
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tricyclic carbohydrate–benzene hybrids as selective inhibitors of galectin-1 and galectin-8 N-terminal domains†
Chunxia Wua,
Can Yonga,
Qiuju Zhonga,
Zhouyu Wang a,
Ulf J. Nilssonb and
Yuanyuan Zhang*a
a,
Ulf J. Nilssonb and
Yuanyuan Zhang*a
aSchool of Science, Xihua University, Jinniu District, 610039 Chengdu, China. E-mail: yuanyuan.zhang@mail.xhu.edu.cn
bCentre for Analysis and Synthesis, Department of Chemistry, Lund University, POB 124, SE-221 00 Lund, Sweden
First published on 22nd May 2020
Abstract
As the galactoside binding family of galectin proteins is involved in many physiological and pathological processes, the inhibitors of these proteins are considered to be of significant interest in the treatment of diseases such as cancer and fibrosis. Herein, fused tricyclic carbohydrate–benzene hybrid core structures are reported to be the selective inhibitors of galectin-1 and the N-terminal domain of galectin-8 by a competitive fluorescence polarization assay. The key intermediates mono- or diiodo tricyclic carbohydrate–benzene hybrids were synthesized from protected 2-bromo-3-O-propargyl-D-galactose via a domino reaction and subsequently utilized for further derivatization by Stille couplings to achieve derivatives carrying substituents at C10 and/or C11. Several compounds showed affinity for the galectin-1 and galectin-8 N-terminal (8N) domains; however, weak or even no binding was observed for galectin-3. Monosubstituted derivatives at C10 or C11 exhibited better affinity for galectin-8N than di-substituted derivatives at C10 or C11. Especially, a benzyl substituent or p-fluorobenzyl substituent at C11 displayed affinity and selectivity for galectin-1 and galectin-8N over galectin-3. This suggests that tricyclic carbohydrate–benzene hybrids are promising scaffolds for the development of selective galectin-1 and galectin-8N inhibitors.
1 Introduction
Galectins are glycan-binding proteins that selectively bind to glycoconjugates containing β-D-galactopyranoside residues. To date, more than 15 members of the galectin family have been identified, purified, isolated, and characterized, out of which, only 12 are found in humans.1,2 These galectins are involved in many biological functions such as cell–cell3 and cell–matrix interactions,4 immune and inflammatory responses,5,6 anti-apoptosis,7 induction of T cell apoptosis, and regulation of cell adhesion and migration.8 Among them, galectin-1 is widely expressed in the human body and is associated with various neurological diseases,9 HIV-1 viral infectivity,10 and cancer progression.11 Moreover, galectin-1 has been highlighted as a diagnostic tumor marker.12 In addition to this, galectin-8 has attracted attention due to its physiological activity. For example, it has been evidenced to regulate autophagy13 and lymphangiogenesis14 and is highly expressed in many clinical diseases such as larynx cancer, prostate cancer, breast cancer, and cutaneous T cell lymphomas.15–19 As a result, due to their intimate connection with diseases, galectins have become promising drug targets.A variety of artificial galectin inhibitors have been synthesized in the past two decades. The structure of the inhibitors were typically based on monosaccharides or oligosaccharides,20 such as D-galactose,21,22 talose,23 lactosamine,24 and thiodigalactoside,25,26 and with modifications mostly at C1 and C3 positions of galactoside. Both 4-OH and 6-OH were always conserved as they were essential for galectin binding. A majority of these inhibitors exhibit good affinity for galectin-3,25–28 whereas fewer show potent affinity for other galectins such as 1 shows potent affinity for galectin-1 (ref. 27) and 2 shows potent affinity for galectin-8N21 (Fig. 1). However, they all show similar or even more potent affinity for galectin-3. This drawback limits the application of the abovementioned compounds as galectin-1 or galectin-8 inhibitors in biological assays or systems. Herein, a series of tricyclic carbohydrate–benzene hybrids was designed and synthesized. The key galectin-binding sites 4-OH and 6-OH of galactose were preserved, and C1–C3 were constrained by two rings to a fused and rigid tricyclic structure as previously reported by Werz et al.29 This ring structure was hypothesized to favorably change the hydrophobic/hydrophilic properties of the inhibitors and enable the incorporation of substituents into them to optimize their affinity and selectivity for selected individual galectins among the panel of galectin-1,3,4 N-terminal (4N), 4 C-terminal (4C), 8N, 9 N-terminal (9N), and 9 C-terminal (9C) evaluated in the study.
 | ||
| Fig. 1 Structures of the compound 1 and 2. | ||
2 Results and discussions
2.1 Chemistry
The tricyclic carbohydrate–benzene hybrids were synthesized by three strategies (Scheme 1).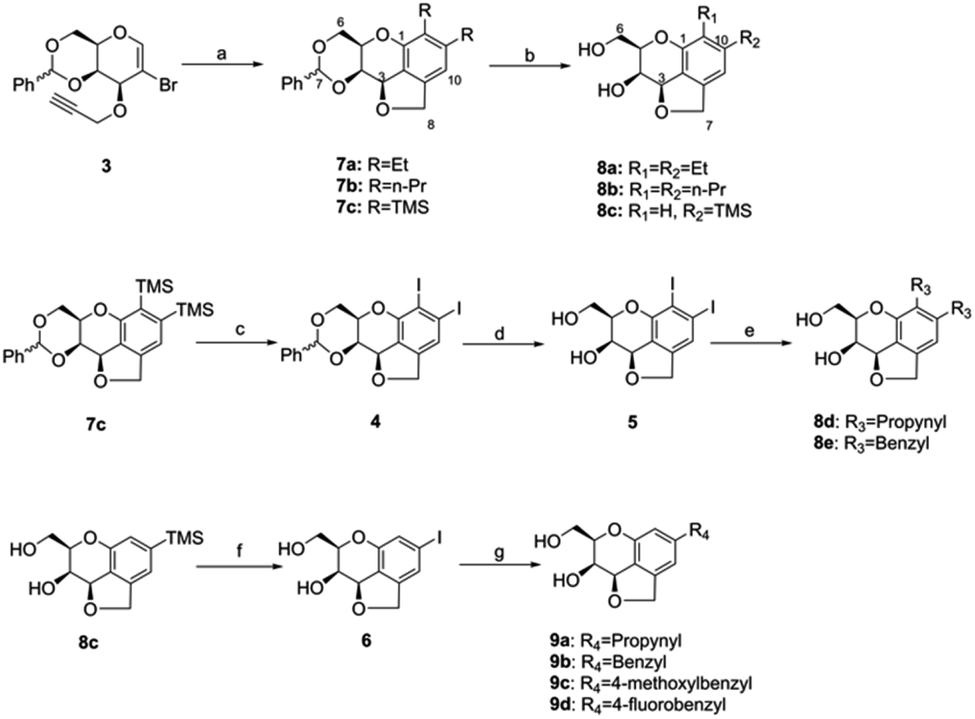 | ||
| Scheme 1 Synthesis of tricyclic carbohydrate–benzene hybrids. Reagents and conditions: (a) substituted alkynes, Pd(PPh3)4, diisopropylamine, [(t-Bu)3PH]BF4, DMF/MeCN/NMP, and 100 °C; (b) 0.2 M HCl, MeOH, and 55 °C; (c) iodine monochloride, DCM, and r.t.; (d) 0.2 M HCl, MeOH, and 55 °C; (e) substituted tributyl stannane, triethylamine, CuI, Pd(PPh3)4, DMF, and 75 °C; (f) iodine monochloride, DCM, and r.t.; and (g) substituted tributyl stannane, triethylamine, CuI, Pd(PPh3)4, DMF, and 95 °C. | ||
At first, the tricyclic carbohydrate–benzene hybrids 8a–c were acquired by the Pd-catalyzed domino reaction of 3 with different alkynes and subsequent deprotection using the method reported by Werz.29 Subsequently, the benzylidene-protected di-trimethylsilyl tricyclic carbohydrate–benzene hybrid 7c was converted to the diiodo-hybrid 4 by iodine monochloride. The benzylidene removal of 4 led to 5, which was converted to the compounds 8d and 8e via palladium-catalyzed Stille couplings using organotins. Herein, four tricyclic carbohydrate–benzene hybrids having same substituents at both C10 and C11 and one compound with the mono-trimethylsilyl group were synthesized. Finally, the deprotected mono-trimethylsilyl-substituted tricyclic carbohydrate–benzene hybrid 8c was utilized to prepare the mono-substituted hybrid molecules 9a–9d through the monoiodo-hybrid intermediate 6 by similar Stille cross-couplings as the third strategy.
2.2 Galectin binding
The binding activities of the compounds 8a–e, 9a–d, 5, and 6 for galectins were tested using a previously described competitive fluorescence polarization assay.30,31 The data of these compounds and three reference ligands, i.e. methyl β-D-galactoside 10, 1,1′-sulfanediyl-bis-{{3-deoxy-3-[4-(thiazol-2-yl)-1H-1,2,3-triazol-1-yl]-β-D-galactopyranoside}} 1, and 3,4-dichlorophenyl 3-O-(7-carboxy-quinolin-2-yl-methyl)-1-thio-α-D-galactopyranoside 2, are provided hereinafter.A majority of the synthesized compounds exhibited obvious affinities for galectin-1 and galectin-8N, but no or very weak affinity for galectin-3. Hence, the selectivity of the tricyclic carbohydrate–benzene hybrids was significantly improved for galectin-1 and galectin-8N over galectin-3, which is rare among the reported inhibitors. Although the compound 1 is the best reported synthetic galectin-1 inhibitor to date, its affinity for galectin-3 was even more potent, which was 10-fold when compared with that for galectin-1. The compound 2 has been reported to be the most potent inhibitor for galectin-8N; however, it has a similar affinity for galectin-3. Compared with the case of the reference methyl β-galactoside 10, the affinities of most of the tricyclic compounds were increased by 3–35 folds and more than 3–71 folds for galectin-8N and galectin-1, respectively, whereas most of the tested compounds exhibited weak or even no affinity for galectin-4C, 4N, 9C, or 9N (Table 1).
| Cpd | Galectin-1 | Galectin-3 | Galectin-4C | Galectin-4N | Galectin-8N | Galectin-9C | Galectin-9N |
|---|---|---|---|---|---|---|---|
| a Not available.b Reference compounds. | |||||||
| 5 | 890 ± 140 | ≫2000 | 840 ± 60 | ≫1000 | 2100 ± 320 | ≫1000 | ≫1000 |
| 6 | 1200 ± 120 | ≫2000 | 1600 ± 220 | 900 ± 87 | 250 ± 22 | ≫2000 | ≫2000 |
| 8a | 2900 ± 580 | ≫8000 | 3700 ± 650 | ≫6000 | ≈10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
≈5000 | ≫6000 |
| 8b | 760 ± 35 | ≫2000 | ≫3000 | ≫3000 | ≫2000 | ≫2000 | ≫2000 |
| 8c | ≫6000 | 2700 ± 440 | ≫6000 | ≈10000 |
330 ± 18 | 2800 ± 290 | ≫6000 |
| 8d | 1100 ± 320 | ≫2000 | ≈3000 | 870 ± 81 | ≫2000 | ≫2000 | ≫2000 |
| 8e | 310 ± 57 | ≫2000 | naa | na | 1500 ± 260 | na | na |
| 9a | 840 ± 85 | ≫2000 | 1100 ± 22 | 1500 ± 200 | 440 ± 47 | ≫2000 | ≫2000 |
| 9b | 140 ± 14 | ≫2000 | na | na | 240 ± 49 | na | na |
| 9c | 1500 ± 11 | ≫2000 | 1700 ± 430 | ≫1000 | 640 ± 58 | ≫1000 | ≫1000 |
| 9d | 420 ± 61 | 4100 ± 370 | ≈3000 | ≫1000 | 180 ± 44 | ≫1000 | ≫1000 |
| 10b | >10000 |
4400 | 10000 |
6600 | 6300 | 8600 | 3300 |
| 1b | <0.01 (ref. 27) | ∼0.0011 (ref. 27) | na | na | na | na | na |
| 2b | 48 ± 4.4 (ref. 21) | 1.27 ± 0.07 (ref. 21) | 43 ± 5.7 (ref. 21) | 43 ± 7.1 (ref. 21) | 1.5 ± 0.08 (ref. 21) | 14 ± 1.3 (ref. 21) | 2.06 ± 0.09 (ref. 21) |
Structure–affinity analysis revealed that the substituents at the C10 and C11 positions of the tricyclic carbohydrate–benzene hybrids play a key role in the affinity of these hybrids for galectins. In general, the mono-substituted derivatives 6, 8c, and 9a–9d showed better galectin-8N binding than the di-substituted compounds 5, 8a, 8b, 8d, and 8e. The affinities of the disubstituted derivatives carrying aliphatic alkyl groups, such as ethyl (8a), n-propyl (8b), or propynyl (8d), for galectin-8N were very weak or could hardly be observed, whereas the affinities of those carrying di-benzyl groups (8e) and di-iodo (5) were increased to around 1500 and 2100 μM, respectively, which was more than 3 times that of the reference galactoside 10. The analysis of the mono-substituted 8c, 9a, 9b, 9c, 9d, and 6 revealed no binding of these derivatives to galectin-3, whereas their affinity for galectin-8N was changed. Compared to the best inhibitor 10,11-disubstituted 8e, all the mono-substituted 8c, 9a, 9b, 9c, 9d, and 6 showed improved affinity for galectin-8N. Similar to di-substituted derivatives, the benzyl derivative 9b exhibited the best affinity for galectin-8N, along with the mono-iodo derivative 6. The evaluation of the derivatives with substituted benzyl groups, i.e. p-methoxybenzyl 9c and p-fluorobenzyl 9d, revealed that the electron-donating group 4-methoxybenzyl at C10 (9c) decreased the affinity; thus, the affinity of 9c was the worst among mono-substituted derivatives; however, the electron-withdrawing group 4-fluorobenzyl at C10 (9d) improved the affinity of the derivative for galectin-8N down to the dissociation constant of around 180 μM. Meanwhile, regardless of being mono-substituted or di-substituted, almost all the tricyclic compounds exhibited affinity for galectin-1. Benzyl group modification exerted positive effects on the binding activities, and 8e and 9b were the best galectin-1 inhibitors. Similar affinity trends were observed for galectin-1; however, for galectin-8N, p-fluorobenzyl 9d was a more potent inhibitor than p-methoxybenzyl 9c. Moreover, the two compounds 5 and 6 containing iodine atoms displayed obvious affinity for galectin-4C and galectin-4N, respectively, with a Kd value below 1000 μM.
2.3 Molecular docking of 9d with galectin-1 and galectin-8N
In order to analyze the possible binding modes of 9d with galectin-1, galectin-3, and galectin-8N, molecular docking was performed.In general, 9d is located in the CRD of galectin-1 and galectin-8N, and its saccharide structure coincides with the galactose part of D-lactose (Fig. 2a and c), respectively. Polar interactions are mainly the hydrogen bonds between the galactopyranose hydroxyl groups at C4 and C6 and the nearby residues (Fig. 2b and d). The C4 hydroxyl group of 9d forms hydrogen bonds with the galectin-1 residues Arg-48 and His-44, whereas the C6 hydroxyl group interacts with the residues Asn-61 and Glu-71. In the case of galectin-8N, the C4 hydroxyl group of 9d forms hydrogen bonds with the galectin-8N residues Arg-45, Arg-69, and His-65, whereas the C6 hydroxyl group interacts with the residues Asn-79 and Glu-89.
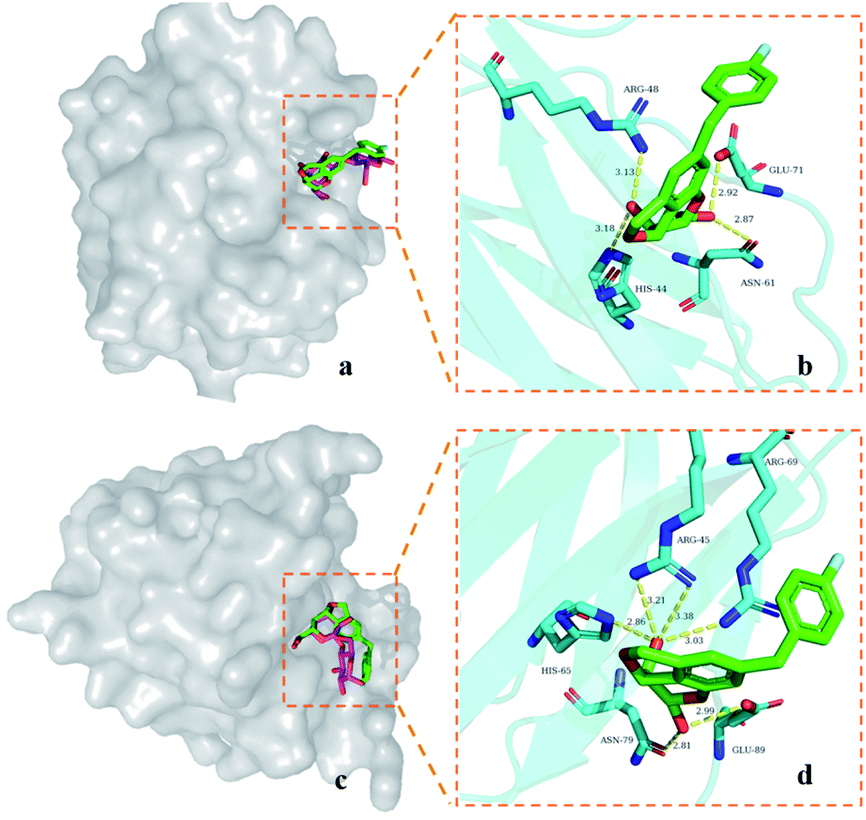 | ||
| Fig. 2 Molecular docking of 9d with galectins. (a) 9d is shown with green carbons, and lactose is shown with magenta carbons in the galectin-1 CRD. (b) Interactions between 9d and CRD of galectin-1. (c) 9d is shown with green carbons and lactose is shown with magenta carbons in the galectin-8N CRD. (d) Interactions between 9d and CRD of galectin-8N. | ||
From the docking poses of 9d in its complex with galectin-1, 3, and 8N (Fig. 3), it can be observed that the benzene parts of the tricyclic skeleton were close to the electropositive arginine-containing regions, which could contribute to the cation–π interactions. The relatively flat surface consisted of Glu-71 and Arg-48, which seemed to favor the stacking of 4-fluorobenzyl in galectin-1 via π–π or cation–π interactions (Fig. 3a). Moreover, a similar trend could be observed in galectin-8N, and the surface was composed of Glu-89 and Arg-69 (Fig. 3c). On the contrary, steric hindrance occurred outside the CRD groove in galectin-3, which might not be conducive to the π-interactions (Fig. 3b).
 | ||
| Fig. 3 Comparison between the complexes of 9d with galectin-1 (a), galectin-3 (b), and galectin-8N (c). | ||
3 Experimental
3.1 Synthesis
All the final compounds were of >95% purity according to the HPLC analysis (Agilent Series 1100 system, Zorbax Eclipse XDB-C18 column, and H2O–MeCN gradient 5–95% with 0.1% TFA).
:ethyl acetate = 3:1) to afford the compounds 7a–7c.
7a. White solid; yield: 44%; 1H NMR (400 MHz, CDCl3): δ = 7.25–7.16 (m, 5H, Ph), 6.68 (s, 1H, 10-H), 5.62 (s, 1H, 7-H), 5.22–5.16 (m, 2H, 8-Ha, 3-H), 4.94 (dd, J = 11.6 Hz, 1.6 Hz, 1H, 8-Hb), 4.60–4.56 (m, 2H, 6-Ha, 4-H), 4.20 (dd, J = 12.0 Hz, 1.2 Hz, 1H, 6-Hb), 4.12 (d, J = 1.4 Hz, 1H, 5-H), 2.76–2.61 (m, 4H, CH2), 1.23 (t, J = 7.6 Hz, 3H, CH3), 1.18 (t, J = 7.6 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): 149.3, 144.2, 137.7, 137.6, 119.2, 113.4, 101.2 (C-Ph, C-1, C-2, C-7, C-9, C-10, C-11, C-12), 129.0, 128.0, 126.6 (C-Ph), 77.3 (C-3), 74.4 (C-8), 72.4 (C-4), 70.2 (C-6), 69.0 (C-5), 26.2 (CH2), 18.5 (CH2), 16.2 (CH3), 14.6 (CH3). HRMS: calculated for C22H24O4 ([M + Na]+): 375.1567; found 375.1570.
7b. White solid; yield: 38%; 1H NMR (400 MHz, CDCl3): δ = 7.25–7.15 (m, 5H, Ph), 6.66 (s, 1H, 10-H), 5.61 (s, 1H, 7-H), 5.22–5.15 (m, 2H, 8-Ha, 3-H), 4.94 (dd, J = 11.6 Hz, 1.8 Hz, 1H, 8-Hb), 4.59 (dd, J = 3.6 Hz, 1.4 Hz, 1H, 4-H), 4.57 (dd, J = 12.3 Hz, 1.8 Hz, 1H, 6-Ha), 4.20 (dd, J = 12.3 Hz, 1.3 Hz, 1H, 6-Hb), 4.10 (d, J = 1.3 Hz, 1H, 5-H), 2.72–2.52 (m, 4H, CH2), 1.66–1.58 (m, 4H, CH2), 0.99 (t, J = 7.4 Hz, 3H, CH3), 0.97 (t, J = 7.4 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): 149.6, 143.2, 137.8, 137.5, 125.0, 119.2, 114.2, 101.3 (C-Ph, C-1, C-2, C-7, C-9, C-10, C-11, C-12), 129.2, 128.2, 126.8 (C-Ph), 77.5 (C-3), 74.5 (C-8), 72.4 (C-4), 70.3 (C-6), 69.1 (C-5), 35.6 (CH2), 27.5 (CH2), 25.1 (CH2), 23.4 (CH2), 14.6 (CH3), 14.3 (CH3). HRMS: calculated for C24H28O4 ([M + Na]+): 403.1880; found: 403.1890.
7c. Colorless oil; yield: 43%; 1H NMR (400 MHz, CDCl3): δ = 7.28–7.22 (m, 5H, Ph), 7.17 (s, 1H, 10-H), 5.64 (s, 1H, 7-H), 5.23–5.18 (m, 2H, 8-Ha, 3-H), 4.94 (dd, J = 13.0 Hz, 3.0 Hz, 1H, 8-Hb), 4.62 (dd, 1H, J = 3.6 Hz, 1.3 Hz, 4-H), 4.58 (dd, J = 12.4 Hz, 1.8 Hz, 1H, 6-Hb), 4.20 (dd, J = 12.4 Hz, 1.4 Hz, 1H, 6-Hb), 4.13 (d, J = 1.4 Hz, 1H, 5-H), 0.46 (s, 9H, CH3), 0.39 (s, 9H, CH3); 13C NMR (100 MHz, CDCl3): 157.3, 149.2, 141.0, 137.7, 121.5 (C-2, C-7, C-9, C-11, C-12), 129.2, 128.8, 128.1, 126.9 (C-Ph), 120.7 (C-10), 101.5 (C-1), 77.6 (C-3), 74.5 (C-8), 72.2 (C-4), 69.9 (C-6), 69.2 (C-5), 3.1 (CH3), 3.0 (CH3). HRMS: calculated for C24H32O4Si2 ([M + Na]+): 463.1731; found: 463.1740.
:ethyl acetate = 3:5) to afford 8a–8c.
8a. White solid; yield: 32%; 1H NMR (400 MHz, DMSO-d6): δ = 6.63 (s, 1H, 9-H), 5.01–4.96 (m, 2H, 7-Ha, 3-H), 4.87 (t, J = 5.6 Hz, 1H, OH), 4.79–4.76 (m, 2H, 7-Hb, OH), 4.19 (t, J = 1.4 Hz, 1H, 4-H), 4.05 (t, J = 6.4 Hz, 1H, 5-H), 3.76–3.70 (m, 2H, 6-Ha, 6-Hb), 2.69–2.67 (m, 2H, CH2), 2.34–2.33 (m, 2H, CH2), 1.14 (t, J = 7.5 Hz, 3H, CH3), 1.04 (t, J = 7.5 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ = 149.2, 143.4, 138.7, 124.9, 118.8 (C-1, C-2, C-8, C-10, C-11), 114.3 (C-9), 77.8, 76.9 (C-3, C-5), 74.2 (C-7), 65.2 (C-4), 63.5 (C-6), 24.9, 18.1 (CH2), 16.8, 14.5 (CH3); HRMS: calculated for C15H20O4 ([M + Na]+): 287.1254; found: 287.1254.
8b. White solid; yield: 40%; 1H NMR (400 MHz, CD3OD): δ = 6.65 (s, 1H, 9-H), 5.10–5.07 (m, 2H, 3-H, 7-Ha), 4.87–4.84 (m, 1H, 7-Hb), 4.33 (dd, J = 3.4 Hz, 0.8 Hz, 1H, 4-H), 4.11 (t, J = 6.4 Hz, 1H, 5-H), 3.92 (d, J = 6.3 Hz, 2H, 6-H), 2.63–2.54 (m, 4H, CH2), 1.59–1.50 (m, 4H, CH2), 0.97 (t, J = 7.4 Hz, 3H, CH3), 0.96 (t, J = 7.4 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ = 150.7, 143.8, 140.0, 125.5, 121.2 (C-1, C-2, C-8, C-10, C-11), 115.0 (C-9), 79.8, 79.4 (C-3, C-5), 74.8 (C-7), 64.8 (C-4), 62.6 (C-6), 36.4, 28.2, 26.2, 24.5 (CH2), 14.6, 14.5 (CH3); HRMS: calculated for C17H24O4 ([M + Na]+): 315.1567; found: 315.1579.
8c. White solid; yield: 43%; 1H NMR (400 MHz, CDCl3): δ = 6.99 (s, 1H, 11-H), 6.90 (s, 1H, 9-H), 5.22–5.18 (m, 1H, 7-Ha), 5.12–5.11 (m, 1H, 3-H), 4.96 (dd, J = 11.9 Hz, 1.8 Hz, 1H, 7-Hb), 4.41 (dd, J = 3.6 Hz, 0.8 Hz, 1H, 4-H), 4.19–4.12 (m, 2H, 5-H, 6-Ha), 4.06 (dd, J = 11.2 Hz, 4.2 Hz, 1H, 6-Hb), 0.25 (s, 9H, CH3); 13C NMR (100 MHz, CDCl3): δ = 151.1, 144.4, 141.8, 122.7 (C-1, C-2, C-8, C-10), 118.2, 117.0 (C-9, C-11), 77.8, 77.2, 65.3 (C-3, C-4, C-5), 74.4 (C-7), 63.5 (C-6), −0.9 (CH3); HRMS: calculated for C14H20O4Si ([M + Na]+): 303.1023; found: 303.1024.
:ethyl acetate = 3:1) as a white solid (72 mg, 39%). 1H NMR (400 MHz, CDCl3): δ = 7.44 (s, 1H, 10-H), 7.28–7.27 (m, 3H, Ph-H), 7.23–7.21 (m, 2H, Ph-H), 5.63 (s, 1H, 7-H), 5.16–5.14 (m, 2H, 3-H, 8-Ha), 4.94 (dd, J = 12.2 Hz, 2.2 Hz, 1H, 8-Hb), 4.66 (dd, J = 12.4 Hz, 1.6 Hz, 1H, 6-Ha), 4.62 (dd, 1H, J = 3.6 Hz, 1.2 Hz, 4-H), 4.26–4.22 (m, 2H, 5-H, 6-Hb); 13C NMR (100 MHz, CDCl3): δ = 151.9, 142.7, 137.2, 129.2, 128.1, 126.5, 124.8, 121.7, 108.8 (C-2, C-7, C-9, C-11, C-12, C-10, C-Ph), 101.2 (C-1), 77.0 (C-3), 73.6 (C-8), 71.9 (C-4), 71.4 (C-5), 69.8 (C-6); HRMS: calculated for C C18H14I2O4 ([M + Na]+): 570.8874; found: 570.8871.:ethyl acetate = 1:2) to obtain the white solid 5 (34 mg, 57%). 1H NMR (400 MHz, DMSO-d6): δ = 7.46 (s, 1H, 9-H), 5.11 (br, 1H, OH), 5.04 (d, J = 2.7 Hz, 1H, 3-H), 4.99–4.95 (m, 2H, OH, 7-Ha), 4.80 (dd, J = 12.7 Hz, 1.8 Hz, 1H, 7-Hb), 4.22–4.19 (m, 2H, 6-Ha, 4-H), 3.77–3.68 (m, 2H, 5-H, 6-Hb); 13C NMR (100 MHz, DMSO-d6): δ = 152.0, 144.1, 124.0, 123.9, 108.7 (C-1, C-2, C-8, C-10, C-11), 92.8, 80.6, 78.1 (C-9, C-3, C-5), 72.4 (C-7), 61.8 (C-4), 59.7 (C-6); HRMS: calculated for C11H10I2O4 ([M + Na]+): 482.8561; found: 482.8562.:ethyl acetate = 3:1) to afford the compounds 8d–8e.
8d. White solid; yield: 43%; 1H NMR (400 MHz, CD3OD): δ = 6.84 (s, 1H, 9-H), 5.09–5.05 (m, 2H, 7-Ha, 3-H), 4.86–4.83 (m, 1H, 7-Hb), 4.35 (dd, J = 3.3 Hz, 0.8 Hz, 1H, 4-H), 4.18 (t, J = 6.4 Hz, 1H, 5-H), 3.99–3.91 (m, 2H, 6-Ha, 6-Hb), 2.09 (s, 3H, CH3), 2.05 (s, 3H, CH3); 13C NMR (100 MHz, CD3OD): δ = 153.2, 142.1, 129.8, 123.8, 112.2 (C-1, C-2, C-8, C-10, C-11), 118.0 (C-9), 94.1, 89.4, 80.5, 74.6 (C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 79.9, 79.7 (C-3, C-5), 74.8 (C-7), 64.4 (C-4), 62.2 (C-6), 4.4, 4.0 (CH3); HRMS: calculated for C17H16O4 ([M + Na]+): 307.0941; found: 307.0948.
C), 79.9, 79.7 (C-3, C-5), 74.8 (C-7), 64.4 (C-4), 62.2 (C-6), 4.4, 4.0 (CH3); HRMS: calculated for C17H16O4 ([M + Na]+): 307.0941; found: 307.0948.
8e. White solid; yield: 42%; 1H NMR (400 MHz, CDCl3): δ = 7.31–7.05 (m, 10H, Ph), 6.63 (s, 1H, 9-H), 5.19–5.16 (m, 2H, 7-Ha, 3-H), 4.94–4.92 (m, 1H, 7-Hb), 4.43–4.42 (m, 1H, 4-H), 4.18 (t, J = 4.8 Hz, 1H, 5-H), 4.06–3.88 (m, 6H, CH2, 6-Ha, 6-Hb); 13C NMR (100 MHz, CDCl3): δ = 149.9, 142.2, 140.8, 140.6, 140.0, 128.9, 128.6, 128.5, 128.3, 126.2, 126.0, 123.4, 120.3 (C-1, C-2, C-8, C-10, C-11, C-Ph), 115.8 (C-9), 78.2 (C-3), 77.5 (C-5), 74.4 (C-7), 65.6 (C-4), 63.7 (C-6), 39.4, 31.1 (CH2); HRMS: calculated for C25H24O4 ([M + Na]+): 411.1567; found: 411.1566.
:ethyl acetate = 3:1) to obtain the white solid 6 (33 mg, 66%). 1H NMR (400 MHz, MeOD): δ = 7.19 (s, 1H, 11-H), 7.02 (s, 1H, 9-H), 5.12–5.05 (m, 2H, 3-H, 7-Ha), 4.90 (m, 1H, 7-Hb), 4.33 (dd, J = 3.6, 0.8 Hz, 1H, 4-H), 4.16 (t, J = 6 Hz, 1H, 5-H), 3.95–3.87 (m, 2H, 6-Ha, 6-Hb); 13C NMR (100 MHz, MeOD): δ = 153.6, 145.5, 124.6 (C-1, C-2, C-8), 123.7, 122.0 (C-9, C-11), 94.2 (C-10), 80.3 (C-5), 79.6 (C-3), 74.3 (C-7), 64.7 (C-4), 62.6 (C-6); HRMS: calculated for C11H11IO4 ([M + Na]+): 356.9594; found: 356.9606.:ethyl acetate = 3:1) to afford the compounds 9a–9d.
9a. White solid; yield: 32%; 1H NMR (400 MHz, CDCl3): δ = 6.87 (s, 1H, 11-H), 6.77 (s, 1H, 9-H), 5.22–5.05 (m, 2H, 3-H, 7-Ha), 4.92 (d, J = 11.9 Hz, 1H, 7-Hb), 4.40 (d, J = 3.2 Hz, 1H, 4-H), 4.24–4.13 (m, 3H, 5-H, 6-Ha, 6-Hb), 2.04 (d, J = 5.6 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ = 151.1, 141.9, 126.4, 121.7 (C-1, C-2, C-8, C-10), 117.3, 115.8 (C-9, C-11), 85.7, 79.8 (C
C), 77.8 (C-3), 77.5 (C-5), 74.2 (C-7), 65.3 (C-4), 63.5 (C-6), 4.5 (CH3); HRMS: calculated for C14H14O4 ([M + Na]+): 269.0784; found: 269.0789.
9b. White solid; yield: 29%; 1H NMR (400 MHz, CDCl3): δ = 7.31–7.27 (m, 2H, Ph), 7.22–7.18 (m, 3H, Ph), 6.68 (s, 1H, 11-H), 6.59 (s, 1H, 9-H), 5.17–5.10 (m, 2H, 3-H, 7-Ha), 4.85 (dd, J = 11.6 Hz, 1.6 Hz, 1H, 7-Hb), 4.40–4.39 (m, 1H, 4-H), 4.17–4.11 (m, 2H, 5-H, 6-Ha), 4.08–4.04 (m, 1H, 6-Hb), 3.96–3.85 (s, 2H, CH2); 13C NMR (101 MHz, MeOD): δ = 151.4, 144.8, 142.3, 129.1, 126.4, 120.5 (C-1, C-2, C-8, C-10, C-Ph), 114.5 (C-11), 113.0 (C-9), 77.7, 77.4 (C-3, C-5), 74.4 (C-7), 65.4 (C-4), 63.6 (C-6), 42.4 (CH2); HRMS: calculated for C18H18O4 ([M + Na]+): 321.1097; found: 321.1101.
9c. White solid; yield: 33%; 1H NMR (400 MHz, CD3OD): δ = 7.11–7.08 (m, 2H, Ph), 6.83–6.79 (m, 2H, Ph), 6.66 (s, 1H, 11-H), 6.47 (s, 1H, 9-H), 5.10–5.07 (m, 2H, 3-H, 7-Ha), 4.85 (m, 1H, 7-Hb), 4.30 (dd, J = 3.3 Hz, 0.8 Hz, 1H, 4-H), 4.12 (t, J = 6.2 Hz, 1H, 5-H), 3.94–3.85 (m, 4H, CH2, 6-Ha, 6-Hb), 3.75 (s, 1H, OCH3); 13C NMR (101 MHz, MeOD): δ = 152.8, 146.1, 143.5, 134.8 (C-1, C-2, C-8, C-Ph), 130.8 (C-Ph), 122.0 (C-10), 114.8 (C-Ph, C-11), 113.4 (C-9), 79.8, 79.6 (C-3, C-5), 74.9 (C-7), 65.0 (C-4), 62.7 (C-6), 55.7 (OCH3), 42.2 (CH2); HRMS: calculated for C19H20O5 ([M + Na]+): 351.1202; found: 351.1202.
9d. White solid; yield: 40%; 1H NMR (500 MHz, MeOD): δ = 7.21–7.18 (m, 2H, Ph), 6.97 (t, J = 8.5 Hz, 2H, Ph), 6.67 (s, 1H, 11-H), 6.48 (s, 1H, 9-H), 5.10–5.08 (m, 2H, 3-H, 7-Ha), 4.31 (d, J = 3.3 Hz, 1H, 4-H), 4.13 (t, J = 6.2 Hz, 1H, 5-H), 3.93–3.86 (m, 4H, CH2, 6-Ha, 6-Hb); 13C NMR (126 MHz, MeOD): δ = 162.8 (C-Ph, J = 244 Hz), 152.9, 145.3, 143.7, 138.8 (C-1, C-2, C-8, C-Ph), 131.5 (C-Ph, J = 7.7 Hz), 122.2 (C-10), 115.9 (C-Ph, J = 21 Hz), 114.9 (C-11), 113.4 (C-9), 79.8, 79.6 (C-3, C-5), 74.9 (C-7), 65.0 (C-4), 62.7 (C-6), 42.1 (CH2); HRMS: calculated for C18H17FO4 ([M + Na]+): 339.1003; found: 339.1009.
3.2 Galectin binding evaluations
Galectin-1 affinities. Experiments were conducted at 20 °C using galectin-1 at 0.50 μM and the fluorescent probe 3,3′-dideoxy-3-[4-(fluorescein-5-yl-carbonylaminomethyl)-1H-1,2,3-triazol-1-yl]-3′-(3,5-dimethoxy-benzamido)-1,1′-sulfanediyl-di-β-D-galactopyranoside at 0.10 μM.
Galectin-3 affinities. Experiments were performed at 20 °C using galectin-3 at 0.20 μM and the fluorescent probe 3,3′-dideoxy-3-[4-(fluorescein-5-yl-carbonylaminomethyl)-1H-1,2,3-triazol-1-yl]-3′-(3,5-dimethoxybenzamido)-1,1′-sulfanediyl-di-β-D-galactopyranoside at 0.02 μM or with galectin-3 at 1.0 μM and 2-(fluorescein-5/6-yl-carbonyl)-aminoethyl 2-acetamido-2-deoxy-α-D-galactopyranosyl-(1–3)-[α-L-fucopyranosyl-(1–2)]-β-D-galactopyranosyl-(1–4)-β-D-glucopyranoside at 0.10 μM.
Galectin-4C affinities. Experiments were conducted at 20 °C using galectin-4C at 0.50 μM and the fluorescent probe 2-(fluorescein-5/6-yl-carbonyl)-aminoethyl 2-acetamido-2-deoxy-α-D-galactopyranosyl-(1–3)-[α-L-fucopyranosyl-(1–2)]-β-D-galactopyranosyl-(1–4)-β-D-glucopyranoside at 0.1 μM.
Galectin-4N affinities. Experiments were carried out at 20 °C using galectin-4N at 3.0 μM and the fluorescent probe 3,3′-dideoxy-3-[4-(fluorescein-5-yl-carbonylaminomethyl)-1H-1,2,3-triazol-1-yl]-3′-(3,5-dimethoxy-benzamido)-1,1′-sulfanediyl-di-β-D-galactopyranoside at 0.10 μM.
Galectin-8N affinities. Experiments were performed at 20 °C using galectin-8N at 0.40 μM and the fluorescent probe 2-(fluorescein-5-yl-carbonylamino) ethyl β-D-galactopyranosyl-(1–4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1–3)-β-D-galactopyranosyl-(1–4)-β-D-glucopyranoside at 0.1 μM.
Galectin-9C affinities. Experiments were conducted at 20 °C using galectin-9C at 2.0 μM and the fluorescent probe 3,3′-dideoxy-3-[4-(fluorescein-5-yl-carbonylaminomethyl)-1H-1,2,3-triazol-1-yl]-3′-(3,5-dimethoxy-benzamido)-1,1′-sulfanediyl-di-β-D-galactopyranoside at 0.10 μM.
Galectin-9N affinities. Experiments were performed at 20 °C using galectin-9N at 1.0 μM and the fluorescent probe 2-(fluorescein-5-yl-carbonylamino)-ethyl β-D-galactopyranosyl-(1–4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1–3)-β-D-galactopyranosyl-(1–4)-β-D-glucopyranoside at 0.1 μM.
3.3 Molecular docking
The X-ray crystal structures of human galectin-1, galectin-3, and N-terminal domain of galectin-8 with D-lactose (PDB ID 1gzw,32 3aye33 and 2yxs) were used to investigate the binding modes of these galectin proteins with the compound 9d. In brief, the proteins were prepared using Autodock Tools 1.5.6 by deleting water molecules, adding polar hydrogens, and assigning Gasteiger charges. The original ligand D-lactose was used to define the binding site, and the box was set using D-lactose as a grid center (galectin-1: center_x = 55.497, center_y = 26.987, center_z = 23.331; size_x = 15, size_y = 15, size_z = 15; galectin-3: center_x = −13.334, center_y = 7.803, center_z = −26.075; size_x = 15, size_y = 15, size_z = 15; galectin-8N: center_x = 14.183, center_y = 24.913, center_z = −7.492; size_x = 15, size_y = 15, size_z = 15). Autodock vina 1.1.2 (ref. 34) was used to perform the docking, and the exhaustiveness value was set to 20.4 Conclusions
Herein, a series of tricyclic carbohydrate–benzene hybrids were synthesized from D-galactose, and derivatizations at C10 and C11 were implemented via iodo-intermediates. Furthermore, their galectin binding activities were evaluated by the competitive fluorescence polarization assay, and the results indicated that most of the hybrid derivatives exhibited selective affinity for galectin-1 and galectin-8N over galectin-3, 4C, 4N, 9C, and 9N. Structure–activity analysis revealed that the C10-mono-substituted compounds displayed better affinity for galectin-8N when compared with the C10, C1-di-substituted, and compounds with (substituted) benzyl groups were good for activity enhancement when compared with other aliphatic alkyl groups. The relatively flat surface near the groove of galectin-1 or galectin-8N CRD was beneficial to the binding since the residues might interact with the benzyl group via cation–π or π–π stackings. Hence, the C10-benzyl tricyclic carbohydrate–benzene hybrids are promising scaffolds for the discovery of selective galectin-1 and galectin-8N inhibitors.Conflicts of interest
UJN is a shareholder in Galecto Biotech AB, Sweden.Acknowledgements
This work was supported by International Cooperation Project of Science and Technology Department of Sichuan Province (2016HH0075), the European Community's Seventh Framework Program (FP7-2007-2013) under the grant agreement no. HEALTH-F2-2011-256986 – project acronym PANACREAS and Ministry of Education Chunhui Project of China (Z2016164). We thank Mrs Barbro Kahl-Knutsson for providing assistance with the fluorescence polarization experiments.Notes and references
- H. Leffler, S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, Glycoconjugate J., 2002, 19, 433–440 CrossRef CAS PubMed.
- S. H. Barondes, V. Castronovo, D. Cooper, R. D. Cummings, K. Drickamer, T. Feizi, M. A. Gitt, J. Hirabayashi, C. Hughes, K.-i. Kasai, H. Leffler, F.-T. Liu, R. Lotan, A. M. Mercurio, M. Monsigny, S. Pillai, F. Poirer, A. Raz, P. W. J. Rigby, J. M. Rini and J. L. Wang, Cell, 1994, 76, 597–598 CrossRef CAS.
- R. C. Hughes, Biochimie, 2001, 83, 667–676 CrossRef CAS PubMed.
- L. Wang and X. Guo, Biomed. Pharmacother., 2016, 78, 165–171 CrossRef CAS PubMed.
- T. Lei, S. Moos, J. Klug, F. Aslani, S. Bhushan, E. Wahle, S. Fröhlich, A. Meinhardt and M. Fijak, Sci. Rep., 2018, 8, 3741–3755 CrossRef PubMed.
- D. Daley, V. R. Mani, N. Mohan, N. Akkad, A. Ochi, D. W. Heindel, K. B. Lee, C. P. Zambirinis, G. S. D. B. Pandian, S. Savadkar, A. Torres-Hernandez, S. Nayak, D. Wang, M. Hundeyin, B. Diskin, B. Aykut, G. Werba, R. M. Barilla, R. Rodriguez, S. Chang, L. Gardner, L. K. Mahal, B. Ueberheide and G. Miller, Nat. Med., 2017, 23, 556–567 CrossRef CAS.
- S. Nakahara, N. Oka and A. Raz, Apoptosis, 2005, 10, 267–275 CrossRef CAS.
- N. L. Perillo, K. E. Pace, J. J. Seilhamer and L. G. Baum, Nature, 1995, 378, 736–739 CrossRef CAS.
- J. Wang, J. Xia, F. Zhang, Y. Shi, Y. Wu, H. Pu, A. K. F. Liou, R. K. Leak, X. Yu, L. Chen and J. Chen, Sci. Rep., 2015, 5, 9621–9630 CrossRef CAS.
- M. Ouellet, S. Mercier, I. Pelletier, S. Bounou, J. Roy, J. Hirabayashi, S. Sato and M. J. Tremblay, J. Immunol., 2005, 174, 4120–4126 CrossRef CAS PubMed.
- J. M. Cousin and M. J. Cloninger, Int. J. Mol. Sci., 2016, 17, 1566–1587 CrossRef PubMed.
- K. Ito, K. Stannard, E. Gabutero, A. M. Clark, S.-Y. Neo, S. Onturk, H. Blanchard and S. J. Ralph, Cancer Metastasis Rev., 2012, 31, 763–778 CrossRef CAS.
- T. L. M. Thurston, M. P. Wandel, N. von Muhlinen, Á. Foeglein and F. Randow, Nature, 2012, 482, 414–418 CrossRef CAS PubMed.
- W.-S. Chen, Z. Cao, S. Sugaya, M. J. Lopez, V. G. Sendra, N. Laver, H. Leffler, U. J. Nilsson, J. Fu, J. Song, L. Xia, P. Hamrah and N. Panjwani, Nat. Commun., 2016, 7, 11302–11320 CrossRef CAS.
- G. Choufani, N. Nagy, S. Saussez, H. Marchant, P. Bisschop, M. Burchert, A. Danguy, S. Louryan, I. Salmon, H. J. Gabius, R. Kiss and S. Hassid, Cancer, 1999, 86, 2353–2363 CrossRef CAS.
- D. Obino, L. Fetler, A. Soza, O. Malbec, J. J. Saez, M. Labarca, C. Oyanadel, F. Del Valle Batalla, N. Goles, A. Chikina, D. Lankar, F. Segovia-Miranda, C. Garcia, T. Léger, A. Gonzalez, M. Espéli, A.-M. Lennon-Duménil and M.-I. Yuseff, Cell Rep., 2018, 25, 3110–3122 CrossRef CAS PubMed.
- F. Ferragut, A. J. Cagnoni, L. L. Colombo, C. Sánchez Terrero, C. Wolfenstein-Todel, M. F. Troncoso, S. I. Vanzulli, G. A. Rabinovich, K. V. Mariño and M. T. Elola, Biochim. Biophys. Acta, 2019, 1866, 1338–1352 CrossRef CAS PubMed.
- E. Pardo, F. Barake, J. A. Godoy, C. Oyanadel, S. Espinoza, C. Metz, C. Retamal, L. Massardo, C. Tapia-Rojas, N. C. Inestrosa, A. Soza and A. González, Mol. Neurobiol., 2019, 56, 7774–7788 CrossRef CAS.
- V. Balan, P. Nangia-Makker and A. Raz, Cancers, 2010, 2, 592–610 CrossRef CAS.
- H. Blanchard, X. Yu, P. M. Collins and K. Bum-Erdene, Expert Opin. Ther. Pat., 2014, 24, 1053–1065 CrossRef CAS PubMed.
- K. B. Pal, M. Mahanti, X. Huang, S. Persson, A. P. Sundin, F. R. Zetterberg, S. Oredsson, H. Leffler and U. J. Nilsson, Org. Biomol. Chem., 2018, 16, 6295–6305 RSC.
- J. Tejler, F. Skogman, H. Leffler and U. J. Nilsson, Carbohydr. Res., 2007, 342, 1869–1875 CrossRef CAS PubMed.
- P. M. Collins, C. T. Öberg, H. Leffler, U. J. Nilsson and H. Blanchard, Chem. Biol. Drug Des., 2012, 79, 339–346 CrossRef CAS PubMed.
- J. Dion, F. Deshayes, N. Storozhylova, T. Advedissian, A. Lambert, M. Viguier, C. Tellier, C. Dussouy, F. Poirier and C. Grandjean, ChemBioChem, 2017, 18, 782–789 CrossRef CAS PubMed.
- I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, Angew. Chem., Int. Ed., 2005, 44, 5110–5112 CrossRef CAS PubMed.
- T. Delaine, P. Collins, A. MacKinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. van Hattum, M. van Scherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, ChemBioChem, 2016, 17, 1759–1770 CrossRef CAS PubMed.
- K. Peterson, R. Kumar, O. Stenstroem, P. Verma, P. R. Verma, M. Haakansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, J. Med. Chem., 2018, 61, 1164–1175 CrossRef CAS PubMed.
- V. K. Rajput, A. MacKinnon, S. Mandal, P. Collins, H. Blanchard, H. Leffler, T. Sethi, H. Schambye, B. Mukhopadhyay and U. J. Nilsson, J. Med. Chem., 2016, 59, 8141–8147 CrossRef CAS PubMed.
- M. Leibeling, B. Milde, D. Kratzert, D. Stalke and D. B. Werz, Chem.–Eur. J., 2011, 17, 9888–9892 CrossRef CAS PubMed.
- P. Sörme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Anal. Biochem., 2004, 334, 36–47 CrossRef PubMed.
- P. Sörme, B. Kahl-Knutson, U. Wellmar, U. J. Nilsson and H. Leffler, Methods Enzymol., 2003, 362, 504–512 Search PubMed.
- M. F. Lopez-Lucendo, D. Solis, S. Andre, J. Hirabayashi, K. Kasai, H. Kaltner, H. J. Gabius and A. Romero, J. Mol. Biol., 2004, 343, 957–970 CrossRef CAS PubMed.
- C. F. Bian, Y. Zhang, H. Sun, D. F. Li and D. C. Wang, PLoS One, 2011, 6, e25007–e25016 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03144e |
| This journal is © The Royal Society of Chemistry 2020 |