
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel catalyzed intramolecular oxidative coupling: synthesis of 3-aryl benzofurans†
Sakshi Aggarwal,
Dasari Srinivas,
Chinnabattigalla Sreenivasulu and
Gedu Satyanarayana *
*
Department of Chemistry, Indian Institute of Technology, Kandi, Sangareddy, Hyderabad, 502 285, Telangana, India. E-mail: gvsatya@iith.ac.in; Fax: +91 40 2301 6003/32; Tel: +91 40 2301 6033
First published on 10th June 2020
Abstract
Recent research has been focused on the transition metal-catalyzed reactions. Herein we have developed nickel-catalyzed synthesis of 3-aryl benzofurans from ortho-alkenyl phenols via intramolecular dehydrogenative coupling. Notably, simple O2 gas served as an oxidant, without using any sacrificial hydrogen acceptor. The strategy enabled the synthesis of 3-aryl benzofurans in good to excellent yields.
Introduction
Benzofuran is a heterocyclic compound made up of benzene ring fused with a furan ring and a prominent structural motif that constitutes naturally occurring compounds, pharmaceuticals, photosensitizers and molecules of biological relevance.1–3 Some of the biologically active compounds containing benzofuran skeleton are fused tricyclic compound (R7000) I, furomollugin II, amiodarone III, Iantheran A IV, viniferifuran V and pterolinus A VI (Fig. 1).3 | ||
| Fig. 1 Representative examples of benzofurans of biological relevance. | ||
Due to their wide occurrence and interesting biological properties, numerous reports have disclosed the synthesis of benzofurans.4–22 In this context, most of the reports mainly centered on the synthesis of 2,3-diaryl benzofurans, either via intermolecular annulation of ortho-halophenols with olefins or via intramolecular annulation of ortho-vinyl phenols promoted by Lewis acids/oxidants/some strong acids/bases.5–9 All of them require stoichiometric amounts of acid/oxidant/base. Arcadi et al. described a first palladium-catalyzed synthesis of 2,3-diaryl benzofuran via intramolecular cyclization of ortho-alkynes substituted phenols.6a Subsequently, a reasonable number of reports have appeared on the synthesis of 2,3-diarlybenzofurans using transition metal catalysts like Au, Ir, Rh, Cu, Pd and Fe etc.10 The best synthetic route to accomplish benzofuran could be the intramolecular cyclization from ortho-alkenyl phenol, unfortunately, this protocol requires a stoichiometric amount of sacrificial hydrogen acceptor-like DDQ.8b Notably, recently, the oxidative C–H functionalization of ortho-alkenyl phenols to generate benzofurans has been accomplished using some transition metals without the need of any sacrificing hydrogen acceptor.9,22b,22d Most of the earlier reports are devoted to the formation of 2,3-diaryl benzofurans/2-aryl benzofurans, whereas the synthesis of 3-aryl benzofurans from ortho-alkenyl phenols was scarcely explored.20,22 Some of the representative examples of the previous study versus the present protocol is described in Scheme 1.
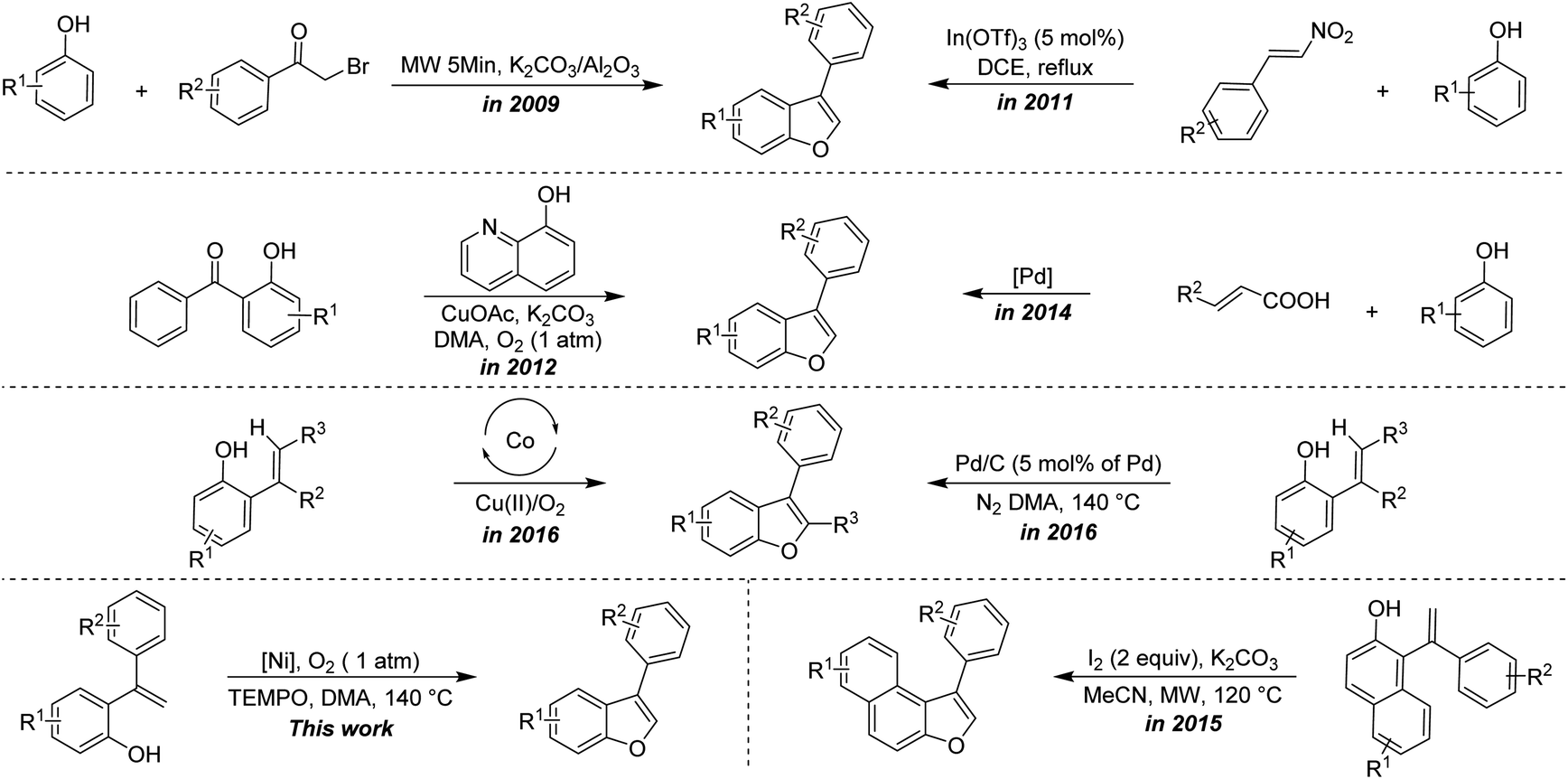 | ||
| Scheme 1 Representative approaches for the synthesis of 3-aryl benzofurans.12,14,20a,20b,22a,22b,22d | ||
We have been interested in the ambitious catalytic nature of late transition metals.23 Recently, we have developed a synthesis of 2,3-diaryl benzofuran by using phenols and internal alkynes11a and also reported a synthesis of 2H/4H-chromenes from phenols with terminal alkynes (aryl/alkyl) using Lewis acidic conditions.11b With this background of phenols and the alkyne chemistry. We intended to develop nickel catalyzed oxidative cross coupling reactions. Herein, we describe an efficient method to cyclize ortho-alkenyl phenols to give benzofurans facilitated by Ni(acac)2 and O2 as an oxidant.
Results and discussion
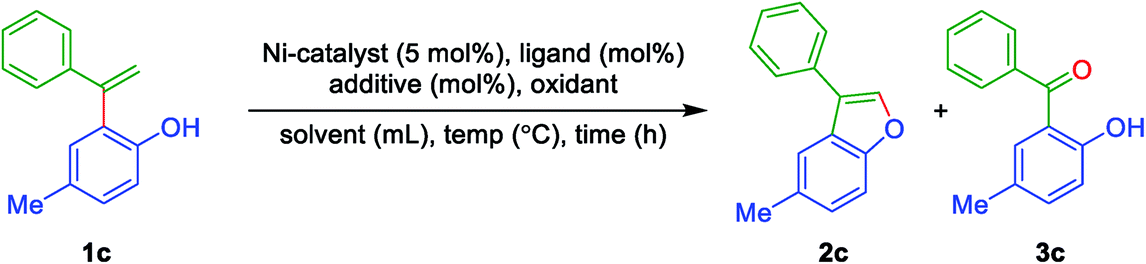
To begin with, it was contemplated that 3-aryl benzofuran can be achieved from ortho-vinyl phenols using intramolecular oxidative coupling feasible by means of a suitable metal catalyst and an oxidant. The required ortho-vinyl phenols have prepared from the reaction of phenols and terminal arylacetylenes using Friedel–Crafts alkenylation induced by a suitable Lewis acid. With the available ortho-alkenyl phenols, it is set for the optimization study to achieve 3-aryl benzofurans. Thus, initially, ortho-alkenyl phenol 1c was chosen as the model compound for the preparation of 3-aryl benzofuran 2c. Various screening conditions (i.e., by varying ligand, additive, oxidant, reaction time and solvent etc.) have explored to find the best-optimized conditions and the outcomes are summarised in Table 1. To begin with, the reaction was performed with Ni(acac)2 (5 mol%), 1,10-phenanthroline (10 mol%) and DMF as solvent under inert conditions (nitrogen atmosphere) at 140 °C for 48 h. The expected 3-aryl benzofuran 2c was obtained albeit in moderate yield along with a minor side product (2-hydroxy-5-methylphenyl)(phenyl)methanone 3c (Table 1, entry 1). Even switching to PPh3 as the ligand, furnished the product 2c in more or less same yield (Table 1, entry 2). Interestingly, under the same reaction conditions (i.e. Table 1, entry 2), but with molecular O2 as the oxidant, there was a drastic change in the yield of 2c along with the minimal amount of ketone 3c (Table 1, entry 3). However, with additive TEMPO and with PPh3 under open air at 140 °C for 48 h, improved the yield of 2c to 80% (Table 1, entry 4). Performing the reaction with TEMPO/PPh3 and under an oxygen atmosphere, gave the product 2c in the same yield but with a reduced amount of time 36 h (Table 1, entry 5). On the other hand, the other additives (DDQ, Oxone, K2S2O8 and TBHP/H2O, under oxygen atmosphere) in the same solvent DMF were not active improve the yields of 2c (Table 1, entries 6 to 9). The reaction was inferior in solvents, such as H2O and ortho-xylene, under oxygen atmosphere (Table 1, entries 10 & 11), while in DMSO under an oxygen atmosphere, gave 2c in 70% yield (Table 1, entry 12). To our delight, the reaction in solvent DMA under oxygen atmosphere afforded the product 2c in 81% yield (Table 1, entry 13). When the reaction conducted using 2 mol% of the catalyst Ni(acac)2, under an oxygen atmosphere, afforded the product 2c but with slightly decreased yield (Table 1, entry 14). Replacing the ligand PPh3 with 1,10-phenanthroline in the open air and an oxygen atmosphere, gave the product 2c in 64% and 60% yields, respectively (Table 1, entries 15 & 16). The reaction without ligand and additive, under oxygen atmosphere, afforded 2c in 45% moderate yield (Table 1, entry 17). On the other hand, treatment of 1c either with TEMPO or DDQ as additive without ligand and in the open air in CH3CN as a solvent found to be slightly inferior (Table 1, entries 18 & 19). While using K2CO3 as the base instead of additive TEMPO, in the open air and in the presence of oxygen atmosphere showed no progress indicating the importance of TEMPO to initiate the reaction (Table 1, entries 20 & 21). On the other hand, by employing NiCl2 as a catalyst, only a 50% yield of the product 2c was obtained (Table 1, entry 22). Further, the reaction at reduced temperatures 120 °C and 80 °C, showed little conversion and no conversion, respectively (Table 1, entries 23 & 24).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Ligand (10 mol%) | Additives (mol%) | Oxidant | Solvent (mL) | Time (h) | Temp. (°C) | Yield 2c (%) |
| a All reactions were carried out using ortho-alkenyl phenol 1c (83 mg, 0.4 mmol), Ni(acac)2 [0.008 mmol (2 mol%) to 0.02 mmol (5 mol%)], ligand (0.04 mmol, 10 mol%).b Reaction was conducted under nitrogen atmosphere.c Very less conversion was observed.d The by-product 3c was formed (up-to 5–20% yields).e Some other volatile by-products also formed along with 2c, which were not isolable. | ||||||||
| 1 | Ni(acac)2 (5) | 1,10-Phen | — | —b | DMF (1.5) | 48 | 140 | 40c,d |
| 2 | Ni(acac)2 (5) | PPh3 | — | —b | DMF (1.5) | 36 | 140 | 42 |
| 3 | Ni(acac)2 (5) | PPh3 | — | O2 | DMF (1.5) | 36 | 140 | 70d |
| 4 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | Open air | DMF (2) | 48 | 140 | 80d |
| 5 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | DMF (2) | 36 | 140 | 80d |
| 6 | Ni(acac)2 (5) | PPh3 | DDQ (10) | O2 | DMF (1.5) | 36 | 140 | —c |
| 7 | Ni(acac)2 (5) | PPh3 | OXONE (10) | O2 | DMF (1.5) | 36 | 140 | 30c,d |
| 8 | Ni(acac)2 (5) | PPh3 | K2S2O8 (10) | O2 | DMF (1.5) | 48 | 140 | 34c,d |
| 9 | Ni(acac)2 (5) | PPh3 | TBHP in H2O (10) | O2 | DMF (1.5) | 48 | 140 | 48c,d |
| 10 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | H2O (1) | 48 | 140 | —c |
| 11 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | o-Xylene (1) | 36 | 140 | —c |
| 12 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | DMSO (1.5) | 36 | 140 | 70c |
| 13 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | DMA (1) | 36 | 140 | 81d |
| 14 | Ni(acac)2 (2) | PPh3 | TEMPO (10) | O2 | DMA (1) | 36 | 140 | 70d |
| 15 | Ni(acac)2 (5) | 1,10-Phen | TEMPO (10) | Open air | DMA (1) | 36 | 140 | 64d |
| 16 | Ni(acac)2 (5) | 1,10-Phen | TEMPO (10) | O2 | DMA (1) | 40 | 140 | 60d |
| 17 | Ni(acac)2 (5) | — | — | O2 | DMA (1) | 40 | 140 | 45 |
| 18 | Ni(acac)2 (5) | — | TEMPO (5) | Open air | CH3CN (2) | 72 | 140 | 60d |
| 19 | Ni(acac)2 (5) | — | DDQ (5) | Open air | CH3CN (2) | 72 | 140 | 50d |
| 20 | Ni(acac)2 (5) | PPh3 | K2CO3 (2 equiv.) | Open air | DMA (1) | 72 | 140 | —c |
| 21 | Ni(acac)2 (5) | PPh3 | K2CO3 (2 equiv.) | O2 | DMA (1) | 72 | 140 | —c |
| 22 | NiCl2 (5) | PPh3 | TEMPO (10) | O2 | DMA (1) | 48 | 140 | 50d,e |
| 23 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | DMA (1) | 36 | 120 | 30c |
| 24 | Ni(acac)2 (5) | PPh3 | TEMPO (10) | O2 | DMA (1) | 36 | 80 | —c |
| 18 | Ni(acac)2 (5) | — | TEMPO (5) | Open air | CH3CN (2) | 72 | 140 | 60d |
| 19 | Ni(acac)2 (5) | — | DDQ (5) | Open air | CH3CN (2) | 72 | 140 | 50d |
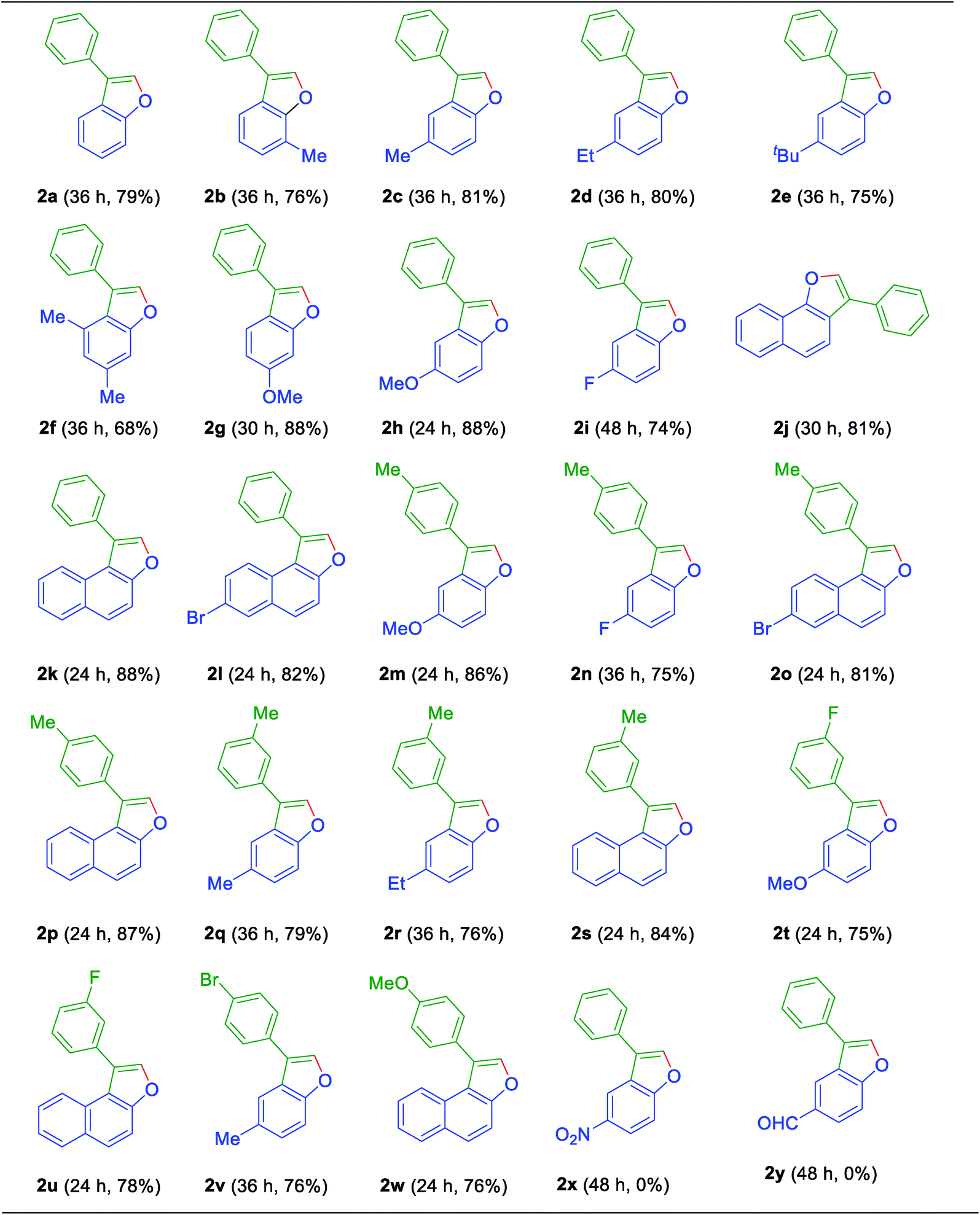
Now to test the scope and applicability of the strategy, best-optimized conditions (Table 1, entry 15) applied to different ortho-alkenyl phenols 1a–w. This protocol was found to be quite successful and afforded the corresponding 3-aryl benzofurans 2a–w, good to very good yields (Table 2). For example, the reaction was amenable with electron-donating groups like Me, Et, OMe substituents on phenol moiety of ortho-alkenyl phenols (Table 2). The reaction also found smooth with the α-naphthol, β-naphthol, and 6-bromo-2-naphthol derived alkenols (Table 2). Moreover, the reaction was obedient with the electron donating (Me and OMe) and the partial electron withdrawing substituents (F and Br) on the ring of terminal alkynes of ortho-alkenyl phenols (Table 2). All synthesized 3-aryl benzofurans 2a–w, are characterized by spectrometric data (1H-NMR and 13C-NMR and Mass Spectrometry) and also with the earlier literature. However, it is essential to note that even alkenylation reaction did not proceed when electron withdrawing groups such as CHO and NO2 flanked to the phenol moiety. And it can be justified based on the fact that electron withdrawing functional groups retard the Friedel–Crafts alkylation/alkenylation reactions. Similarly, the attempt made to synthesis ortho-alkenyl phenols were not successful with meta-amino functionality on the aromatic ring of the alkyne. This could be due to the more reactive nature of the amino group under Lewis acid conditions. Moreover, aliphatic alkynes could not make compatible to give ortho-alkenylation products as well.
Plausible mechanism
Though the exact mechanism is not very certain at this stage, based on the present observations and literature reports,12,20,23 we have attempted to propose a plausible reaction mechanism as depicted in Scheme 2. Initially, Ni(II) could combine with phosphine ligand PPh3 converted into its reduced Ni(0)-catalyst, which may act as an active catalyst. On the other hand, in an independent path, ortho-alkenyl phenol 1 would be transformed into its oxy-radical A under probable catalytic oxidative conditions of TEMPO/O2 system. Now coupling of oxy-radical A with Ni(0)-catalyst would lead to the formation of intermediate B. Subsequently, intramolecular π-complexation with olefinic double bond followed by olefinic C–H activation could generate a six-membered oxa-nickelacycle C via the removal of H-radical. Finally, reductive elimination of catalyst from C gives 3-aryl benzofuran 2 and regenerates the Ni(0)-catalyst. Thus, completes the catalytic cycle.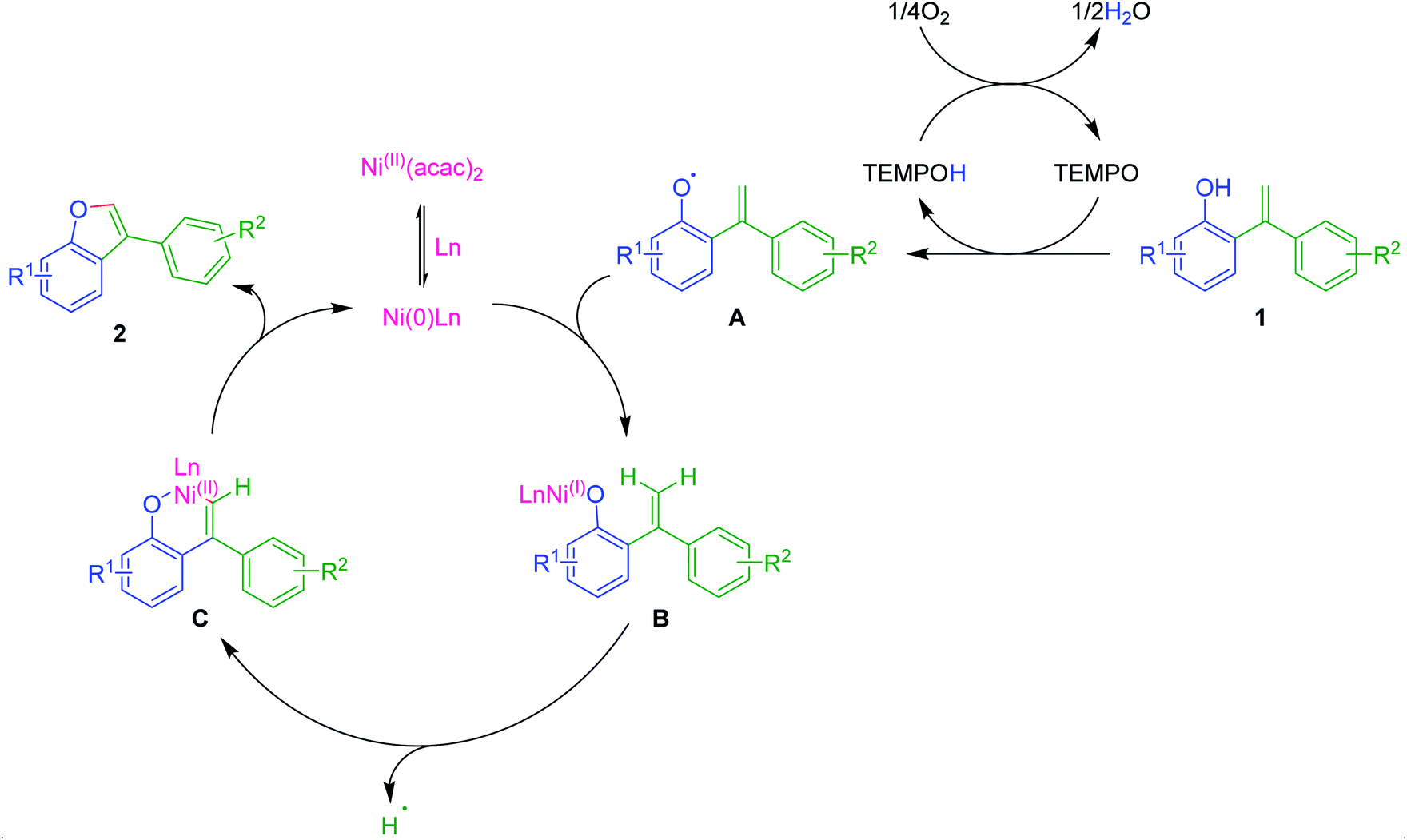 | ||
| Scheme 2 Plausible mechanism for the formation of 3-aryl benzofurans 2 from 1. | ||
Conclusion
In summary, we have established a nickel-catalyzed synthesis of 3-aryl benzofurans via intramolecular oxidative cyclization of ortho-alkenyl phenols. Notably, simple oxygen served as the sole oxidant and precludes the use of sacrificial hydrogen acceptor. This methodology found viable for accomplishing several different 3-arylbenzofuran derivatives in good to very good yields.Experimental section
IR spectra recorded on a Bruker Tensor 37 (FT-IR) spectrophotometer. 1H-NMR spectra recorded on Bruker Avance 400 (400 MHz) spectrometer at 295 K in CDCl3; chemical shifts (δ in ppm) and coupling constants (J in Hz) reported in standard fashion with reference to either internal standard tetramethylsilane (TMS) (δH = 0.00 ppm) or CHCl3 (δH = 7.25 ppm). 13C-NMR spectra recorded on Bruker Avance 400 (100 MHz) spectrometer at RT in CDCl3; chemical shifts (δ in ppm) are reported relative to CDCl3 [δC = 77.00 ppm (central line of the triplet)]. In the 13C-NMR, the nature of carbons (C, CH, CH2 and CH3) was determined by recording the DEPT-135 spectra, and is given in parentheses and noted as s = singlet (for C), d = doublet (for CH), t = triplet (for CH2) and q = quartet (for CH3). In the 1H-NMR, the following abbreviations were used throughout: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. The assignment of signals confirmed by 1H, 13C CPD and DEPT spectra. High-resolution mass spectra (HR-MS) were recorded on an Agilent 6538 UHD Q-TOF using multimode source. Reactions were monitored by TLC on silica gel coated on alumina plate or glass plate using a mixture of petroleum ether and ethyl acetate as eluents. Reactions carried out under oxygen atmosphere.Materials
All solvents distilled before using; petroleum ether with a boiling range of 60 to 80 °C, dichloromethane (DCM), ethyl acetate, dry DMA (boiling range 160 to 170 °C; with purity 99%) were purchased from Sigma Aldrich & locally available commercial sources used. Acme's silica gel (100–200 mesh) used for column chromatography.GP (general procedure for the synthesis of 3-aryl benzofurans)
To an oven dry Schlenk tube was equipped with a magnetic stir bar, were added Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol), ortho-alkenyl phenols 1a–w (79–110.0 mg, 0.4 mmol), and DMA (1 mL). Then a balloon filled with O2 was attached to the Schlenk tube. The reaction mixture stirred at 140 °C for 24 to 36 h. TLC monitored the progress of the reaction. The reaction mixture was then cooled to room temperature and extracted by using ethyl acetate (3 × 20 mL). The organic layers were washed with saturated NH4Cl solution, dried by Na2SO4 and then filtered. Evaporation of the solvent(s) under reduced pressure and refinement of the crude mixture by silica gel column chromatography (petroleum ether/ethyl acetate), gave the 3-aryl benzofurans (68–88%) as semi-solid or liquid.GP was carried out with 1d (89 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 36 h for the product 2d formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
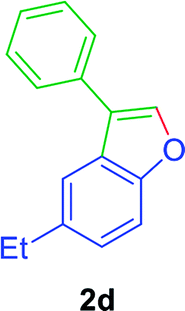
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :01), furnished the product 2d (71 mg, 80%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1d) = 0.10, Rf(2d) = 0.60, UV detection]. 1H NMR (CDCl3, 400 MHz): δ = 7.77 (s, 1H, Ar–O–CH), 7.70–7.57 (m, 3H, Ar–H), 7.56–7.44 (m, 3H, Ar–H), 7.43–7.28 (m, 1H, Ar–H), 7.27–7.13 (m, 1H, Ar–H), 2.79 (q, 2H, J = 7.6 Hz, Ar–CH2CH3), 1.31 (t, 3H, J = 7.6 Hz, Ar–CH2CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 154.3 (s, Ar–C), 141.5 (d, Ar–O–CH), 139.2 (s, Ar–C), 132.3 (s, Ar–C), 128.9 (d, 2C, 2 × Ar–CH), 127.5 (d, 2C, 2 × Ar–CH), 127.3 (d, Ar–CH), 126.5 (s, Ar–C), 124.8 (d, Ar–CH), 122.1 (s, Ar–C), 119.0 (d, Ar–CH), 111.4 (d, Ar–CH), 29.01 (t, Ar–CH2), 16.44 (q, Ar–CH2–CH3) ppm. HR-MS (ESI+) m/z calculated for [C16H14OK]+ = [M + K]+: 261.0676; found 261.0859.
:01), furnished the product 2d (71 mg, 80%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1d) = 0.10, Rf(2d) = 0.60, UV detection]. 1H NMR (CDCl3, 400 MHz): δ = 7.77 (s, 1H, Ar–O–CH), 7.70–7.57 (m, 3H, Ar–H), 7.56–7.44 (m, 3H, Ar–H), 7.43–7.28 (m, 1H, Ar–H), 7.27–7.13 (m, 1H, Ar–H), 2.79 (q, 2H, J = 7.6 Hz, Ar–CH2CH3), 1.31 (t, 3H, J = 7.6 Hz, Ar–CH2CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 154.3 (s, Ar–C), 141.5 (d, Ar–O–CH), 139.2 (s, Ar–C), 132.3 (s, Ar–C), 128.9 (d, 2C, 2 × Ar–CH), 127.5 (d, 2C, 2 × Ar–CH), 127.3 (d, Ar–CH), 126.5 (s, Ar–C), 124.8 (d, Ar–CH), 122.1 (s, Ar–C), 119.0 (d, Ar–CH), 111.4 (d, Ar–CH), 29.01 (t, Ar–CH2), 16.44 (q, Ar–CH2–CH3) ppm. HR-MS (ESI+) m/z calculated for [C16H14OK]+ = [M + K]+: 261.0676; found 261.0859.GP was carried out with 1f (89 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 36 h for the product 2f formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
 :01), furnished the product 2f (60 mg, 68%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1f) = 0.10, Rf(2f) = 0.60, UV detection]. 1H NMR (CDCl3, 400 MHz): δ = 7.47 (s, 1H, Ar–O–CH), 7.46–7.30 (m, 5H, Ar–H), 7.19 (s, 1H, Ar–H), 6.84 (s, 1H, Ar–H), 2.44 (s, 3H, CH3), 2.21 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 155.9 (s, Ar–C), 141.4 (d, Ar–O–CH), 134.6 (s, Ar–C), 133.1 (s, Ar–C), 131.4 (s, Ar–C), 130.1 (d, 2C, 2 × Ar–CH), 128.0 (s, 2C, Ar–CH), 127.4 (d, Ar–CH), 125.9 (d, Ar–CH), 123.5 (s, Ar–C), 123.2 (s, 1Ar–C), 109.4 (d, Ar–CH), 21.4 (q, CH3) 19.7 (q, CH3) ppm. HR-MS (ESI+) m/z calculated for [C16H18OFN]+ = [M + NH4]+: 240.1383; found 240.2062.
:01), furnished the product 2f (60 mg, 68%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1f) = 0.10, Rf(2f) = 0.60, UV detection]. 1H NMR (CDCl3, 400 MHz): δ = 7.47 (s, 1H, Ar–O–CH), 7.46–7.30 (m, 5H, Ar–H), 7.19 (s, 1H, Ar–H), 6.84 (s, 1H, Ar–H), 2.44 (s, 3H, CH3), 2.21 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 155.9 (s, Ar–C), 141.4 (d, Ar–O–CH), 134.6 (s, Ar–C), 133.1 (s, Ar–C), 131.4 (s, Ar–C), 130.1 (d, 2C, 2 × Ar–CH), 128.0 (s, 2C, Ar–CH), 127.4 (d, Ar–CH), 125.9 (d, Ar–CH), 123.5 (s, Ar–C), 123.2 (s, 1Ar–C), 109.4 (d, Ar–CH), 21.4 (q, CH3) 19.7 (q, CH3) ppm. HR-MS (ESI+) m/z calculated for [C16H18OFN]+ = [M + NH4]+: 240.1383; found 240.2062.GP was carried out with 1n (90 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 36 h for the product 2n formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
 :01), furnished the product 2n (68 mg, 75%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1n) = 0.10, Rf(2n) = 0.60, UV detection]. 1H NMR (CDCl3, 400 MHz): δ = 7.78 (s, 1H, Ar–O–CH), 7.54–7.41 (m, 4H, Ar–H), 7.28 (d, 2H, J = 7.8 Hz, Ar–H), 7.05 (td, 1H, J = 9.1, 2.45 Hz, Ar–H), 2.41 (s, 3H, Ar–CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 160.6 (s, Ar–C), 158.3 (s, Ar–C), 152.0 (s, Ar–C), 142.7 (d, Ar–O–CH), 137.5 (s, Ar–C), 129.8 (d, 2C, 2 × Ar–CH), 128.5 (s, Ar–C), 127.5 (s, Ar–C), 127.4 (s, Ar–C) 127.2 (d, 2C, 2 × Ar–CH), 122.5 (s, Ar–C), 122.4 (s, Ar–C), 112.4 (d, Ar–CH), 112.3 (d, Ar–CH), 112.1 (d, Ar–CH), 106.2 (d, Ar–CH), 105.9 (d, Ar–CH), 21.23 (q, Ar–CH3) ppm. HR-MS (ESI+) m/z calculated for [C15H10F]+ = [(M + H) + (–H2O)]+: 209.0761; found 209.0765.
:01), furnished the product 2n (68 mg, 75%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1n) = 0.10, Rf(2n) = 0.60, UV detection]. 1H NMR (CDCl3, 400 MHz): δ = 7.78 (s, 1H, Ar–O–CH), 7.54–7.41 (m, 4H, Ar–H), 7.28 (d, 2H, J = 7.8 Hz, Ar–H), 7.05 (td, 1H, J = 9.1, 2.45 Hz, Ar–H), 2.41 (s, 3H, Ar–CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 160.6 (s, Ar–C), 158.3 (s, Ar–C), 152.0 (s, Ar–C), 142.7 (d, Ar–O–CH), 137.5 (s, Ar–C), 129.8 (d, 2C, 2 × Ar–CH), 128.5 (s, Ar–C), 127.5 (s, Ar–C), 127.4 (s, Ar–C) 127.2 (d, 2C, 2 × Ar–CH), 122.5 (s, Ar–C), 122.4 (s, Ar–C), 112.4 (d, Ar–CH), 112.3 (d, Ar–CH), 112.1 (d, Ar–CH), 106.2 (d, Ar–CH), 105.9 (d, Ar–CH), 21.23 (q, Ar–CH3) ppm. HR-MS (ESI+) m/z calculated for [C15H10F]+ = [(M + H) + (–H2O)]+: 209.0761; found 209.0765.GP was carried out with 1q (89 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 36 h for the product 2q formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
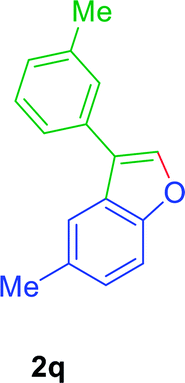 :01), furnished the product 2q (70 mg, 79%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1q) = 0.10, Rf(2q) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3): δ = 7.73 (s, 1H Ar–O–CH), 7.61 (s, 1H, Ar–H), 7.47–7.33 (m, 4H, Ar–H), 7.23–7.08 (m, 2H, Ar–H), 2.47 (s, 3H, Ar–CH3), 2.44 (s, 3H, Ar–CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 154.2 (s, Ar–C), 141.4 (d, Ar–O–CH), 138.6 (s, Ar–C), 132.4 (s, Ar–C), 132.1 (s, Ar–C), 128.8 (d, Ar–CH), 128.2 (d, Ar–CH), 128.1 (d, Ar–CH), 126.6 (s, Ar–C), 125.7 (d, Ar–CH), 124.6 (d, Ar–CH), 122.0 (s, Ar–C), 120.2 (d, Ar–CH), 111.2 (d, Ar–CH), 21.5 (q, Ar–CH3), 21.5 (q, Ar–CH3) ppm. HR-MS (ESI+) m/z calculated for [C16H18NO]+ = [M + NH4]+: 240.1383; found 240.1373.
:01), furnished the product 2q (70 mg, 79%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1q) = 0.10, Rf(2q) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3): δ = 7.73 (s, 1H Ar–O–CH), 7.61 (s, 1H, Ar–H), 7.47–7.33 (m, 4H, Ar–H), 7.23–7.08 (m, 2H, Ar–H), 2.47 (s, 3H, Ar–CH3), 2.44 (s, 3H, Ar–CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 154.2 (s, Ar–C), 141.4 (d, Ar–O–CH), 138.6 (s, Ar–C), 132.4 (s, Ar–C), 132.1 (s, Ar–C), 128.8 (d, Ar–CH), 128.2 (d, Ar–CH), 128.1 (d, Ar–CH), 126.6 (s, Ar–C), 125.7 (d, Ar–CH), 124.6 (d, Ar–CH), 122.0 (s, Ar–C), 120.2 (d, Ar–CH), 111.2 (d, Ar–CH), 21.5 (q, Ar–CH3), 21.5 (q, Ar–CH3) ppm. HR-MS (ESI+) m/z calculated for [C16H18NO]+ = [M + NH4]+: 240.1383; found 240.1373.GP was carried out with 1r (94 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 36 h for the product 2r formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
 :01), furnished the product 2r (71 mg, 76%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1r) = 0.10, Rf(2r) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3): δ = 7.74 (s, 1H, Ar–O–CH), 7.62 (d, 1H, J = 0.1 Hz, Ar–H), 7.51–7.41 (m, 3H, Ar–H), 7.39–7.33 (m, 1H, Ar–H), 7.21–7.15 (m, 2H, Ar–H), 2.77 (q, 2H, J = 7.3 Hz, Ar–CH2CH3), 2.44 (s, 3H, CH3), 1.28 (t, 3H, J = 7.6 Hz, Ar–CH2CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 154.3 (s, Ar–C), 141.5 (d, Ar–O–CH), 139.1 (s, Ar–C), 138.6 (s, Ar–C), 132.2 (s, Ar–C), 128.8 (d, Ar–CH), 128.2 (d, Ar–CH), 128.2 (d, Ar–CH), 126.6 (s, Ar–C), 124.7 (d, Ar–CH), 124.6 (d, Ar–CH), 122.2 (s, Ar–C), 119.0 (d, Ar–C), 111.3 (d, Ar–C), 29.0 (t, CH2), 21.5 (q, CH3), 16.5 (q, CH3) ppm. HR-MS (ESI+) m/z calculated for [C17H19NO]+ = [M + NH4]+: 253.1461; found: 254.2219.
:01), furnished the product 2r (71 mg, 76%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1r) = 0.10, Rf(2r) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3): δ = 7.74 (s, 1H, Ar–O–CH), 7.62 (d, 1H, J = 0.1 Hz, Ar–H), 7.51–7.41 (m, 3H, Ar–H), 7.39–7.33 (m, 1H, Ar–H), 7.21–7.15 (m, 2H, Ar–H), 2.77 (q, 2H, J = 7.3 Hz, Ar–CH2CH3), 2.44 (s, 3H, CH3), 1.28 (t, 3H, J = 7.6 Hz, Ar–CH2CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 154.3 (s, Ar–C), 141.5 (d, Ar–O–CH), 139.1 (s, Ar–C), 138.6 (s, Ar–C), 132.2 (s, Ar–C), 128.8 (d, Ar–CH), 128.2 (d, Ar–CH), 128.2 (d, Ar–CH), 126.6 (s, Ar–C), 124.7 (d, Ar–CH), 124.6 (d, Ar–CH), 122.2 (s, Ar–C), 119.0 (d, Ar–C), 111.3 (d, Ar–C), 29.0 (t, CH2), 21.5 (q, CH3), 16.5 (q, CH3) ppm. HR-MS (ESI+) m/z calculated for [C17H19NO]+ = [M + NH4]+: 253.1461; found: 254.2219.GP was carried out with 1t (98 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 24 h for the product 2t formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
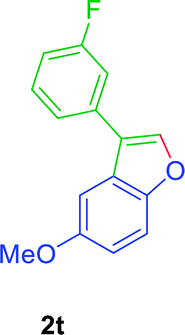 :01), furnished the product 2t (74 mg, 75%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1t) = 0.10, Rf(2t) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3) δ = 7.77 (s, 1H, Ar–O–CH), 7.49–7.43 (m, 2H, Ar–H), 7.41 (dd, J = 3.4, 1.9 Hz, 1H, Ar–H), 7.32 (ddd, J = 9.8, 2.4, 1.6 Hz, 1H, Ar–H), 7.24 (d, J = 2.6 Hz, 1H, Ar–H), 7.11–7.03 (m, 1H, Ar–H), 6.98 (dd, J = 8.9, 2.6 Hz, 1H, Ar–H), 3.88 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 163.2 (d, Jc–f = 294 Hz, Ar–CF), 156.38 (s, Ar–C), 150.76 (s, Ar–C), 142.57 (d, Ar–CH), 134.3 (d, Jc–f = 8 Hz, Ar–C), 130.5 (d, Jc–f = 9 Hz, Ar–CH), 126.57 (s, Ar–C), 123.02 (d, Jc–f = 2 Hz, Ar–CH), 121.4 (s, Ar–C), 114.2 (d, Jc–f = 21 Hz, Ar–CH), 114.2 (d, Jc–f = 21 Hz, Ar–CH), 113.50 (d, Ar–CH), 112.31 (d, Ar–CH), 102.65 (d, Ar–CH), 56.03 (q, OCH3) ppm. HR-MS (ESI+) m/z calculated for [C15H13FKO3]+ = [(M + K) + (–H2O)]+: 263.0269; found 263.0273.
:01), furnished the product 2t (74 mg, 75%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1t) = 0.10, Rf(2t) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3) δ = 7.77 (s, 1H, Ar–O–CH), 7.49–7.43 (m, 2H, Ar–H), 7.41 (dd, J = 3.4, 1.9 Hz, 1H, Ar–H), 7.32 (ddd, J = 9.8, 2.4, 1.6 Hz, 1H, Ar–H), 7.24 (d, J = 2.6 Hz, 1H, Ar–H), 7.11–7.03 (m, 1H, Ar–H), 6.98 (dd, J = 8.9, 2.6 Hz, 1H, Ar–H), 3.88 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 163.2 (d, Jc–f = 294 Hz, Ar–CF), 156.38 (s, Ar–C), 150.76 (s, Ar–C), 142.57 (d, Ar–CH), 134.3 (d, Jc–f = 8 Hz, Ar–C), 130.5 (d, Jc–f = 9 Hz, Ar–CH), 126.57 (s, Ar–C), 123.02 (d, Jc–f = 2 Hz, Ar–CH), 121.4 (s, Ar–C), 114.2 (d, Jc–f = 21 Hz, Ar–CH), 114.2 (d, Jc–f = 21 Hz, Ar–CH), 113.50 (d, Ar–CH), 112.31 (d, Ar–CH), 102.65 (d, Ar–CH), 56.03 (q, OCH3) ppm. HR-MS (ESI+) m/z calculated for [C15H13FKO3]+ = [(M + K) + (–H2O)]+: 263.0269; found 263.0273.GP was carried out with 1u (105 mg, 0.4 mmol) using Ni(acac)2 (5.2 mg, 0.02 mmol), PPh3 (10.5 mg, 0.04 mmol), TEMPO (6.3 mg, 0.04 mmol) and allowed the reaction mixture to stirred at 140 °C for 24 h for the product 2u formation. Purification of the crude material by silica gel column chromatography (petroleum ether/ethyl acetate, 100 to 99
 :01), furnished the product 2u (82 mg, 88%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1u) = 0.10, Rf(2u) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3) δ = 7.86 (d, J = 8.2 Hz, 1H, Ar–H), 7.80 (d, J = 8.0 Hz, 1H, Ar–H), 7.63 (d, J = 9.0 Hz, 1H, Ar–H), 7.55 (d, J = 9.8 Hz, 2H, Ar–H), 7.37–7.29 (m, 2H, Ar–H), 7.25 (t, J = 7.5 Hz, 2H, Ar–H), 7.18 (d, J = 9.6 Hz, 1H, Ar–H), 7.09–6.98 (m, 1H, Ar–H) ppm. 13C NMR (100 MHz, CDCl3) δ = 162.8 (d, Jc–f = 245 Hz, Ar–C), 153.2 (s, Ar–C), 141.8 (d, Ar–CH), 135.3 (d, Jc–f = 8 Hz, Ar–C), 130.8 (s, Ar–C), 130.0 (d, Jc–f = 9 Hz, Ar–CH), 129.0 (d, Ar–CH), 128.1 (s, Ar–C), 126.1 (d, Jc–f = 5 Hz, Ar–CH), 125.6 (d, Jc–f = 3 Hz, Ar–CH), 124.5 (d, Ar–CH), 123.3 (d, Jc–f = 2 Hz, Ar–C), 123.2 (d, Ar–CH), 120.3 (s, Ar–C), 116.8 (d, Jc–f = 22 Hz, Ar–CH), 114.8 (d, Jc–f = 21 Hz, Ar–CH), 112.6 (d, Ar–CH) ppm. HR-MS (ESI+) m/z calculated for [C18H12FO]+ = [M + H]+: 263.0867; found 263.0862.
:01), furnished the product 2u (82 mg, 88%) as pale yellow viscous liquid. [TLC control (petroleum ether/ethyl acetate 99:01), Rf(1u) = 0.10, Rf(2u) = 0.60, UV detection]. 1H NMR (400 MHz, CDCl3) δ = 7.86 (d, J = 8.2 Hz, 1H, Ar–H), 7.80 (d, J = 8.0 Hz, 1H, Ar–H), 7.63 (d, J = 9.0 Hz, 1H, Ar–H), 7.55 (d, J = 9.8 Hz, 2H, Ar–H), 7.37–7.29 (m, 2H, Ar–H), 7.25 (t, J = 7.5 Hz, 2H, Ar–H), 7.18 (d, J = 9.6 Hz, 1H, Ar–H), 7.09–6.98 (m, 1H, Ar–H) ppm. 13C NMR (100 MHz, CDCl3) δ = 162.8 (d, Jc–f = 245 Hz, Ar–C), 153.2 (s, Ar–C), 141.8 (d, Ar–CH), 135.3 (d, Jc–f = 8 Hz, Ar–C), 130.8 (s, Ar–C), 130.0 (d, Jc–f = 9 Hz, Ar–CH), 129.0 (d, Ar–CH), 128.1 (s, Ar–C), 126.1 (d, Jc–f = 5 Hz, Ar–CH), 125.6 (d, Jc–f = 3 Hz, Ar–CH), 124.5 (d, Ar–CH), 123.3 (d, Jc–f = 2 Hz, Ar–C), 123.2 (d, Ar–CH), 120.3 (s, Ar–C), 116.8 (d, Jc–f = 22 Hz, Ar–CH), 114.8 (d, Jc–f = 21 Hz, Ar–CH), 112.6 (d, Ar–CH) ppm. HR-MS (ESI+) m/z calculated for [C18H12FO]+ = [M + H]+: 263.0867; found 263.0862.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the Department of Science and Technology-Science and Engineering Research Board (DST-SERB) [No. EMR/2017/005312], New Delhi, for financial support. C. B. S thanks to UGC and D. S thanks to DST-SERB for the research fellowship.References
-
(a) S. O. Simonetti, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Nat. Prod. Rep., 2013, 30, 941 RSC
; (b) J. A. Builla, J. J. Vaquero and J. Barluenga, Mod. Heterocycl. Chem., 2011, 1, 238 Search PubMed
-
(a) R. R. Rodrigues, W. G. D. Santos, A. B. Oliveira, V. Snieckus, C. L. Zani and A. J. Romanha, Bioorg. Med. Chem. Lett., 1995, 5, 1509 CrossRef
-
(a) B. Carlsson, B. N. Singh, M. Temciuc, S. Nilsson, Y. L. Li, C. Mellin and J. Malm, J. Med. Chem., 2002, 45, 623 CrossRef CAS PubMed
-
(a) K. W. Anderson, T. Ikawa, T. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694 CrossRef CAS PubMed
-
(a) X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387 CrossRef CAS PubMed
-
(a) A. Arcadi, S. Cacchi, M. Del Rosario, G. Fabrizi and F. Marinelli, J. Org. Chem., 1996, 61, 9280 CrossRef CAS
-
(a) D. N. Nicolaides, K. C. Fylaktakidou, K. E. Litinas and S. G. Adamopoulos, J. Heterocycl. Chem., 1998, 35, 91 CrossRef CAS
-
(a) S. N. Aslam, P. Stevenson, S. J. Phythian, N. C. Veitch and D. R. Hall, Tetrahedron, 2006, 62, 4214 CrossRef CAS
-
(a) W. Chen, P. Li, T. Miao, L. G. Meng and L. Wang, Org. Biomol. Chem., 2013, 11, 420 RSC
-
(a) S. Wang, P. Li, L. Yu and L. Wang, Org. Lett., 2011, 13, 5968 CrossRef CAS PubMed
-
(a) C. Sreenivasulu, A. G. K. Reddy and G. Satyanarayana, Org. Chem. Front., 2017, 4, 972 RSC
- D. Yang, Y. Zhu, N. Yang, Q. Jiang and R. Liua, Adv. Synth. Catal., 2016, 358, 1731 CrossRef CAS
- V. K. Rao, G. M. Shelke, R. Tiwari, K. Parang and A. Kumar, Org. Lett., 2013, 15, 9 Search PubMed
- D. Kundu, M. D. Samim, A. Majee and A. Hajra, Chem.–Asian J., 2011, 6, 406 CrossRef CAS PubMed
- D. G. A. Kraus and J. D. Schroeder, Synlett, 2005, 2504 CrossRef
- U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669 CrossRef CAS PubMed
-
(a) R. Adams and L. Whitaker, J. Am. Chem. Soc., 1956, 78, 8 Search PubMed
- T. Yanagi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582 CrossRef CAS PubMed
- B. Wang, J. Zhang, J. Liao, Y. Peng and H. Zheng, Hetrocycles, 2016, 92, 1468 CrossRef CAS
-
(a) J. Ghorai, A. C. S. Reddy and P. Anbarasan, Chem.–Eur. J., 2016, 22, 16042 CrossRef CAS PubMed
- H. Zheng, B. Wang, J. Hu and F. Zhang, Hetrocycles, 2016, 92, 103 CrossRef
-
(a) V. K. Rao, P. Kaswan, G. M. Shelke, A. Ryan, M. Jha and A. Kumar, Synthesis, 2015, 47, 3990 CrossRef CAS
-
(a) L. Mahendar, J. Krishna, A. G. K. Reddy, B. V. Ramulu and G. Satyanarayana, Org. Lett., 2012, 14, 628 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03071f |
| This journal is © The Royal Society of Chemistry 2020 |