DOI:
10.1039/D0RA03314F
(Review Article)
RSC Adv., 2020,
10, 21662-21685
Recent advances in the design of cathode materials for Li-ion batteries
Received
13th April 2020
, Accepted 2nd June 2020
First published on 8th June 2020
Abstract
The Li-ion battery (LIB) industry has rapidly developed and dominates the market of electric vehicles and portable electronic devices. Special attention is devoted to achieving higher power and energy densities, along with enhancing safety and reducing cost. Therefore, critical insights should be made on the understanding of the behavior of the components of LIBs under working conditions in order to direct future research and development. The present review discusses the literature on the properties and limitations of different cathode materials for LIBs, including layered transition metal oxides, spinels, and polyanionic positive electrode materials, with critical insights on the structural, thermal, and electrochemical changes that take place during cycling. Besides, the strategies and techniques capable of overcoming current limitations are highlighted.
 Nourhan Mohamed | Nourhan Mohamed received her M.Sc. degree in Sciences, Technologies, Health from Picardie Jules Verne University. Her master's program was a joint Erasmus program in materials for energy storage and conversion “MESC”. She joined the Energy Materials Laboratory at AUC for a year as a Research Assistant. Her research is focused on positive electrode materials for Li-ion batteries. |
 Nageh K. Allam | Nageh Allam received his PhD in Materials Science and Engineering from The Pennsylvania State University. He joined Georgia Institute of Technology as a postdoctoral fellow then Massachusetts Institute of Technology as a Research Scholar. He moved to The American University in Cairo in September 2011 where he is now a professor of Materials Science and Engineering. His research focuses on the fabrication of well-designed nanostructured materials with composition, size, and shape control for use in energy conversion and storage, electrochemical sensors, and biomedical applications, among others. His research comprises both experimental and theoretical activities. |
1 Introduction
The pressing demand to introduce alternative energy sources to fossil fuels and minimize CO2 emissions generates considerable recent research interest in the development of renewables along with energy storage systems. Shortly, the Earth is expected to run out of petroleum resources as they are being depleted at a fast rate. In addition, avoiding petroleum resources is necessary because of their negative environmental impacts, such as the water and air pollution that led to global warming problems. At the global level, renewable sources show a great theoretical ability to cover the whole human need for energy. For instance, Earth receives 1.2 × 105 TW of solar power, which can cover the annual worldwide need for energy in almost one hour. Despite the current limitations in the properties of materials that lead to inefficient collection and conversion of light, the primary current focus is on how to progress rapidly towards highly functional materials to satisfy the ever-increasing energy demand.
Energy storage technologies are divided into stationary and portable systems; both are of great importance, and each type is at a different stage of development. In multiple stationary applications, grid-connected energy storage systems have been deployed to facilitate the integration of renewable power sources due to their variable nature. Energy storage systems are connected to the grid to provide sustainability by storing energy during the low-energy demand time and supply it during the high-energy demand time. Nowadays, the global energy storage is dominated by one technology, the pumped hydropower, for grid applications, which accounts for 95% of the worldwide energy storage. The rest 5% comprise mainly of thermal, mechanical, and electrochemical storage. In comparison, a project of Li-ion batteries contributes 24 MW that is as low as 0.01% of the overall storage, as shown in Fig. 1.
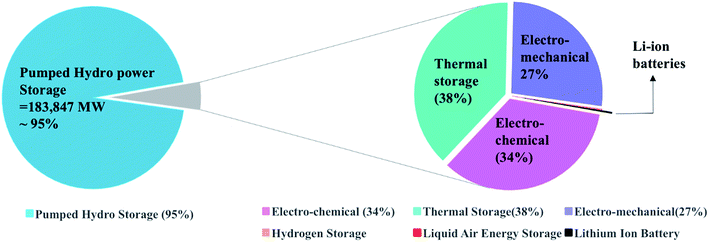 |
| | Fig. 1 Worldwide energy storage projects contribution to the grid applications. Data is extracted from US-DOE.3 | |
Advances in renewable energy require modernization of the electricity storage systems, including electrochemical capacitors, lead-acid batteries, high-temperature sodium batteries, lithium-ion batteries, and redox flow batteries. In this review, we focus mainly on Li-ion batteries because of their superior performance in comparison with other conventional battery types. Li-ion batteries dominate the portable devices industry due to their prime properties such as high-energy-density, high charge/discharge rate, low maintenance, and long-lifetime, which make them the most suitable candidates for a plethora of applications. Therefore, they met the market requirements, and their sales increased significantly between 2008 and 2014 from 0.1 to 22.5 billion dollars in the electric vehicles sector and from 9.1 to 31 billion dollars in the total market.1,2
Li-batteries have been widely adopted because Li is the lightest metal (6.94 g mol−1) with the smallest radius that can provide high gravimetric and volumetric capacity, leading to a significant weight and volume reduction of the battery. Furthermore, Li exhibits the lowest reduction potential (−3.04 V versus SHE), allowing the cell to deliver the highest possible potential resulting in a decrease in the number of cells required to operate a device. However, several safety problems caused the withdrawal of Li metal batteries from the market after some catastrophic accidents because of the high reactivity of the Li metal and the growth of Li dendrite, which results in short circuits. Therefore, Li metal anode was replaced by safer alternatives such as intercalation, alloying, and conversion materials. Consequently, much research has focused on the chemical, thermal, and structural stability of the materials for Li-ion batteries and how to introduce practical solutions to their well-recognized safety problems. These problems are related to the reactions that can take place under extreme conditions such as high over-potential, electrolyte decomposition, SEI decomposition, cathode material decomposition, and generation of flammable gases. The safety stands as an obstacle to the introduction of new high energy cathode materials because they work outside the stability window of the available electrolytes. Improving the performance, along with the cost and safety, are the key factors to expand the Li-ion battery applications significantly. A significant interest in Li-ion batteries is given to the cathode materials and how to improve its electrochemical performance along with preserving the mechanical, electrochemical, and chemical stability of the materials upon cycling. Because the performance of the battery, including cell potential, energy density, power density, lifetime, and safety in addition to cost, depends to a great extent on the cathode material. For instance, high energy can be obtained from a battery by increasing the intercalation voltage (cathode material type) or the amount of Li+ that can participate in the electrochemical reaction (capacity). In this review paper, we focus on different types of cathode materials and discuss their electrochemical properties considering their structure and morphology as well as thermal and electrochemical performance.
2 Li-ion batteries (LIBs)
A Li-ion module consists of several cells connected into packs to deliver the required voltage, power, and energy. LIBs convert chemical energy into electricity via a reversible electrochemical reaction. They consist mainly of anode and cathode, which are electronically isolated by an electrolyte. The anode is the negative electrode, which is oxidized during the electrochemical reaction to donating electrons to the external circuit. The cathode is the positive electrode, which is reduced during the electrochemical reaction and accepts electrons from the external circuit. The electrolyte is a solid or a liquid medium that allows the Li ions to move between the anode and cathode and force the electrons to move through the external circuit. Therefore, electrolyte should exhibit high ionic and low electronic conductivity. Moreover, the Fermi level of the anode should be at lower energy than the LUMO of the electrolyte to prevent the electrolyte reduction, while the Fermi level of the cathode should be at higher energy than the HUMO of the electrolyte to prevent the electrolyte oxidation. During the first charge, the electrolyte undergoes a reduction reaction at the surface of the anode forming an irreversible passive layer, known as solid electrolyte interphase (SEI). It is a complicated layer composed of organic and inorganic components that prevent the further decomposition of the electrolyte upon cycling, but it results in an irreversible capacity loss. The performance of the battery is dependent on the morphology, composition, and thickness of this layer.4 If this protective layer is damaged, the decomposition of the electrolyte will continue again, leading to fast heating and undesirable chemical reactions that are hazardous. There are several reported mechanisms for the formation and stabilization of the SEI on the surface of both anode and cathode.5–8
3 Challenges and trend in cathode materials for Li-ion batteries
The development of the cathode materials for Li-ion batteries remains challenging because the existing materials such as layered transition metals oxides, olivines, or spinel all show upsides and downsides. For example, the layered oxides such as LiCoO2 suffers from the instability that limits their potential window and capacity. Moreover, the toxicity and the high production cost of the Co-based materials are undesirable. On the other hand, Li-rich layered oxides (Li1+xM1−xO2), where M is a mixture of transition metals (Ni, Mn, and Co), is promising as they can work at high discharge voltages of >4.5 V and deliver high specific capacities. However, they suffer from large voltage decay during cycling and high irreversible capacity loss at the first cycles, which limit their use. Moreover, olivine materials such as LiFePO4 are competitive candidates surpassing the stability of layered oxides at elevated temperatures due to their high thermal and structural stability. However, their low electronic and ionic conductivity stand as an obstacle to expand their use for high energy Li-ion batteries. Several steps forward have been made in the development of cathode materials for Li-ion batteries to overcome current challenges. First, investigating the changes in the electrochemical performance inside the cathode during cycling will lead to understanding the origin of the malfunction of the materials under working conditions. The strategies considered to improve these materials should address the problems related to the electrochemical performance side by side with the safety and cost issues. These strategies mainly include designing nanostructured materials with controlled morphology, introducing artificial defects in the nanostructured materials, using nanostructured complexes by doping or surface coating, and surface modification that is indispensable to minimize the undesirable side reaction at the electrolyte–cathode interface. Those are critical factors in jeopardizing the electrode performance. Also, there is a great interest in identifying new cathode materials that can fulfill the required balance between the performance and environmental requirements. In this review, we compare the different cathode materials that have been widely used in Li-ion batteries and analyze their performance.
4 Layered transition metal oxides positive electrode materials
The reason for the continued attention to transition metal oxides LiMO2 (M = Co, Mn, Ni) as electrode materials is their high Li+ mobility in 2-dimensional space, thanks to its layered structure, as shown in Fig. 2. The crystallographic framework of Layered LiMO2 (O3 type) is α-NaFeO2-like structure, which consists of a cubic close-packed oxygen array occupying the 6c sites, where Li and M ions occupy the 3a and 3b sites, respectively. M and Li ions occupy alternate layers in the octahedral sites along the (111) plane forming ABC stacking sequence. The cell parameters of LiMO2 (M = Co and Ni) are shown in Table 1.
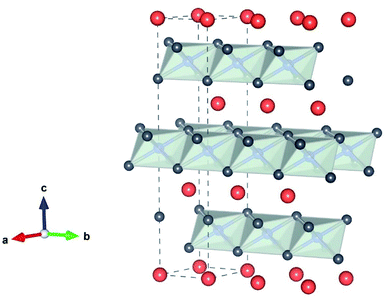 |
| | Fig. 2 The crystallographic framework of the layered LiMO2 (M is Co or Ni) consisting of MO6 octahedra (gray) and Li+ (orange) “by VESTA”. | |
Table 1 Cell parameters of LiMO2 (M is Co or Ni)
| LiMO2 |
a (Å) |
c (Å) |
c/a |
V (Å3) |
Ref. |
| LiCoO2 |
2.8157(3) |
14.0517(3) |
4.99 |
96.48(3) |
9 |
| LiNiO2 |
2.8898(2) |
14.2212(2) |
4.92 |
102.85(6) |
4.1 LiCoO2
LiCoO2 represents a significant advance in the history of rechargeable Li-ion batteries, as it was the first commercialized positive electrode material by Sony in 1991. Sony combined the LiCoO2 cathode and carbon anode to produce the first successful rechargeable Li-ion battery. From the electrochemistry point of view, LixCoO2 is a very attractive material because it exhibits a high theoretical capacity of 274 mA h g−1 and high energy density. However, its performance is strongly affected by structural and thermal instability at high voltage, and high temperature, as well as fast capacity, fade at the high current rate or deep cycling. During charging, several phase transitions take place at different stages of the de-intercalation of Li+. Fig. 3 shows a typical charge/discharge behavior of LixCoO2 as a function of the amount of x. The large plateau at 3.93 V vs. Li+/Li0 corresponds to a first-order transition (H1 ↔ H2). The coexistence of two phases between the lithium concentrations x = 0.75 and 0.93 is accompanied by the transition from a semiconductor to the metal, which facilitates the charge transfer. In the x range between 0.75 and 0.25, two order/disorder transitions were observed from hexagonal to monoclinic, then from monoclinic to hexagonal, which correspond to the plateaus b and c. Therefore, almost one-half of the lithium ions can be cycled reversibly in and out of the compound corresponding to the composition range between x = 1 and 0.5. Consequently, a practical specific capacity of 140 mA h g−1 is obtained at a charging voltage of 4.2 V vs. Li/Li+ to avoid the structural rearrangement of the material.10,11
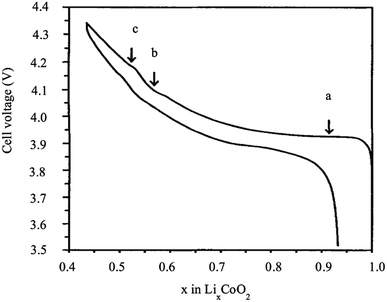 |
| | Fig. 3 Cell voltage of LixCoO2 as a function of x.12 | |
It was traditionally accepted that the positive charge decrease by Li ions removal from LixCoO2 is compensated by the increased charge of transition metal ions. At the same time, O2− electronic state is fixed to keep the electro-neutrality of the material. But sensitive techniques to the oxidation state, spin state, bond covalence, and the electronic structures such as EELS and XAS suggest that both oxygen and transition metal ions experience an electronic change, and both participate to the electrochemical reaction. The oxygen K-edge XAS for LixCoO2 shows significant change with respect to Li-ion content compared to the Co LII,III-edge XAS, suggesting that the electronic structure of the oxygen ions is more influenced than Co ions by the Li+ removal. In addition, the capacity fading upon cycling is explained with the change in the Co–O covalence that changes with the Li content as a result of the local structural distortion of CoO6 octahedra that accompanies oxygen and cobalt orbitals re-hybridization.13
Degradation has a significant contribution to the performance decay of LiCoO2. Bulk structure degradation of LiCoO2 is the main reason for capacity loss at high voltage. Jiang et al. observed that by increasing the cutoff potential from 4.2 to 4.7 V, Li concentration was significantly decreased in the bulk.14 Besides, the samples with low Li content suffered from thermal instability when cycled to 4.7 V. The STEM-HAADF images, Fig. 4, show bright strips, where in the middle, the O1 phase is identified, and the density of these bright strips increases with increasing the cutoff voltage and the number of cycles. This reveals that there are uneven lithiation and de-lithiation behaviors in the bulk material. Besides structure degradation, interfacial degradation is one of the mechanisms assigned to the failure of layered cathode materials. It originates from the surface phase transformation layer and the cathode electrolyte interface (CEI) layer. The surface degradation is enhanced by increasing the cutoff potential. Fig. 5 shows that the thickness of the degradation layer after 50 cycles is about four times higher in the samples cycled to a cutoff voltage of 4.7 V compared to that cycled to 4.2 V.
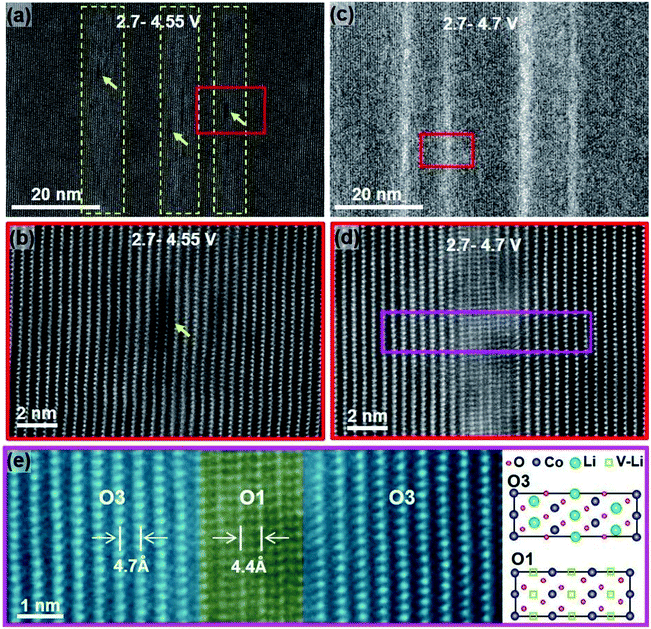 |
| | Fig. 4 STEM-HAADF images showing the degradation induced from cycling up to high cutoff voltages for 50 cycles. (a) STEM-HAADF image showing the bright stripes (highlighted by dashed yellow frames) and the dark pits (yellow arrows) on sample cycled between 2.7–4.5 V, (b) High-resolution lattice image to show lattice images in (a), (c) STEM-HAADF image showing the bright stripes formed in the sample cycled between 2.7–4.7 V, (d) lattice image from the region marked in (c), and (e) enlarged lattice image from the pink region in (d).14 | |
 |
| | Fig. 5 Effect of high charge cutoff voltage on the thickness of the surface degradation layer.14 | |
Tremendous efforts have been devoted to improving the functional performance of Li1−xCoO2 at high voltage and elevated temperatures. Surface coating, doping with foreign elements, and synthesis by wet chemistry methods such as sol–gel are traditional strategies that have been explored to improve the materials' properties. There are several methods for surface coating, including wet chemistry (sol–gel), chemical polymerization routes, and deposition techniques (sputtering, pulsed laser deposition, atomic layer deposition, chemical vapor deposition).15 The properties of the coated material depend on the homogeneity and thickness of the coating layer. Insulator metals oxides, such as AlO2, ZrO2, and AlPO4, show outstanding improved performance in terms of capacity retention and rate capability at high voltage compared to the bare LiCoO2. The enhanced electrochemical performance is attributed to (1) the suppression of cobalt dissolution in the electrolyte, (2) the prevention of the direct contact between the cathode and the liquid electrolyte by the inactive coating layer,16 and (3) cation diffusion to form Li–M–O (M = Al, Co) on the surface during calcination that can suppress the phase transitions and hence decrease the capacity fading especially at high potentials.17 Recently, Yano et al.18 synthesized 0.5 wt% AlO2-coated LiCoO2 by the sol–gel method. It shows electrochemical activity at potential > 4.2 V vs. Li/Li+ and an initial capacity of 247 mA h g−1 at the first cycle and preserved ∼82.6% of its initial capacity after 20 cycles. The improved performance is attributed to the formation of a solid solution surface phase of LiAlO2 and LiCoO2 by the diffusion during the calcination after the sol–gel coating. The coated LiCoO2 shows very small phase transition upon cycling as Al3+ is electrochemically inactive and shares the octahedral sites with Co ions in the MO6, resulting in a difficultly for both Li and O ions to leave the LiAlO2 framework and thereby preserving the surface structure. Table 2 lists several examples of the improved electrochemical performance at high cutoff voltage by coating. Moreover, conductive oxides or Li ion conductors that show low interfacial resistance and maintain high ionic conductivity such as Al–ZnO, lithium phosphorus oxynitride and lithium tungsten oxide (LWO) are excellent alternatives to the insulator metal oxide coating.19,20
Table 2 The performance of coated LiCoO2 by different materials and different techniques
| Surface coating material |
Coating technique |
Electrochemical performance |
Ref. |
| Al2O3 |
Vapor-assisted hydrolysis |
165.5 mA h g−1 @ 360 mA g−1, voltage range (3–4.5 V vs. Li/Li+), capacity retention 98.6% (180 cycle) |
21 |
| Al2O3 |
Atomic layer deposition |
175.72 voltage range (2.5–4.5 V vs. Li/Li+), capacity retention 94.9% (50 cycle) |
22 |
| AlWxFy |
Atomic layer deposition |
180.62 voltage range (2.5–4.5 V vs. Li/Li+), capacity retention 94.99% (50 cycle) |
| AlF3 |
2 ALD (LCO-2% MWCNT-5% NCF) paper electrode |
216 mA h g−1, cutoff voltage 4.7 V, capacity retention (75.7%, 70% after 100 and 160 cycle) |
23 |
| (1.0 wt%) AlPO4 |
Solution |
233 mA h g−1 (cutoff voltage = 4.8), capacity retention ∼64% (50 cycle) |
24 |
| (2 wt%) AlPO4 |
Freeze drying method |
∼210 mA h g−1 (2.75–4.55 V), capacity retention 86.1% (50 cycle) |
19 |
| (2 wt%) LiAlPO3.93F1.07 |
Freeze drying method |
206 mA h g−1 (2.75–4.55 V), capacity retention 91.7% (50 cycle) |
| (20 nm) Al2O3-doped ZnO |
Magnetron sputtering coating |
193 mA h g−1 voltage range (3–4.5), capacity retention 90% (150 cycle) |
25 |
| (2 wt%) CeO2 |
Sol–gel |
138.7 mA h g−1 (3–4.4 V), ∼89% (50 cycle) |
26 |
| (2 wt%) CeO2 |
Sol–gel |
150.37 mA h g−1 (3–4.5), capacity retention ∼76% (50 cycle) |
4.2 LiNiO2
LiNiO2 is considerably less expensive, shows an outstanding attainable capacity of 240 mA h g−1,27 and with less toxicity than LiCoO2. However, it is relatively difficult to synthesize stoichiometric LiNiO2 because of the preferable cation mixing between Li and Ni ions to form Li1zNi1+zO2.28,29 The excess of Ni ions sitting in the lithium layers hinders the smooth motion of Li ions during cycling, resulting in poor performance. Moreover, the material goes through several structural transformations upon cycling. For the range 1 ≥ x ≥ 0.25, LixNiO2 shows three single-phases; a rhombohedral phase (H1) when 1 > x > 0.75, a monoclinic phase when 0.75 ≥ x ≥ 0.45, a rhombohedral phase (H2) when 0.45 > x > 0.25, then a two-phase region (H2 + H3).30,31 In addition, LixNiO2 is known for its high thermal instability as it decomposes and liberates oxygen at low temperatures compared to LixCoO2 as shown in Fig. 6. The O2 evolution was found to be dependent on the degree of de-lithiation (x). Lee et al.32 synthesized and studied [Li0.948Ni0.052]3a[Li0.003Ni0.997]3bO2. When x ≥ 0.5, the material decomposes into layered LiNiO2 and spinel LiNi2O4 (Fd3m) in the range between 180 and 250 °C. In the composition range 0.5 > x ≥ 0.2, the material is completely transformed into spinel. Further increasing the temperature, the material turns into rock salt structure (Fm3m), with all transformations accompanied by oxygen evolution. It has been suggested that the exothermic reaction mechanism is driven by the phase transformations and oxygen evolution, which are originally due to the random mixing between Li and Ni ions.33,34 Recently, Yoon et al.27 successfully suppressed the H1 to H2 phase transition by preparing a compact geometry of LiNiO2 in which particles of 200 nm are packed tightly in a spherical particle of 10 microns. Such geometry minimized the contact between the electrolyte and the surface of the electrode resulting in better structure stability and capacity of 179 at 0.1 °C rate, 95% of the capacity was retained after 100 cycles when the cutoff voltage was 4.1 V. Similar to LiCoO2, better thermal, structural, and electrochemical stability is obtained by surface coating with ZrO2 or SiO2, Co and (Mn, Co) as shown in Table 3.
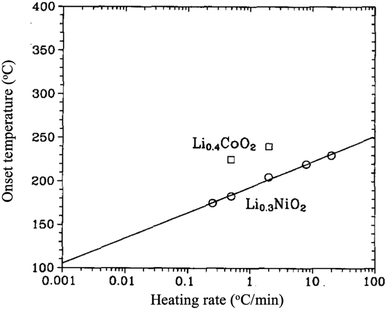 |
| | Fig. 6 The onset temperature for oxygen release for Li0.3NiO2 and Li0.4CoO2 as a function of heating rate.35 | |
Table 3 LiNiO2 improved performance by coating Co and (Mn, Co) coating found to form a solid solution
| Surface coating material |
Technique |
Electrochemical performance |
Ref. |
| ZrO2 |
Sol–gel |
190 mA h g−1 @ 36 mA g−1, voltage range (4.3–2.75 V) |
36 |
| Capacity retention 98% (70 cycle) |
| SiO2 (2 wt%) |
Sol–gel |
212 mA h g−1, @ 1C (3–4.5 V) |
37 |
| Capacity retention (92.02% after 60 cycles) |
| Co (content = 0.05) |
Solid state synthesis |
180.62 mA h g−1 @ 1C voltage range (2.75–4.2 V) |
38 |
| Capacity retention 96.8% (50 cycle) |
| Co and Mn |
Solid state synthesis |
181.3 mA g−1 voltage (3–4.35 V vs. Li/Li+) |
39 |
| Capacity retention (91.1 after 50 cycles) |
4.3 LiCo1−xNixO2
LiCo1−xNixO2 was suggested as an alternative to LiNiO2 to merge the relatively better stability of LiCoO2 and the high capacity of LiNiO2. The LiCo1−xNixO2 solid solution can be formed in the whole x range. The lattice parameters and cell volume increase linearly while c/a decreases obeying Vegard's law when Ni ions are partially substituted for Co ions.40 The higher the Co ions content in LiCo1−xNixO2, the more stable the structure is. Co substitution suppresses the non-stoichiometry of Li1−zNi1+zO2 from z = 0.04 to 0.01 when Co is increased from 0 to 0.2, and a pure stoichiometric 2D structure of LiCo1−xNixO2 was observed when Co content reached 0.3.41 The best performance of LiCo1−xNixO2 was obtained for 0.1 ≥ x ≥ 0.3.42 Differential scanning calorimetry for LiCo1−xNixO2 proved relatively higher safety and thermal stability than LiNiO2 at x = 0.1 ≥ x ≥ 0.3 at cutoff voltages of 4.2 and 4.3 V vs. Li/Li+.43 Upon increasing the Co content, the exothermic reaction shifts toward higher temperatures, and its enthalpy decreases. In addition, when the cell is charged to higher potential, the exothermic event takes place at a lower temperature in agreement with the fact that the lower the Li+ content, the lower the thermal stability of these oxide materials is. Recently, Xie et al.44 doped Li(Co,Ni)O2 with Mg ions to obtain highly structural, thermal, and electrochemical stable structure. Note that 80.1% of its initial capacity (214 mA h g−1) was maintained after 500 cycles compared to 56.3% without Mg ions. The Mg2+ prevented the Li+/Ni2+ site mixing, suppressed the migration of Ni ions to the Li layers, and showed a pillar effect in the position of the Li ions, which improved the thermal stability significantly even at such a high voltage as 4.7 V vs. Li+/Li.45 Table 4 summarizes the thermal behavior of different transition metal oxides.
Table 4 Thermal behavior of different transition metal oxides
| Composition |
Decomposition temperature (°C) |
Cutoff voltage (V) |
Heat generated (J g−1) |
Ref. |
| LiNiO2 |
181.2 |
4.3 |
1965 |
46 |
| LiCoO2 |
212 |
4.5 |
2785 |
47 |
| LiNi0.9Co0.07Mg0.03O2 |
243.7 |
4.3 |
543.2 |
45 |
| LiNi0.9Co0.07Mg0.03O2 |
237.2 |
4.5 |
596.8 |
45 |
| LiNi0.9Co0.07Mg0.03O2 |
211.6 |
4.7 |
667.5 |
45 |
| Li[Ni0.95Co0.025 Mn0.025]O2 |
185.5 |
4.3 |
1609 |
46 |
| LiNi0.81Co0.19O2 |
212.3 |
4.3 |
1095.2 |
48 |
| LiNi0.80Co0.19Mg0.01O2 |
219.6 |
4.3 |
845.7 |
48 |
| LiNi0.78Co0.19Mg0.01Al0.02O2 |
221.9 |
4.3 |
394.6 |
48 |
| Li[Ni0.6Co0.2Mn0.2]O2 |
201 |
4.3 |
1670 |
49 |
| Li[Ni0.9Co0.05Mn0.05]O2 |
272 |
4.3 |
769 |
49 |
| Li[Ni1/3Co1/3Mn1/3]O2 |
261.5 |
4.5 |
1998 |
47 |
4.4 LiMnO2
Manganese oxides are promising cathode materials due to their less toxicity and lower cost than cobalt and nickel oxides. Ion exchange is a successful method to synthesize layered LiMnO2 by exchanging Na in NaMnO2 with Li.50 Truly layered LiMnO2 with α-NaFeO2 like structure is thermodynamically unstable because of the Jahn–Teller effect that distorts the structure to less symmetry orthorhombic (o-LiMnO2) with the space group Pmmn or monoclinic (m-LiMnO2) with the space group C2/m.51 Monoclinic LiMnO2 is thermodynamically less stable than the spinel and orthorhombic lithium manganese oxides because of the antiferromagnetic interactions between Mn3+ distorting the MnO6 octahedra.52 This results in a transition from the initial monoclinic to hexagonal at the first charge then to spinel gradually upon cycling.53 This transformation takes place through the migration of 25% of Mn ions from the octahedral sites in the transition metal layer to the lithium layer, and the Li ions move to tetrahedral sites. Stabilization of the layered structure can be obtained by partially substituting Mn ions by antiferromagnetic elements such as Al or Cr.54,55 Many studies have been carried out on the structural and electrochemical properties of the Li–Ni–Mn–O and Li–Ni–Mn–Co–O systems as alternatives to the single oxides.
4.5 LiNi0.5Mn0.5O2
LiNi0.5Mn0.5O2 is a 4 V cathode material with a theoretical capacity of 280 mA h g−1. During charging, Ni2+ is oxidized to Ni4+ through two stages, while Mn4+ is electrochemically inactive and remains unchanged.56,57 The presence of the stable Mn4+ octahedral ions stabilizes the LiNi0.5Mn0.5O2 structure during Li+ removal, thus avoiding the Jahn–Teller distortion that is associated with the presence of Mn3+. There are different structural models that explain the composition of the transition metal layer in LiNi0.5Mn0.5O2; the most stable and lowest energy structures are shown in Fig. 7. The zigzag ordering of Ni2+ and Mn4+ is preferred if there is no Li+/Ni2+ mixing, while the flower order is preferred if there is Li+/Ni2+ mixing. The flower structure is ordered in such a way that the Li+ is located in the center of an Mn4+ hexagon surrounded by Ni2+.58,59 LiNi0.5Mn0.5O2 is indexed in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, with around 8 to 10% anti-cite mixing between Li and Ni usually takes place.60 Li+ prefers to occupy positions close to Mn4+ ions than Ni2+ in the transition metal layer.58 Therefore, Li, Ni, and Mn ions are not randomly distributed in the transition metal layer. Single crystal selected area electron diffraction patterns of pristine LiNi0.5Mn0.5O2 reveal the presence of superlattice reflections indicating the long-range ordering of Li-rich and Li-deficient sites in the transition metal layer. The superlattice reflections are indexed in
m space group, with around 8 to 10% anti-cite mixing between Li and Ni usually takes place.60 Li+ prefers to occupy positions close to Mn4+ ions than Ni2+ in the transition metal layer.58 Therefore, Li, Ni, and Mn ions are not randomly distributed in the transition metal layer. Single crystal selected area electron diffraction patterns of pristine LiNi0.5Mn0.5O2 reveal the presence of superlattice reflections indicating the long-range ordering of Li-rich and Li-deficient sites in the transition metal layer. The superlattice reflections are indexed in 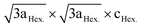 and correspond to a trigonal structure with the space group P3112.61,62 Investigation of the local structure upon charging suggested the disappearance of Li+ located in the transition metal layer very early during the first charge.63 Li+ migrates to the tetrahedral sites that share faces with vacant octahedral sites in the transition metal layer. The presence of Li in the tetrahedral sites reflects negatively on the electrochemical performance of the material. Because Litet+ is very energetically stable and requires high potential to be removed, which decreased the capacity obtained in the practical voltage range.64 Regarding the Ni ions in the Li layer, by charging above 4.6 V, 75% of NiLi ions migrate to vacant Li positions in the transition metal (TM) layer.65 The migration of Ni ions to the TM layer is partly a reversible process and reflects on the weaken or disappearance of the
and correspond to a trigonal structure with the space group P3112.61,62 Investigation of the local structure upon charging suggested the disappearance of Li+ located in the transition metal layer very early during the first charge.63 Li+ migrates to the tetrahedral sites that share faces with vacant octahedral sites in the transition metal layer. The presence of Li in the tetrahedral sites reflects negatively on the electrochemical performance of the material. Because Litet+ is very energetically stable and requires high potential to be removed, which decreased the capacity obtained in the practical voltage range.64 Regarding the Ni ions in the Li layer, by charging above 4.6 V, 75% of NiLi ions migrate to vacant Li positions in the transition metal (TM) layer.65 The migration of Ni ions to the TM layer is partly a reversible process and reflects on the weaken or disappearance of the 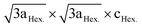 c supercell. Simply, in other words, the Ni2+/Li+ mixing was decreased by cycling compared to pristine LiNi0.5Mn0.5O2.
c supercell. Simply, in other words, the Ni2+/Li+ mixing was decreased by cycling compared to pristine LiNi0.5Mn0.5O2.
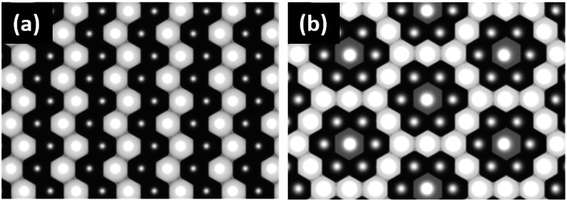 |
| | Fig. 7 (a) TM layer ordering of the flower structure and (b) transition metal layer ordering of the zigzag structure.59 | |
4.6 LiNixCoyMn1−x−yO2
The mixed transition metal oxides family LiNixCo1−2xMnxO2 (ref. 66) was proposed to avoid the shortcoming of each individual oxide such as low stability, safety, and specific capacity. The cation mixing in this compound is lower than at LiNi0.5Mn0.5O2, and it increases by increasing the Ni content. LiNi1/3Co1/3Mn1/3O2 was found to be promising among the series of materials in the composition range 0 ≤ x ≤ ½. LiNi1/3Co1/3Mn1/3O2 showed a specific capacity of 200 mA h g−1 when charged up to 4.6 V vs. Li/Li+. It crystallizes in the typical layered structure of α-NaFeO2 with Rm space group with the electronic charge of Co, Ni, and Mn being +3, +2, and +4, respectively. Several authors debated its electronic structure upon the de-intercalation of lithium. Tsai et al. and Yoon et al. reported that its electrochemical performance is attributed to the redox reactions Ni2+/Ni3+ at the surface and Ni3+/Ni4+ in bulk, while the oxidation states of Mn4+ and Co3+ remain unchanged.67,68 In addition, K-edge XAS of the oxygen ions shows that it plays an essential role in the charge compensation in its site during Li+ de-intercalation. On the other hand, Kim et al.69 reported that the charge compensation during charging is due to the redox Ni2+/Ni4+ and Co3+/Co4+ while Mn4+ is stable.
4.7 Li-rich cathode materials
4.7.1 Li2MnO3. Li2MnO3 is one of the xLi2MnO3·(1 − x)LiMO2 family of materials when x = 0. It possesses layered structure Li3a[Li1/3Mn2/3]3bO2 with C2/m space group. In Li2MnO3, the Mn4+ is electrochemically inactive because it cannot be oxidized to a higher oxidation state. Surprisingly, Li+ could be inserted and extracted from Li2MnO3 when activated at 4.5 V.70–72 This material has promoted many debates on the mechanism of the electrochemical behavior and Li+ extraction. During the first charge, the Li+ extraction was found to be accompanied by oxygen evolution, leading to the removal of Li2O, resulting in electrochemical activation of Li2MnO3.73 Robertson et al. suggested that there is a proton exchange with Li+ to form Li2−xHxMnO3,74 which can take place after the production of hydrogen by electrolyte oxidation. The presence of H in the oxide layers forming O–H–O bond leads to a change to a more stable stacking sequence from O3(ABCABC) to P3(ABBCCA). Another proposed mechanism based on DFT calculations shows that the electrochemical behavior is attributed to the charge compensation by the oxidation of anion “oxygen”.75 However, the thermodynamically unstable localized holes at the beginning of the oxidation result in the evolution of oxygen. This study showed that the structural transformation to spinel is driven by the oxygen dimerization, which facilitates the Mn ions migration onto octahedral sites in the vacated lithium layers. Rana et al. suggested that both oxygen oxidation and proton exchange are responsible for the electrochemical behavior of Li2MnO3.76 Oxygen and Li+ are removed from the material, leaving layered Mn2O and the structure change from O3 to P3 due to the proton–Li+ exchange. This structural change is reversible, and it turns to O3 during the following discharge, but there is an irreversible oxygen loss due to the material activation during the first cycle. The repeated Li+–H+ exchange, which leads to the change from O3 to P3 and the reverse upon cycling, was found to be responsible for the poor electrochemical performance of Li2MnO3. The repeated shearing of oxygen layers results in permeant damage of the structure upon cycling.
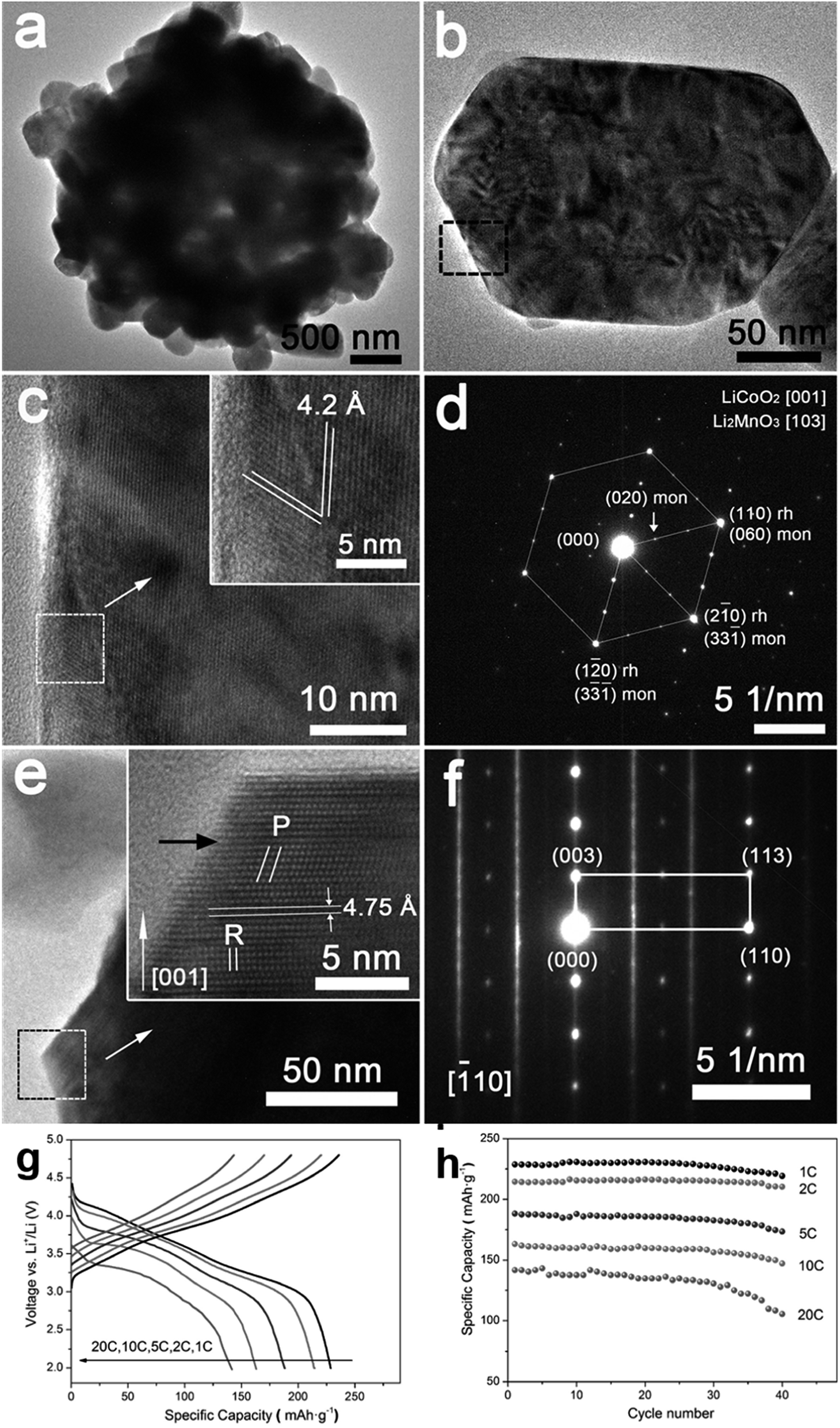
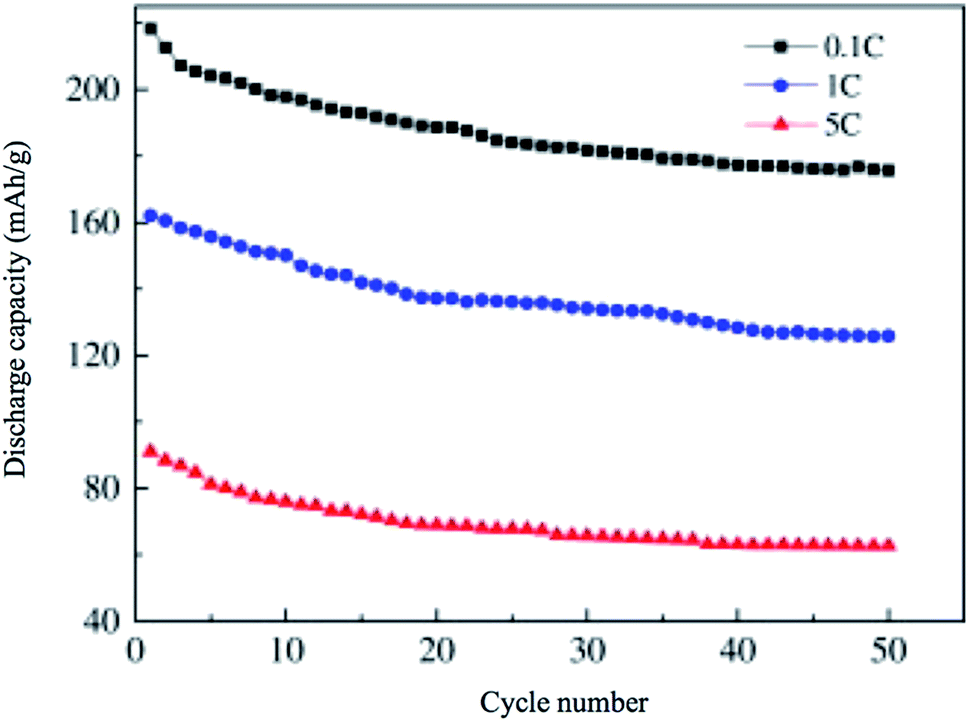
4.7.2 xLi2MnO3·(1 − x)LiMO2. xLi2MnO3·(1 − x)LiMO2 (M = Ni, Co, Cr) are attractive materials due to their high-energy density and low cost. Li[Li1/3−2x/3NixMn2/3−x/3]O2 is a layered oxide composed of two phases: monoclinic xLi2MnO3 and hexagonal (1 − x)LiMO2 integrated into a single nanoparticle.77,78 In the range 0 ≤ x ≤ 1/3, the Li and transition metals are ordered in the transition metal layer on a 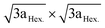 super structure.79 It exhibits high voltage during charging (>4.5 V), high specific capacity (>250 mA h g−1), and high energy density.70,80,81 The electrochemical behavior of Li-rich, layered oxides is divided into two stages. The first is before 4.4 V when the Li+ is extracted from LiMO2. The second starts at 4.5 V when the Li2O is extracted to activate Li2MnO3. The Li-rich, layered oxides suffer from some drawbacks such as an irreversible capacity loss due to the irreversible extraction of Li2O and the reaction with electrolyte at high potential, poor rate capability due to high charge transfer resistance and a voltage fade upon cycling. The origin of the voltage fade was suggested to be the transition from layered to spinel, which is accompanied by an irreversible transition metals migration to the Li-ions layers.82–85 Despite the fact that the nanomaterials with the high surface area are known for their undesired side reactions with the electrolyte, reduction of particle size of Li-rich materials to the nanoscale shows a better rate capability and a decrease in the diffusion path of Li ions.86,87 Moreover, decreasing the interface area between the electrode and the electrolyte by controlling the morphology of the nanomaterials could suppress the erosion from the electrolyte and improve the cycling performance. Taking Li1.2Ni0.2Mn0.6O2 as an example, hierarchical nanoplates structure shows outstanding rate capability as shown in Fig. 8, with specific capacity of ∼231, 216.5, 188, 163 and 142 mA h g−1 at as high rates as 1C, 2C, 5C, 10C, and 20C.87 Furthermore, it shows high capacity retention of 95.5% and 86.6% after 60 cycles for 1C and 2C rates, respectively. The classical voltage decay of Li-rich material was still observed due to the transition to spinel. An expected significant decrease in capacity retention to 68% occurred when the hierarchical structure was damaged by ultrasonic treatment.
super structure.79 It exhibits high voltage during charging (>4.5 V), high specific capacity (>250 mA h g−1), and high energy density.70,80,81 The electrochemical behavior of Li-rich, layered oxides is divided into two stages. The first is before 4.4 V when the Li+ is extracted from LiMO2. The second starts at 4.5 V when the Li2O is extracted to activate Li2MnO3. The Li-rich, layered oxides suffer from some drawbacks such as an irreversible capacity loss due to the irreversible extraction of Li2O and the reaction with electrolyte at high potential, poor rate capability due to high charge transfer resistance and a voltage fade upon cycling. The origin of the voltage fade was suggested to be the transition from layered to spinel, which is accompanied by an irreversible transition metals migration to the Li-ions layers.82–85 Despite the fact that the nanomaterials with the high surface area are known for their undesired side reactions with the electrolyte, reduction of particle size of Li-rich materials to the nanoscale shows a better rate capability and a decrease in the diffusion path of Li ions.86,87 Moreover, decreasing the interface area between the electrode and the electrolyte by controlling the morphology of the nanomaterials could suppress the erosion from the electrolyte and improve the cycling performance. Taking Li1.2Ni0.2Mn0.6O2 as an example, hierarchical nanoplates structure shows outstanding rate capability as shown in Fig. 8, with specific capacity of ∼231, 216.5, 188, 163 and 142 mA h g−1 at as high rates as 1C, 2C, 5C, 10C, and 20C.87 Furthermore, it shows high capacity retention of 95.5% and 86.6% after 60 cycles for 1C and 2C rates, respectively. The classical voltage decay of Li-rich material was still observed due to the transition to spinel. An expected significant decrease in capacity retention to 68% occurred when the hierarchical structure was damaged by ultrasonic treatment.
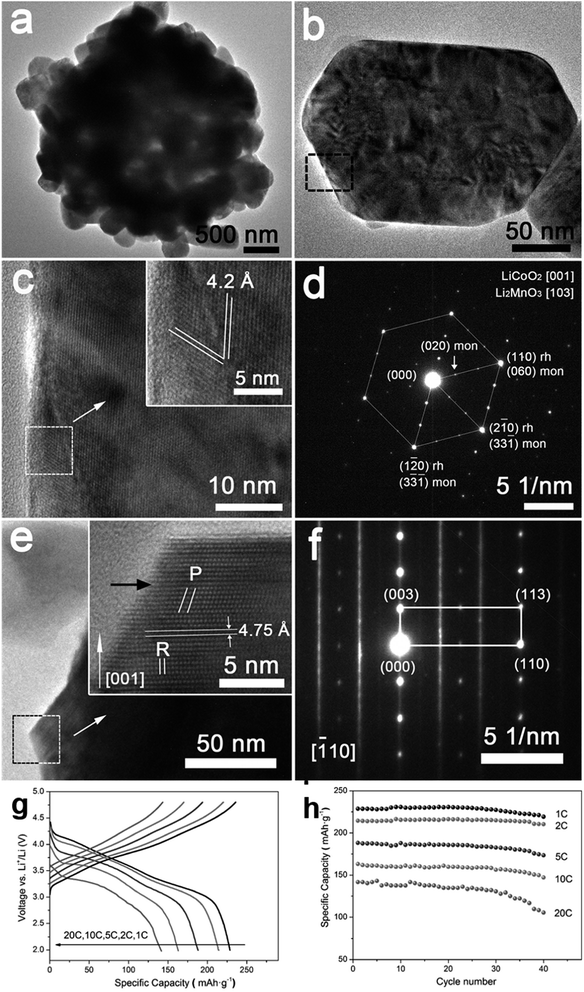 |
| | Fig. 8 (a–f) Hierarchical Li1.2Ni0.2Mn0.6O2 nanoplates with exposed {010} planes as high-performance cathode material for lithium-ion batteries, (g) discharge curves of half cells based on the Li1.2Ni0.2Mn0.6O2 hierarchical structure nanoplates at 1C, 2C, 5C, 10C, and 20C rates after charging at C/10 rate to 4.8 V, and (h) the rate capability at 1C, 2C, 5C, 10C, and 20C rates.88 | |
5 Spinel positive electrode materials
5.1 LiMn2O4
Spinel LiMn2O4 is one of the most attractive energy storage materials due to its low cost and reversible and fast intercalation and de-intercalation of Li ions.89–91 The LiMn2O4 spinel structure (space-group: Fd3m), consists of oxygen ions close-packed array occupy the 32e position, Mn ions occupy the octahedral site 16d, and Li+ occupies the tetrahedral 8a site. In LixMn2O4 (0 < x ≤ 2), Mn2O4 provides a 3-dimensional host network for Li+. When the voltage is ∼4 V, Li ions are hosted in the 8a site in the range 0 < x ≤ 1. However, at 3 V, the inserted Li+ ions occupy the octahedral 16c sites, which share faces with the 8a tetrahedral. The Li-ions at the 8a sites move to the empty 16c sites, resulting in a first-order phase transition to Li2Mn2O4. In addition, when Mn3+ concentration increases, Jahn–Teller distortion occurs to decrease the crystal symmetry from cubic to tetragonal Li2Mn2O4. Therefore, it is believed that the electrochemical performance of spinel LiMn2O4 is better at 4 V than 3 V. However, it is still possible to observe the presence of Li2Mn2O4 on the surface of LiMn2O4 in a 4 V cell at the end of the discharge process as an over-discharge product.92 Then, MnO could be dissolved from Li2Mn2O4, resulting in the formation of Li2MnO3 on the surface of LiMn2O4.93 MnO dissolution is attributed to the formation of HF acid from the electrolyte.| | |
Li2Mn2O4 → Li2MnO3 + MnO
| (1) |
Generally, LiMn2O4 suffers from a capacity fading problem, especially at a temperature of 55 °C due to the phase transitions discussed earlier as well as the chemical instability of the spinel LiMn2O4 with the electrolyte, resulting in the dissolution of Mn ions.94 The improvement of the electrode and electrolyte interface region by providing a protective coating on the material surface to decrease the direct contact with the electrolyte is one of the most extensively studied methods to avoid the previously mentioned problems. Electrochemically active materials coatings, such as LiNi0.5Mn1.5O2, LiCoO2, Li4Ti5O12, and metal oxide coating by B2O3, ZnO, or TiO2, show improved performance of the material at room temperature and 55 °C.95–99 Doping with Al, Co, Cr, or Ni improved the cyclability but at the expense of the initial capacity.100,101 Furthermore, the anionic substitution by F, along with the cationic substitution, show improved performance.102,103
5.2 LiNi0.5Mn1.5O4
The substitution of 25% of Mn for Ni ions in the spinel LiMn2O4 results in one of the most promising materials for high energy applications (LiNi0.5Mn1.5O4) because it shows one dominant voltage plateau at 4.7 V, while another doped LiMn2O4 show two plateaus between 4 and 5 V. Electrochemical performance of spinel LiNi0.5Mn1.5O2 is attributed to the Ni2+/Ni4+ redox at 4.7 V with Mn4+ remains unchanged. LiNi0.5Mn1.5O2 orders in two different space groups Fd3m or P4332. The spinel type that exhibits Fd3m space group is a disordered phase that has a face centered cubic structure in which Li ions occupy the 8a sites, the transition metals Ni and Mn ions randomly occupy the 16d sites, and the ccp oxygen array occupies the 32e sites. The spinel type that exhibits P4332 space group is an ordered phase that has a primitive cubic structure in which the Li ions occupy the 8c positions, Mn and Ni are regularly ordered in the 12d and 4a sites, respectively, while the oxygen ions occupy both the 24e and 8c positions.104 The disordered structure exhibits better performance than the ordered spinel structure.105 Despite its voltage is considered as one of the highest among other available materials, this voltage is outside the stability window of the electrolyte, which leads to the formation of SEI that affects the kinetics of the electrochemical insertion and de-insertion of Li ions. The stability of the SEI can be improved by surface modification via coating.106 In addition, coating plays a beneficial role in suppressing the dissolution of Mn ions from Li Ni0.5Mn1.5O4.
6 Polyanionic positive electrode materials
6.1 Phosphates-based cathode materials
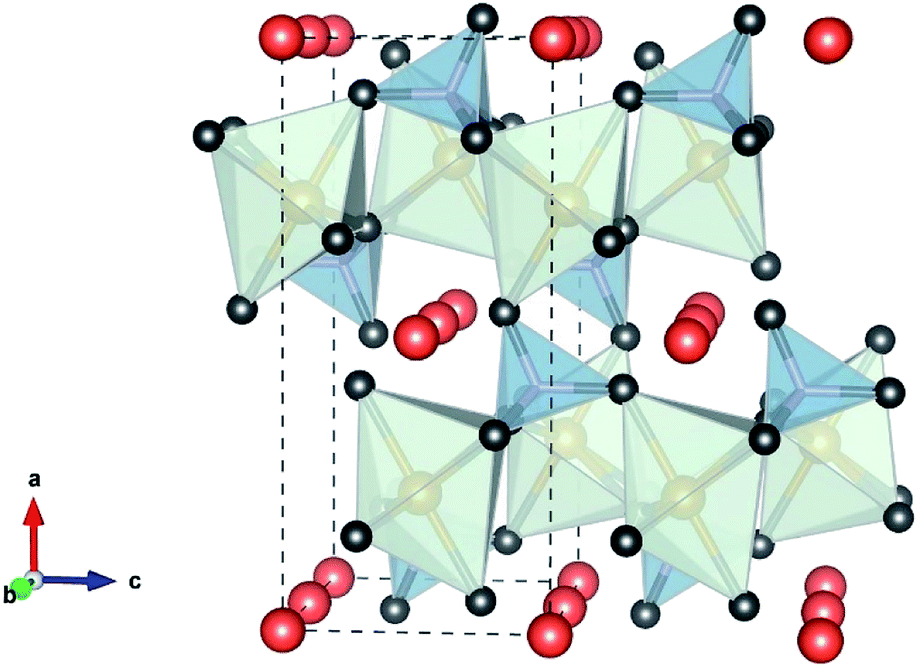
Polyanionic olivine LiMPO4 (M = Fe, Mn, Co or Ni) are very attractive as cathode materials for LIBs. LiMPO4 has an olivine structure, Fig. 9, and crystallizes in the orthorhombic Pnma space group in which the oxygen ions form a hexagonal close-packed framework, Li occupies the octahedral M1 site (4a) forming a tunnel along the direction [010], the transition metal (Fe, Ni, Mn, or Co) ion is located in the M2 site (4c) forming a zigzag chain of corner shared octahedra linked to the phosphate tetrahedra. The MO6 octahedral and PO4 tetrahedral share their edges. The strong P–O covalent bond provides high thermodynamic stability and prevents oxygen evolution at high temperatures compared to LiCoO2 and LiNiO2. This strong PO43− shows lower redox energy of 3d orbital of the metal than the Fermi level. Therefore, the P–O covalent bond is responsible for the lower potential in olivine than oxide materials. The cell parameters of LiMPO4 (M = Fe, Mn, Co, or Ni) are given in Table 5.
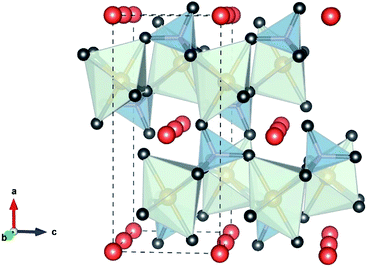 |
| | Fig. 9 Polyhedral representation of the olivine structure. FeO6 (light green), PO4 (gray), and the Li+ (orange) “by VESTA”. | |
Table 5 Cell parameters of LiMPO4 (M = Fe, Mn, Co or Ni)
| LiMPO4 |
a |
b |
c |
Ref. |
| LiFePO4 |
10.3377(5) |
6.0112(2) |
4.6950(2) |
107 |
| LiMnPO4 |
10.4472(5) |
6.11049 |
4.7459(2) |
108 |
| LiCoPO4 |
10.20 |
5.92 |
4.70 |
109 |
| LiNiPO4 |
10.060 |
5.776 |
4.683 |
110 |
6.1.1 LiFePO4. LiFePO4 is the most attractive commercialized cathode material because of its desirable safety features, high theoretical capacity (170 mA h g−1) at moderate current densities, stable Fe3+/Fe4+ redox potential of 3.5 V vs. Li+/Li, flat voltage plateau, thermal stability even at high temperatures, stable electrochemical and chemical properties, excellent cycling performance, low cost, and abundance of iron. During the insertion and removal of Li+, it was observed that there are two coexisting phases, LiFePO4 and FePO4. Both phases have the same space group (Pnma). Padhi et al. suggested that the insertion and extraction of Li+ is associated with the motion of the two-phase interface.111 They suggested the shrinkage core model in which the Li ions move from the surface of a particle inward through the phase boundary. Upon lithiation, the surface area of the interface decreases until it reaches a critical surface area when the rate of Li insertion is not able to sustain the applied current. This makes LiFePO4 a suitable material for low power applications as it influences the performance of the material in terms of capacity and rate capability at high current density. Srinivasan et al. assumed a juxtaposition of the LiFePO4 and FePO4 phases and suggested a shrinking core model in which the shell of one phase covers the core of the other phase by considering that the Li-ion diffusion drives the phase boundary motion.112The battery cycling and calendar age influence the cycle life, capacity, and power capability of LIB. The factors that influence the performance can be a function of either the operation conditions, such as temperature or working potential window, or the type of the cathode material. For example, Sun et al. showed that the high temperature to have a tremendous negative impact on the performance of the LiFePO4 battery.113 The battery lifetime at room temperature is about seven times higher than that at 55 °C as presented in Fig. 10. The reason for capacity loss at room temperature is assigned to the irreversible loss of Li by the formation of a solid electrolyte interface (SEI) layer. Upon increasing the ambient temperature, the interplanar spacing between the crystallographic planes of the structure becomes larger, and the surface layer (SEL) becomes thicker, which is a sign of degradation. Also, the SEM images of the surface of the aged cathodes at temperatures higher than 45 °C showed a rounded-shaped particle rich in fluorine with the particle size increased from 0.7–0.8 μm to 1.3–1.5 μm with increasing the temperature, which is accompanied by the accelerated decomposition of the electrolyte.
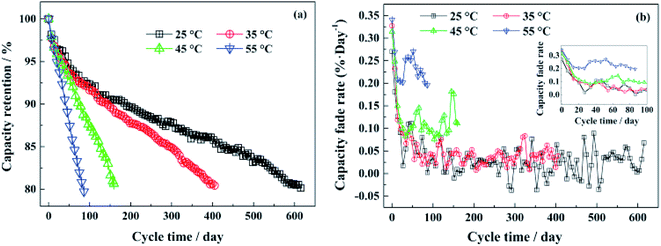 |
| | Fig. 10 (a) The capacity retentions of the LiFePO4/graphite full cells at different temperatures with continuous cycling days, (b) the capacity fade rate at each test temperature during cycling.113 | |
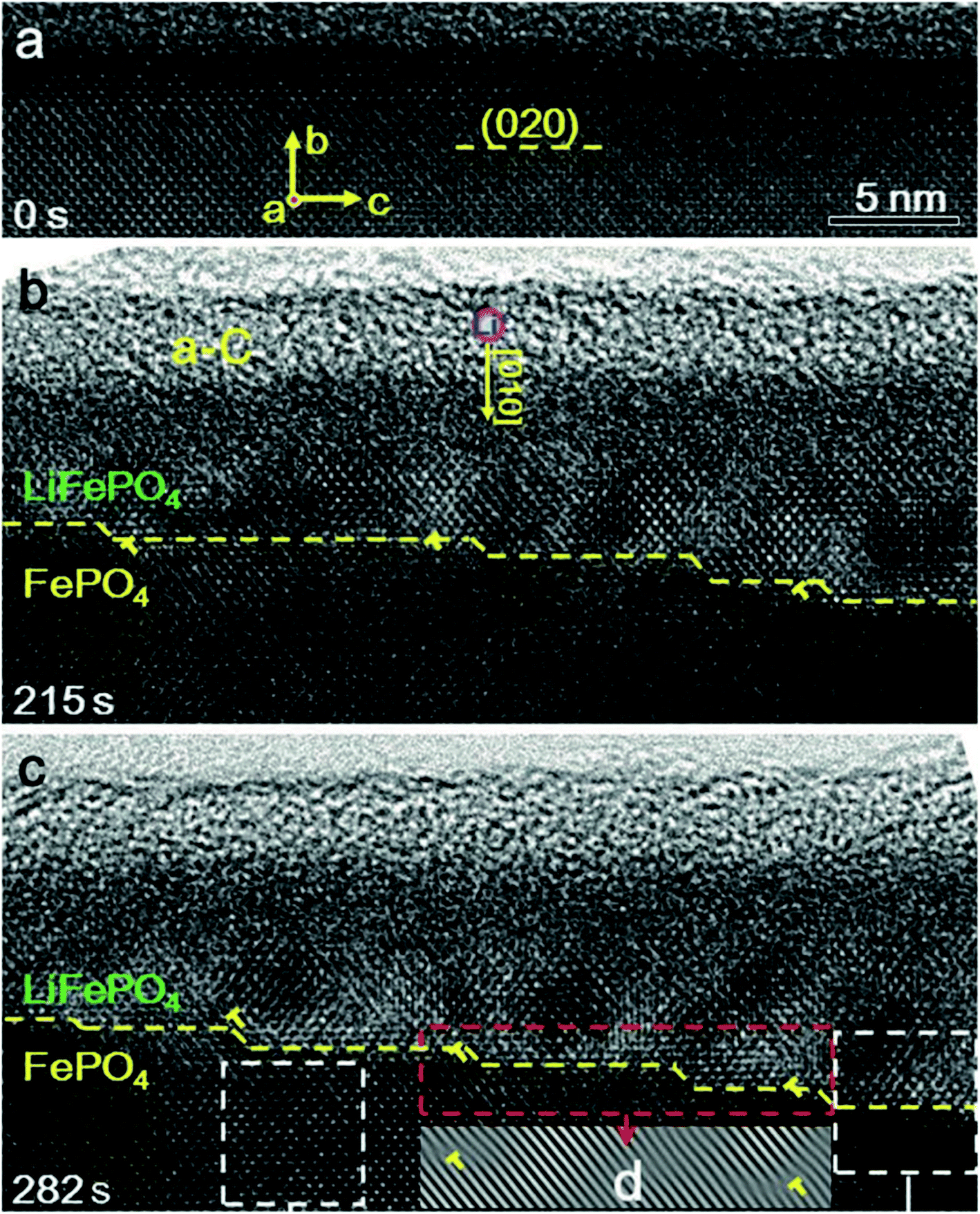
There is a structural instability that usually occurs during the intercalation and de-intercalation process of Li, which results in fast capacity fading upon cycling. A study used in situ TEM and HRTEM analyses showed that the lithiation and de-lithiation in LiFePO4 occur by two phase reactions (LiFePO4 and FePO4).114 Upon applying potential to FePO4, Li ions inserted into the structure to form a layer of LiFePO4 that gets thicker with time, which was evident via the migration of the step shape phase boundary along the b axis or [010] direction as shown in Fig. 11. This study proves that there is a lattice mismatch that induced dislocation at the phase boundary, where the transformation between the two phases produced an array of dislocations in the FePO4 side. Also, the orientation of LiFePO4 changed slightly compared to FePO4 as a result of the elastic deformation that can accommodate the transformation strain. These dislocations accumulate during the cycling and can cause further defects/cracks that cause degradation. Moreover, the ageing of C6/LiFePO4 (LFP) batteries was studied theoretically by the electron-tunneling-based model for lithium immobilization.115 The SEI formation model presumes the existence of a porous outer and dense inner SEI layers. Electron tunneling through the inner SEI layer is considered to be the rate-determined step in the SEI formation process and its initial thickness after activation process controls the degradation rate. The outer SEI layer grows much faster than the inner layer. During cycling, cracks formation takes place, exposing the new surface of graphite to the electrolyte directly resulting in the formation of SEI, hence loss in capacity. The formed SEI was found to be dependent on the number of cycles.
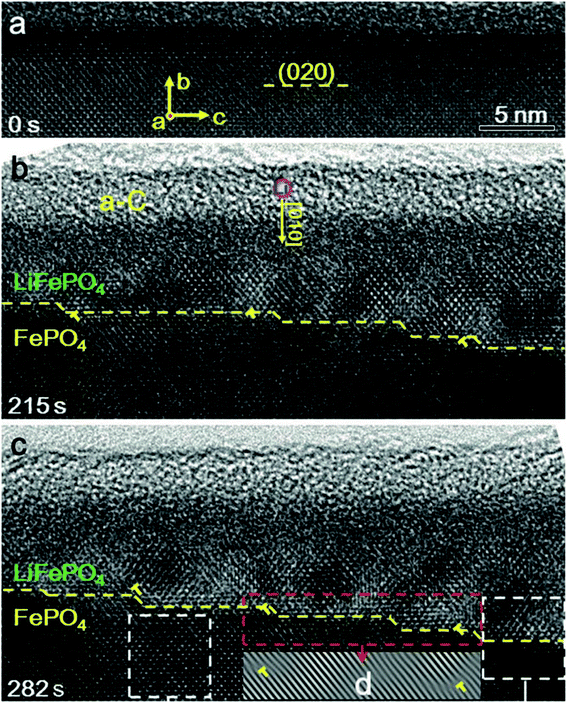 |
| | Fig. 11 Phase boundary between FePO4 and LiFePO4 and its migration along the [010] direction during lithiation. (a) A HRTEM image of the pristine FePO4 (b) after 215 second from applying the voltage, a step-like phase boundary was formed between FePO4 and LiFePO4, as pointed out by the yellow dashed line. (c) At 282 seconds, the thickness of the LiFePO4 layer increased as the step-like phase boundary propagating along the [010] direction.114 | |
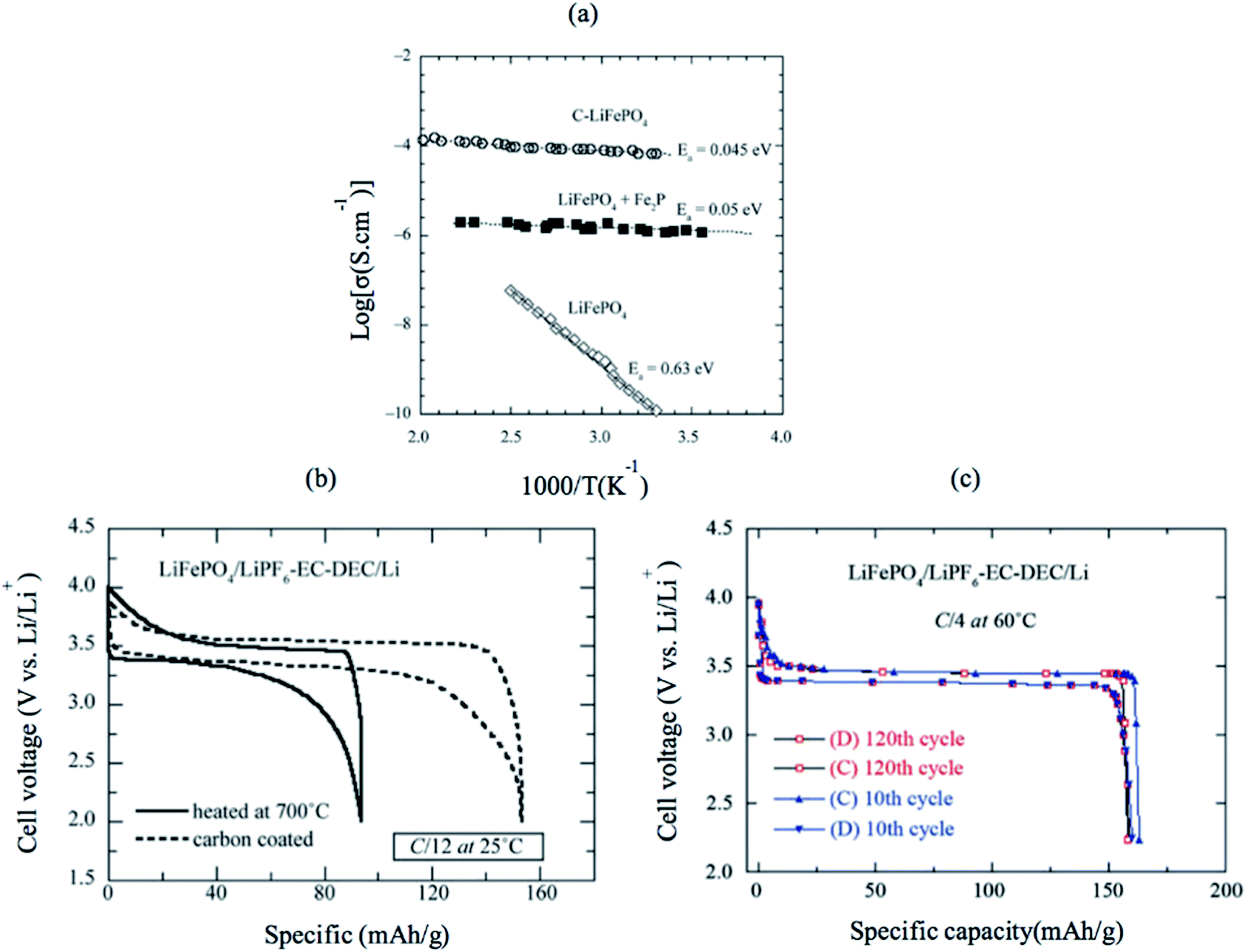
LiFePO4 has a low intrinsic electronic conductivity of 10−9 S cm−1 and low ionic conductivity. Several attempts were carried out to increase both its ionic and electronic conductivity and, consequently, its electrochemical performance. First, conductive surface coating by carbon was found to improve the electronic conductivity due to the improved electric contact between the electrode particles, as shown in Fig. 12a.116 Several groups tailored the C/LiFePO4 composite electrode and obtained specific capacity close to the theoretical limits of LiFePO4 (170 mA h g −1). Furthermore, C-coating prevents particle growth during heat treatment resulting in the synthesis of smaller particle size and higher surface area LiFePO4.117 Fig. 12b shows a comparison between the electrochemical profile of the bare and C-coated LiFePO4 at C/12 rate; both have the same particle size (40 nm). The C-coated sample showed high capacity that is close to the theoretical limit, while bare LiFePO4 showed only 55% of the theoretical capacity. Even at higher rate of C/4, C/LiFePO4 shows excellent Li ions extraction kinetics and outstanding ability to maintain 100% of its initial capacity (160 mA h g−1) after 120 cycles. However, the addition of additives such as C would increase the conductivity, but it decreases the energy density of the cathode because it is counted as electrochemical inactive material. Carbon-free LiFePO4 small particles with narrow size distribution can shorten the ionic and electronic diffusion length and improve the conductivity of the material.118,119 Delacourzt et al. prepared 100–200 nm LiFePO4, which showed a high reversible specific capacity of 145 mA h g−1 at C/2. Third, the electronic conductivity and Li ion diffusion in LiFePO4 structure would be improved by doping at the anionic and/or cationic sites. Chung et al. reported that multivalent ions-doped LiFePO4 showed outstanding electronic conductivity of eight orders of magnitude higher than pure LiFePO4.120 However, the reason for the improved conductivity is questioned since Herle et al.121 showed that the iron salt precursor in Chung's experiment was the source of C, which can reduce Fe and P to Fe2P and/or Fe3P (conductivity of LiFePO4 + Fe2P is shown in Fig. 12a). Therefore, the metal-rich phosphides are believed to be responsible for the improved conductivity. A combination of C and Zn4+ doping showed a synergetic effect that facilitated electron transport.122
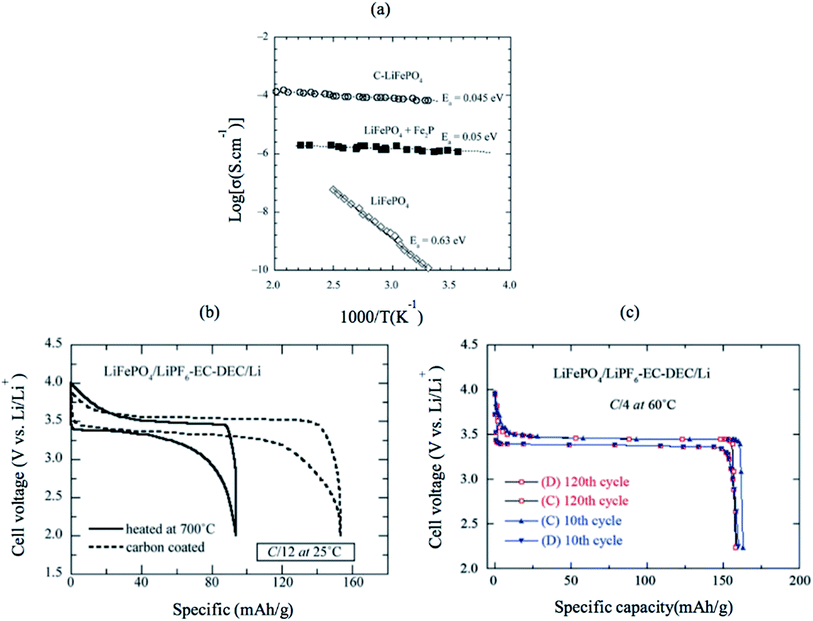 |
| | Fig. 12 (a) Electronic conductivity of pure LiFePO4, Fe2P-containing sample and C/LiFePO4. (b) The electrochemical profiles at C/12 of non-coated and heated at 700 °C LiFePO4//Li cells (c) electrochemical performance of the C–LiFePO4//Li cell operating at 60 °C. Charge–discharge cycling was conducted at the C/4 rate (about 35 mA g−1) in the voltage range 2.2–4.0 V vs. Li0/Li+.123 | |
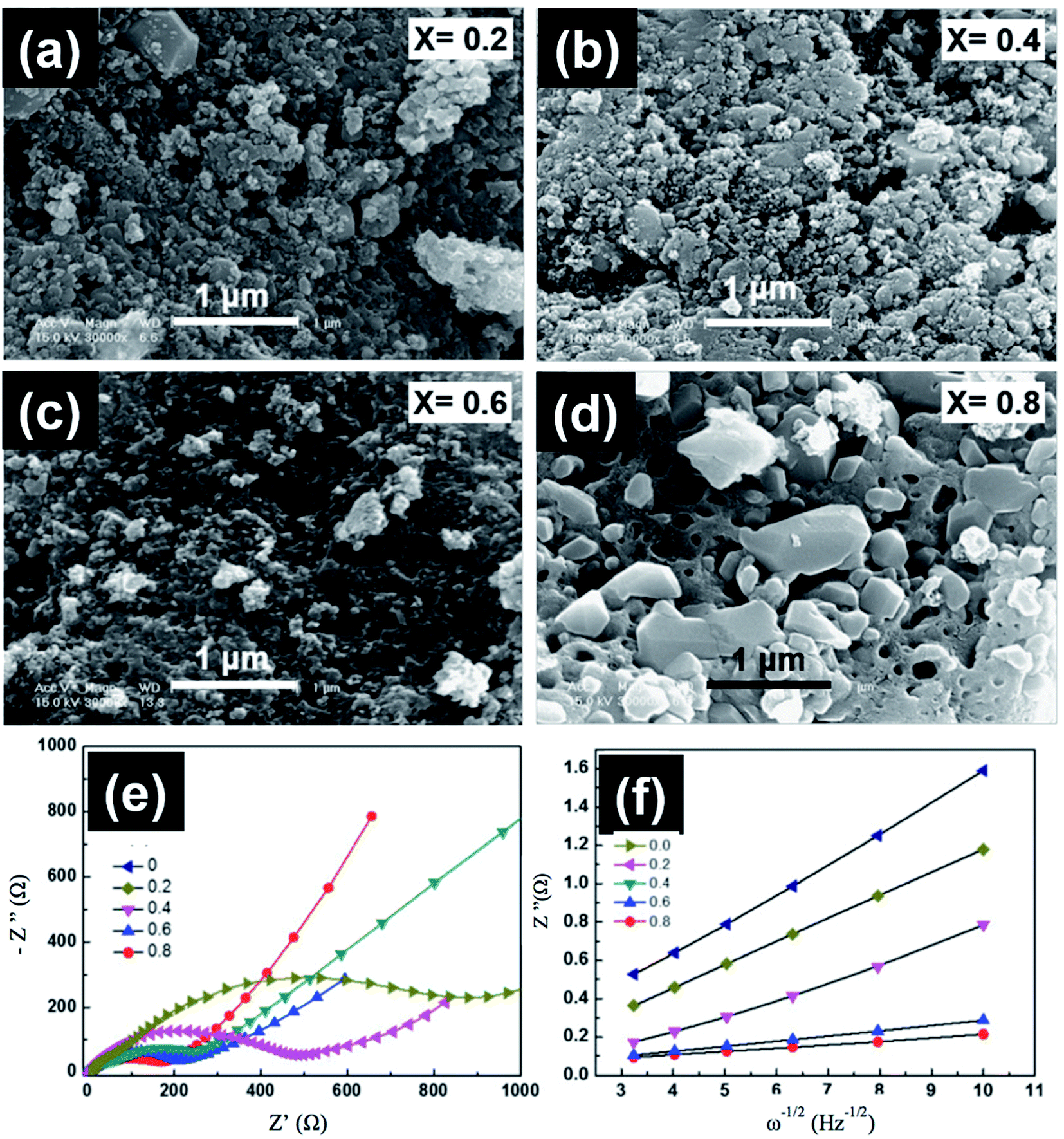
6.1.2 LiMnPO4. The low energy density of LiFePO4 restricts their use in the automotive industry. The study and development of other members from the olivine family with higher discharge voltage and energy density, such as LiMnPO4, are critical. LiMnPO4 showed higher potential than LiFePO4 (4.1 V vs. Li0/Li+) and a high energy density of ∼700 W h kg−1.124 However, cyclic stability and rate capability of LiMnPO4 are hindered by the poor kinetics and low ionic and electronic conductivity. Furthermore, the presence of Mn3+ ions in an unstable position in the lattice leads to cell distortion, which is controlled by the Jahn–Teller effect.125 Similar to LiFePO4, several strategies were used to improve the conductivity of LiMnPO4, such as the synthesis of smaller particle size, carbon coating, and cation substitution by Fe, Mg, or Zn.126–128 The solid solution of LiMnPO4 and LiFePO4 operates in the voltage range between 3.4 and 4 V vs. (Li/Li+), which is not very high to prevent the electrolyte decomposition and at the same time offers high energy density with respect to LiFePO4 and better rate capability than LiMnPO4.129 A common defect of anti-site mixing between Fe/Mn and Li ions in the mixed LMFP is dependent on the synthesis method, and it affects the Li ions motion along the 010 channel upon cycling. Sun et al.130 studied the effect of Fe substitution on the conductivity and Mn ions dissolution. They prepared nanostructured LiFexMn1−xPO4 (x = 0 and 0.15) by spray pyrolysis and suggested that the performance was improved because the conductivity increased, and Mn dissolution decreased from 103.7 to 87.2 ppm. Seo et al.131 studied the charge transfer resistance (Rct) and the electrochemical performance of LiFexMn1−xPO4 at x = 0, 0.2, 0.4, 0.6, 0.8. The best electrochemical performance was observed when x = 0.8 and 0.6, which corresponded to the highest discharge capacity of almost 154 and 149 mA h g−1 and retained 100% and 101% of its initial capacity after 50 cycles, respectively. The impedance patterns of all compositions showed a single semicircle in the high frequency region as shown in Fig. 13. The semicircle at high frequencies is due to the combination between double layer capacitance and Rct. The Rct decreased by increasing the Fe content and reached its lowest value of 170 Ω when x = 0.8. The Warburg impedance at low frequency is related to the diffusion of Li ions. The Li ions diffusion increased with decreasing the slope of the line and reached its highest value of 7.33 × 10−14 when x = 0.8. Dai et al. reported that the co-substitution of Mn by Fe and Mg could significantly improve the cycling performance, where the optimal composition was found to be LiMn0.8Fe0.19Mg0.01PO4.132 Moreover, they studied the effect of C content on the electrochemical performance of this composition. The material with 10.5 wt% carbon showed the best rate capability and capacity of 143.1 mA h g−1, 130 mA h g−1, and 125.7 mA h g−1 at 1C, 5C, and 10C rates, respectively. Lower C content of 4.1 wt% showed a slight decrease in the rate capability. However, further decrease or increase in the C content to 2 wt% or 27.4 wt% resulted in fast capacity fade at high C rate.133 This better performance is explained by the lowest charge transfer resistance of ∼27 Ω at 10 wt% C electrodes.
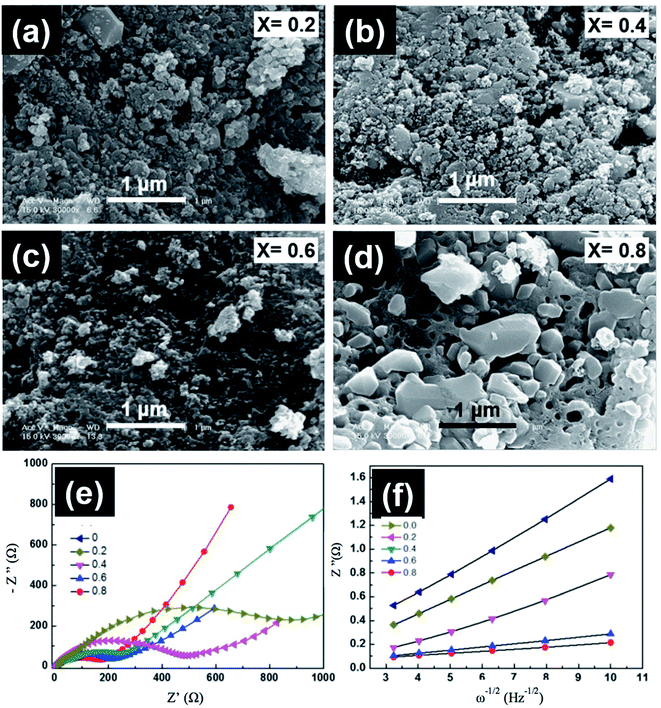 |
| | Fig. 13 (a–d) FESEM images, (e) impedance spectra, and (f) the relationship between Z′′ and the square root of frequency (ω−1/2) in the low-frequency region for LiMn1−xFexPO4 at different compositions.131 | |
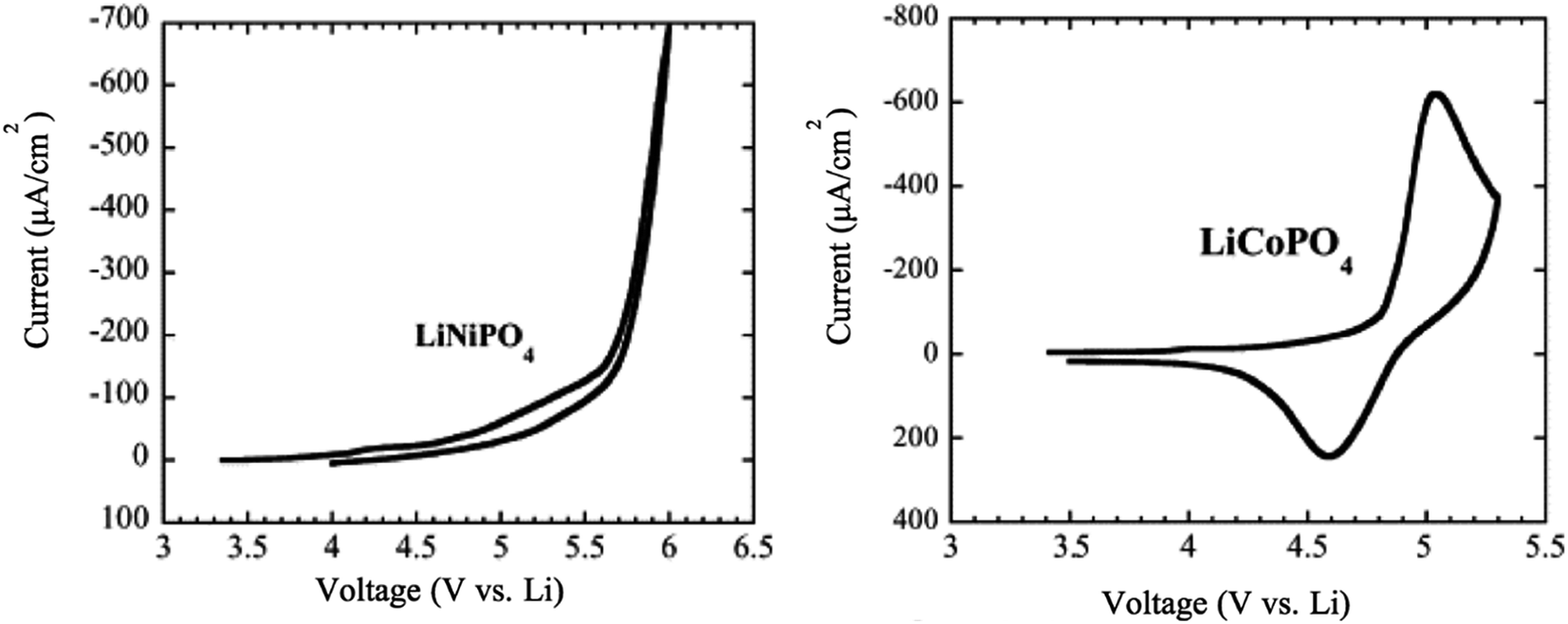
6.1.3 LiCoPO4 and LiNiPO4. The LiCoPO4 and LiNiPO4 materials exhibit a high potential of 4.8 V and 5.2–5.4 V, respectively.134 It is challenging to extract lithium from LiNiPO4 within the stable voltage window of the available electrolytes. Moreover, both compounds showed much lower electronic conductivity than LFP and LMP. The cyclic voltammetry of LiCoPO4 showed that there is a reduction peak at 4.6 V and an oxidation peak at 5.1 V, while there are no significant reduction peaks for LiNiPO4 in the range between 3.5 to 6 V vs. Li/Li+ as shown in Fig. 14.135
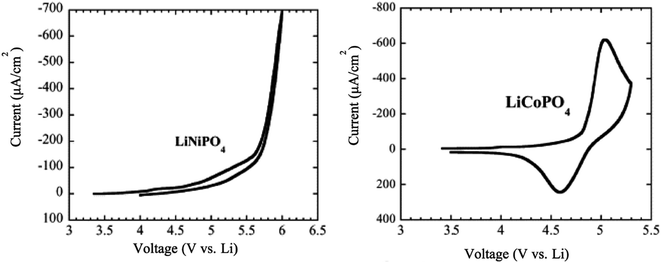 |
| | Fig. 14 Cyclic voltammetry of LiNiPO4 and LiCoPO4 at a scan rate of 0.2 mV s−1.135 | |
6.2 Silicate-based cathode materials
The replacement of phosphate by silicate was a crucial step in the search for new high energy cathode materials. Orthosilicate cathode materials (Li2MSiO4, M = Fe, Mn) are of particular interest due to their superior electrochemical performance, safety, raw material abundance, and low cost. Li2MSiO4 has a high theoretical specific capacity (∼333 mA h g−1) if the two Li ions are fully extracted per formula unit. Several temperatures and pressure-dependent polymorphs of Li2MSiO4 exist.136 The framework consists of distorted hexagonal close backed oxygen array, and all cations occupy tetrahedral sites. The different possible cations ordering produce different polymorphs. These polymorphs can be divided into low or high temperature, which corresponds to β or γ Li3PO4 like structures, respectively. Each type could be explained in several space groups such as orthorhombic βI (Pbn21, Pna21) and βII (Pmn21), monoclinic γs/γ0 (P21/n), and orthorhombic γII (Pmna, Pmnb).
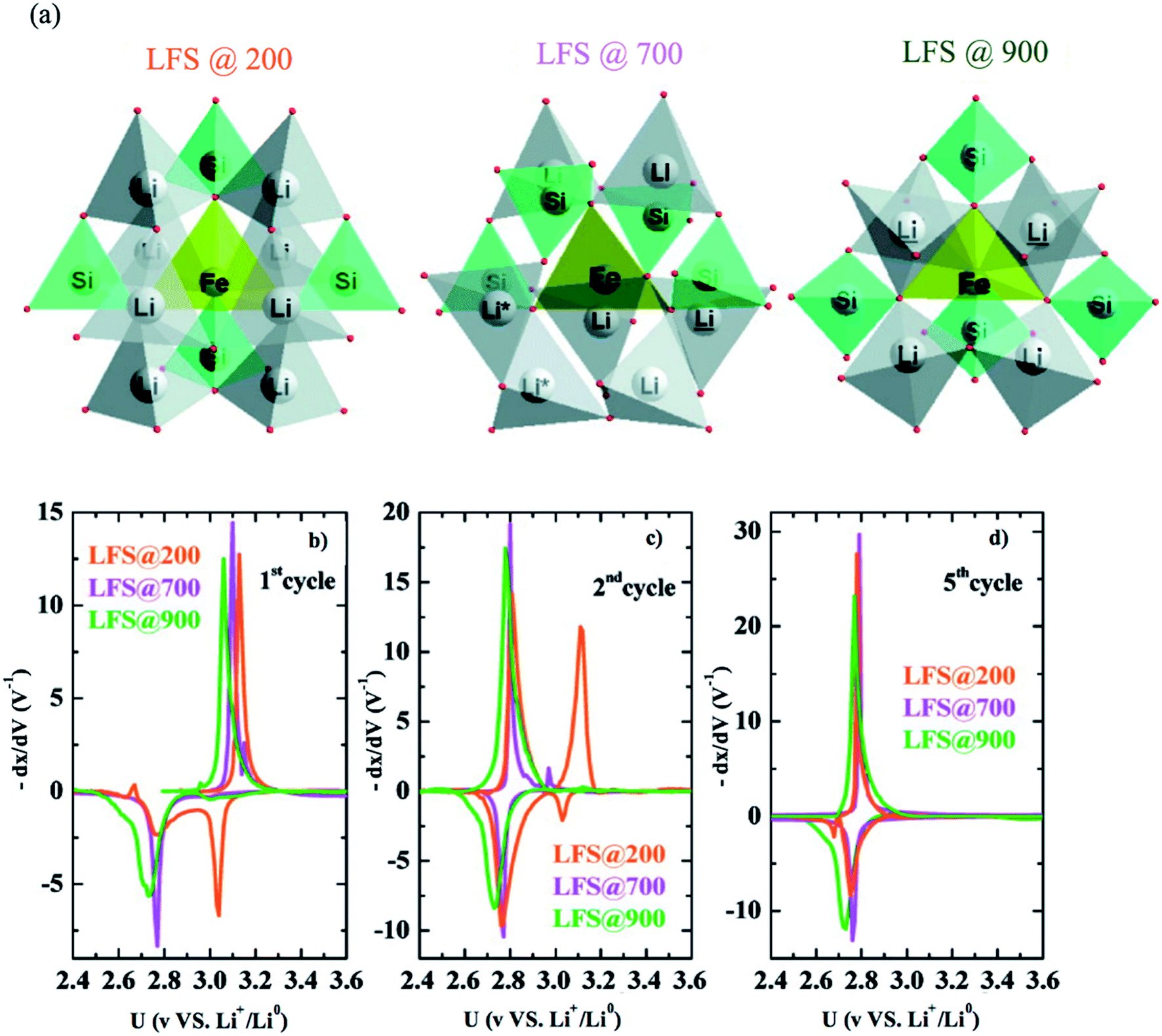
6.2.1 Li2FeSiO4. Li2FeSiO4 exhibits a specific capacity of 166 mA h g−1 since only one Li+ can be extracted due to the redox couple Fe3+/Fe2+ at 3.1 V vs. Li+/Li. The lower electronegativity of Si compared to P results in a lowering of the Fe3+/Fe2+ redox couple in Li2FeSiO4 than in LiFePO4. The other redox couple Fe4+/Fe3+ takes place at a high voltage of 4.85, which is outside the stability window of the electrolyte; therefore, it is not reached. Li2FeSiO4 exhibits complex structural defects that make it difficult to analyze its structure. Nytén et al.137 reported Li2FeSiO4 in the orthorhombic system with Pmn21 space group and explained two additional peaks in its diffractogram as an unidentifiable impurity. Its potential shifts irreversibly from 3.1 V to 2.8 V in the second cycle. This voltage drop was suggested to be due to site mixing between Li ions and Fe ions since the Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ratio in the 4b site changed from 96:4 to 40:60 during the first cycle.138 Nishimura et al.139 reported Li2FeSiO4 in the monoclinic system and P21 space group. Sirisopanaporn et al.140 synthesized a polymorph, which is an iso-structural with Li2CdSiO4 and is described by the Pmnb space group. Sirisopanaporn et al.141 compared the electrochemical behavior of Li2FeSiO4 polymorphs, which were explained in the Pmn21, P21/n, and Pmnb space groups by synthesis under hydrothermal conditions at 200, 700, and 900 °C, respectively. The structural difference of these polymorphs is shown in Fig. 15a. Each polymorph showed different first oxidation potential. The Pmnb polymorph exhibited the lowest potential (2.9 V vs. Li+/Li0), P21/n showed an intermediate potential of 2.97 vs. Li+/Li0, while Pmn21 showed the highest oxidation potential (3.1 V vs. Li+/Li0). Derivative plots obtained from PITT of the three polymorphs showed that they all experience the voltage fade from 3 to 2.8 V upon cycling due to the transformation to Li-poor phase as shown in Fig. 15b–d. More precisely, it showed that the high temperature Pmnb and P21/n, which have high degree of disorder, transformed faster than the low temperature Pmn21. The first discharge reduction potential of Pmnb and P21/n was ∼2.76 V vs. Li+/Li0, while the discharge of Pmn21 showed two reduction peaks at ∼2.76 and ∼3.04 V vs. Li+/Li0 indicating the presence of initial Li2FeSiO4. The high-temperature Pmnb and P21/n experienced a phase transformation at the first cycle faster than Pmn21, which is fully transformed at the fifth discharge.
:Fe ratio in the 4b site changed from 96:4 to 40:60 during the first cycle.138 Nishimura et al.139 reported Li2FeSiO4 in the monoclinic system and P21 space group. Sirisopanaporn et al.140 synthesized a polymorph, which is an iso-structural with Li2CdSiO4 and is described by the Pmnb space group. Sirisopanaporn et al.141 compared the electrochemical behavior of Li2FeSiO4 polymorphs, which were explained in the Pmn21, P21/n, and Pmnb space groups by synthesis under hydrothermal conditions at 200, 700, and 900 °C, respectively. The structural difference of these polymorphs is shown in Fig. 15a. Each polymorph showed different first oxidation potential. The Pmnb polymorph exhibited the lowest potential (2.9 V vs. Li+/Li0), P21/n showed an intermediate potential of 2.97 vs. Li+/Li0, while Pmn21 showed the highest oxidation potential (3.1 V vs. Li+/Li0). Derivative plots obtained from PITT of the three polymorphs showed that they all experience the voltage fade from 3 to 2.8 V upon cycling due to the transformation to Li-poor phase as shown in Fig. 15b–d. More precisely, it showed that the high temperature Pmnb and P21/n, which have high degree of disorder, transformed faster than the low temperature Pmn21. The first discharge reduction potential of Pmnb and P21/n was ∼2.76 V vs. Li+/Li0, while the discharge of Pmn21 showed two reduction peaks at ∼2.76 and ∼3.04 V vs. Li+/Li0 indicating the presence of initial Li2FeSiO4. The high-temperature Pmnb and P21/n experienced a phase transformation at the first cycle faster than Pmn21, which is fully transformed at the fifth discharge.
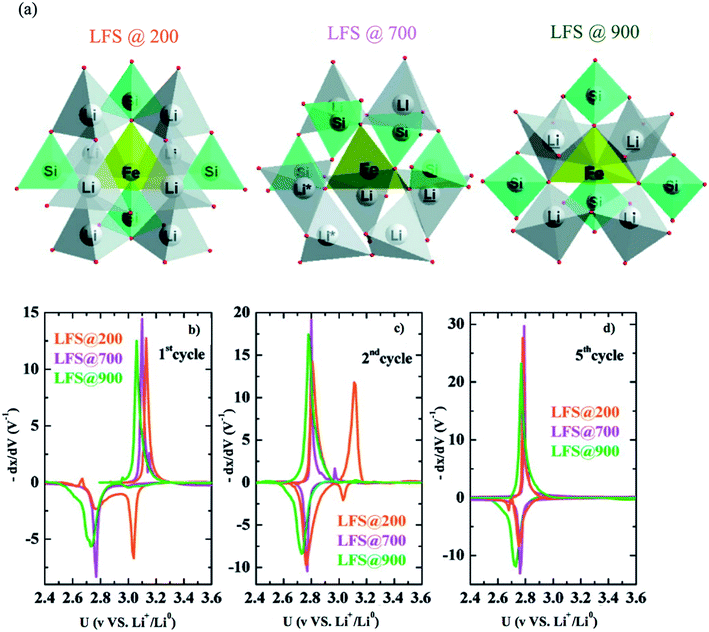 |
| | Fig. 15 (a) The three polymorphs of Li2FeSiO4, derivative plots obtained from PITT measurements in the (b) first, (c) second, and (d) fifth cycles for all three polymorphs.141 | |
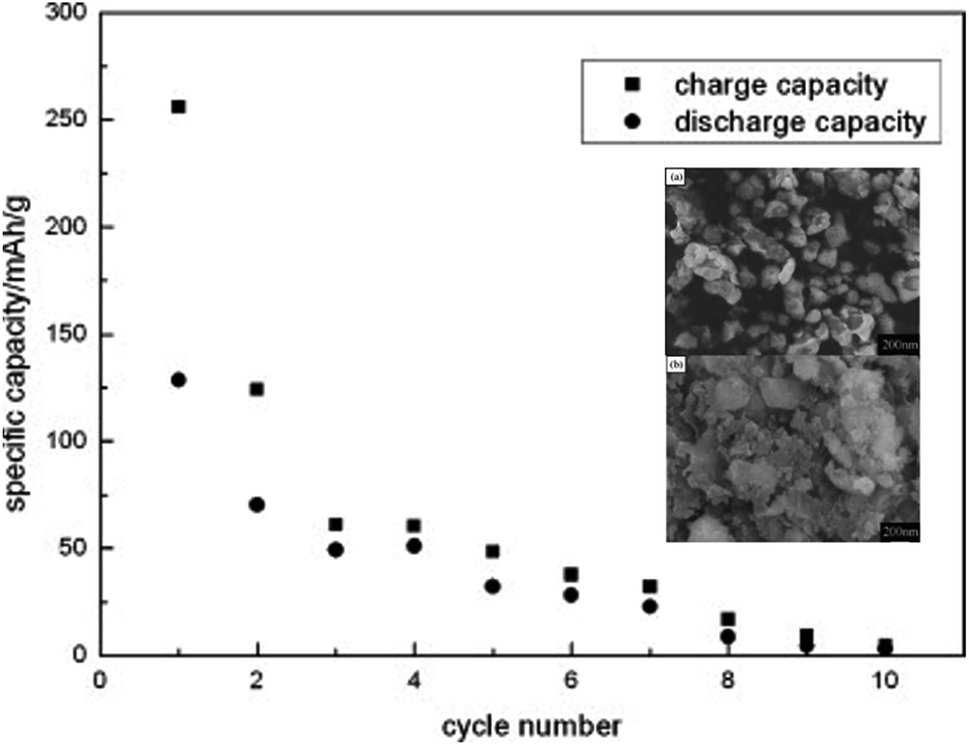
6.2.2 Li2MnSiO4. Li2MnSiO4 is a more interesting material than Li2FeSiO4 as it offers higher redox potential and specific capacity. Li2MnSiO4 possesses a theoretical capacity of 333 mA h g−1 because of the contribution of two Li ions to the electrochemical reaction. This is because Mn ions can be reversibly oxidized and reduced from the +2 state to +4 state with the redox couples of Mn2+/Mn3+ and Mn3+/Mn4+ at 4.1 and 4.5 V vs. Li+/Li, respectively. At 0 K, the low-temperature β-Li3PO4 is more thermodynamically stable than the high-temperature γ-Li3PO4. The transformation from β to γ structures is exothermic and can take place at higher temperatures. Also, the γ to β transformation can happen under high-pressure conditions.142 The separation of polymorphs is possible by applying the preferred conditions of each structure. P21/n is obtained by synthesis at high temperatures, followed by fast quenching. The synthesis of Pmn21 polymorph is possible by controlling both the pressure and temperature at 2–8 GPa and 600–900 °C, respectively.142 Among these polymorphs, the low-temperature orthorhombic Pmn21 is the best polymorph as it shows favorable thermodynamic stability and exhibits the lowest Li diffusion energy barrier. Li2MnSiO4 suffers from poor performance and fast capacity fading problems upon cycling, as shown in Fig. 16. This is because of the low ionic and electronic conductivity (∼5 × 10−16 S cm−1 at RT)143 and the structural instability due to the presence of Mn ions in the tetrahedral sites. Several strategies were proposed to improve its structural stability upon cycling. The synthesis of new polymorphs in which the Mn ions are sitting in stable octahedral sites was tested under high pressure up to 15 GPa. However, it was not obtained because of the strong cationic repulsion in the dense structures.144,145 The addition of conductive carbon, such as acetylene black, showed significantly improved performance.146 Nano C/Li2MnSiO4 prepared by ionothermal method showed improved performance and capacity retention even at high C rates as shown in Fig. 17. Cation substitution by Mg, Ca, Cr, Ti, V, Ni, Na, Cu or P improved the intrinsic conductivity and increased the Li ion diffusion coefficient.147–151
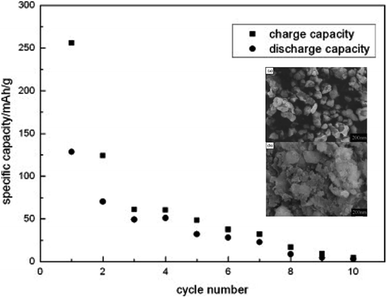 |
| | Fig. 16 Cyclic performance of Li2MnSiO4 synthesized by solid state method tested at C/16 rate to a cut-off voltage between 1.5 and 4.8 V.152 The inset shows FESEM micrographs of (a) Li2MnSiO4 and (b) Li2MnSiO4/C. | |
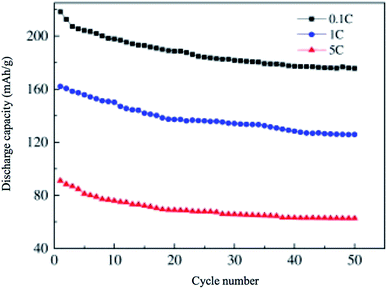 |
| | Fig. 17 Cyclic performance of nano C/Li2MnSiO4 synthesized by ionothermal method, the material is tested in the potential range of 2.5–4.5 V.153 | |
7 Go green with Li-ion batteries
Following the growth of the Li-ion batteries industry, the environmental impact of such a technology is critical and must be carefully considered. Cathode materials are the key components in the battery in terms of cost and active weight. Their toxic byproducts during the manufacturing process as well as inadequate disposal ways, require urgent green approaches to avoid their severe health and environmental problems. One of the proposed methods in literature is to switch from conventional to green synthesis routs. To avoid environmental pollution and avert additional costs associated with processing the harmful byproducts that contain anions such as Cl−, SO42−, NO3−, and gases like NxOy, CO, and NH3.154 The electrochemical performance is one of the main factors that direct the industry, therefore, considering it side by side with the environmental issues is logical and crucial. As an example, Bolloju et al. obtained 165 mA g−1 at 0.1C from LiFePO4 prepared using Fe powder as a precursor, which is comparable to that obtained by conventional chemical synthesis.155 Fe powder is very advantageous over other precursors because it does not produce harmful anions. Also, there is a 100% atom economy delivered by the iron metal, which makes it cost-effective as well. Liu et al. prepared multi-doped LiFePO4/C with Mn, Co, and Ni using Fe powder and spent electroless nickel plating solution.156 The product shows comparable performance to that obtained by the expensive ferrous oxalate or ferric oxide as well as to that prepared by conventional chemical routs.
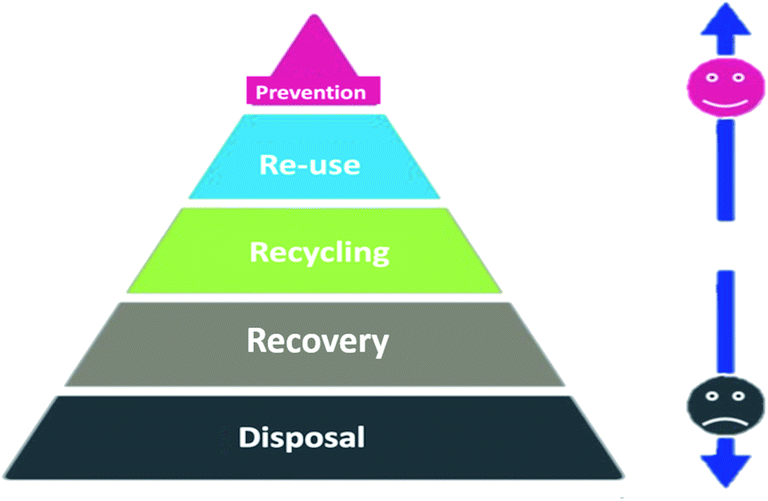
Waste management is a process that considers the protection of the environment with various strategies in a hierarchy order (disposal, recovery, recycling, reuse, and prevention), as illustrated in Fig. 18. Accordingly, reusing a battery is cleaner than recycling because recycling needs to spend energy on transportation and processing. Thus, it is possible, for instance, to collect the batteries that retain 85% to 80% of their initial capacity from electric vehicles to reuse in stationary grid applications.157 Recycling can still help in decreasing pollution and satisfy the need for raw materials. Recycling relies first on the recovery of different streams of raw materials followed by a pyrometallurgical or hydrometallurgical process to extract metals (Li, Ni, Mn, Al, Cu or Co). Pyrometallurgical is simple; the battery is smelted in a high-temperature furnace to reduce the active materials into metallic alloy form and also produce slag and gases.158 This method requires filters to trap the emitted toxic gases.
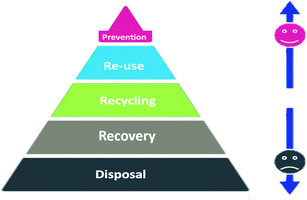 |
| | Fig. 18 The waste management hierarchy and range of recycling options.158 | |
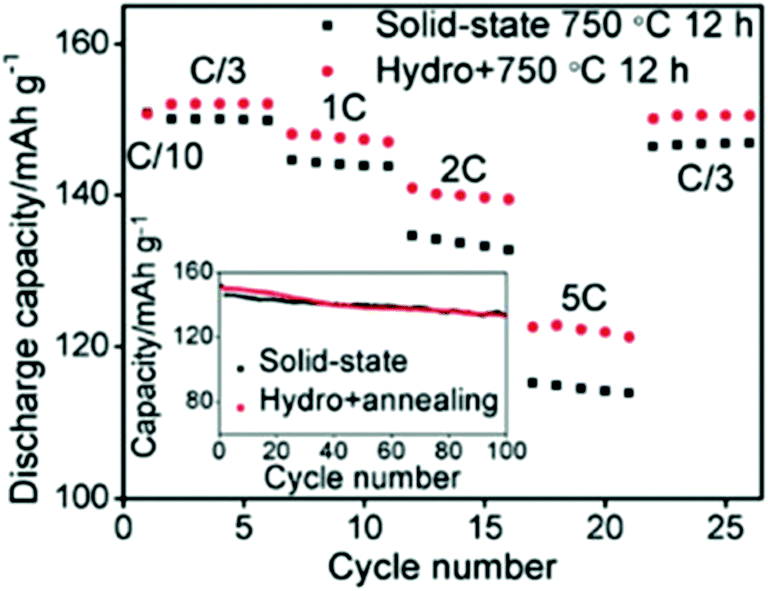
On the other hand, the hydrometallurgy process involves the leaching of the metal from the cathode by the use of aqueous solutions, such as a mixture of acid and hydrogen peroxide. Each metal can be obtained by extraction, precipitation, or electrodeposition. Shi et al. combined hydrothermal and annealing method to efficiently obtain LiCoO2 with controlled microstructure and composition and found that its rate capability is higher than that obtained by conventional solid-state synthesis (Fig. 19).159 Transition metals oxides and hydroxides were collected from commercial spent Li-ion batteries and used as a potential material for supercapacitors.160,161 The battery cathode tapes were recycled and used as a catalyst for the degradation of the toxic methylene blue dye.162
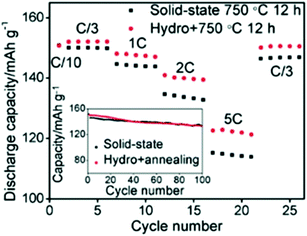 |
| | Fig. 19 Comparison of two materials, one was sintered for 12 h at 750 °C and the other was prepared by hydrothermal method then sintered for 12 h at 750 °C.159 | |
8 Summary and outlook
The use of different cathode materials, such as layered oxides, olivines, and spinels, for Li-ion batteries is summarized and discussed. The classification of these cathodes materials is based on the Li ion diffusion pathway in different structures. The principle challenge for Li-ion batteries is the development of functional materials that can offer higher energy, power, and lifetime than the currently existing materials. In order to achieve this goal, huge efforts were explored, including the development of Li-rich oxides or mixed transition metal layered oxides. Moreover, the origin of the factors affecting the battery lifetime, such as degradation of the materials upon cycling and aging, are of great importance as outlined in this review. Improving the performance of existing materials is possible through different strategies, including the creation of defects in the material, manipulation of nanostructured complexes, substitution with different transition metals, doping with foreign elements, and smart coatings. Furthermore, continued progress to safer batteries, clean green materials, and lower cost is required. To this end, modelling is a great tool to identify new cathode materials that can satisfy the balance between the electrochemical performance and environmental requirements. Moreover, recycling Li-ion batteries and recovery of their components to construct new devices should be adapted and widely investigated.
Conflicts of interest
There are no conflicts to declare.
References
- K. Kariatsumari, H. Kume, H. Yomogita and P. Keys, Nikkei Electronics Asia, February, 2010 Search PubMed.
- J. Molenda, in Li-ion Batteries for Electric Vehicles, Annales Universitatis Mariae Curie-Sklodowska, De Gruyter Open Sp. z oo, 2011, p. 23 Search PubMed.
- http://www.energystorageexchange.org/projects/data_visualization, accessed May 10, 2020.
- I. Yoon, S. Jurng, D. P. Abraham, B. L. Lucht and P. R. Guduru, Measurement of mechanical and fracture properties of solid electrolyte interphase on lithium metal anodes in lithium ion batteries, Energy Storage Materials, 2020, 25, 296–304 CrossRef.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling, Carbon, 2016, 105, 52–76 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries, Electrochim. Acta, 2010, 55(22), 6332–6341 CrossRef CAS.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J.-G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer and Y. Du, Real-time mass spectrometric characterization of the solid–electrolyte interphase of a lithium-ion battery, Nat. Nanotechnol., 2020, 15(3), 224–230 CrossRef CAS PubMed.
- G. M. Hobold, A. Khurram and B. M. Gallant, Operando Gas Monitoring of Solid Electrolyte Interphase Reactions on Lithium, Chem. Mater., 2020, 32(6), 2341–2352 CrossRef CAS.
- D. S. Adipranoto, T. Ishigaki, A. Hoshikawa, K. Iwase, M. Yonemura, K. Mori, T. Kamiyama, Y. Morii and M. Hayashi, Neutron diffraction studies on structural effect for Ni-doping in LiCo1−xNixO2, Solid State Ionics, 2014, 262, 92–97 CrossRef CAS.
- M. S. Whittingham, Lithium batteries and cathode materials, Chem. Rev., 2004, 104(10), 4271–4302 CrossRef CAS PubMed.
- J. Graetz, A. Hightower, C. C. Ahn, R. Yazami, P. Rez and B. Fultz, Electronic Structure of Chemically-Delithiated LiCoO2 Studied by Electron Energy-Loss Spectrometry, J. Phys. Chem. B, 2002, 106(6), 1286–1289 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, Electrochemical and In Situ X-Ray Diffraction Studies of Lithium Intercalation in LixCoO2, J. Electrochem. Soc., 1992, 139(8), 2091–2097 CrossRef CAS.
- W.-S. Yoon, K.-B. Kim, M.-G. Kim, M.-K. Lee, H.-J. Shin, J.-M. Lee, J.-S. Lee and C.-H. Yo, Oxygen Contribution on Li-Ion Intercalation−Deintercalation in LiCoO2 Investigated by O K-Edge and Co L-Edge X-ray Absorption Spectroscopy, J. Phys. Chem. B, 2002, 106(10), 2526–2532 CrossRef CAS.
- Y. Jiang, C. Qin, P. Yan and M. Sui, Origins of capacity and voltage fading of LiCoO2 upon high voltage cycling, J. Mater. Chem. A, 2019, 7(36), 20824–20831 RSC.
- S. Kalluri, M. Yoon, M. Jo, S. Park, S. Myeong, J. Kim, S. X. Dou, Z. Guo and J. Cho, Surface Engineering Strategies of Layered LiCoO2 Cathode Material to Realize High-Energy and High-Voltage Li-Ion Cells, Adv. Energy Mater., 2017, 7(1), 1601507 CrossRef.
- Y. J. Kim, H. Kim, B. Kim, D. Ahn, J.-G. Lee, T.-J. Kim, D. Son, J. Cho, Y.-W. Kim and B. Park, Electrochemical Stability of Thin-Film LiCoO2 Cathodes by Aluminum-Oxide Coating, Chem. Mater., 2003, 15(7), 1505–1511 CrossRef CAS.
- Y. Zhao, J. Li and J. R. Dahn, Interdiffusion
of Cations from Metal Oxide Surface Coatings into LiCoO2 During Sintering, Chem. Mater., 2017, 29(12), 5239–5248 CrossRef CAS.
- A. Yano, M. Shikano, A. Ueda, H. Sakaebe and Z. Ogumi, LiCoO2 Degradation Behavior in the High-Voltage Phase Transition Region and Improved Reversibility with Surface Coating, J. Electrochem. Soc., 2017, 164(1), A6116–A6122 CrossRef CAS.
- B. Shen, Q. Liu, L. Wang, G. Yin, P. Zuo, Y. Ma, X. Cheng, C. Du and Y. Gao, Mixed lithium ion and electron conducting LiAlPO3.93F1.07-coated LiCoO2 cathode with improved electrochemical performance, Electrochem. Commun., 2017, 83, 106–109 CrossRef CAS.
- M. Kato, T. Hayashi, G. Hasegawa, X. Lu, T. Miyazaki, Y. Matsuda, N. Kuwata, K. Kurihara and J. Kawamura, Electrochemical properties of LiCoO2 thin film surface modified by lithium tantalate and lithium niobate coatings, Solid State Ionics, 2017, 308, 54–60 CrossRef CAS.
- F. Zhao, Y. Tang, J. Wang, J. Tian, H. Ge and B. Wang, Vapor-assisted synthesis of Al2O3-coated LiCoO2 for high-voltage lithium ion batteries, Electrochim. Acta, 2015, 174, 384–390 CrossRef CAS.
- J. S. Park, A. U. Mane, J. W. Elam and J. R. Croy, Atomic Layer Deposition of Al–W–Fluoride on LiCoO2 Cathodes: Comparison of Particle- and Electrode-Level Coatings, ACS Omega, 2017, 2(7), 3724–3729 CrossRef CAS PubMed.
- Y. Zhou, Y. Lee, H. Sun, J. M. Wallas, S. M. George and M. Xie, Coating Solution for High-Voltage Cathode: AlF3 Atomic Layer Deposition for Freestanding LiCoO2 Electrodes with High Energy Density and Excellent Flexibility, ACS Appl. Mater. Interfaces, 2017, 9(11), 9614–9619 CrossRef CAS PubMed.
- J.-G. Lee, B. Kim, J. Cho, Y.-W. Kim and B. Park, Effect of AlPO4-Nanoparticle Coating Concentration on High-Cutoff-Voltage Electrochemical Performances in LiCoO2, J. Electrochem. Soc., 2004, 151(6), A801–A805 CrossRef CAS.
- X. Dai, A. Zhou, J. Xu, B. Yang, L. Wang and J. Li, Superior electrochemical performance of LiCoO2 electrodes enabled by conductive Al2O3-doped ZnO coating via magnetron sputtering, J. Power Sources, 2015, 298, 114–122 CrossRef CAS.
- H.-W. Ha, N. J. Yun, M. H. Kim, M. H. Woo and K. Kim, Enhanced electrochemical and thermal stability of surface-modified LiCoO2 cathode by CeO2 coating, Electrochim. Acta, 2006, 51(16), 3297–3302 CrossRef CAS.
- C. S. Yoon, D.-W. Jun, S.-T. Myung and Y.-K. Sun, Structural stability of LiNiO2 cycled above 4.2 V, ACS Energy Lett., 2017, 2(5), 1150–1155 CrossRef CAS.
- J. B. Goodenough, D. G. Wickham and W. J. Croft, Some magnetic and crystallographic properties of the system Li+xNi++1−2xNi+++xO, J. Phys. Chem. Solids, 1958, 5(1), 107–116 CrossRef CAS.
- L. D. Dyer, B. S. Borie and G. P. Smith, Alkali Metal-Nickel Oxides of the Type MNiO2, J. Am. Chem. Soc., 1954, 76(6), 1499–1503 CrossRef CAS.
- T. Ohzuku, A. Ueda and M. Nagayama, Electrochemistry and Structural Chemistry of LiNiO2 (Rm) for 4 Volt Secondary Lithium Cells, J. Electrochem. Soc., 1993, 140(7), 1862–1870 CrossRef CAS.
- W. Li, J. N. Reimers and J. R. Dahn, In situ x-ray diffraction and electrochemical studies of Li1−xNiO2, Solid State Ionics, 1993, 67(1), 123–130 CrossRef CAS.
- K.-K. Lee, W.-S. Yoon, K.-B. Kim, K.-Y. Lee and S.-T. Hong, Thermal behavior and the decomposition mechanism of electrochemically delithiated Li1−xNiO2, J. Power Sources, 2001, 97–98, 321–325 CrossRef CAS.
- H. Arai, S. Okada, Y. Sakurai and J.-i. Yamaki, Thermal behavior of Li1−yNiO2 and the decomposition mechanism, Solid State Ionics, 1998, 109(3), 295–302 CrossRef CAS.
- L. de Biasi, A. Schiele, M. Roca-Ayats, G. Garcia, T. Brezesinski, P. Hartmann and J. Janek, Phase Transformation Behavior and Stability of LiNiO2 Cathode Material for Li-Ion Batteries from In Situ Pressure and Gas Analysis and Operando X-Ray Diffraction, ChemSusChem, 2019, 12, 2240–2250 CrossRef CAS PubMed.
- J. R. Dahn, E. W. Fuller, M. Obrovac and U. von Sacken, Thermal stability of LixCoO2, LixNiO2 and λ-MnO2 and consequences for the safety of Li-ion cells, Solid State Ionics, 1994, 69(3), 265–270 CrossRef CAS.
- J. Cho, T.-J. Kim, Y. J. Kim and B. Park, High-performance ZrO2-coated LiNiO2 cathode material, Electrochem. Solid-State Lett., 2001, 4(10), A159–A161 CrossRef CAS.
- P. Mohan and G. P. Kalaignan, Electrochemical behaviour of surface modified SiO2-coated LiNiO2 cathode materials for rechargeable lithium-ion batteries, J. Nanosci. Nanotechnol., 2013, 13(4), 2765–2770 CrossRef CAS PubMed.
- S.-W. Zhong, Y.-J. Zhao, L. Fang, L. Yan, H. Yang, P.-Z. Li, M. Jia and Q.-G. Liu, Characteristics and electrochemical performance of cathode material Co-coated LiNiO2 for Li-ion batteries, Trans. Nonferrous Metals Soc. China, 2006, 16(1), 137–141 CrossRef CAS.
- X.-r. Deng, G.-r. Hu, K. Du, Z.-d. Peng, X.-G. Gao and Y.-N. Yang, Synthesis and electrochemical properties of Co, Mn-coated LiNiO2 lithium-ion battery cathode materials, Mater. Chem. Phys., 2008, 109(2–3), 469–474 CrossRef CAS.
- C. M. Julien, A. Amdouni, S. Castro-Garcia, M. Selmane and S. Rangan, LiCo1−yMyO2 positive electrodes for rechargeable lithium batteries: II. Nickel substituted materials grown by the citrate method, Materials Science and Engineering: B, 2006, 128(1), 138–150 CrossRef CAS.
- A. Rougier, I. Saadoune, P. Gravereau, P. Willmann and C. Delmasa, Effect of cobalt substitution on cationic distribution in LiNi1−yCoyO2 electrode materials, Solid State Ionics, 1996, 90(1), 83–90 CrossRef CAS.
- D. Caurant, N. Baffier, B. Garcia and J. P. Pereira-Ramos, Synthesis by a soft chemistry route and characterization of LiNixCo1−xO2 (0 ≤ x ≤ 1) cathode materials, Solid State Ionics, 1996, 91(1), 45–54 CrossRef CAS.
- J. Cho, H. Jung, Y. Park, G. Kim and H. S. Lim, Electrochemical Properties and Thermal Stability of Li a Ni1−xCOxO2 Cathode Materials, J. Electrochem. Soc., 2000, 147(1), 15–20 CrossRef CAS.
- Q. Xie, W. Li and A. Manthiram, A Mg-doped High-nickel Layered Oxide Cathode Enabling Safer, High-energy-density Li-ion Batteries, Chem. Mater., 2019, 31(3), 938–946 CrossRef CAS.
- H. Li, P. Zhou, F. Liu, H. Li, F. Cheng and J. Chen, Stabilizing nickel-rich layered oxide cathodes by magnesium doping for rechargeable lithium-ion batteries, Chem. Sci., 2019, 10(5), 1374–1379 RSC.
- C. S. Yoon, H.-H. Ryu, G.-T. Park, J.-H. Kim, K.-H. Kim and Y.-K. Sun, Extracting maximum capacity from Ni-rich Li[Ni0.95Co0.025Mn0.025]O2 cathodes for high-energy-density lithium-ion batteries, J. Mater. Chem. A, 2018, 6(9), 4126–4132 RSC.
- K.-S. Lee, S.-T. Myung, D.-W. Kim and Y.-K. Sun, AlF3-coated LiCoO2 and Li [Ni1/3Co1/3Mn1/3]O2 blend composite cathode for lithium ion batteries, J. Power Sources, 2011, 196(16), 6974–6977 CrossRef CAS.
- M.-H. Liu, C.-W. Jen, J.-M. Chen and S.-C. Liao, Improved Electrochemical Performance of Ni-rich Cathode Materials via Al Gradient Doping, Int. J. Electrochem. Sci., 2018, 13(5), 4350–4366 CrossRef CAS.
- H.-H. Sun and A. Manthiram, Impact of microcrack generation and surface degradation on a nickel-rich layered Li[Ni0.9Co0.05Mn0.05]O2 cathode for lithium-ion batteries, Chem. Mater., 2017, 29(19), 8486–8493 CrossRef CAS.
- J. M. Paulsen, C. L. Thomas and J. R. Dahn, Layered Li-Mn-Oxide with the O2 Structure: A Cathode Material for Li-Ion Cells which does not Convert to Spinel, J. Electrochem. Soc., 1999, 146(10), 3560–3565 CrossRef CAS.
- A. R. Armstrong and P. G. Bruce, Synthesis of layered LiMnO2 as an electrode for rechargeable lithium batteries, Nature, 1996, 381, 499 CrossRef CAS.
- G. Ceder and S. K. Mishra, The Stability of Orthorhombic and Monoclinic-Layered LiMnO2, Electrochem. Solid-State Lett., 1999, 2(11), 550–552 CrossRef CAS.
- A. R. Armstrong, N. Dupre, A. J. Paterson, C. P. Grey and P. G. Bruce, Combined Neutron Diffraction, NMR, and Electrochemical Investigation of the Layered-to-Spinel Transformation in LiMnO2, Chem. Mater., 2004, 16(16), 3106–3118 CrossRef CAS.
- B. Ammundsen, J. Desilvestro, T. Groutso, D. Hassell, J. B. Metson, E. Regan, R. Steiner and P. J. Pickering, Formation and Structural Properties of Layered LiMnO2 Cathode Materials, J. Electrochem. Soc., 2000, 147(11), 4078–4082 CrossRef CAS.
- R. Saroha, A. Gupta and A. K. Panwar, Electrochemical performances of Li-rich layered-layered Li2MnO3-LiMnO2 solid solutions as cathode material for lithium-ion batteries, J. Alloys Compd., 2017, 696, 580–589 CrossRef CAS.
- W.-S. Yoon, M. Balasubramanian, X.-Q. Yang, Z. Fu, D. A. Fischer and J. McBreen, Soft X-Ray Absorption Spectroscopic Study of a LiNi0.5Mn0.5O2 Cathode during Charge, J. Electrochem. Soc., 2004, 151(2), A246–A251 CrossRef CAS.
- A. Deb, U. Bergmann, S. P. Cramer and E. J. Cairns, Local structure of LiNi0.5Mn0.5O2 cathode material probed by in situ x-ray absorption spectroscopy, J. Appl. Phys., 2006, 99(6), 063701 CrossRef.
- W.-S. Yoon, S. Iannopollo, C. P. Grey, D. Carlier, J. Gorman, J. Reed and G. Ceder, Local Structure and Cation Ordering in O3 Lithium Nickel Manganese Oxides with Stoichiometry Li[NixMn(2−x)/3Li(1−2x)/3]O2: NMR Studies and First Principles Calculations, Electrochem. Solid-State Lett., 2004, 7(7), A167–A171 CrossRef CAS.
- Y. Hinuma, Y. S. Meng, K. Kang and G. Ceder, Phase Transitions in the LiNi0.5Mn0.5O2 System with Temperature, Chem. Mater., 2007, 19(7), 1790–1800 CrossRef CAS.
- H. Kobayashi, H. Sakaebe, H. Kageyama, K. Tatsumi, Y. Arachi and T. Kamiyama, Changes in the structure and physical properties of the solid solution LiNi1-xMnxO2 with variation in its composition, J. Mater. Chem., 2003, 13(3), 590–595 RSC.
- H. H. Li, N. Yabuuchi, Y. S. Meng, S. Kumar, J. Breger, C. P. Grey and Y. Shao-Horn, Changes in the Cation Ordering of Layered O3 LixNi0.5Mn0.5O2 during Electrochemical Cycling to High Voltages: An Electron Diffraction Study, Chem. Mater., 2007, 19(10), 2551–2565 CrossRef CAS.
- Y. S. Meng, G. Ceder, C. P. Grey, W.-S. Yoon and Y. Shao-Horn, Understanding the Crystal Structure of Layered LiNi0.5Mn0.5O2 by Electron Diffraction and Powder Diffraction Simulation, Electrochem. Solid-State Lett., 2004, 7(6), A155–A158 CrossRef CAS.
- W.-S. Yoon, Y. Paik, X.-Q. Yang, M. Balasubramanian, J. McBreen and C. P. Grey, Investigation of the Local Structure of the LiNi0.5Mn0.5O2 Cathode Material during Electrochemical Cycling by X-Ray Absorption and NMR Spectroscopy, Electrochem. Solid-State Lett., 2002, 5(11), A263–A266 CrossRef CAS.
- A. Van der Ven and G. Ceder, Ordering in Lix(Ni0.5Mn0.5)O2 and its relation to charge capacity and electrochemical behavior in rechargeable lithium batteries, Electrochem. Commun., 2004, 6(10), 1045–1050 CrossRef CAS.
- J. Bréger, Y. S. Meng, Y. Hinuma, S. Kumar, K. Kang, Y. Shao-Horn, G. Ceder and C. P. Grey, Effect of High Voltage on the Structure and Electrochemistry of LiNi0.5Mn0.5O2: A Joint Experimental and Theoretical Study, Chem. Mater., 2006, 18(20), 4768–4781 CrossRef.
- D. D. MacNeil, Z. Lu and J. R. Dahn, Structure and Electrochemistry of Li[NixCo1−2xMnx]O2 (0≤x≤1/2), J. Electrochem. Soc., 2002, 149(10), A1332–A1336 CrossRef CAS.
- Y. W. Tsai, B. J. Hwang, G. Ceder, H. S. Sheu, D. G. Liu and J. F. Lee, In-Situ X-ray Absorption Spectroscopic Study on Variation of Electronic Transitions and Local Structure of LiNi1/3Co1/3Mn1/3O2 Cathode Material during Electrochemical Cycling, Chem. Mater., 2005, 17(12), 3191–3199 CrossRef CAS.
- W.-S. Yoon, M. Balasubramanian, K. Y. Chung, X.-Q. Yang, J. McBreen, C. P. Grey and D. A. Fischer, Investigation of the Charge Compensation Mechanism on the Electrochemically Li-Ion Deintercalated Li1-xCo1/3Ni1/3Mn1/3O2 Electrode System by Combination of Soft and Hard X-ray Absorption Spectroscopy, J. Am. Chem. Soc., 2005, 127(49), 17479–17487 CrossRef CAS PubMed.
- M. G. Kim, H. J. Shin, J.-H. Kim, S.-H. Park and Y.-K. Sun, XAS Investigation of Inhomogeneous Metal-Oxygen Bond Covalency in Bulk and Surface for Charge Compensation in Li-Ion Battery Cathode Li[Ni1/3Co1/3Mn1/3]O2 Material, J. Electrochem. Soc., 2005, 152(7), A1320–A1328 CrossRef CAS.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, The significance of the Li2MnO3 component in ‘composite’ xLi2MnO3·(1−x)LiMn0.5Ni0.5O2 electrodes, Electrochem. Commun., 2004, 6(10), 1085–1091 CrossRef CAS.
- P. Kalyani, S. Chitra, T. Mohan and S. Gopukumar, Lithium metal rechargeable cells using Li2MnO3 as the positive electrode, J. Power Sources, 1999, 80(1), 103–106 CrossRef CAS.
- M. M. Thackeray, Manganese oxides for lithium batteries, Prog. Solid State Chem., 1997, 25(1), 1–71 CrossRef CAS.
- S. Hy, F. Felix, J. Rick, W.-N. Su and B. J. Hwang, Direct In situ Observation of Li2O Evolution on Li-Rich High-Capacity Cathode Material, Li[NixLi(1–2x)/3Mn(2–x)/3]O2 (0 ≤ x ≤0.5), J. Am. Chem. Soc., 2014, 136(3), 999–1007 CrossRef CAS PubMed.
- A. D. Robertson and P. G. Bruce, Mechanism of Electrochemical Activity in Li2MnO3, Chem. Mater., 2003, 15(10), 1984–1992 CrossRef CAS.
- H. Chen and M. S. Islam, Lithium Extraction Mechanism in Li-Rich Li2MnO3 Involving Oxygen Hole Formation and Dimerization, Chem. Mater., 2016, 28(18), 6656–6663 CrossRef CAS.
- J. Rana, M. Stan, R. Kloepsch, J. Li, G. Schumacher, E. Welter, I. Zizak, J. Banhart and M. Winter, Structural Changes in Li2MnO3 Cathode Material for Li-Ion Batteries, Adv. Energy Mater., 2014, 4(5), 1300998 CrossRef.
- M. Gu, A. Genc, I. Belharouak, D. Wang, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, Nanoscale Phase Separation, Cation Ordering, and Surface Chemistry in Pristine Li1.2Ni0.2Mn0.6O2 for Li-Ion Batteries, Chem. Mater., 2013, 25(11), 2319–2326 CrossRef CAS.
- Z. Lu, D. D. MacNeil and J. R. Dahn, Layered Cathode Materials Li[NixLi(1/3−2x/3)Mn(2/3−x/3)]O2 for Lithium-Ion Batteries, Electrochem. Solid-State Lett., 2001, 4(11), A191–A194 CrossRef CAS.
- Z. Lu, Z. Chen and J. R. Dahn, Lack of Cation Clustering in Li[NixLi1/3-2x/3Mn2/3-x/3]O2 (0 < x ≤ 1/2) and Li[CrxLi(1-x)/3Mn(2-2x)/3]O2 (0 < x < 1), Chem. Mater., 2003, 15(16), 3214–3220 CrossRef CAS.
- R. Wang, X. He, L. He, F. Wang, R. Xiao, L. Gu, H. Li and L. Chen, Atomic Structure of Li2MnO3 after Partial Delithiation and Re-Lithiation, Adv. Energy Mater., 2013, 3(10), 1358–1367 CrossRef CAS.
- S. Guo, H. Yu, P. Liu, X. Liu, D. Li, M. Chen, M. Ishida and H. Zhou, Surface coating of lithium-manganese-rich layered oxides with delaminated MnO2 nanosheets as cathode materials for Li-ion batteries, J. Mater. Chem. A, 2014, 2(12), 4422–4428 RSC.
- M. N. Ates, Q. Jia, A. Shah, A. Busnaina, S. Mukerjee and K. M. Abraham, Mitigation of Layered to Spinel Conversion of a Li-Rich Layered Metal Oxide Cathode Material for Li-Ion Batteries, J. Electrochem. Soc., 2014, 161(3), A290–A301 CrossRef CAS.
- F. Wu, N. Li, Y. Su, H. Shou, L. Bao, W. Yang, L. Zhang, R. An and S. Chen, Spinel/Layered Heterostructured Cathode Material for High-Capacity and High-Rate Li-Ion Batteries, Adv. Mater., 2013, 25(27), 3722–3726 CrossRef CAS PubMed.
- D. Mohanty, J. Li, D. P. Abraham, A. Huq, E. A. Payzant, D. L. Wood and C. Daniel, Unraveling the Voltage-Fade Mechanism in High-Energy-Density Lithium-Ion Batteries: Origin of the Tetrahedral Cations for Spinel Conversion, Chem. Mater., 2014, 26(21), 6272–6280 CrossRef CAS.
- Y. Xu, E. Hu, F. Yang, J. Corbett, Z. Sun, Y. Lyu, X. Yu, Y. Liu, X.-Q. Yang and H. Li, Structural integrity—Searching the key factor to suppress the voltage fade of Li-rich layered cathode materials through 3D X-ray imaging and spectroscopy techniques, Nano Energy, 2016, 28, 164–171 CrossRef CAS.
- Y. Xiang, Z. Sun, J. Li, X. Wu, Z. Liu, L. Xiong, Z. He, B. Long, C. Yang and Z. Yin, Improved electrochemical performance of Li1.2Ni0.2Mn0.6O2 cathode material for lithium ion batteries synthesized by the polyvinyl alcohol assisted sol-gel method, Ceram. Int., 2017, 43(2), 2320–2324 CrossRef CAS.
- Y.-X. Yin, S. Xin and Y.-G. Guo, Nanoparticles Engineering for Lithium-Ion Batteries, Part. Part. Syst. Charact., 2013, 30(9), 737–753 CrossRef CAS.
- L. Chen, Y. Su, S. Chen, N. Li, L. Bao, W. Li, Z. Wang, M. Wang and F. Wu, Hierarchical Li1.2Ni0.2Mn0.6O2 Nanoplates with Exposed {010} Planes as High-Performance Cathode Material for Lithium-Ion Batteries, Adv. Mater., 2014, 26(39), 6756–6760 CrossRef CAS PubMed.
- C. Yang, Y. Deng, M. Gao, X. Yang, X. Qin and G. Chen, High-rate and long-life performance of a truncated spinel cathode material with off-stoichiometric composition at elevated temperature, Electrochim. Acta, 2017, 225, 198–206 CrossRef CAS.
- K. R. Ragavendran, H. Xia, G. Yang, D. Vasudevan, B. Emmanuel, D. Sherwood and A. K. Arof, On the theory of high rate capability of LiMn2O4 with some preferred orientations: insights from the crystal shape algorithm, Phys. Chem. Chem. Phys., 2014, 16(6), 2553–2560 RSC.
- C. M. Sim, S. H. Choi and Y. C. Kang, Superior electrochemical properties of LiMn2O4 yolk-shell powders prepared by a simple
spray pyrolysis process, Chem. Commun., 2013, 49(53), 5978–5980 RSC.
- M. M. Thackeray, Y. Shao-Horn, A. J. Kahaian, K. D. Kepler, E. Skinner, J. T. Vaughey and S. A. Hackney, Structural Fatigue in Spinel Electrodes in High Voltage (4 V) Li/LixMn2O4 Cells, Electrochem. Solid-State Lett., 1998, 1(1), 7–9 CrossRef CAS.
- J. Choa and M. M. Thackeray, Structural Changes of LiMn2O4 Spinel Electrodes during Electrochemical Cycling, J. Electrochem. Soc., 1999, 146(10), 3577–3581 CrossRef.
- M. Hirayama, H. Ido, K. Kim, W. Cho, K. Tamura, J. i. Mizuki and R. Kanno, Dynamic Structural Changes at LiMn2O4/Electrolyte Interface during Lithium Battery Reaction, J. Am. Chem. Soc., 2010, 132(43), 15268–15276 CrossRef CAS PubMed.
- S. Lee, Y. Cho, H.-K. Song, K. T. Lee and J. Cho, Carbon-Coated Single-Crystal LiMn2O4 Nanoparticle Clusters as Cathode Material for High-Energy and High-Power Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2012, 51(35), 8748–8752 CrossRef CAS PubMed.
- J. Cho, G. B. Kim, H. S. Lim, C. S. Kim and S. I. Yoo, Improvement of Structural Stability of LiMn2O4 Cathode Material on 55°C Cycling by Sol-Gel Coating of LiCoO2, Electrochem. Solid-State Lett., 1999, 2(12), 607–609 CrossRef CAS.
- C. Zhang, X. Liu, Q. Su, J. Wu, T. Huang and A. Yu, Enhancing Electrochemical Performance of LiMn2O4 Cathode Material at Elevated Temperature by Uniform Nanosized TiO2 Coating, ACS Sustainable Chem. Eng., 2017, 5(1), 640–647 CrossRef CAS.
- Y.-K. Sun, K.-J. Hong and J. Prakash, The Effect of ZnO Coating on Electrochemical Cycling Behavior of Spinel LiMn2O4 Cathode Materials at Elevated Temperature, J. Electrochem. Soc., 2003, 150(7), A970–A972 CrossRef CAS.
- D.-Q. Liu, X.-Q. Liu and Z.-Z. He, The elevated temperature performance of LiMn2O4 coated with Li4Ti5O12 for lithium ion battery, Mater. Chem. Phys., 2007, 105(2), 362–366 CrossRef CAS.
- L. Xiao, Y. Zhao, Y. Yang, Y. Cao, X. Ai and H. Yang, Enhanced electrochemical stability of Al-doped LiMn2O4 synthesized by a polymer-pyrolysis method, Electrochim. Acta, 2008, 54(2), 545–550 CrossRef CAS.
- J. H. Lee, J. K. Hong, D. H. Jang, Y. K. Sun and S. M. Oh, Degradation mechanisms in doped spinels of LiM0.05Mn1.95O4 (M=Li, B, Al, Co, and Ni) for Li secondary batteries, J. Power Sources, 2000, 89(1), 7–14 CrossRef CAS.
- S.-J. Bao, Y.-Y. Liang, W.-J. Zhou, B.-L. He and H.-L. Li, Enhancement of the electrochemical properties of LiMn2O4 through Al3+ and F− co-substitution, J. Colloid Interface Sci., 2005, 291(2), 433–437 CrossRef CAS PubMed.
- G. Amatucci, A. Du Pasquier, A. Blyr, T. Zheng and J. M. Tarascon, The elevated temperature performance of the LiMn2O4/C system: failure and solutions, Electrochim. Acta, 1999, 45(1), 255–271 CrossRef CAS.
- G. Q. Liu, L. Wen and Y. M. Liu, Spinel LiNi0.5Mn1.5O4 and its derivatives as cathodes for high-voltage Li-ion batteries, J. Solid State Electrochem., 2010, 14(12), 2191–2202 CrossRef CAS.
- J. H. Kim, S. T. Myung, C. S. Yoon, S. G. Kang and Y. K. Sun, Comparative Study of LiNi0.5Mn1.5O4-δ and LiNi0.5Mn1.5O4 Cathodes Having Two Crystallographic Structures: Fdm and P4332, Chem. Mater., 2004, 16(5), 906–914 CrossRef CAS.
- R. Santhanam and B. Rambabu, Research progress in high voltage spinel LiNi0.5Mn1.5O4 material, J. Power Sources, 2010, 195(17), 5442–5451 CrossRef CAS.
- G. Rousse, J. Rodriguez-Carvajal, S. Patoux and C. Masquelier, Magnetic Structures of the Triphylite LiFePO4 and of its Delithiated Form FePO4, Chem. Mater., 2003, 15(21), 4082–4090 CrossRef CAS.
- J. Hong, F. Wang, X. Wang and J. Graetz, LiFexMn1−xPO4: a cathode for lithium-ion batteries, J. Power Sources, 2011, 196(7), 3659–3663 CrossRef CAS.
- Y.-M. Kang, Y.-I. Kim, M.-W. Oh, R.-Z. Yin, Y. Lee, D.-W. Han, H.-S. Kwon, J. H. Kim and G. Ramanath, Structurally stabilized olivine lithium phosphate cathodes with enhanced electrochemical properties through Fe doping, Energy Environ. Sci., 2011, 4(12), 4978–4983 RSC.
- R. Qing, M.-C. Yang, Y. S. Meng and W. Sigmund, Synthesis of LiNixFe1−xPO4 solid solution as cathode materials for lithium ion batteries, Electrochim. Acta, 2013, 108, 827–832 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries, J. Electrochem. Soc., 1997, 144(4), 1188–1194 CrossRef CAS.
- V. Srinivasan and J. Newman, Discharge Model for the Lithium Iron-Phosphate Electrode, J. Electrochem. Soc., 2004, 151(10), A1517–A1529 CrossRef CAS.
- S. Sun, T. Guan, B. Shen, K. Leng, Y. Gao, X. Cheng and G. Yin, Changes of degradation mechanisms of LiFePO4/graphite batteries cycled at different ambient temperatures, Electrochim. Acta, 2017, 237, 248–258 CrossRef CAS.
- Y. Zhu, J. W. Wang, Y. Liu, X. Liu, A. Kushima, Y. Liu, Y. Xu, S. X. Mao, J. Li and C. Wang, In situ atomic-scale imaging of phase boundary migration in FePO4 microparticles during electrochemical lithiation, Adv. Mater., 2013, 25(38), 5461–5466 CrossRef CAS PubMed.
- D. Li, D. L. Danilov, B. Zwikirsch, M. Fichtner, Y. Yang, R.-A. Eichel and P. H. Notten, Modeling the degradation mechanisms of C6/LiFePO4 batteries, J. Power Sources, 2018, 375, 106–117 CrossRef CAS.
- M. Herstedt, M. Stjerndahl, A. Nytén, T. Gustafsson, H. Rensmo, H. Siegbahn, N. Ravet, M. Armand, J. O. Thomas and K. Edström, Surface Chemistry of Carbon-Treated LiFePO4 Particles for Li-Ion Battery Cathodes Studied by PES, Electrochem. Solid-State Lett., 2003, 6(9), A202–A206 CrossRef CAS.
- H. Huang, S.-C. Yin and L. F. Nazar, Approaching Theoretical Capacity of LiFePO4 at Room Temperature at High Rates, Electrochem. Solid-State Lett., 2001, 4(10), A170–A172 CrossRef CAS.
- C. M. Julien, A. Mauger, A. Ait-Salah, M. Massot, F. Gendron and K. Zaghib, Nanoscopic scale studies of LiFePO4 as cathode material in lithium-ion batteries for HEV application, Ionics, 2007, 13(6), 395–411 CrossRef CAS.
- C. Delacourt, P. Poizot, S. Levasseur and C. Masquelier, Size Effects on Carbon-Free LiFePO4 Powders: The Key to Superior Energy Density, Electrochem. Solid-State Lett., 2006, 9(7), A352–A355 CrossRef CAS.
- S.-Y. Chung, J. T. Bloking and Y.-M. Chiang, Electronically conductive phospho-olivines as lithium storage electrodes, Nat. Mater., 2002, 1, 123 CrossRef CAS PubMed.
- P. S. Herle, B. Ellis, N. Coombs and L. F. Nazar, Nano-network electronic conduction in iron and nickel olivine phosphates, Nat. Mater., 2004, 3, 147 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, J. Ma, L. Yang, X. Li, E. Zhao, S. Fan, G. Xu, S. Yang and C. Yang, Synthesis of LiFePO4 Nanocomposite with Surface Conductive Phase by Zr Doping with Li Excess for Fast Discharging, J. Electrochem. Soc., 2019, 166(2), A410–A415 CrossRef CAS.
- C. M. Julien, K. Zaghib, A. Mauger and H. Groult, Enhanced electrochemical properties of LiFePO4 as positive electrode of Li-ion batteries for HEV application, Adv. Chem. Eng. Sci., 2012, 2(03), 321 CrossRef CAS.
- V. Aravindan, J. Gnanaraj, Y.-S. Lee and S. Madhavi, LiMnPO4 - a next generation cathode material for lithium-ion batteries, J. Mater. Chem. A, 2013, 1(11), 3518–3539 RSC.
- H. Wang, Y. I. Jang and Y. M. Chiang, Origin of Cycling Stability in Monoclinic- and Orthorhombic-Phase Lithium Manganese Oxide Cathodes, Electrochem. Solid-State Lett., 1999, 2(10), 490–493 CrossRef CAS.
- T. Shiratsuchi, S. Okada, T. Doi and J.-i. Yamaki, Cathodic performance of LiMn1−xMxPO4 (M=Ti, Mg and Zr) annealed in an inert atmosphere, Electrochim. Acta, 2009, 54(11), 3145–3151 CrossRef CAS.
- D. Wang, C. Ouyang, T. Drézen, I. Exnar, A. Kay, N.-H. Kwon, P. Gouerec, J. H. Miners, M. Wang and M. Grätzel, Improving the Electrochemical Activity of LiMnPO4 via Mn-Site Substitution, J. Electrochem. Soc., 2010, 157(2), A225–A229 CrossRef CAS.
- C. Hu, H. Yi, H. Fang, B. Yang, Y. Yao, W. Ma and Y. Dai, Improving the electrochemical activity of LiMnPO4 via Mn-site co-substitution with Fe and Mg, Electrochem. Commun., 2010, 12(12), 1784–1787 CrossRef CAS.
- S. K. Martha, J. Grinblat, O. Haik, E. Zinigrad, T. Drezen, J. H. Miners, I. Exnar, A. Kay, B. Markovsky and D. Aurbach, LiMn0.8Fe0.2PO4: An Advanced Cathode Material for Rechargeable Lithium Batteries, Angew. Chem., Int. Ed., 2009, 48(45), 8559–8563 CrossRef CAS PubMed.
- S.-M. Oh, H.-G. Jung, C. S. Yoon, S.-T. Myung, Z. Chen, K. Amine and Y.-K. Sun, Enhanced electrochemical performance of carbon–LiMn1−xFexPO4 nanocomposite cathode for lithium-ion batteries, J. Power Sources, 2011, 196(16), 6924–6928 CrossRef CAS.
- I. Seo, B. Senthilkumar, K.-H. Kim, J.-K. Kim, Y. Kim and J.-H. Ahn, Atomic structural and electrochemical impact of Fe substitution on nano porous LiMnPO4, J. Power Sources, 2016, 320, 59–67 CrossRef CAS.
- H. Fang, E. Dai, B. Yang, Y. Yao and W. Ma, LiMn0.8Fe0.19Mg0.01PO4/C as a high performance cathode material for lithium ion batteries, J. Power Sources, 2012, 204, 193–196 CrossRef CAS.
- S. Liu, H. Fang, E. Dai, B. Yang, Y. Yao, W. Ma and Y. Dai, Effect of carbon content on properties of LiMn0.8Fe0.19Mg0.01PO4/C composite cathode for lithium ion batteries, Electrochim. Acta, 2014, 116, 97–102 CrossRef CAS.
- X. Wu, M. Meledina, H. Tempel, H. Kungl, J. Mayer and R.-A. Eichel, Morphology-controllable synthesis of LiCoPO4 and its influence on electrochemical performance for high-voltage lithium ion batteries, J. Power Sources, 2020, 450, 227726 CrossRef CAS.
- J. Wolfenstine and J. Allen, LiNiPO4–LiCoPO4 solid solutions as cathodes, J. Power Sources, 2004, 136(1), 150–153 CrossRef CAS.
- H. N. Girish and G. Q. Shao, Advances in high-capacity Li2MSiO4 (M[space] = Mn, Fe, Co, Ni, …) cathode materials for lithium-ion batteries, RSC Adv., 2015, 5(119), 98666–98686 RSC.
- A. Nytén, A. Abouimrane, M. Armand, T. Gustafsson and J. O. Thomas, Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material, Electrochem. Commun., 2005, 7(2), 156–160 CrossRef.
- A. Nyten, S. Kamali, L. Haggstrom, T. Gustafsson and J. O. Thomas, The lithium extraction/insertion mechanism in Li2FeSiO4, J. Mater. Chem., 2006, 16(23), 2266–2272 RSC.
- S.-i. Nishimura, S. Hayase, R. Kanno, M. Yashima, N. Nakayama and A. Yamada, Structure of Li2FeSiO4, J. Am. Chem. Soc., 2008, 130(40), 13212–13213 CrossRef CAS PubMed.
- C. Sirisopanaporn, A. Boulineau, D. Hanzel, R. Dominko, B. Budic, A. R. Armstrong, P. G. Bruce and C. Masquelier, Crystal Structure of a New Polymorph of Li2FeSiO4, Inorg. Chem., 2010, 49(16), 7446–7451 CrossRef CAS PubMed.
- C. Sirisopanaporn, C. Masquelier, P. G. Bruce, A. R. Armstrong and R. Dominko, Dependence of Li2FeSiO4 Electrochemistry on Structure, J. Am. Chem. Soc., 2011, 133(5), 1263–1265 CrossRef CAS PubMed.
- M. E. Arroyo-deDompablo, R. Dominko, J. M. Gallardo-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik and E. Morán, On the Energetic Stability and Electrochemistry of Li2MnSiO4 Polymorphs, Chem. Mater., 2008, 20(17), 5574–5584 CrossRef CAS.
- R. Dominko, Li2MSiO4 (M=Fe and/or Mn) cathode materials, J. Power Sources, 2008, 184(2), 462–468 CrossRef CAS.
- D. Santamaría-Pérez, U. Amador, J. Tortajada, R. Dominko and M. E. Arroyo-de Dompablo, High-Pressure Investigation of Li2MnSiO4 and Li2CoSiO4 Electrode Materials for Lithium-Ion Batteries, Inorg. Chem., 2012, 51(10), 5779–5786 CrossRef PubMed.
- M. E. Arroyo y de Dompablo, J. M. Gallardo-Amores, J. García-Martínez, E. Morán, J. M. Tarascon and M. Armand, Is it possible to prepare olivine-type LiFeSiO4?: a joint computational and experimental investigation, Solid State Ionics, 2008, 179(27), 1758–1762 CrossRef CAS.
- P. Ghosh, S. Mahanty and R. N. Basu, Improved Electrochemical Performance of Li2MnSiO4/C Composite Synthesized by Combustion Technique, J. Electrochem. Soc., 2009, 156(8), A677–A681 CrossRef CAS.
- Y. Feng, R. Ji, Z. Ding, D. Zhang, C. Liang, L. Chen, D. G. Ivey and W. Wei, Understanding the Improved Kinetics and Cyclability of a Li2MnSiO4 Cathode with Calcium Substitution, Inorg. Chem., 2018, 57(6), 3223–3231 CrossRef CAS PubMed.
- H.-M. Cheng, S.-X. Zhao, X. Wu, J.-W. Zhao, L. Wei and C.-W. Nan, Synthesis and structural stability of Cr-doped Li2MnSiO4/C cathode materials by solid-state method, Appl. Surf. Sci., 2018, 433, 1067–1074 CrossRef CAS.
- X. Wu, S.-X. Zhao, L. Wei, H. Deng and C.-W. Nan, Improving the structure stability and electrochemical performance of Li2MnSiO4/C cathode materials by Ti-doping and porous microstructure, J. Alloys Compd., 2018, 735, 1158–1166 CrossRef CAS.
- S.-N. Lee, S. Baek, S. Amaresh, V. Aravindan, K. Y. Chung, B. W. Cho, W.-S. Yoon and Y.-S. Lee, Cu–Li2MnSiO4-polyaniline composite hybrids as high performance cathode for lithium batteries, J. Alloys Compd., 2015, 630, 292–298 CrossRef CAS.
- C. Wang, Y. Xu, B. Zhang and X. Ma, Dual-site magnesium doping in Li2MnSiO4/C/rGO cathode material for lithium-ion batteries, Solid State Ionics, 2019, 338, 39–46 CrossRef CAS.
- W. Liu, Y. Xu and R. Yang, Synthesis, characterization and electrochemical
performance of Li2MnSiO4/C cathode material by solid-state reaction, J. Alloys Compd., 2009, 480(2), L1–L4 CrossRef CAS.
- X. Li, Y. Liu, Z. Xiao, W. Guo and R. Zhang, Ionothermal synthesis and characterization of Li2MnSiO4/C composites as cathode materials for lithium-ion batteries, Ceram. Int., 2014, 40(1, part A), 289–296 CrossRef CAS.
- R. Liu, J. Chen, Z. Li, Q. Ding, X. An, Y. Pan, Z. Zheng, M. Yang and D. Fu, Preparation of LiFePO4/C Cathode Materials via a Green Synthesis Route for Lithium-Ion Battery Applications, Materials, 2018, 11(11), 2251 CrossRef PubMed.
- S. Bolloju, R. Rohan, S.-T. Wu, H.-X. Yen, G. D. Dwivedi, Y. A. Lin and J.-T. Lee, A green and facile approach for hydrothermal synthesis of LiFePO4 using iron metal directly, Electrochim. Acta, 2016, 220, 164–168 CrossRef CAS.
- W. Liu, Q. Liu, M. Qin, L. Xu and J. Deng, Inexpensive and green synthesis of multi-doped LiFePO4/C composites for lithium-ion batteries, Electrochim. Acta, 2017, 257, 82–88 CrossRef CAS.
- M. Pagliaro and F. Meneguzzo, Lithium battery reusing and recycling: a circular economy insight, Heliyon, 2019, 5(6), e01866 CrossRef PubMed.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich and S. Lambert, Recycling lithium-ion batteries from electric vehicles, Nature, 2019, 575(7781), 75–86 CrossRef CAS PubMed.
- Y. Shi, G. Chen and Z. Chen, Effective regeneration of LiCoO2 from spent lithium-ion batteries: a direct approach towards high-performance active particles, Green Chem., 2018, 20(4), 851–862 RSC.
- E. A. Aboelazm, G. A. Ali, H. Algarni, H. Yin, Y. L. Zhong and K. F. Chong, Magnetic electrodeposition of the hierarchical cobalt oxide nanostructure from spent lithium-ion batteries: its application as a supercapacitor electrode, J. Phys. Chem. C, 2018, 122(23), 12200–12206 CrossRef CAS.
- Y. I. Mesbah, N. Ahmed, B. A. Ali and N. K. Allam, Recycling of Li−Ni−Mn−Co Hydroxide from Spent Batteries to Produce High-Performance Supercapacitors with Exceptional Stability, ChemElectroChem, 2020, 7(4), 975–982 CrossRef CAS.
- M. C. A. Gonçalves, E. M. Garcia, H. A. Taroco, H. F. Gorgulho, J. O. Melo, R. R. Silva and A. G. Souza, Chemical recycling of cell phone Li-ion batteries: application in environmental remediation, Waste Manag., 2015, 40, 144–150 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*


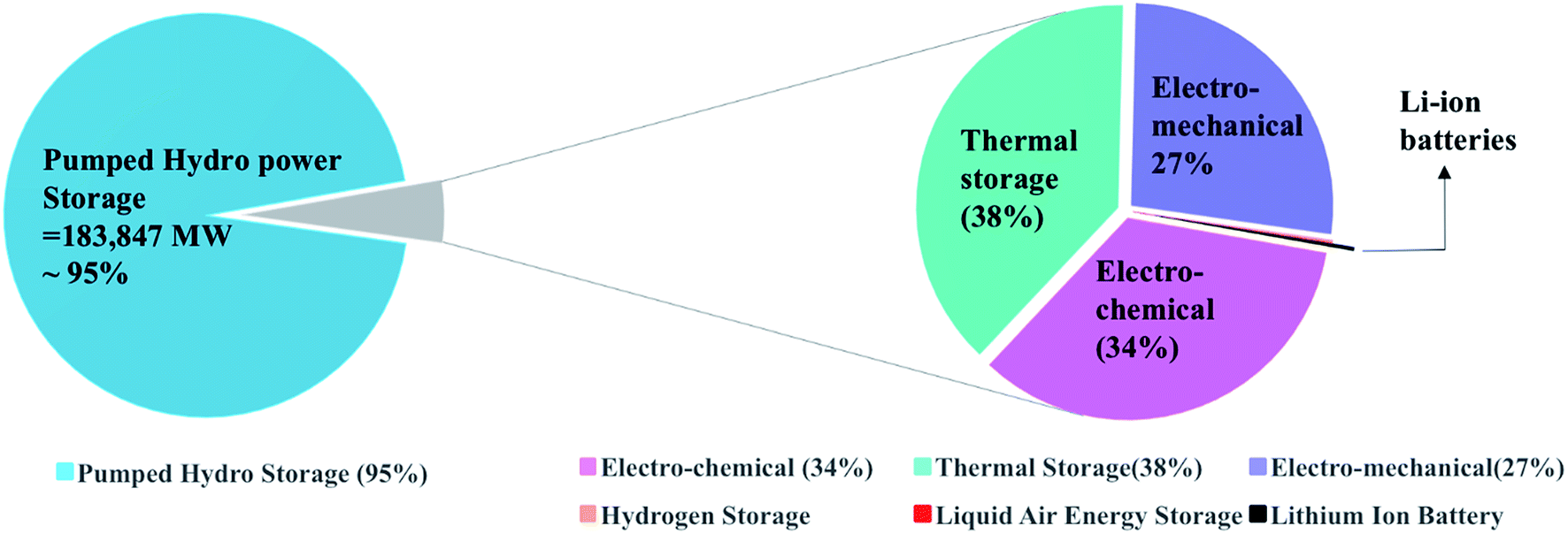
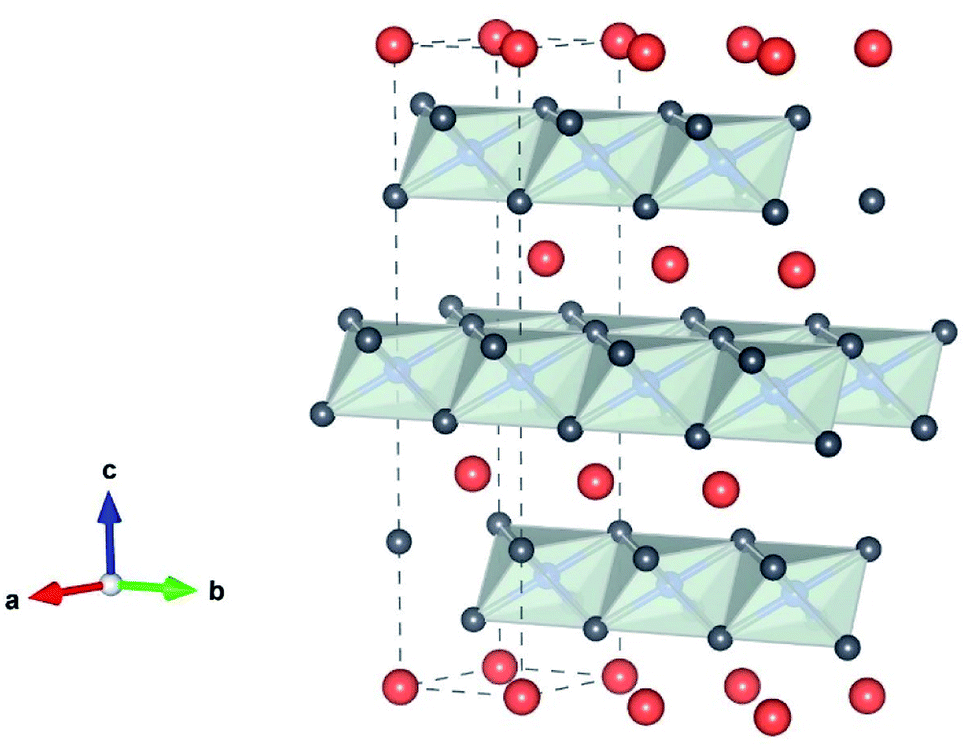
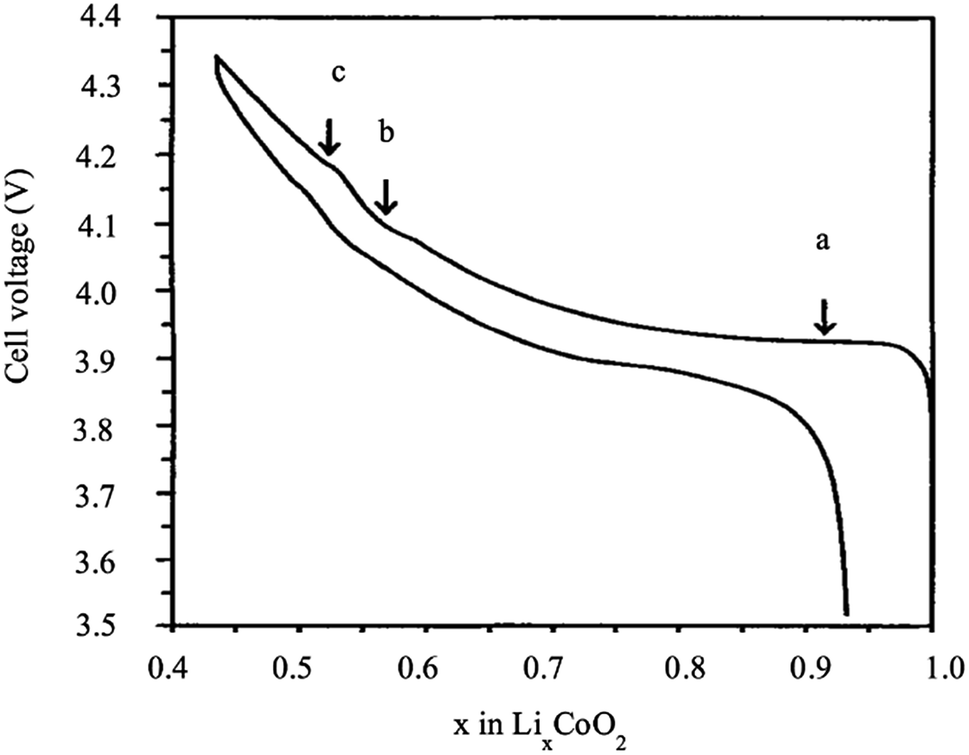
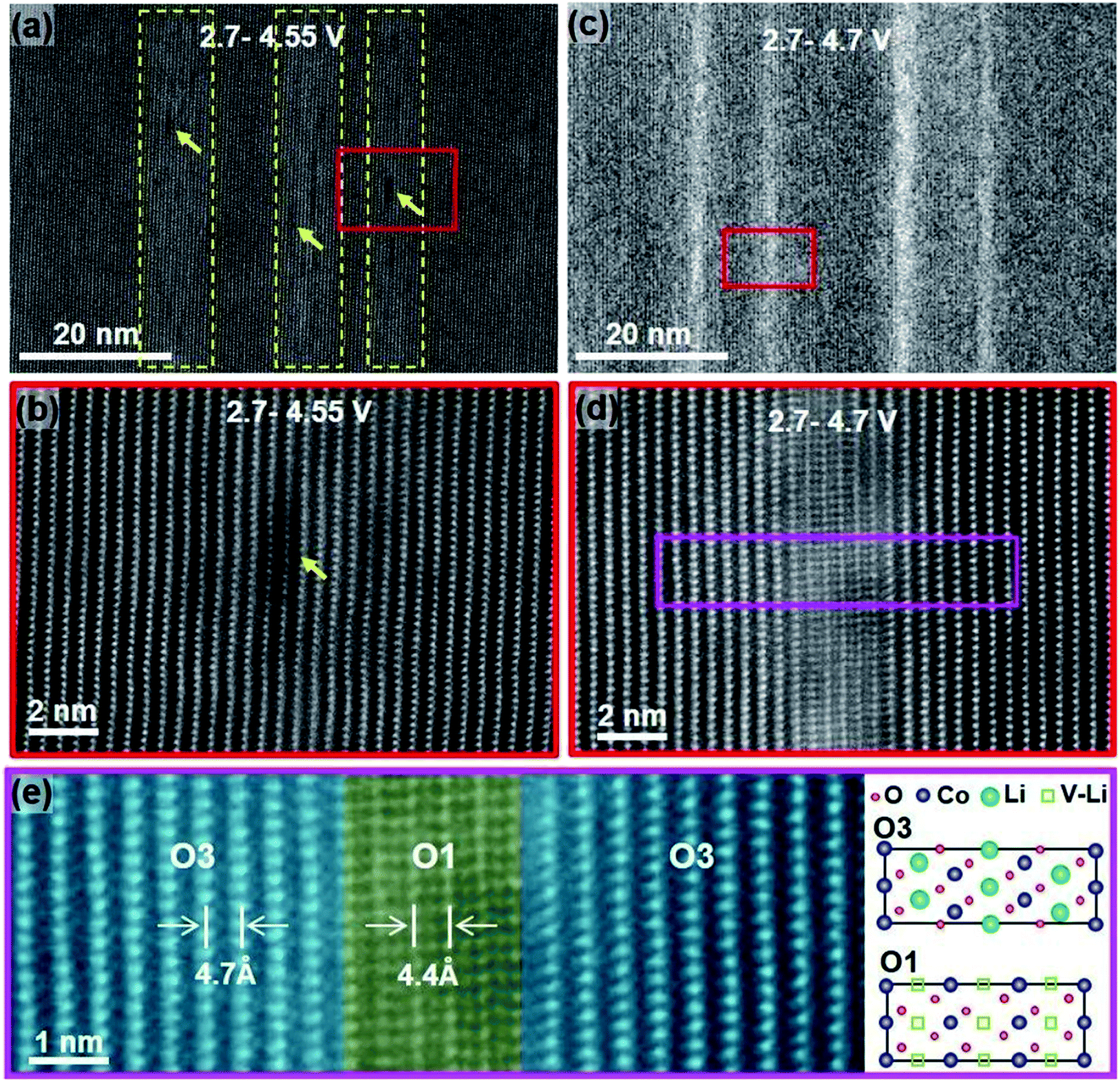

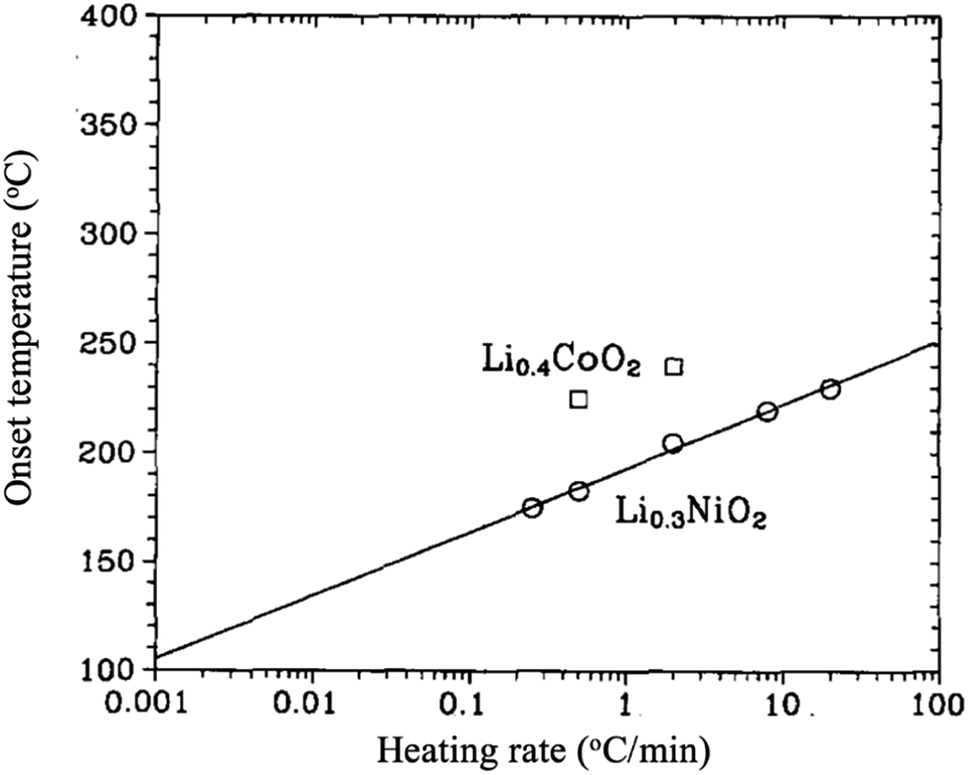
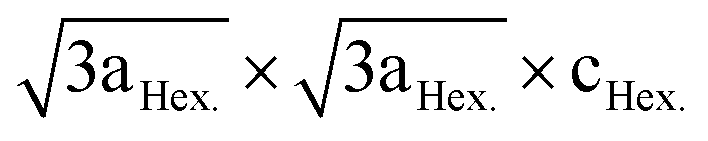 and correspond to a trigonal structure with the space group P3112.61,62 Investigation of the local structure upon charging suggested the disappearance of Li+ located in the transition metal layer very early during the first charge.63 Li+ migrates to the tetrahedral sites that share faces with vacant octahedral sites in the transition metal layer. The presence of Li in the tetrahedral sites reflects negatively on the electrochemical performance of the material. Because Litet+ is very energetically stable and requires high potential to be removed, which decreased the capacity obtained in the practical voltage range.64 Regarding the Ni ions in the Li layer, by charging above 4.6 V, 75% of NiLi ions migrate to vacant Li positions in the transition metal (TM) layer.65 The migration of Ni ions to the TM layer is partly a reversible process and reflects on the weaken or disappearance of the
and correspond to a trigonal structure with the space group P3112.61,62 Investigation of the local structure upon charging suggested the disappearance of Li+ located in the transition metal layer very early during the first charge.63 Li+ migrates to the tetrahedral sites that share faces with vacant octahedral sites in the transition metal layer. The presence of Li in the tetrahedral sites reflects negatively on the electrochemical performance of the material. Because Litet+ is very energetically stable and requires high potential to be removed, which decreased the capacity obtained in the practical voltage range.64 Regarding the Ni ions in the Li layer, by charging above 4.6 V, 75% of NiLi ions migrate to vacant Li positions in the transition metal (TM) layer.65 The migration of Ni ions to the TM layer is partly a reversible process and reflects on the weaken or disappearance of the 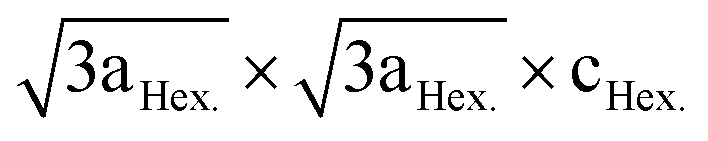 c supercell. Simply, in other words, the Ni2+/Li+ mixing was decreased by cycling compared to pristine LiNi0.5Mn0.5O2.
c supercell. Simply, in other words, the Ni2+/Li+ mixing was decreased by cycling compared to pristine LiNi0.5Mn0.5O2.
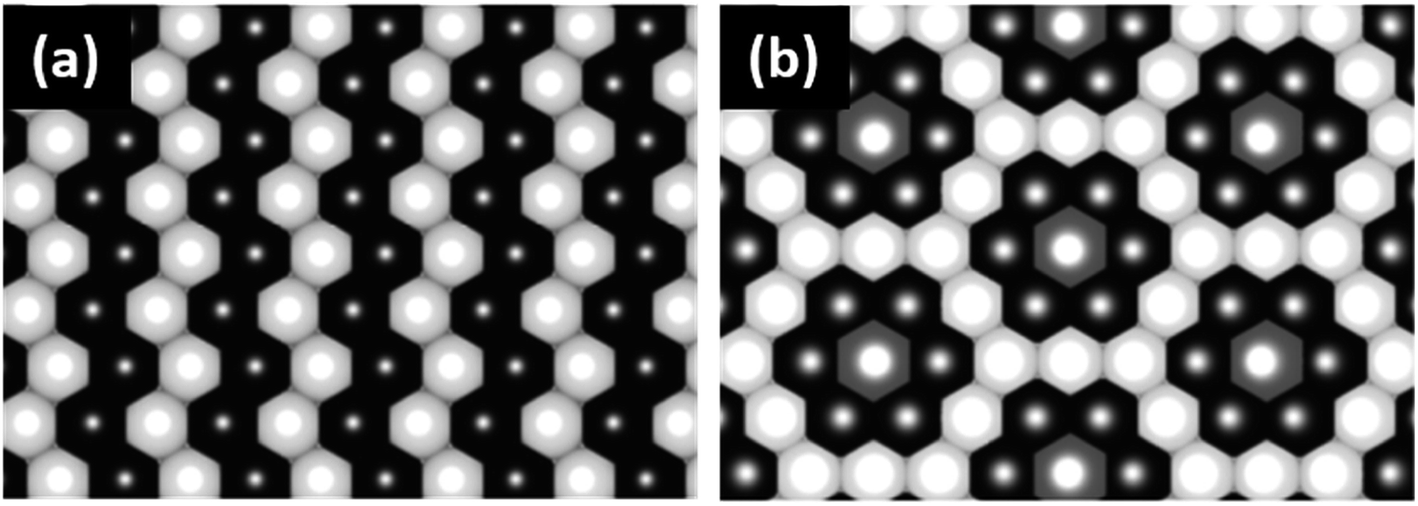
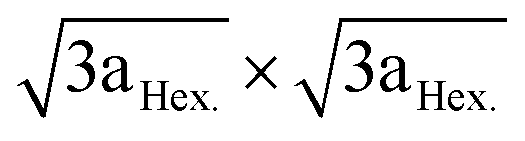 super structure.79 It exhibits high voltage during charging (>4.5 V), high specific capacity (>250 mA h g−1), and high energy density.70,80,81 The electrochemical behavior of Li-rich, layered oxides is divided into two stages. The first is before 4.4 V when the Li+ is extracted from LiMO2. The second starts at 4.5 V when the Li2O is extracted to activate Li2MnO3. The Li-rich, layered oxides suffer from some drawbacks such as an irreversible capacity loss due to the irreversible extraction of Li2O and the reaction with electrolyte at high potential, poor rate capability due to high charge transfer resistance and a voltage fade upon cycling. The origin of the voltage fade was suggested to be the transition from layered to spinel, which is accompanied by an irreversible transition metals migration to the Li-ions layers.82–85 Despite the fact that the nanomaterials with the high surface area are known for their undesired side reactions with the electrolyte, reduction of particle size of Li-rich materials to the nanoscale shows a better rate capability and a decrease in the diffusion path of Li ions.86,87 Moreover, decreasing the interface area between the electrode and the electrolyte by controlling the morphology of the nanomaterials could suppress the erosion from the electrolyte and improve the cycling performance. Taking Li1.2Ni0.2Mn0.6O2 as an example, hierarchical nanoplates structure shows outstanding rate capability as shown in Fig. 8, with specific capacity of ∼231, 216.5, 188, 163 and 142 mA h g−1 at as high rates as 1C, 2C, 5C, 10C, and 20C.87 Furthermore, it shows high capacity retention of 95.5% and 86.6% after 60 cycles for 1C and 2C rates, respectively. The classical voltage decay of Li-rich material was still observed due to the transition to spinel. An expected significant decrease in capacity retention to 68% occurred when the hierarchical structure was damaged by ultrasonic treatment.
super structure.79 It exhibits high voltage during charging (>4.5 V), high specific capacity (>250 mA h g−1), and high energy density.70,80,81 The electrochemical behavior of Li-rich, layered oxides is divided into two stages. The first is before 4.4 V when the Li+ is extracted from LiMO2. The second starts at 4.5 V when the Li2O is extracted to activate Li2MnO3. The Li-rich, layered oxides suffer from some drawbacks such as an irreversible capacity loss due to the irreversible extraction of Li2O and the reaction with electrolyte at high potential, poor rate capability due to high charge transfer resistance and a voltage fade upon cycling. The origin of the voltage fade was suggested to be the transition from layered to spinel, which is accompanied by an irreversible transition metals migration to the Li-ions layers.82–85 Despite the fact that the nanomaterials with the high surface area are known for their undesired side reactions with the electrolyte, reduction of particle size of Li-rich materials to the nanoscale shows a better rate capability and a decrease in the diffusion path of Li ions.86,87 Moreover, decreasing the interface area between the electrode and the electrolyte by controlling the morphology of the nanomaterials could suppress the erosion from the electrolyte and improve the cycling performance. Taking Li1.2Ni0.2Mn0.6O2 as an example, hierarchical nanoplates structure shows outstanding rate capability as shown in Fig. 8, with specific capacity of ∼231, 216.5, 188, 163 and 142 mA h g−1 at as high rates as 1C, 2C, 5C, 10C, and 20C.87 Furthermore, it shows high capacity retention of 95.5% and 86.6% after 60 cycles for 1C and 2C rates, respectively. The classical voltage decay of Li-rich material was still observed due to the transition to spinel. An expected significant decrease in capacity retention to 68% occurred when the hierarchical structure was damaged by ultrasonic treatment.