
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuEChERS pretreatment combined with high-performance liquid chromatography-tandem mass spectrometry for determination of aristolochic acids I and II in Chinese herbal patent medicines
Jinghe
Zhang
ab,
Yinan
Wang
 *a,
Jing
Sun
b,
Guowei
Zhou
a,
Xiaojie
Jiang
a and
Xikui
Wang
*bc
*a,
Jing
Sun
b,
Guowei
Zhou
a,
Xiaojie
Jiang
a and
Xikui
Wang
*bc
aKey Laboratory of Fine Chemicals in Universities of Shandong, School of Chemistry and Pharmaceutical Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250100, China. E-mail: wangyn@qlu.edu.cn; Tel: +86-531-89631696
bSchool of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, China
cSchool of Environmental Science and Engineering, Shandong Agriculture and Engineering University, Jinan 250100, China
First published on 3rd July 2020
Abstract
Aristolochic acid I and II (AA I and II), a kind of nephrotoxic and carcinogenic compound, are widely added in Chinese herbal patent medicines though they have been banned due to their toxicity. However, the traditional sample pre-treatment combined with the LC-MS analysis system is not effective to determine AAs in such complicated patent medicines. The QuEChERS pretreatment method possesses some merits such as being quick and effective. In this work, the modified QuEChERS method was first used to determine AA I and II in Chinese herbal patent medicines combined with the HPLC-MS/MS analysis system. Extraction and removal of target analytes from powder, tablet, and capsule samples were conducted using the modified QuEChERS pretreatment. The liquid extracts of Chinese herbal patent medicines could be analyzed directly. The method optimization results show that average recoveries ranged from 96.6% to 110.3% with relative standard deviations ranging from 4.2% to 13.0%. The quantization limits of the three selected matrices are estimated as follows (AA I/II): 2.8/6.5 ng mL−l in liquid herbal extract, 6.5/12.5 ng g−1 in tablets, and 22.1/42.1 ng g−1 in capsules. This method was conducted to investigate the presence of AAs, which are a type of nephrotoxic and carcinogenic carboxylic acid, in 30 herbal products sold through the Internet in China. AA I and II were detected in 53% and 20%, respectively, of tested samples.
1 Introduction
Aristolochic acids (AAs) are a type of nephrotoxic and carcinogenic carboxylic acid produced naturally by Aristolochia and Asarum plants, which are classified as Group I human carcinogens by the International Agency for Research on Cancer.1,2 The major components of AAs are 8-methoxy-6-nitro-phenanthro-(3,4-d)-1,3-dioxolo-5-carboxylic acid (AA I) and 6-nitro-phenanthro-(3,4-d)-1,3-dioxolo-5-carboxylic acid (AA II), which are derived from herbaceous Chinese medicines, such as Aristolochia manshuriensis and Aristolochia fructus (Fig. 1).3,4 The application of AA-containing herbs has been banned for decades because of their reported strong nephrotoxicity and carcinogenicity in rats.5–8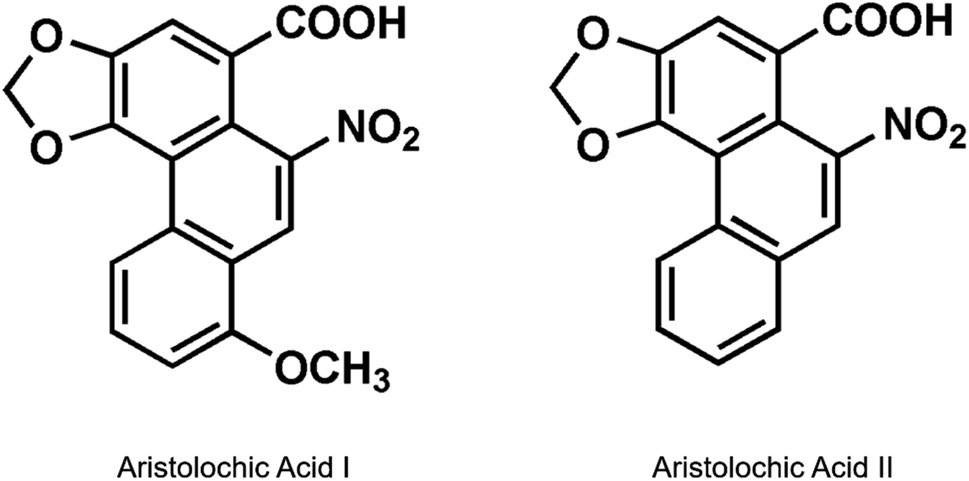 | ||
| Fig. 1 Chemical structures of aristolochic acid I and II. | ||
Contemporary medicines extracted from Aristolochia plants caused approximately 100 cases of renal disease in Belgium in 1991.3,9 The tragedy could be attributed to the accidental substitution of Stephania tetrandra (Hanfangji or Fengangji) with Aristolochia fangchi (Guangfangji) because both are called Fangji in Chinese.9 Patients who took this pill for a prolonged period suffered from a unique type of rapid progressive renal fibrosis, and most of them needed renal replacement therapy, such as kidney transplantation.10 This disease was ultimately designated as Chinese herb nephropathy or AA nephropathy.11,12 Apart from renal failure, AA could lead to a chronic dietary poisoning called Balkan endemic nephropathy and urothelial cancer in humans.13–16 Furthermore, AAs and its derivatives are believed to be widely associated with liver cancer in Taiwan and even in the whole Asia. Recently, another study provided supporting evidence for the mutation process of AA induced liver cancer in the process of malignant clone evolution, and laid a solid foundation for the prevention and diagnosis of AA related human cancers.17,18 AAs have been listed as a Group I carcinogen by the International Agency for Research on Cancer, which is classified as a member of the most potent carcinogens in the Carcinogenic Potency Database because of its strong toxicity. Consequently, medicines containing AAs have been prohibited from sale and adoption in several countries.19,20
However, some Chinese herbal patent medicines containing Aristolochia, Asarum or other ingredients are still sold in websites, which may be incorrectly identified as aristolochic acid or replaced by aristolochic acid. Meanwhile, the traditional sample pre-treatment combining with LC-MS analysis system is not effective to determine AAs in such a complicated patent medicines which contain plenty of ingredients as pill or tablet. Therefore, a sensitive and selective method is needed to analyze the potential existence of aristolochic acid in suspicious products.
In this contribution, a quick, easy, cheap, effective, rugged, and safe (QuEChERS) method 21–26 was modified and was first used to pre-treat various Chinese herbal patent medicines for quantitating AA I and II. Firstly, acetonitrile was used as the extraction solution, and then sodium chloride/anhydrous magnesium sulfates were added to facilitate the separation of solvent and water phase. The solvent is then purified with anhydrous magnesium sulfate and used for UPLC-MS analysis directly.27–29 Compared with most traditional methods, the modified QuEChERS method has the following advantages. (1) This method has a high spike recovery (>85%) for most polar and volatile compounds, and (2) internal standard materials are used for calibration to solve the differences in matrix composition and moisture content, so as to ensure the accuracy and precision of the results. (3) It can reduce the amount of solvent needed, save the cost, do not need the solvent containing chloride, and reduce the emission of pollutants. (4) Finally, the operator's exposure to organic reagents is reduced.23,30–32 Then, the optimized method was used to detect 30 kinds of products sold through the Internet to provide data for the content of AAs in this kind of Chinese herbal patent medicines. These data may help to assess the risks associated with taking these proprietary Chinese herbal patent medicines. And this work could serve as a modest spur to extend the application of QuEChERS method to induce more valuable forward investigation.
2 Materials and methods
2.1 Chemicals
All chemicals used in this work are of the highest purity without further purification unless noted otherwise. Analytical grade AA I (≥97% purity) and II (≥95% purity) were obtained from Sigma-Aldrich. Primary and secondary amine (PSA) sorbents were purchased from Agela Technologies. MgSO4 and NaCl are of analytical grade and brought from Sigma-Aldrich. High-performance liquid chromatography grade CH3CN and CH3OH were purchased from Tedia. Water used in this work was purified by a Milli-Q ultrapure water purification system.2.2 Sample preparation
All the samples of Chinese herbal patent medicines supplied in the form of tablets, capsules, liquid extracts, and powdered extracts were collected via the internet or from pharmacies in China in 2019. All samples were processed with the QuEChERS sample extraction method as follows. A dispersive solid-phase extraction (d-SPE) system was developed and used in this study. The shell of capsules was removed before homogenization. Tablets, powder, and pills were homogenized directly using a pulverizer. In brief, 1.00 ± 0.01 g of homogenized samples were weighed in a 50 mL tube and added with 10 mL acetonitrile. For liquid samples, 1 mL of electuary was transferred to a 50 mL tube and diluted to 10 mL with acetonitrile. Then, the dispersion was ultrasonicated at room temperature for 30 min. Thereafter, 1 g NaCl and 4 g MgSO4 were added into the dispersion and mixed thoroughly using a vortex mixer. Then, d-SPE clean-up was conducted as follows. The dispersion was centrifuged at 3800 rpm for 5 min, and 1 mL of the supernatant was transferred into a 1.5 mL centrifuge tube containing 30 mg PSA sorbent and 150 mg MgSO4 powder. The mixture was vortex-mixed for 1 min and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 rpm for 10 min to settle the PSA. The supernatant was isolated and dried under nitrogen flow, and then internal standard benz[cd]indol-2(1H)-one (0.3 μg mL−1 in methanol, 50 μL) and methanol (450 μL) were added to the residues for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
800 rpm for 10 min to settle the PSA. The supernatant was isolated and dried under nitrogen flow, and then internal standard benz[cd]indol-2(1H)-one (0.3 μg mL−1 in methanol, 50 μL) and methanol (450 μL) were added to the residues for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
2.3 LC-MS/MS analysis
LC separation of AAs was performed on a Prevail C18 column (150 × 4.6 mm inner diameter, 5 μm; HiCHROM) powered by a SHIMADZU LCMS-8040 Tandem Quadrupole Mass Spectrometer, equipped with an ESI source. The sample extract (5 μL) was injected into a 20 mM ammonium acetate in water (A) and acetonitrile (B) mobile phase and eluted at 0.4 mL min−1 with the following gradient: 0–10 min, 10 to 100% (B, v/v); 10–13 min, 100% (B, v/v); and 13–18 min, 10% (B, v/v). The LC eluent was directed to SHIMADZU LCMS-8040 system, under positive electrospray ionization mode. Using the optimized instrument parameters, the quantitative analysis is carried out by the MRM mode of MS/MS system. The m/z values for MRM transitions for the target analytes and internal standard are listed in Table 1, where the residence time for each transition is set to 100 ms.| Ion monitored | MRM transition | Declustering potential (V) | Entrance potential (V) | Collision energy | Collision cell exit potential (V) | |
|---|---|---|---|---|---|---|
| a Most abundance MRM transition and used for quantification. The ESI parameters are as follows: ion spray voltage, 5500 V; drying gas temperature, 550 °C; curtain gas, 40 psi; nebulizing gas, 30 psi; drying gas, 40 psi. | ||||||
| AAI | [M + NH4]+ | 359 → 298a | 55 | 9 | 10 | 18 |
| 359 → 324 | 55 | 9 | 13 | 18 | ||
| AAII | [M + NH4]+ | 329 → 268a | 55 | 9 | 7 | 14 |
| 329 → 294 | 55 | 9 | 20 | 17 | ||
2.4 Calibration
Stock solutions of individual analytes were prepared at 200 μg mL−1 in methanol and combined into mixed standards at 10000 and 1000 ng mL−1 in acetonitrile. The extract was further diluted with acetonitrile or a blank liquid sample to obtain a solvent and matrix matching standard with a concentration range of 0.1–1000 ng mL−1. Internal standard benz[cd]indol-2(1H)-one (50 μL, 0.3 μg mL−1) was added into 450 μL of the working standard solution mixtures with concentrations ranging from 0.1 ng mL−1 to 1000 ng mL−1. After vortex mixing, the standard solutions were analyzed via LC-MS/MS method. The calibration curves were established by plotting the peak area ratio of each AAs to that of the internal standard against the concentration of AAs in working standards.
2.5 Method validation
The sensitivity, precision and accuracy of the optimized method are also verified. The limit of detection (LOD) and limit of quantification (LOQ) are set as the amount of analytes in the extract of blank sample, and the signal-to-noise ratio generated is 3 and 10 times of the signal-to-noise ratio.33 Method precision was evaluated by analyzing herbal extracts spiked with AAs at two different concentrations (50 and 500 ng g−1) on the same day (n = 5) and over separate days in a month (n = 5). Method recovery was evaluated by spiking AAs (50 and 500 ng g−1) into samples and extracting AAs via QuEChERS method and then analyzed through HPLC-MS/MS. Real samples may have lower extractability than spiked samples; thus, the samples were mixed for 1 h before being analyzed after spiking the analytes.3 Results and discussion
The extraction of AAs from each sample was conducted with methanol under sonication for 30 min, followed by d-SPE clean-up to isolate AAs from the extraction system.34,35 We developed the aforementioned extraction process instead of using the simpler one-step extraction method due to the discrimination of co-extractants in the final extract. The QuEChERS sample preparation method is mainly used for qualitative analysis;36 therefore, we expected this approach to be successfully used in the quantitative analysis of AAs in Chinese herbal patent medicines. PSA, used in this modified method as sorbent to remove colored impurities efficiently in sample extracts, was favorable for subsequent chromatographic analysis.We added different amounts of PSA (0, 10, 20, 30, and 40 mg) into sample extracts of AAs to confirm the optimized dosage of PSA and investigate their effect on analytical signals. The PSA dosages of 30 and 40 mg were the most efficient in removing colored impurities in sample extracts without sacrificing extraction recovery. Therefore, 30 mg of PSA was selected because of its sorbent-saving and clean-up performances.37 This modified QuEChERS method was less expensive, less time-consuming, and less labor-intensive for sample clean-up than the SPE method.
LC-MS/MS method was used to analyze the working standard solution, and the calibration curve was established by the peak area ratio of AAs and internal standard, which was found to be linear in the concentration range of 0.1–1000 ng mL−1 for AAs quantification. The results could be fitted into linear regression yielded lines with equations y = 0.0048x + 0.0468 (r2 = 0.9994) and y = 0.0013x − 0.0023 (r2 = 0.9997) for AAs I and II, respectively.
Method accuracy was confirmed by quantifying AA-fortified samples at two different concentrations (50 and 500 ng g−1) in blank samples, and the concentration of AA recovered was compared with that of the spiked content to evaluate the method recovery. Method recovery was calculated to be less than 13.0% deviation from the spiked values for all analytes at the tested concentrations. This value demonstrated that the developed QuEChERS extraction method is highly quantitative (Table 2). Meanwhile, method precision was investigated by analyzing blank samples spiked with AAs at two different concentrations (50 and 500 ng g−1) on the same day (n = 5) and over seven separate days for two weeks. Intraday precision of this method showed a standard deviation (peak area) of less than 9.0% for AA I and II at the two spiked dosages (Table 2). Reproducibility of this developed method for AA determination varied by less than 10.1% (n = 5) in a two-week period. Thus, data on the accuracy and precision (Table 2) of the method indicated that the analytical method combining d-SPE-based QuEChERS extraction and LC-MS/MS analysis has a sensitive and reliable performance in the quantification of AAs in Chinese herbal patent medicines.
| Matrix | Precision | LOD ng g−1 | LOQ ng g−1 | Accuracy | ||||
|---|---|---|---|---|---|---|---|---|
| Intra-day *(%RSD) | Inter-day *(%RSD) | Spike (ng g−1) | Measured concentration (ng g−1) | Recovery, % | ||||
| AA I | Liquid extract (L2) | 4.7 | 10.1 | 1.0 | 2.8 | 50 | 52.8 ± 4.8 | 110.3 ± 6.1 |
| 3.3 | 7.6 | 500 | 492.6 ± 21.2 | 98.5 ± 4.2 | ||||
| Tablets (T12) | 5.6 | 8.7 | 2.3 | 6.5 | 50 | 48.5 ± 3.7 | 97.1 ± 7.4 | |
| 4.3 | 7.9 | 500 | 479.8 ± 28.7 | 96.0 ± 5.7 | ||||
| Capsules (C5) | 6.4 | 9.5 | 7.6 | 22.1 | 50 | 48.3 ± 6.5 | 96.6 ± 13.0 | |
| 6.8 | 8.8 | 500 | 521.4 ± 33.9 | 104.3 ± 6.8 | ||||
| AA II | Liquid extract (L2) | 9.0 | 9.0 | 2.2 | 6.5 | 50 | 51.6 ± 2.6 | 103.1 ± 5.3 |
| 5.9 | 6.7 | 500 | 483.1 ± 3.6 | 96.6 ± 7.3 | ||||
| Tablets (T12) | 5.5 | 5.9 | 4.5 | 12.5 | 50 | 50.7 ± 3.6 | 101.3 ± 7.2 | |
| 4.2 | 6.0 | 500 | 494.2 ± 6.1 | 98.8 ± 12.2 | ||||
| Capsules (C5) | 7.4 | 7.2 | 15.0 | 42.1 | 50 | 50.8 ± 5.1 | 101.5 ± 10.2 | |
| 3.7 | 5.4 | 500 | 488.4 ± 2.3 | 97.7 ± 4.6 | ||||
We used the established method to test the presence of AAs in actual medicine samples. A total of 30 samples of medicines for rheumatism and urinary tract infections were purchased via the Internet or from pharmacies in China. Commercial medicines collected in this work were known or suspected to contain AAs from Aristolochia spp. The determination of AAs in the tested samples was conducted using the developed LC-MS/MS method, and the results are summarized in Table 3. Among all tested samples, AA I was detected in 16 of the 30 products at concentrations ranging from 19.4 ng g−1 to 1409.5 ng g−1, whereas the occurrence of AA II was much lower with only 6 samples and with a concentration range of 17.5–329.9 ng g−1. This finding is in accordance with previous reports that AA I has a higher concentration than AA II in Aristolochia spp.38,39
| Product number | Form | Label ingredients | Concentration (ng g−1) | Daily dose (μg day−1) | ||
|---|---|---|---|---|---|---|
| AA I | AA II | AA I | AA II | |||
| a The name of tested Chinese herbal patent medicines are displaced in serial number as L: liquid samples, P: powder samples, T: tablet samples and C: capsule samples. b ND: not detected. c NBPL: no banned plants listed. | ||||||
| L1 | Liquid extract | Clematis armandii Franch. | 1409.5 | 329.9 | 84.6 | 19.8 |
| P1 | Powder | Clematis armandii Franch. | 1190.8 | 274.8 | 7.9 | 1.8 |
| T1 | Tablet | Akebia stem | <6.5 | NDb | — | — |
| T2 | Tablet | Akebia stem | 22.3 | ND | 0.3 | — |
| T3 | Tablet | Akebia stem | 35.4 | ND | 0.2 | — |
| T4 | Tablet | Akebia stem | 42.0 | <12.5 | 0.2 | — |
| T5 | Tablet | Akebia stem | ND | ND | — | — |
| T6 | Tablet | Akebia stem | ND | ND | — | — |
| T7 | Tablet | Akebia stem | ND | ND | — | — |
| T8 | Tablet | Akebia stem | ND | ND | — | — |
| T9 | Tablet | Akebia stem | ND | ND | — | — |
| T10 | Tablet | Akebia stem | ND | ND | — | — |
| T11 | Tablet | Akebia stem | ND | ND | — | — |
| T12 | Tablet | Akebia stem | ND | ND | — | — |
| C1 | Capsule | Asarum sieboldii, Aristolochiae mollissima | 103.3 | <42.1 | 0.6 | — |
| T13 | Tablet | Asarum sieboldii, Aristolochiae mollissima | 45.6 | <12.5 | 0.3 | — |
| T14 | Tablet | Aristolochiae mollissima | 54.6 | 17.5 | 2.0 | 0.6 |
| C2 | Capsule | Aristolochiae moupinensis | 246.5 | 45.9 | 0.6 | 0.1 |
| T15 | Tablet | Aristolochiae moupinensis | 61.1 | 20.2 | 0.2 | 0.1 |
| T16 | Tablet | Aristolochiae moupinensis | ND | ND | — | — |
| T17 | Tablet | Aristolochiae moupinensis | 45.9 | <12.5 | 0.08 | — |
| T18 | Tablet | Aristolochiae moupinensis | 36.9 | ND | 0.03 | — |
| T19 | Tablet | Aristolochia contorta | 87.3 | 30.4 | 1.1 | 0.4 |
| C3 | Capsule | Asarum sieboldii, Radix Aristolochiae | 58.7 | ND | 0.2 | — |
| T20 | Tablet | Asarum sieboldii | ND | ND | — | — |
| L2 | Liquid extract | NBPLc | ND | ND | — | — |
| P2 | Powder | NBPL | ND | ND | — | — |
| C4 | Capsule | NBPL | 23.3 | ND | 0.03 | — |
| T21 | Tablet | NBPL | 19.4 | ND | 0.2 | — |
| C5 | Capsule | NBPL | ND | ND | — | — |
As shown in Table 3, the sample L1, which is a kind of common medicines in China to treat urinary tract infections, had the highest AAs content. Fig. 2 shows the typical chromatograms of AA I and AA II in prepared standard solutions. Meanwhile, LC-MS/MS analysis of the tested Chinese herbal medicine samples in Fig. 3 agree with the authentic standards in retention times. The extract was diluted with acetonitrile to ensure that the response of the analytes was within the linear dynamic range of MS as the concentration of AAs in these samples was relatively high. Then, we performed a LC-MS/MS detection. The concentrations of AAs detected in the medicines evaluated in this work were lower than those previously reported. Schaneberg and Khan38 analyzed twenty-five products that are suspected of containing Aristolochia or Asarum spp. and proved that AA I is present in 6 samples with concentrations of 40–280 mg kg−1 and AA II is detected in 5 samples with concentrations of 90–140 mg kg−1. In another work, Martena39 reported that more than 10% of the herbal preparations collected in the Dutch market were contaminated with AA I and II with concentrations ranging from 12 mg kg−1 to 1676 mg kg−1. Interestingly, the substitution of Aristolochia, Xungufeng, and Mutong led to different results in AAs determination. All samples labeled Xungufeng were contaminated with AAs, whereas medicines that contained Mutong had no AAs, except the sample T1. Surprisingly, some medicines, such as sample C4 and T21, which claimed to have no Aristolochia spp., contained AAs.
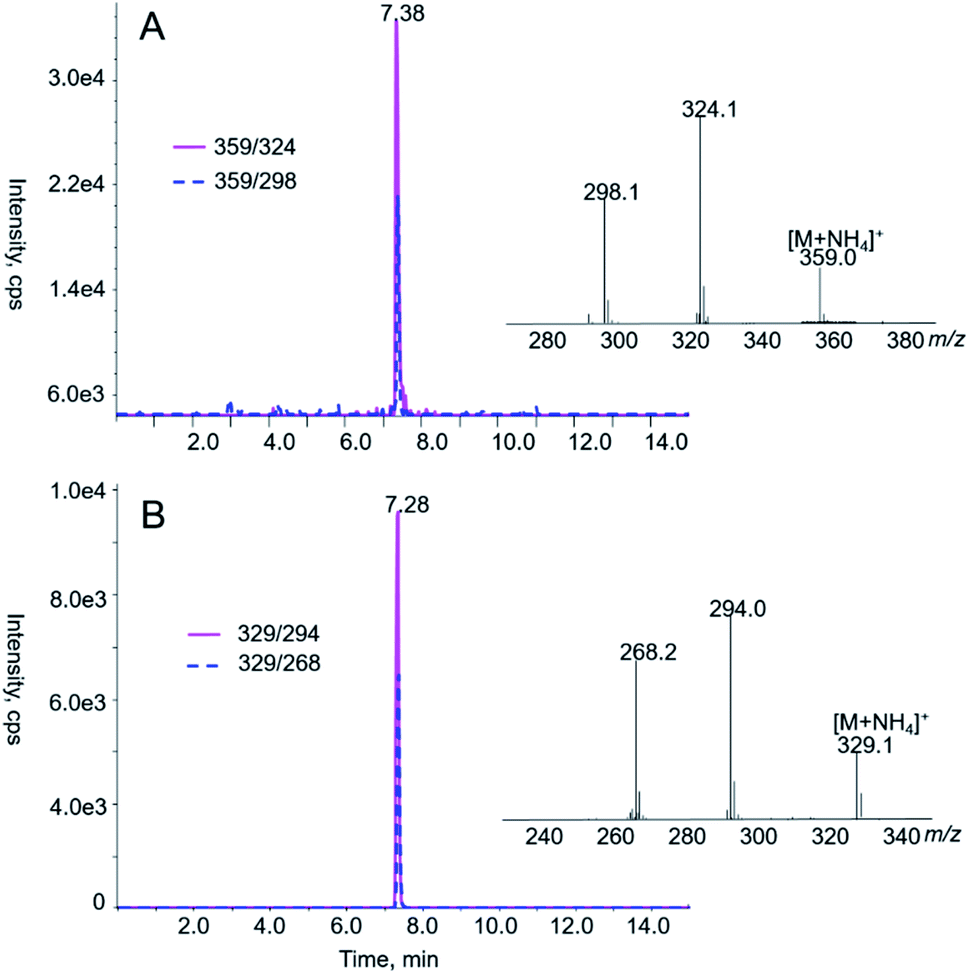 | ||
| Fig. 2 LC-MS/MS analyses of AA I (A) and AA II (B) in prepared standard solutions. AA I and AA II were eluted at 7.38 and 7.28 min, respectively. Shown in the insets are the product ion spectra of the [M + NH4]+ ion of AA I (A, m/z 359) and AA II (B, m/z 329). | ||
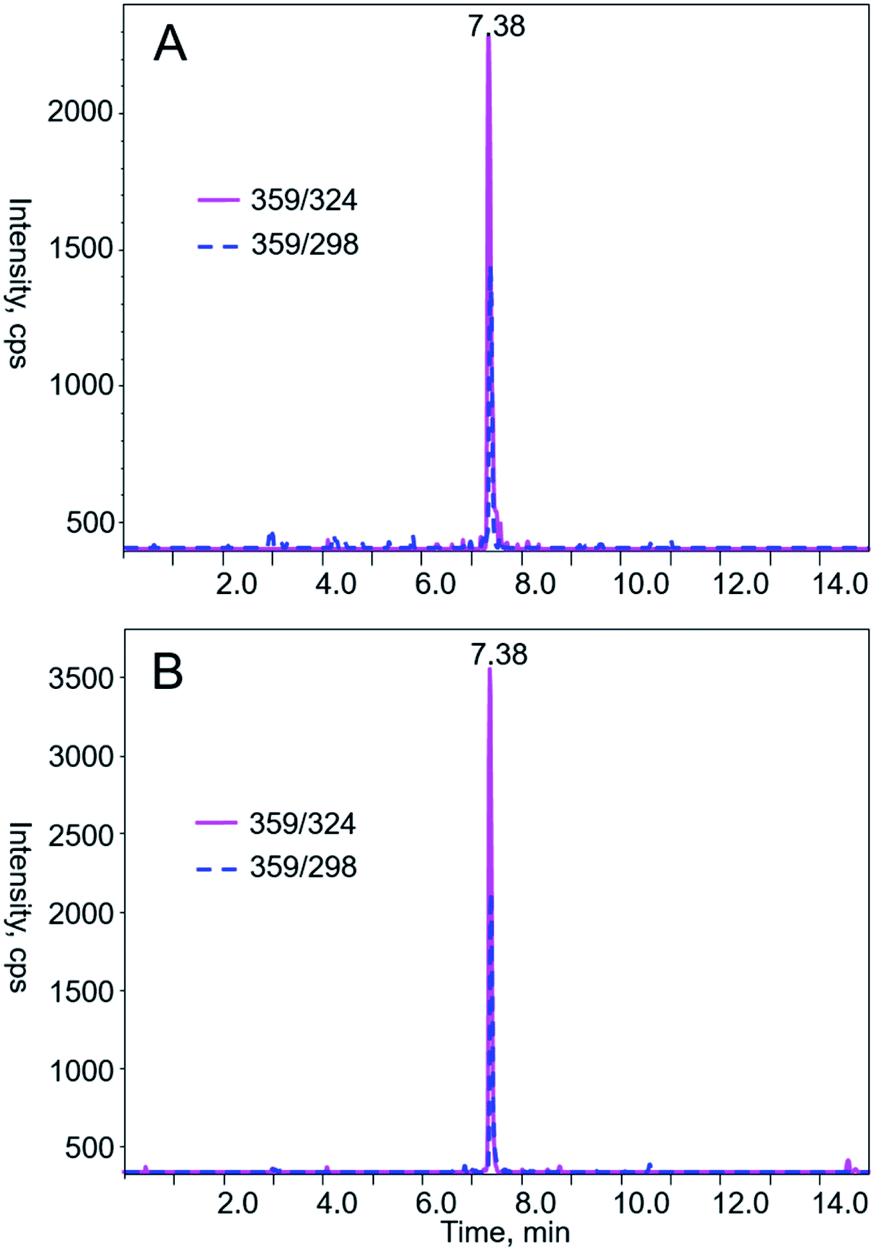 | ||
| Fig. 3 Typical LC-MS/MS chromatograms from MRM of AA I (m/z 359 → 324 and 359 → 298) in (A) liquid sample and (B) solid sample purchased from Alibaba of China. AA I was eluted at 7.38 min under the chromatographic condition described in the Materials and methods section. AA II is with concentration below the method detection limit. | ||
China Food and Drug Administration (CFDA) has cancelled the medicinal usage standards of Aristolochia manshuriensis, radix Stephania tetrandra, and radix Aristolochia. Although CFDA has deleted AA-containing herbs in the Chinese codex, marketing and application of AA-containing Chinese herb patent medicines has not been thoroughly banned. The daily doses of the 22 products positive for AA I and/or II were compared to determine the maximum amount of AA I and II that a person would consume in one day (Table 3). Sample T18 and C4 gave the lowest daily dose (0.03 μg AA I), whereas sample L1 gave the highest daily dose (84.6 μg AA I and 19.8 μg AA II). AA content in some Chinese herb patent medicines has declined compared with previously reported results, indicating that warnings on AAs have been raised.
4 Conclusion
As a summary, this work proposed the modification and application of a QuEChERS method for the determination of AAs in Chinese herbal patent medicine samples. The simple extraction with an acetonitrile–water system and followed by d-SPE clean-up step enabled the effective isolation of target analytes because background signal was remarkably reduced and interferences were eliminated, thereby improving the method sensitivity. The results of quantitative limit analysis showed that AA I and II could be easily detected by this method even at a very low content. A small-scale investigation of thirty herbal products showed that AA I and II existed in 53% and 20% of the samples respectively. The goal of this study was not to quantify the amount of AA I and II found in products, but to detect AA I and II that were still present in products in the market. Although FDA has advised the industry to remove products that contain AAs since 2001, these products remain in the market for consumption.Caution
AAs are carcinogenic and should be handled carefully.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the Project of the National Natural Science Foundation of China (Grant No. 41907315), and the Program for Scientific Research Innovation Team in Colleges and Universities of Jinan (Grant No. 2018GXRC006), Key Research & Development Project of Shandong Province (Grant No. 2019GGX102070).References
- V. Kumar, A. K. Prasad and V. S. Parmar, Nat. Prod. Rep., 2003, 20, 565–583 RSC.
- N. M. Pavlović, V. Maksimović, J. D. Maksimović, W. H. Orem, C. A. Tatu, H. E. Lerch, J. E. Bunnell, E. N. Kosti, D. N. Szilagyi and V. Paunescu, Environ. Geochem. Health, 2013, 35, 215–226 CrossRef.
- J. L. Vanherweghem, C. Tielemans, D. Abramowicz, M. Depierreux, R. Vanhaelen-Fastre, M. Vanhaelen, M. Dratwa, C. Richard, D. Vandervelde, D. Verbeenlen and M. Jadoul, Lancet, 1993, 341, 387–391 CrossRef CAS.
- H. Gao, F. Guo, F. Feng, J. Yin, M. Song and H. Wang, J. Environ. Sci., 2009, 21, 1769–1776 CrossRef CAS.
- U. Mengs, Arch. Toxicol., 1987, 59, 328–331 CrossRef CAS.
- U. Mengs, W. Lang and J. A. Poch, Arch. Toxicol., 1982, 51, 107–119 CrossRef CAS.
- U. Mengs, Arch. Toxicol., 1983, 52, 209–220 CrossRef CAS.
- N. Mei, V. M. Arlt, D. H. Phillips, R. H. Heflich and T. Chen, Mutat. Res., 2006, 602, 83–91 CrossRef CAS.
- V. M. Arlt, M. Stiborova and H. H. Schmeiser, Mutagenesis, 2002, 17, 265–277 CrossRef CAS.
- J. L. Vanherweghem, J Altern Complement Med., 1998, 4, 9–13 CrossRef.
- G. Gillerot, M. Jadoul, V. M. Arlt, C. V. Y. de Strihou, H. H. Schmeiser, P. P. But, C. A. Bieler and J. P. Cosyns, Am. J. Kidney Dis., 2001, 38, E26 CrossRef CAS.
- D. M. Bhowmik, V. Jha and J. P. Cosyns, Am. J. Kidney Dis., 2001, 38, 1141–1142 Search PubMed.
- V. M. Arlt, M. Stiborová, J. vom Brocke, M. L. Simoes, G. M. Lord, J. L. Nortier, M. Hollstein, D. H. Phillips and H. H. Schmeiser, Carcinogenesis, 2007, 28, 2253–2261 CrossRef CAS.
- A. P. Grollman, S. Shibutani, M. Moriya, F. Miller, L. Wu, U. Moll, N. Suzuki, A. Fernandes, T. Rosenquist, Z. Medverec, K. Jakovina, B. Brdar, N. Slade, R. J. Turesky, A. K. Goodenough, R. Rieger, M. Vukelic and B. Jelakovic, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12129–12134 CrossRef CAS.
- J. P. Cosyns, M. Jadoul, J. P. Squifflet, F. X. Wese and C. V. Y. de Strihou, Am. J. Kidney Dis., 1999, 33, 1011–1017 CrossRef CAS.
- J. L. Nortier, M. C. M. Martinez, H. H. Schmeiser, V. M. Arlt, C. A. Bieler, M. Petein, M. F. Depierreux, L. de Pauw, D. Abramowicz, P. Vereerstraeten and J. Vanherweghem, N. Engl. J. Med., 2000, 342, 1686–1692 CrossRef CAS.
- A. W. Ng, S. L. Poon, M. N. Huang, J. Q. Lim, A. Boot, W. Yu, Y. Suzuki, S. Thangaraju, C. C. Y. Ng, P. Tan, S. T. Pang, H. Y. Huang, M. C. Yu, P. H. Lee, S. Y. Hsieh, A. Y. Chang, B. T. The and S. G. Rozen, Sci. Transl. Med., 2017, 9, E6446 CrossRef.
- Z. N. Lu, Q. Luo, L. N. Zhao, X. B. Su and Z. G. Han, bioRxiv, 2018, 507301 Search PubMed.
- L. S. Gold and E. Zeiger, Handbook of carcinogenic potency and genotoxicity databases, CRC Press, 1996 Search PubMed.
- Aristolochic Acid: Safety Alert. FDA concerned about botanical products, including dietary supplements, containing aristolochic acid, http://www.fda.gov/Food/RecallsOutbreaksEmergencies/SafetyAlertsAdvisories/ucm095272.html Search PubMed.
- U. Koesukwiwat, S. J. Lehotay, K. Mastovska, K. J. Dorweiler and N. Leepipatpiboon, J. Agric. Food Chem., 2009, 58, 5950–5958 CrossRef.
- S. J. Lehotay, K. A. Son, H. Kwon, U. Koesukwiwat, W. Fu, K. Mastovska, H. Eunha and L. Natchanun, J. Chromatogr. A, 2010, 1217, 2548–2560 CrossRef CAS.
- S. C. Cunha, S. J. Lehotay, K. Mastovska, J. O. Fernandes, M. Beatriz and P. P. Oliveira, J. Sep. Sci., 2007, 30, 620–632 CrossRef CAS.
- S. Ma, P. Han, A. Li, J. Wang, X. Feng and M. Wang, Chromatographia, 2018, 81, 435–445 CrossRef CAS.
- V. Tripathy, A. Saha and J. Kumar, J. Sci. Food Agric., 2017, 54, 458–468 CAS.
- U. Koesukwiwat, S. J. Lehotay, S. Miao and N. Leepipatpiboon, J. Chromatogr. A, 2010, 1217, 6692–6703 CrossRef CAS.
- V. G. Amelin, N. M. Volkova, A. A. Timofeev and A. V. Tret'yakov, J. Anal. Chem., 2015, 70, 1076–1084 CrossRef CAS.
- C. G. Pinto, M. E. F. Laespada, S. H. Martín, A. M. C. Ferreira, J. L. P. Pavón and B. M. Cordero, Talanta, 2010, 81, 385–391 CrossRef CAS.
- A. C. Valese, G. A. Oliveira, C. R. Kleemann, L. Molognoni and H. A. Daguer, J. Food Compos. Anal., 2016, 47, 38–44 CrossRef CAS.
- P. Calatayud-Vernich, F. Calatayud, E. Simó and Y. Picó, MethodsX, 2016, 3, 452–458 CrossRef.
- L. Pareja, V. Cesio, H. Heinzen and A. R. Fernández-Alba, Talanta, 2011, 83, 1613–1622 CrossRef CAS.
- F. Vela-Soria, L. M. Iribarne-Durán, V. Mustieles, I. Jiménez-Díaz, M. F. Fernández and N. Olea, J. Chromatogr. A, 2018, 1546, 1–9 CrossRef CAS.
- Y. Wang and W. Chan, J. Agric. Food Chem., 2016, 64, 2802–2808 CrossRef CAS.
- Y. Wang and W. Chan, J. Agric. Food Chem., 2014, 62, 5859–5864 CrossRef CAS.
- S. A. Chan, M. J. Chen, T. Y. Liu, M. R. Fuh, J. F. Deng, M. L. Wu and S. J. Hsieh, Talanta, 2003, 60, 679–685 CrossRef CAS.
- H. G. J. Mol, R. C. J. Van Dam, P. Zomer and P. P. Mulder, Food Addit. Contam., Part A, 2011, 28, 1405–1423 CrossRef CAS.
- W. Li, Q. Hu and W. Chan, J. Agric. Food Chem., 2015, 64, 107–112 CrossRef.
- B. T. Schaneberg and I. A. Khan, J. Ethnopharmacol., 2004, 94, 245–249 CrossRef CAS.
- M. J. Martena, J. C. van der Wielen, L. F. van de Laak, E. J. Konings, H. N. de Groot and I. M. Rietjens, Anal. Bioanal. Chem., 2007, 389, 263–275 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |