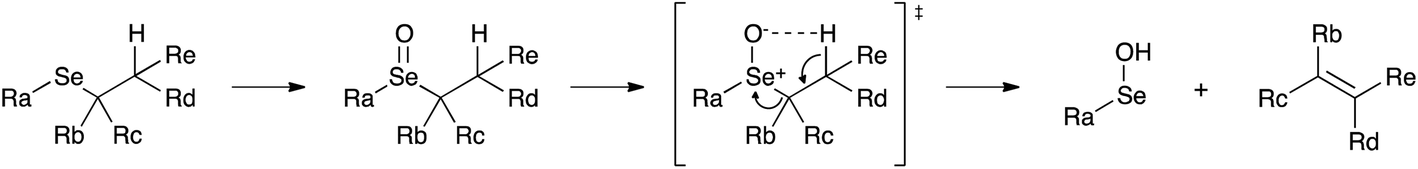 | ||
| Scheme 4 Mechanistic details of selenoxide elimination leading to the formation of selenenic acid and olefin. Selenenic acid undergoes disproportionation and only seleninic acid was experimentally observed. | ||
The above described selenoxide elimination was studied considering the selenium-based analogues of fluoxetine as models. As anticipated, fluoxetine was modified with the aim of improving its antioxidant properties through the substitution of its oxygen atom with selenium, leading to compound 1-CF3. In this connection, compound 1-H was designed as a further simplified model for our investigation aiming at drawing widely general conclusions. Finally, due to synthetic feasibility and to improve water solubility, both analogues differ from fluoxetine also for the presence of a tertiary amine instead of a secondary amine. To explore the potential application of these derivatives in the field of green chemistry and to better mimic a biologically relevant environment, the oxidation of 1-H was performed in water. Indeed, although selenium and its oxidised species were reported to show toxic effect at high concentrations,78 growing attention is recently being paid to organoselenides catalysing organic reactions in environmentally friendly conditions.79 In particular, this holds true when aqueous medium is considered.80 In this context, good water solubility was achieved for 1-H and 1-CF3, since the compounds were prepared as hydrochloride salts. Firstly, to gain insight into the reaction mechanism and to fully characterise intermediates and products, the whole process was followed by 1H-NMR spectroscopy according to the procedure reported in the Experimental section. Compounds were dissolved in deuterated water and the oxidation was carried out at room temperature using 1 equivalent of H2O2. During the reaction of 1-H, prompt disappearance of the signal corresponding to the α position proton of the starting material (4.42 ppm) occurred, accompanied by the appearance of the signals of the two selenoxide diastereoisomers (4.48 ppm and 4.23 ppm) (Fig. 2). The same behaviour was observed for 1-CF3, but the signal of one of the two selenoxide diastereoisomers unluckily overlap to the signals of the starting material (approximately 4.55 ppm), thus complicating the mechanistic investigation (Fig. S23–S25†).
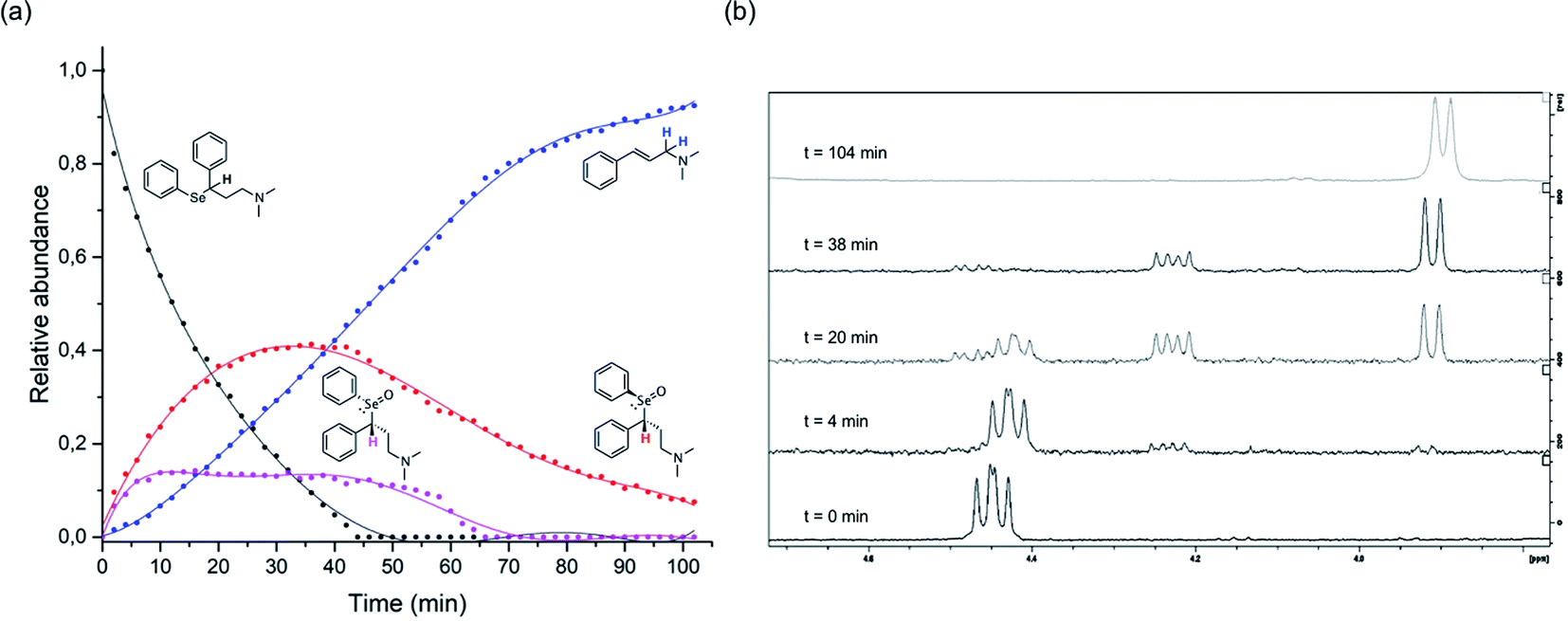 | ||
Fig. 2 1H-NMR mechanistic study of the reaction of 1-H with H2O2. The graph reported in (a) was obtained by integration of the α-hydrogen signals for the starting material and the two diastereoisomers and the allylic-hydrogens signals for the cinnamylamine in the 1H-NMR spectra acquired at different time points ( starting material, starting material,  R–R (and its enantiomer), R–R (and its enantiomer),  R–S (and its enantiomer) R–S (and its enantiomer)  cinnamylamine 6). In (b), representative 1H-NMR spectra are reported showing the variation over time during the oxidation-reaction of compound 3, focusing on the region between 4.7 and 3.8 ppm (area of interest to plot the graph; see Fig. S31† for additional spectra). cinnamylamine 6). In (b), representative 1H-NMR spectra are reported showing the variation over time during the oxidation-reaction of compound 3, focusing on the region between 4.7 and 3.8 ppm (area of interest to plot the graph; see Fig. S31† for additional spectra). | ||
When a chiral centre is present in the starting compound, since in the transition state the carbon–hydrogen and carbon–selenium bonds are co-planar, the two selenoxide diastereoisomer products may react with different formation and subsequent different elimination rate (Fig. 2). This phenomenon will be discussed more in detail in the computational section, since even slight differences in the activation energies for the elimination reactions that are strictly related to the structure of the transition states may affect the reaction speed.
As the reaction proceeded, it was possible to observe the disappearance of the selenoxide diastereoisomers (depicted in Fig. 2(a)) and the formation of the elimination products: (E)-N,N-dimethyl-cinnamyl amine 6, which was characterised by NMR, and the corresponding highly oxidised Se-containing species, i.e. seleninic acids 7 and 8. At this stage, it must be noted that seleninic acid itself is endowed with synthetic value in the context of organic catalysis. It is indeed generally referred to as a pre-catalyst,81 which can be converted to the corresponding benzeneperoxyseleninic acid, a known oxygen-transfer agent, in presence of an excess of peroxide.82,83 For the studied compounds, the overall reaction demonstrated to be relatively fast. Particularly, a 90% conversion was observed for 1-CF3 after 36 minutes, whereas for 1-H the same conversion was achieved after 95 minutes (Fig. S26† and 2).
Importantly, it has to be pointed out that selenenic acid is involved in the chemical equilibrium reported in Scheme 5,77 and according to this mechanism it disproportionates providing seleninic acid, diphenyl diselenide and water.
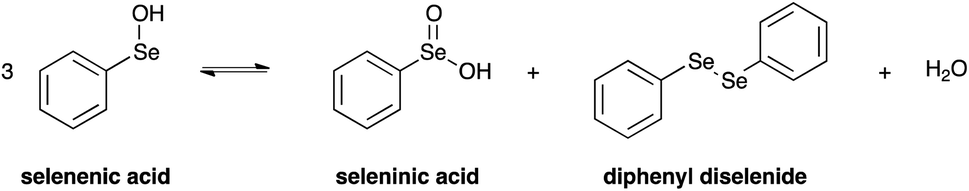 | ||
| Scheme 5 Redox equilibrium involving differently oxidised selenium species. | ||
On the other hand, also diphenyl diselenide can be oxidised by H2O2 giving seleninic acid.84 The formation of these less water soluble species was also suggested by the fact that a solid precipitate was observed in the NMR sample after the reaction study. Moreover, a signal at 1172.0 ppm was detected in the 77Se NMR spectrum acquired at the end of the reaction, likely due to the presence of seleninic acid 8 (Fig. S32†).
Thus, to better clarify this behaviour and confirm the identity of the products, a more focused 1H–77Se HMBC NMR experiment was carried out. A mid-reaction sample from the oxidation study carried out on 1-CF3 was diluted 1:1 in MeOD to completely dissolve the precipitate. The NMR analysis demonstrated the presence of p-trifluoromethyl seleninic acid 7 and 1,2-bis(4-(trifluoromethyl)phenyl)diselane 5, respectively testified by the 77Se signals detected at 447.9 and 1211.8 ppm (Fig. S29†). These findings are in agreement with previous observations by Wang and colleagues.85 The authors studied the selenoxide elimination reaction in a simple model system by reacting a phenylalkylselenide with 2 equivalents of H2O2 in CD3CN, observing the appearance of a signal at 1175.1 ppm in the 77Se NMR spectrum, which is consistent with the presence of seleninic acid. Our results were further confirmed by ESI-MS experiments, as described in the following.
Investigation of the reaction mechanism by ESI-MS
Mass spectrometry, and ESI-MS in particular, can be used to identify the species in solution and complete the information obtained from NMR studies, even if few examples of its application to the chemistry and biochemistry of selenium are available.39,86,87 In this study, mass spectra analysis was used as an independent technique for the identification and characterisation of the species involved in the transformation of the fluoxetine analogues and the compound identity was confirmed by collision-induced dissociation (CID) studies.The monitoring of the reaction with H2O2 was carried out under the same experimental conditions (solvent, concentration, temperature) used to investigate the mechanistic details by NMR, working on independently prepared samples and according to the procedure reported in the Experimental section. Briefly, the reaction was carried out in water and time point samples were diluted 1:1000 in methanol before ESI-MS analysis. In analogy with the NMR protocol, the reaction of 1-H was studied more in detail. In particular, the conversion of the starting material into the products was followed by sampling the reaction at the same time points used during the NMR experiments, and the two analytical techniques showed that the oxidation/elimination process was accomplished in a similar time frame (approximately 70 min, see Fig. S43†). Upon addition of H2O2, the signals corresponding to the selenoxide (m/z = 334) and cinnamylamine 6 (m/z = 162) were detected in the mass spectra obtained in positive ionisation mode. Moreover, MS analyses performed in negative ionisation mode highlighted the presence of the signal corresponding to seleninic acid 8 (m/z = 189, see Fig. S33–S36† for representative mass spectra), in agreement with the data reported by Wang and colleagues.85
Finally, MS analysis was also performed on the NMR samples used in the mechanistic study above described. Positive and negative ionisation mode MS spectra acquired on the completely reacted sample (t = 24 h) diluted in methanol confirmed the presence of seleninic acid 8 (m/z = 189), diphenyl diselenide (m/z = 316) and cinnamylamine 6 (m/z = 162) as reaction products, that were identified using CID experiments (Fig. S37–S42†). A similar behaviour was observed for 1-CF3. Again, MS analysis on the sample reacted with H2O2 unambiguously confirmed the presence in solution of 6 (m/z = 162) and 7 (m/z = 255) as final products (Fig. S44 and S45†).
Mechanistic DFT analysis
The radical scavenging activity via HAT of fluoxetine has been recently investigated in silico by some of us.17 HAT occurs from the non-aromatic sites C7, C14, C15, N16 and C17. We have considered the corresponding sites of selenofluoxetine (Scheme 1) and focused on the scavenging of HO˙, which is the most reactive and electrophilic oxygen centred radical, of the peroxyl radical HOO˙ and of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHOO˙, which mimics larger unsaturated peroxyl radicals.
CHOO˙, which mimics larger unsaturated peroxyl radicals. 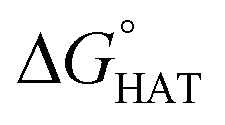 values are shown in Fig. 3 and listed in Table S1.†
values are shown in Fig. 3 and listed in Table S1.†
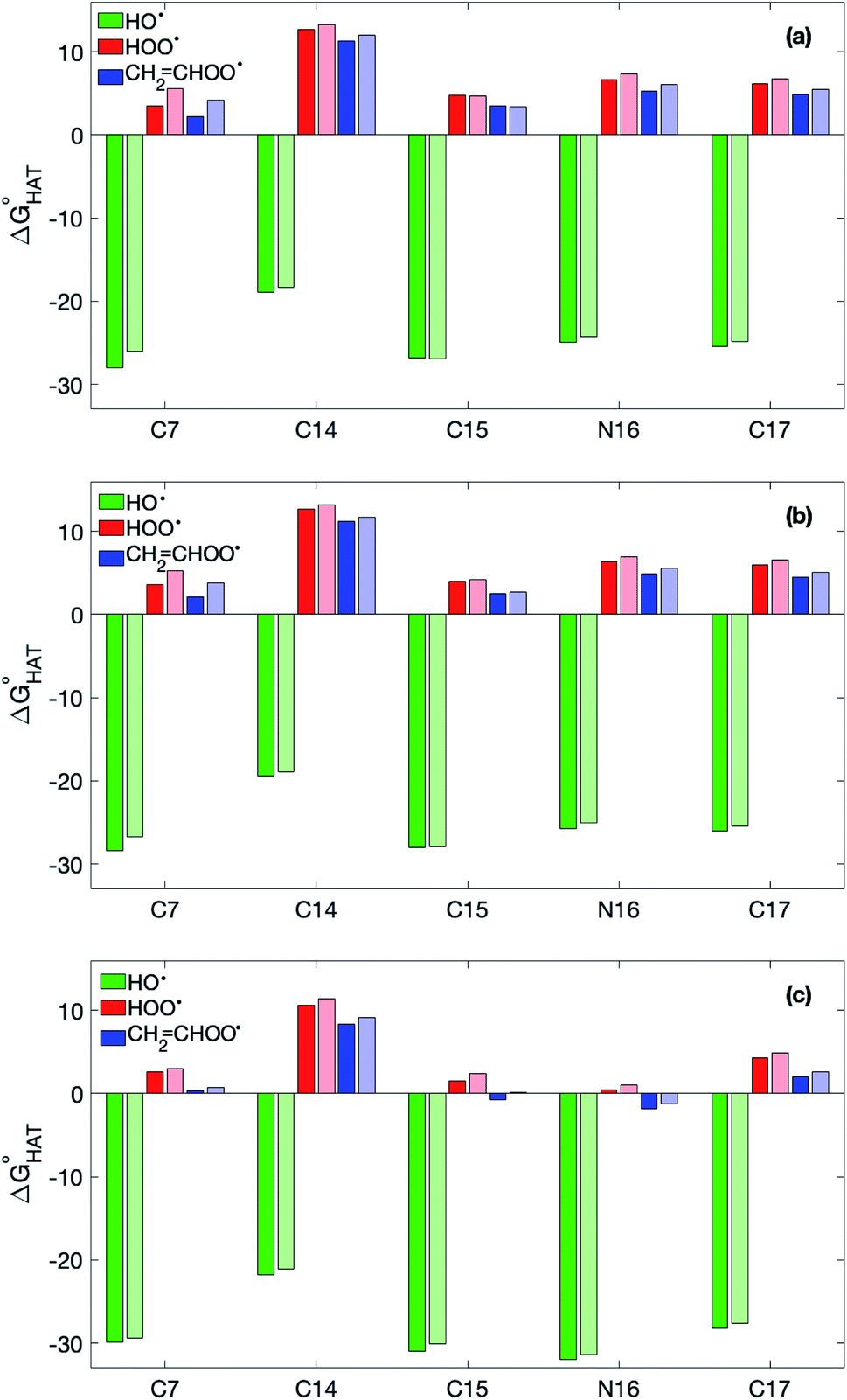 | ||
Fig. 3 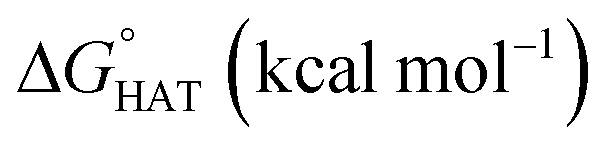 in the gas phase (a), in benzene (b), and in water (c) for the scavenging of HO˙, HOO˙ and CH2CHOO˙ from non-aromatic sites of selenofluoxetine. Values for fluoxetine taken from Muraro et al.17 are showed in a lighter shade for comparison. Level of theory: (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d). in the gas phase (a), in benzene (b), and in water (c) for the scavenging of HO˙, HOO˙ and CH2CHOO˙ from non-aromatic sites of selenofluoxetine. Values for fluoxetine taken from Muraro et al.17 are showed in a lighter shade for comparison. Level of theory: (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d). | ||
As reported for fluoxetine, selenofluoxetine is not selective for peroxyl radicals; in fact, all HATs from all the five sites to HOO˙ and most HATs to CH2CHOO˙ are endergonic in gas-phase as well as in solvent. Focusing on HAT to HO˙, in gas phase, it is thermodynamically most favoured from C7, which is close to Se, and more favoured from C15 and N16 than from C14 and C17. 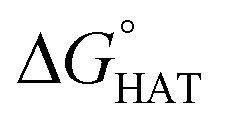 values become more negative when going from the gas to the condensed phase and with increasing solvent polarity. In benzene, the same trend of gas-phase is found; conversely, in water, HAT from the amino site is the most favoured, as found for fluoxetine.
values become more negative when going from the gas to the condensed phase and with increasing solvent polarity. In benzene, the same trend of gas-phase is found; conversely, in water, HAT from the amino site is the most favoured, as found for fluoxetine. 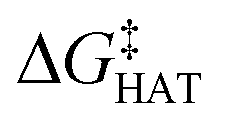 were computed for all five non-aromatic sites to HO˙, i.e. those from C7, C14, C15, N16 and C17 and are reported in Table 1. In gas-phase, the lowest activation energy is computed for HAT from C7, which is also the most exergonic process. i.e. 2.8 kcal mol−1. This value slightly increases in benzene where HAT from C7 becomes also less thermodynamically favoured process and decreases to 0.9 kcal mol−1 in water where
were computed for all five non-aromatic sites to HO˙, i.e. those from C7, C14, C15, N16 and C17 and are reported in Table 1. In gas-phase, the lowest activation energy is computed for HAT from C7, which is also the most exergonic process. i.e. 2.8 kcal mol−1. This value slightly increases in benzene where HAT from C7 becomes also less thermodynamically favoured process and decreases to 0.9 kcal mol−1 in water where 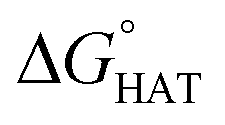 reaches the lowest negative value, i.e. −32.0 kcal mol−1. Notably, in selenofluoxetine, HAT from all the five sites to HO˙ is thermodynamically as well as kinetically more favoured than in fluoxetine, denoting a better radical scavenging potential.
reaches the lowest negative value, i.e. −32.0 kcal mol−1. Notably, in selenofluoxetine, HAT from all the five sites to HO˙ is thermodynamically as well as kinetically more favoured than in fluoxetine, denoting a better radical scavenging potential.
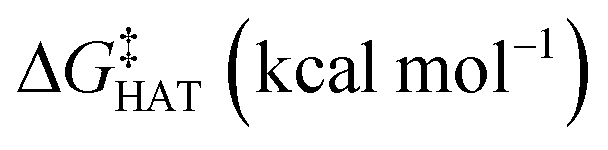 in the gas phase (a), in benzene (b), and in water (c) for the scavenging of HO˙, from non-aromatic sites of selenofluoxetine. Level of theory: (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d)
in the gas phase (a), in benzene (b), and in water (c) for the scavenging of HO˙, from non-aromatic sites of selenofluoxetine. Level of theory: (SMD)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d)
| HO˙ | |||
|---|---|---|---|
| Site | Gas phasea | Benzenea | Watera |
| a Values referring to the analogous sites of fluoxetine,17 computed at the same level of theory, are reported in parentheses for comparison. | |||
| C7 | 2.8 (6.6) | 3.8 (8.6) | 4.1 (10.0) |
| C14 | 8.9 (7.5) | 10.2 (8.9) | 11.0 (9.5) |
| C15 | 4.6 (5.4) | 5.3 (5.9) | 3.0 (4.2) |
| N16 | 3.5 (3.7) | 3.8 (4.0) | 0.9 (0.8) |
| C17 | 6.1 (7.8) | 4.1 (8.3) | 6.4 (5.5) |
The oxidation of the fluoxetine analogues by H2O2 was investigated in silico at ZORA-OLYP/TZ2P level of theory. In Fig. 4, the computed intermediates and transition states for selenofluoxetine are shown, while energy data are reported in Table S2.†
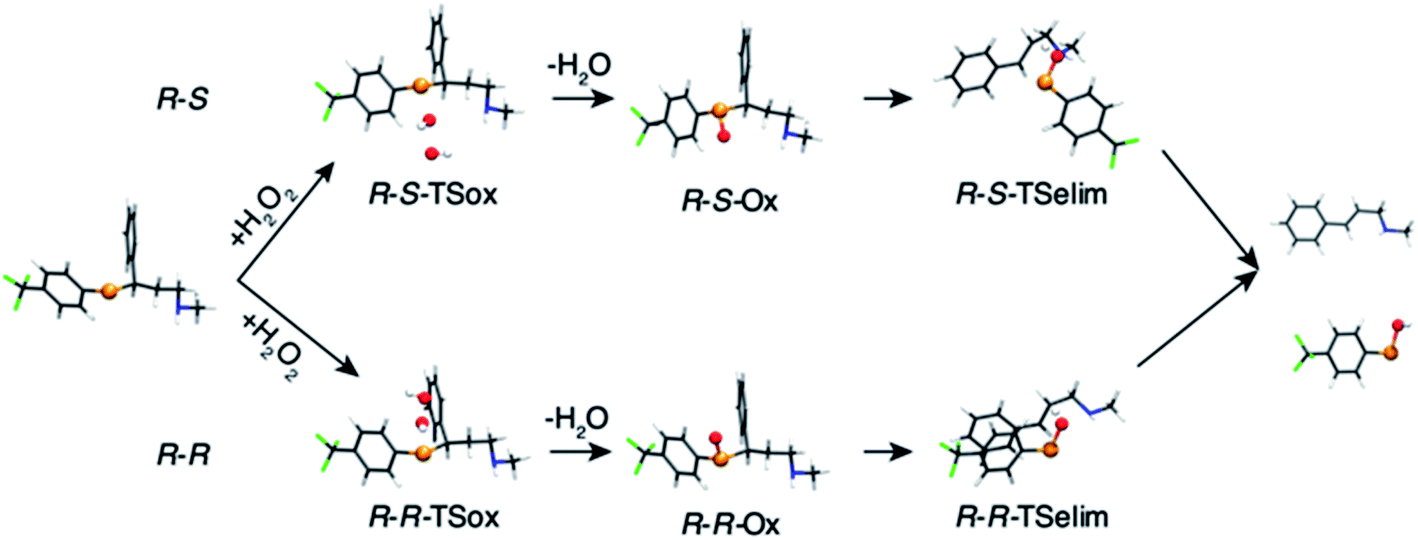 | ||
| Fig. 4 Fully optimised geometries of intermediates and transition states of the oxidation of selenofluoxetine shown in Scheme 3 (R = H, R′ = CF3). Reactant and product complexes are omitted for clarity. Level of theory: ZORA-OLYP/TZ2P. | ||
As can be seen from the energy profile shown in Fig. 5(a), in gas phase, the transition states are preceded and followed by the formation of a reactant complex (RCox) and a product complex (PCox), which are stabilised with respect to the free reactants and products, respectively. Upon reaction with H2O2, the selenide is oxidised to selenoxide. Since the attack of the peroxide may occur from two opposite sides, two paths are envisioned and two diastereoisomeric products form.
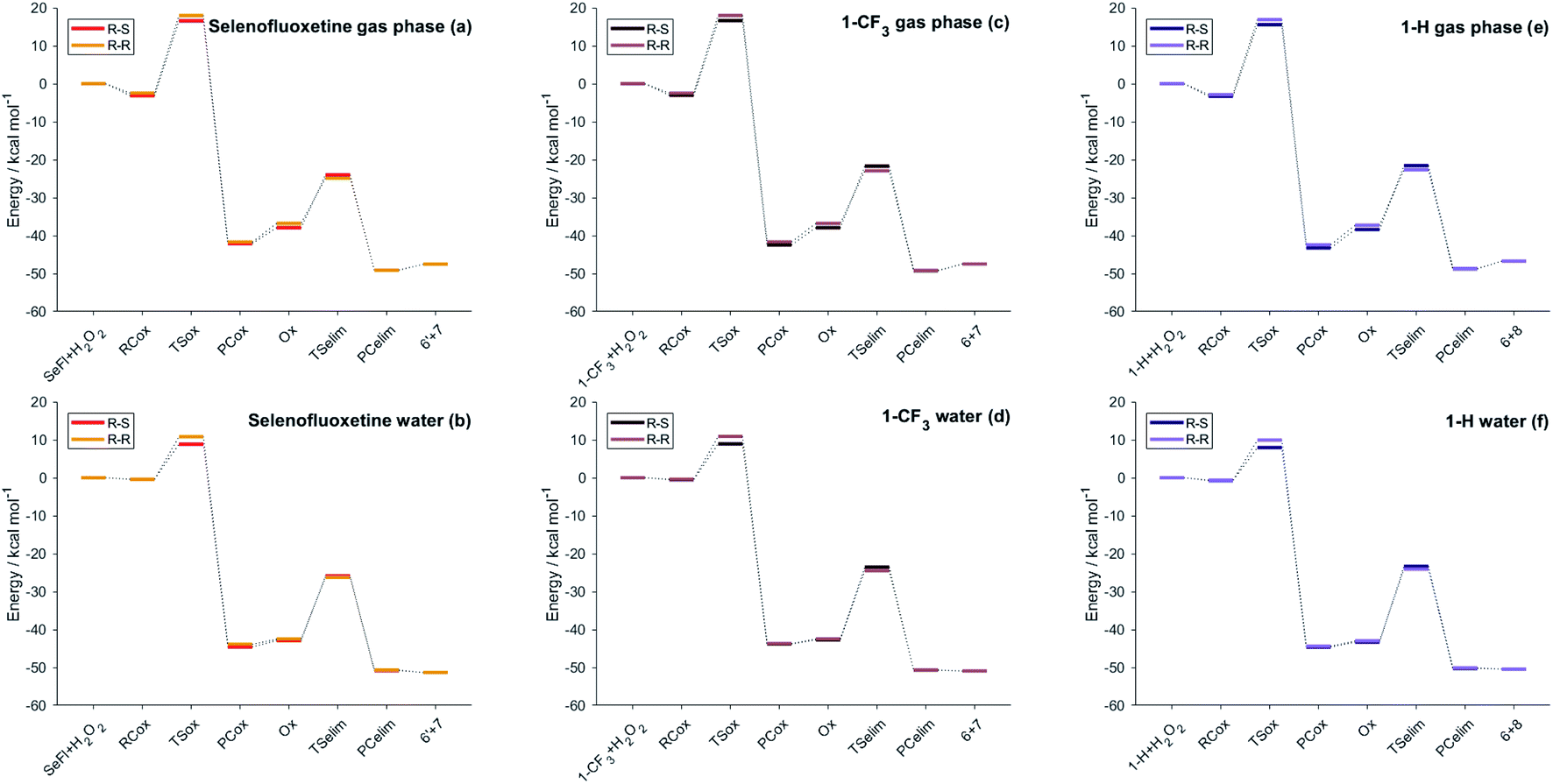 | ||
| Fig. 5 Energies of the stationary points of the investigated reactions (Scheme 3) for different selenofluoxetine derivatives in the gas-phase (left column) and in water (right column). Level of theory: (COSMO)-ZORA-OLYP/TZ2P. | ||
The oxidation occurring at selenium keeps the stereochemistry of C7 intact (R), therefore the two diastereoisomeric oxidised product differ only in the stereochemistry of selenium and are labelled R–R and R–S (Fig. 4). Despite sterically more hindered, there are no large differences in the energetics along the R–R path and R–S path, and the former is predicted to be slightly more stabilised. From the two products, i.e. R–R-Ox and R–S-Ox, elimination also occurs via two distinct paths with rather similar energetics, leading to the seleninic acid 7 and the alkene 6. The energy profiles shown in Fig. 5(c) and (d) are those of 1-CF3. No large mechanistic and energy differences are computed between fluoxetine and 1-CF3, suggesting that small geometry modifications do not affect significantly the reactivity. Calculations have been carried out also for 1-H. Considering the oxidation step, the barrier decreases when decreasing the electron-withdrawing character of the substituent on the phenyl ring R′, i.e. ΔE‡(R′ = CF3) > ΔE‡(R′ = H) (Table S2†). Conversely, in the elimination step, selenofluoxetine (R′ = CF3) has the lowest activation energy. The energy profiles of fluoxetine and 1-CF3 have been recalculated also in solvent using the COSMO continuum approach for water. They are shown in Fig. 5(b) and (d). As reported for similar reactions,88 we assist to the disappearance of the reactant and product complexes, which leads to a significant decrease of the activation energy for the oxidation step. Consequently, the highest barrier in water is the one computed for the elimination.
Conclusions
The results of our multi-approach experimental and theoretical investigation on the reaction of selenofluoxetine derivatives with H2O2 in water demonstrate that the substrates undergo an oxidation–elimination process, providing cinnamylamine and seleninic acid as final products. The model Se-based compounds here presented are closely related, from a structural point of view, to fluoxetine. Thus, they are expected to maintain the pharmacokinetic properties, such as CNS permeation, and biological activities of the original drug. Analogues of fluoxetine bearing tertiary amines were demonstrated to bind SERT with similar affinity.89 Consequently, toxicological and medical research is prompted to further explore this aspect by probing the antidepressant potential of selenofluoxetine and its derivatives. Moreover, the studied organoselenides are novel tandem antioxidants, combining the radical scavenging properties of fluoxetine, which are mainly related to C7 and N16 sites, and serotonin-mediated redox activity, with the GPx-like reactivity, due to the presence of the selenium nucleus. In addition, the here reported reactions are also endowed with synthetic value, since the basis for a novel approach for the synthesis of cinnamylamine, a pharmacologically-relevant scaffold are presented. More in general, the here described procedure provides an efficient alternative, on the green chemistry side, for the preparation of the allylamine. The oxidation step can indeed be carried out in water, only requiring H2O2 and affording a quantitative conversion. Notably, the reaction is highly selective providing the trans products. Moreover, we verified that 1-CF3 and 1-H react following the same mechanism to give cinnamylamine. This prompted us to further investigate the oxidation–elimination reaction on other substrates in our ongoing investigation. Our perspective goal, supported by preliminary results, consists in probing the potential application of this methodology to a broader range of substrates for the production of unsaturated amines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Università degli Studi di Padova, thanks to the P-DiSC (BIRD2018-UNIPD) project MAD3S (Modelling Antioxidant Drugs: Design and Development of computer-aided molecular Systems); P. I. L. O. All the calculations were carried out on Galileo (CINECA: Casalecchio di Reno, Italy) thanks to the ISCRA Grant REBEL2 (REdox state role in Bio-inspired ELementary reactions 2), P. I.: L. O. L.O. contributed to this research as part of the scientific activity of the international multidisciplinary network ‘SeS Redox and Catalysis’.References
- D. T. Wong, J. S. Horng, F. P. Bymaster, K. L. Hauser and B. B. Molloy, Life Sci., 1974, 15, 471–479 CrossRef CAS PubMed.
- D. T. Wong, K. W. Perry and F. P. Bymaster, Nat. Rev. Drug Discovery, 2005, 4, 764–774 CrossRef CAS PubMed.
- J. A. Coleman and E. Gouaux, Nat. Struct. Mol. Biol., 2018, 25, 170–175 CrossRef CAS PubMed.
- M. G. Scordo, E. Spina, M.-L. Dahl, G. Gatti and E. Perucca, Basic Clin. Pharmacol. Toxicol., 2005, 97, 296–301 CrossRef CAS PubMed.
- C. Mancuso, E. Esposito, M. G. Morgese, F. Caraci, F. Tascedda, S. Merlo, C. Benatti, S. F. Spampinato, A. Munafò, G. M. Leggio, F. Nicoletti, N. Brunello, F. Drago, M. A. Sortino and A. Copani, Front. Pharmacol., 2016, 7, 389 Search PubMed.
- K. Manton, S. Volovik and A. Kulminski, Curr. Alzheimer Res., 2004, 1, 277–293 CrossRef CAS PubMed.
- P. R. Angelova and A. Y. Abramov, FEBS Lett., 2018, 592, 692–702 CrossRef CAS PubMed.
- K. Bisht, K. Sharma and M.-È. Tremblay, Neurobiol. Stress, 2018, 9, 9–21 CrossRef PubMed.
- M. C. W. Oswald, N. Garnham, S. T. Sweeney and M. Landgraf, FEBS Lett., 2018, 592, 679–691 CrossRef CAS PubMed.
- M. Bortoli, M. Dalla Tiezza, C. Muraro, C. Pavan, G. Ribaudo, A. Rodighiero, C. Tubaro, G. Zagotto and L. Orian, Comput. Struct. Biotechnol. J., 2019, 17, 311–318 CrossRef CAS PubMed.
- O. M. E. Abdel-Salam, S. M. Y. Morsy and A. A. Sleem, EXCLI J., 2011, 10, 290–302 Search PubMed.
- G. A. Behr, J. C. F. Moreira and B. N. Frey, Oxid. Med. Cell. Longev., 2012, 2012, 609421 Search PubMed.
- M. Herbet, M. Gawrońska-Grzywacz, M. Izdebska and I. Piątkowska-Chmiel, Exp. Ther. Med., 2016, 12, 3440–3444 CrossRef CAS PubMed.
- V. Caiaffo, B. D. R. Oliveira, F. B. de Sá and J. Evêncio Neto, Pharmacol. Res. Perspect., 2016, 4, e00231 CrossRef PubMed.
- M. M. Safhi, H. M. Qumayri, A. U. M. Masmali, R. Siddiqui, M. F. Alam, G. Khan and T. Anwer, Arch. Physiol. Biochem., 2019, 125, 150–155 CrossRef CAS PubMed.
- N. Kolla, Z. Wei, J. S. Richardson and X.-M. Li, J. Psychiatry Neurosci., 2005, 30, 196–201 Search PubMed.
- C. Muraro, M. Dalla Tiezza, C. Pavan, G. Ribaudo, G. Zagotto and L. Orian, Appl. Sci., 2019, 9, 3631 CrossRef CAS.
- S. Sarikaya and I. Gulcin, Curr. Bioact. Compd., 2013, 9, 143–152 CrossRef CAS.
- O. Vašíček, A. Lojek and M. Číž, J. Physiol. Biochem., 2020, 76, 49–60 CrossRef PubMed.
- A. Tutakhail, Q. A. Nazari, S. Khabil, A. Gardier and F. Coudore, Life Sci., 2019, 232, 116508 CrossRef CAS PubMed.
- O. Dalmizrak, K. Teralı, E. B. Asuquo, I. H. Ogus and N. Ozer, Protein J., 2019, 38, 515–524 CrossRef CAS PubMed.
- E. Byeon, J. C. Park, A. Hagiwara, J. Han and J.-S. Lee, Aquat. Toxicol., 2020, 218, 105337 CrossRef CAS PubMed.
- L. Orian and S. Toppo, Free Radical Biol. Med., 2014, 66, 65–74 CrossRef CAS PubMed.
- L. P. Wolters and L. Orian, Curr. Org. Chem., 2015, 20, 189–197 CrossRef.
- M. D. Tiezza, G. Ribaudo and L. Orian, Curr. Org. Chem., 2019, 23, 1381–1402 CrossRef CAS.
- J. T. Rotruck, A. L. Pope, H. E. Ganther, A. B. Swanson, D. G. Hafeman and W. G. Hoekstra, Science, 1973, 179, 588–590 CrossRef CAS PubMed.
- L. Orian, P. Mauri, A. Roveri, S. Toppo, L. Benazzi, V. Bosello-Travain, A. De Palma, M. Maiorino, G. Miotto, M. Zaccarin, A. Polimeno, L. Flohé and F. Ursini, Free Radical Biol. Med., 2015, 87, 1–14 CrossRef CAS PubMed.
- M. Bortoli, M. Torsello, F. M. Bickelhaupt and L. Orian, ChemPhysChem, 2017, 18, 2990–2998 CrossRef CAS PubMed.
- L. Orian, G. Cozza, M. Maiorino, S. Toppo, F. Ursini, M. Maiorino, S. Toppo and F. Ursini, in Glutathione, Series Oxidative stress and disease, ed. L. Flohé, CRC Press, 2018, pp. 53–66 Search PubMed.
- M. Maiorino, V. Bosello-Travain, G. Cozza, G. Miotto, L. Orian, A. Roveri, S. Toppo, M. Zaccarin and F. Ursini, in Selenium: its molecular biology and role in human health, ed. Springer, 4th edn, 2016, pp. 223–234 Search PubMed.
- M. T. Zimmerman, C. A. Bayse, R. R. Ramoutar and J. L. Brumaghim, J. Inorg. Biochem., 2015, 145, 30–40 CrossRef CAS PubMed.
- A. Wendel, M. Fausel, H. Safayhi, G. Tiegs and R. Otter, Biochem. Pharmacol., 1984, 33, 3241–3245 CrossRef CAS PubMed.
- G. Mugesh, W. W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2179 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1419 CrossRef CAS PubMed.
- L. S. Galant, M. M. Braga, D. de Souza, A. F. de Bem, L. Sancineto, C. Santi and J. B. T. da Rocha, Free Radical Res., 2017, 51, 657–668 CAS.
- A. F. de Bem, M. Farina, R. de L. Portella, C. W. Nogueira, T. C. P. Dinis, J. A. N. Laranjinha, L. M. Almeida and J. B. T. Rocha, Atherosclerosis, 2008, 201, 92–100 CrossRef CAS PubMed.
- T. Posser, M. B. Moretto, A. L. Dafre, M. Farina, J. B. T. da Rocha, C. W. Nogueira, G. Zeni, J. dos S. Ferreira, R. B. Leal and J. L. Franco, Chem.-Biol. Interact., 2006, 164, 126–135 CrossRef CAS PubMed.
- G. Ghisleni, L. O. Porciúncula, H. Cimarosti, J. B. T. Rocha, C. G. Salbego and D. O. Souza, Brain Res., 2003, 986, 196–199 CrossRef CAS PubMed.
- G. Ribaudo, M. Bellanda, I. Menegazzo, L. P. Wolters, M. Bortoli, G. Ferrer-Sueta, G. Zagotto and L. Orian, Chem.–Eur. J., 2017, 23, 2405–2422 CrossRef CAS PubMed.
- C. Santi, S. Santoro, C. Tomassini, F. Pascolini, L. Testaferri and M. Tiecco, Synlett, 2009, 2009, 743–746 CrossRef.
- M. Tiecco, L. Testaferri, A. Temperini, F. Marini, L. Bagnoli and C. Santi, Synth. Commun., 1999, 29, 1773–1778 CrossRef CAS.
- M. Tiecco, L. Testaferri, A. Temperini, L. Bagnoli, F. Marini and C. Santi, Synlett, 2001, 1767–1771 CrossRef CAS.
- C. Santi, R. G. Jacob, B. Monti, L. Bagnoli, L. Sancineto and E. J. Lenardão, Molecules, 2016, 21(11), 1482 CrossRef PubMed.
- C. Santi, C. Tomassini and L. Sancineto, Chimia, 2017, 71, 592–595 CrossRef CAS PubMed.
- C. Santi and L. Bagnoli, Molecules, 2017, 22, 2124 CrossRef PubMed.
- P. Frøyen and P. Juvvik, Tetrahedron Lett., 1995, 36, 9555–9558 CrossRef.
- F. D. Lewis, G. D. Reddy, S. Schneider and M. Gahr, J. Am. Chem. Soc., 1991, 113, 3498–3506 CrossRef CAS.
- C. Chen, Y. Huang, Z. Zhang, X.-Q. Dong and X. Zhang, Chem. Commun., 2017, 53, 4612–4615 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. Galano, Phys. Chem. Chem. Phys., 2011, 13, 7178–7188 RSC.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- E. J. Baerends, T. Ziegler, A. J. Atkins, J. Autschbach, D. Bashford, O. Baseggio, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerritger, L. Cavallo, C. Daul, D. P. Chong, D. V. Chulhai, L. Deng, R. M. Dickson, J. M. Dieterich, D. E. Ellis, M. van Faassen, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. Goez, A. W. Götz, S. Gusarov, F. E. Harris, P. van den Hoek, Z. Hu, C. R. Jacob, H. Jacobsen, L. Jensen, L. Joubert, J. W. Kaminski, G. van Kessel, C. König, F. Kootstra, A. Kovalenko, M. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, S. M. Morton, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, M. Pavanello, C. A. Peeples, P. H. T. Philipsen, D. Post, C. C. Pye, H. Ramanantoanina, P. Ramos, W. Ravenek, J. I. Rodríguez, P. Ros, R. Rüger, P. R. T. Schipper, D. Schlüns, H. van Schoot, G. Schreckenbach, J. S. Seldenthuis, M. Seth, J. G. Snijders, M. Solà, M. Stener, M. Swart, D. Swerhone, G. te Velde, V. Tognetti, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and A. L. Yakovlev, ADF2018, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2018 Search PubMed.
- N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS.
- N. C. Handy and A. J. Cohen, J. Chem. Phys., 2002, 116, 5411–5418 CrossRef CAS.
- F. Zaccaria, L. P. Wolters, C. Fonseca Guerra and L. Orian, J. Comput. Chem., 2016, 37, 1672–1680 CrossRef CAS PubMed.
- M. Bortoli, L. P. Wolters, L. Orian and F. M. Bickelhaupt, J. Chem. Theory Comput., 2016, 12, 2752–2761 CrossRef CAS PubMed.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 CrossRef CAS.
- M. Franchini, P. H. T. Philipsen and L. Visscher, J. Comput. Chem., 2013, 34, 1819–1827 CrossRef CAS PubMed.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS.
- M. Abid and A. Azam, Bioorg. Med. Chem., 2005, 13, 2213–2220 CrossRef CAS PubMed.
- K. Tóth, N. I. Wenzel, N. Chavain, Y. Wang, W. Friebolin, L. Maes, B. Pradines, M. Lanzer, V. Yardley, R. Brun, C. Herold-Mende, C. Biot and E. Davioud-Charvet, J. Med. Chem., 2010, 53, 3214–3226 CrossRef PubMed.
- D. M. Perrine, N. R. Sabanayagam and K. J. Reynolds, J. Chem. Educ., 1998, 75, 1266 CrossRef CAS.
- J. C. Liou, S. S. Badsara, Y. T. Huang and C. F. Lee, RSC Adv., 2014, 40, 41237–41244 RSC.
- A. Clemenceau, Q. Wang and J. Zhu, Chem.–Eur. J., 2016, 22, 18368–18372 CrossRef CAS PubMed.
- K. B. Sharpless, M. W. Young and R. F. Lauer, Tetrahedron Lett., 1973, 14, 1979–1982 CrossRef.
- A. Krief, W. Dumont and A. F. De Mahieu, Tetrahedron Lett., 1988, 29, 3269–3272 CrossRef CAS.
- A. J. Waring and J. H. Zaidi, J. Chem. Soc., Perkin Trans. 1, 1985, 631 RSC.
- H. J. Reich, S. Wollowitz, J. E. Trend, F. Chow and D. F. Wendelborn, J. Org. Chem., 1978, 43, 1697–1705 CrossRef CAS.
- K. L. Nuttall, Ann. Clin. Lab. Sci., 2006, 36, 409–420 CAS.
- F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073–1091 RSC.
- D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409–8411 CrossRef CAS PubMed.
- B. Cerra, F. Mangiavacchi, C. Santi, A. M. Lozza and A. Gioiello, React. Chem. Eng., 2017, 2, 467–471 RSC.
- L. Syper and J. Młochowski, Tetrahedron, 1987, 43, 207–213 CrossRef CAS.
- J. Młochowski and L. Syper, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2001 Search PubMed.
- D. De Filippo and F. Momicchioli, Tetrahedron, 1969, 25, 5733–5744 CrossRef CAS.
- L. Wang, K. Zhu, W. Cao, C. Sun, C. Lu and H. Xu, Polym. Chem., 2019, 10, 2039–2046 RSC.
- A. Tepecik, Z. Altin and S. Erturan, J. Autom. Methods Manage. Chem., 2008, 2008, 1–6 CrossRef PubMed.
- K. Bierla, S. Godin, R. Lobinski and J. Szpunar, TrAC, Trends Anal. Chem., 2018, 104, 87–94 CrossRef CAS.
- M. Bortoli, F. Zaccaria, M. Dalla Tiezza, M. Bruschi, C. Fonseca Guerra, F. M. Bickelhaupt and L. Orian, Phys. Chem. Chem. Phys., 2018, 20, 20874–20885 RSC.
- J. Andersen, N. Stuhr-Hansen, L. G. Zachariassen, H. Koldsø, B. Schiøtt, K. Strømgaard and A. S. Kristensen, Mol. Pharmacol., 2014, 85, 703–714 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03509b |
| This journal is © The Royal Society of Chemistry 2020 |