
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA quick and versatile one step metal–organic chemical deposition method for supported Pt and Pt-alloy catalysts†
Colleen Jacksona,
Graham T. Smithb,
Nobuhle Mpofuc,
Jack M. S. Dawsona,
Thulile Khozad,
Caelin Septembere,
Susan M. Taylorf,
David W. Inwood g,
Andrew S. Leachh,
Denis Krameri,
Andrea E. Russellj,
Anthony R. J. Kucernaka and
Pieter B. J. Levecque*c
g,
Andrew S. Leachh,
Denis Krameri,
Andrea E. Russellj,
Anthony R. J. Kucernaka and
Pieter B. J. Levecque*c
aDepartment of Chemistry, Imperial College London, Imperial College Road, London, SW7 2AZ, UK
bNational Physical Laboratory, Hampton Road, Teddington, TW11 0LW, UK
cHySA/Catalysis, Centre for Catalysis Research, Department of Chemical Engineering, University of Cape Town, Rondebosch, 7701, South Africa
dSINTEF Industry, SINTEF, Trondheim, 7034, Norway
eUnilever, Boksburg East Industrial, Boksburg, 1459, South Africa
fRD Graphene, 34-35 Stirling Enterprise Park, Stirling, FK7 7RP, UK
gZapGo Ltd. Rutherford Appleton Laboratory, Harwell, Oxford, OX11 0QX, UK
hElectrochemical Innovation Lab, University College London, London, WC1E 7JE, UK
iFaculty of Engineering and the Environment, University of Southampton, University Road, Southampton, SO17 1BJ, UK
jDepartment of Chemistry, University of Southampton, University Road, Southampton, SO17 1BJ, UK. E-mail: Pieter.Levecque@uct.ac.za
First published on 27th May 2020
Abstract
A simple, modified Metal–Organic Chemical Deposition (MOCD) method for Pt, PtRu and PtCo nanoparticle deposition onto a variety of support materials, including C, SiC, B4C, LaB6, TiB2, TiN and a ceramic/carbon nanofiber, is described. Pt deposition using Pt(acac)2 as a precursor is shown to occur via a mixed solid/liquid/vapour precursor phase which results in a high Pt yield of 90–92% on the support material. Pt and Pt alloy nanoparticles range 1.5–6.2 nm, and are well dispersed on all support materials, in a one-step method, with a total catalyst preparation time of ∼10 hours (2.4–4× quicker than conventional methods). The MOCD preparation method includes moderate temperatures of 350 °C in a tubular furnace with an inert gas supply at 2 bar, a high pressure (2–4 bar) compared to typical MOCVD methods (∼0.02–10 mbar). Pt/C catalysts with Pt loadings of 20, 40 and 60 wt% were synthesised, physically characterised, electrochemically characterised and compared to commercial Pt/C catalysts. TEM, XRD and ex situ EXAFS show similar Pt particle sizes and Pt particle shape identifiers, namely the ratio of the third to first Pt coordination numbers modelled from ex situ EXAFS, between the MOCD prepared catalysts and commercial catalysts. Moreover, electrochemical characterisation of the Pt/C MOCD catalysts obtained ORR mass activities with a maximum of 428 A gPt−1 at 0.9 V, which has similar mass activities to the commercial catalysts (80–160% compared to the commercial Pt/C catalysts).
Introduction
Platinum metal and alloy nanoparticles supported on high surface area carbon (Pt/C and Pt-M/C) are the typical electrocatalysts used for both the anode and cathode of the hydrogen polymer electrolyte membrane fuel cell (PEMFC),1–6 as platinum is the most active metal for the hydrogen oxidation (HOR on the anode)1,3,4 and oxygen reduction reactions (ORR on the cathode).2,4–6 Similarly, Pt/C catalysts are used to catalyse the hydrogen evolution reaction (HER)1,3,7,8 in PEM water electrolysers, as well as in electrochemical hydrogen compressors which utilise the hydrogen oxidation and evolution reactions to purify and compress hydrogen9 for fuel cell electric vehicle re-fuelling stations. Furthermore, alternative Pt support materials to carbon, in order to prevent the loss of Pt surface area due to carbon corrosion causing dissolution/agglomeration of Pt nanoparticles, is a prevalent research topic.10–16 Additionally, strong metal support interactions (SMSIs) between the Pt and alternative support materials have been shown to increase the electrochemical activity of Pt through electronic, charge transfer effects.10,11,17–23As the infrastructure for the hydrogen economy grows, due to a recent emphasis on renewable, clean energy sources, so does the need for large scale-up and optimisation of the Pt nanoparticle synthesis method. Moreover, alternative support materials require considerable efforts to improve the Pt utilisation, conductivity, porosity, surface area and corrosion resistance of the support24 in order to increase the commercialisation of these catalysts. Thus, a reliable, versatile and quick Pt deposition method is desirable for the current, commonly used catalysts (Pt/C) as well as for Pt deposited on emerging alternative support materials.
Wet chemistry methods, namely the sodium borohydride/formate reduction14,15,25–28 and ethylene glycol reduction (polyol method)16,26,28–32 methods are the most commonly used synthesis methods for supported Pt nanoparticle preparation. However, these methods are time consuming (∼24–40 hours per batch) and frequently involve five steps, including (1) impregnation with mixture of precursors to form a homogenous mixture, (2) chemical reduction with a reducing agent, (3) washing, (4) drying and (5) calcination/activation at temperature.28 An extensive washing step (3) is typically required with large volumes of de-ionised water, as contaminants are left on the catalyst surface, which may deactivate the catalyst.
Organometallic Chemical Vapour Deposition (OMCVD), also known as Metal–Organic Vapour-Phase Epitaxy (MOVPE) or Metal–Organic Chemical Vapour Deposition (MOCVD), is a vacuum deposition method which produces a thin solid film on a substrate material by a chemical reaction of vapour-phase metal–organic or organometallic precursors.33 The precursors used in the CVD method dictate the naming of the technique, as organometallic (OM-) compounds contain one or more chemical bonds between a metal and a carbon in an organic molecule and metal–organic (MO-) compounds contain a chemical bond between a metal and an organic ligand. CVD has many advantages over wet chemical methods since it is a one-stage process, removing synthesis steps such as impregnation, washing, drying and calcination/activation. Additionally, thin-film platinum deposition using this method has been extensively studied and shown to produce materials which show good electrochemical properties for a range of electrochemical devices.34–38
The procedure of a traditional OMCVD/MOCVD method of metal deposition to a thin film is performed by either the injection of the vaporised precursor with an inert gas into a reactor or a liquid delivery system of the precursor and a solvent which is sprayed onto the substrate. Another method of CVD involves the precursors' decomposition in the vapour phase, contained in a reactor with an inert gas, allowing for the metal to deposit on the surface of a substrate.39 Simply put, this CVD mechanism consists of (1) a physical van der Waals interaction between the precursor and the substrate, (2) excitation of the precursor by interactions with the atoms in the substrate and through collisions with the surface or other precursors, after repeat collisions then sufficient energy is obtained to break chemical bonds in the precursor and (3) chemisorption of the precursor reactive intermediates occurs onto the surface of the substrate.40 The mean dissociation energy required to break the metal–oxygen bond in a β-diketonate compound is 418–481 kJ mol−1.41 After the production of reactive intermediates, now absorbed to the surface, and gaseous by-products, (4) further decomposition of the precursor occurs40 followed by (5) surface diffusion to growth sites and nucleation leading to film formation.33 The remaining ligand fragment by-products from the decomposition process then desorb and are transported away from the deposition substrate33 and out of the reactor.
One experimental challenge of the OMCVD/MOCVD method, particularly for scale-up to industrial scale, is the need for a vacuum in order for the precursor to sublime or vaporise at low temperatures. The OMCVD/MOCVD conditions typically used are ∼0.02–10 mbar with temperature ranges of 100–700 °C.33–38 The necessity of low vacuum pressures requires specialised equipment to achieve and maintain a vacuum throughout the experiment, which creates practical difficulties. One way to address this challenge is to increase the operating pressure to higher pressures, such as atmospheric pressure or above. One study by Kulbakov et al.42 describes a two-step non-isothermal decomposition of platinum acetylacetonate (Pt(acac)2) at atmospheric pressure under air to prepare size controlled Pt nanoparticles deposited on carbon which were shown to be active for ethanol electro-oxidation.
This study describes the experimental setup and description of a one-step metal–organic chemical deposition (MOCD) method, wherein which Pt deposition, to form nanoparticles, occurs from a mixed solid–liquid–vapour phase rather than purely from the vapour phase. This system operates with a 2 bar inert gas (operation pressure is between 4.6–15.3 bar if the temperature and ligand pressure is included, assuming the ideal gas law) at 350 °C, with a total preparation time of ∼10 hours, to produce Pt and Pt alloy nanoparticles dispersed on high surface area carbon and other support materials such as carbides, a nitride, borides and carbon nanofibers. Here, the versatility of this MOCD method to deposit Pt nanoparticles on carbon and on other support substrates is shown. Additionally, direct physical and electrochemical comparisons for ORR in acidic media will be made between Pt/C catalysts produced via this MOCD method and commercially available Pt/C catalysts.
Experimental
Catalyst synthesis
In order to remove any surface contaminants, the reactor and reactor end tubing were rinsed with 18.2 MΩ cm DI water (Milli-Q) and acetone (AR Grade Kimix), then sonicated in DI water for 30 minutes and left to dry in an oven at 90 °C for 1 hour.Pt(acac)2 (97% Sigma-Aldrich), Ru(acac)2 (97% Sigma-Aldrich) and/or Co(acac)3 (98% Sigma-Aldrich) was physically mixed with the desired support material. The support materials, used as-received, were Vulcan XC-72R (ElectroChem, Inc.), Silicon Carbide (NaBond Technologies Co., Ltd.),43 LaB6 (American Elements), TiB2 (US Research Nanomaterials),44,45 TiN (US Research Nanomaterials), ceramic/carbon nanofiber (Stellenbosch Nanofiber Company).45 Boron carbide/graphite composite (BC) (NaBond Technologies Co., Ltd.) was treated with nitric acid to increase the number of nucleation sites10,11,43 and boron doped SiC (B:SiC) and boron doped B4C (B:B4C) were synthesised in-house using adaptations of previously reported carbothermal synthesis methods.46,47 Briefly, the B:SiC and B:B4C preparation involved either co-polymerisation or co-precipitation of carbon, boron and silicon precursors to yield a solid homogenous precursor that was finely powdered using a bead-mill and reduced to ceramic powders by heating in argon.
After mixing, by shaking (no drying or grinding), the desired Pt(acac)2 (with Ru(acac)2 or Co(acac)3) mass with the support material to produce a total catalyst mass, of the Pt and support ignoring the acetylacetonate contribution, of ∼250 mg for the ∼8 cm3 (main reactor chamber is 4.2 cm in length with an inner diameter of 1.6 cm) volume reactor, the precursors were placed in the reactor48 and the reactor was placed into a tubular furnace (Fig. 1). For the nanofibers, these were placed into a graphite weighing boat and the precursor was uniformly distributed over the fibers before being placed in the reactor and then the reactor into the furnace. Argon was fed into the reactor at a pressure of 2 bar, controlled by a pressure regulator, at room temperature while the tubular furnace was ramped to 100 °C in 30 minutes and held for 30 minutes at 100 °C. This initial temperature ramp and Ar purge removed the air from the reactor, allowed for dynamic mixing of the precursors in the reactor and also removed excess moisture from the precursors. After the initial temperature ramp and hold (1 hour), the two manual control valves, placed before (CV1-Fig. 1) and after the furnace (CV2-Fig. 1), were closed and the temperature of the furnace was ramped to 350 °C over 1 hour (∼4 °C min−1) and held at 350 °C for 2 hours. The reactor was then removed from the furnace, while still closed and under Ar atmosphere, and left to cool for at least 5 hours at room temperature and then placed on ice for 20 minutes before opening and removing the synthesised catalyst. The cooling of the reactor under ice before opening was a safety precaution as the catalyst, mostly when synthesising PtRu catalysts, may spontaneously combust when first exposed to air. This is due to the initial surface oxidation/passivation of Pt and Ru or oxidation of the acetylacetone ligand, an exothermic process, which may provide enough heat to combust a support material, particularly carbon. Thus, cooling down of the reactor and catalyst under ice reduced the reaction rate of the surface passivation or oxidation process. Alternatively, passivation with humidified N2 at 70 °C or dilute O2 in N2 (humidified) could be used. When O2/N2 was used, the O2 concentration was slowly increased from 5–20% and kept at each combination for 10 minutes at room temperature.
 | ||
| Fig. 1 Image of reactor and furnace setup, under argon atmosphere with a feed of 2 bar at room temperature, when heated to 350 °C. The manual control valves (CV1 and CV2) are open during purging (∼1 h) and closed after initial purge. The quartz wool in the reactor outlet is to contain the precursors in the reactor and a beaker filled with water on the exit flow is placed while flowing Ar to monitor the gas flow and prevent air from entering the reactor. | ||
The commercial Pt/C catalysts used in this study are 20 wt% Pt/C (HiSPEC 3000, Alfa Aesar and Premetek Co.), 40 wt% Pt/C (HiSPEC 4000, Alfa Aesar) and 60 wt% Pt/C (HiSPEC 9100, Alfa Aesar).
Physical characterisation
Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) carried out on a Mettler Toledo TGA/SDTA851 under air and under N2. Under air the temperature was increased with a ramp rate of 5 °C min−1 from 20–800 °C and under the inert atmosphere (N2) the reactor conditions were mimicked by using a ramp rate of ∼3 °C min−1 to 100 °C then a ramp rate of ∼4 °C min−1 to 350 °C while measuring the weight and temperature of the sample.Transmission Electron Microscopy (TEM) was performed on a Tecnai F20-FEI 200 kV + G2 electron microscope operating at 200 kV. Pt particle size was determined using an average of at least 100 Pt particle diameters from TEM images for each catalyst using ImageJ software. X-ray diffraction (Bruker D8 Advance Co Kα) was used to verify the Pt crystallite size and ICP-OES (Varian 730 ES) to confirm the Pt loading on the support.
Ex situ X-ray adsorption spectroscopy on the B18 line at the diamond light source was used to investigate the morphology of Pt on the surface of the support. The Pt L3 (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564 eV) edge was measured on the beamline which operated with a ring energy 3 GeV and at a current of 300 mA. The monochromator used Si(111) crystals operating in Quick EXAFS (QEXAFS) mode. A total of three spectra were averaged for each sample. The measurements were collected using the ionization chambers in transmission mode at 298 K. Calibration of the monochromator was carried out at both edges using Pt foils. These catalysts were prepared using boron nitride to bind pellets of the catalysts, the pellets were then placed under a H2 atmosphere to reduce surface oxides and all spectra were collected at room temperature. The absorption spectra were modelled with Demeter by Bruce Ravel using Ifeffit by Matt Newville49 to solve the EXAFS equation for the first four Pt shell co-ordination shells, as described by Frenkel et al.50
564 eV) edge was measured on the beamline which operated with a ring energy 3 GeV and at a current of 300 mA. The monochromator used Si(111) crystals operating in Quick EXAFS (QEXAFS) mode. A total of three spectra were averaged for each sample. The measurements were collected using the ionization chambers in transmission mode at 298 K. Calibration of the monochromator was carried out at both edges using Pt foils. These catalysts were prepared using boron nitride to bind pellets of the catalysts, the pellets were then placed under a H2 atmosphere to reduce surface oxides and all spectra were collected at room temperature. The absorption spectra were modelled with Demeter by Bruce Ravel using Ifeffit by Matt Newville49 to solve the EXAFS equation for the first four Pt shell co-ordination shells, as described by Frenkel et al.50
Electrochemical characterisation
A glassy carbon (GC) electrode (Pine Instruments) with 5 mm diameter was polished using 0.1, followed by 0.05 μm alumina polish (Buehler) and sonicated for 1 minute in 18.2 MΩ cm water (Millipore), 1 minute in isopropanol (VWR Chemicals) and again in 18.2 MΩ cm water (Millipore). Catalyst inks for RDE experiments were prepared with 5 mg of catalyst in 5 ml 18.2 MΩ cm water (Millipore) and 1.5 ml isopropanol (VWR Chemicals), sonicated for 30 minutes, followed by 2:1 ratio of carbon:Nafion® using 20 wt% Nafion® (Sigma Aldrich), and sonicated for a further 45 minutes. Finally, 10 μL of the ink was placed on the GC electrode. The GC electrode was rotated at 700 rpm in air while the catalyst ink dried.
All glassware was cleaned in NOCHROMIX® and boiled 6 times in 18.2 MΩ cm water before testing. Electrochemical characterisations were measured in 0.1 M HClO4 (Merck Suprapur®) electrolyte solutions at room temperature and atmospheric pressure in a three-electrode setup, utilising a hydrogen reference electrode and gold counter electrode. Cyclic voltammograms were measured in N2 (6.8 N purity, Air Products BIP® Plus) saturated electrolytes. Electrochemical cleaning cycles were firstly done with 50 cycles and a scan rate of 100 mV s−1 followed by 2 cycles at 20 mV s−1 in the potential range of 0.05–1.2 V vs. RHE. Linear sweep voltammetry was then used to measure ORR activity in O2 (5.8 N purity Air Products UltraPure Plus) saturated electrolyte with a potential range of −0.01 to 1 V vs. RHE and a scan rate of 20 mV s−1, a rotation rate of 1600 rpm, for oxygen reduction activity the anodic scan was reported after iR and baseline corrections in a N2 saturated electrolyte, as well as corrected for mass transport limitations of the setup using the Koutecký–Levich equation (eqn (1)).51 CO stripping voltammetry was used to determine the electrochemically active surface area (ECSA), the potential was held at 0.1 V vs. RHE for 20 minutes in pure CO (BOC Gases) saturated electrolyte and 20 minutes in a N2 saturated 0.1 M HClO4 electrolyte, and cyclic voltammetry was measured with a potential range of 0.05–1.2 V vs. RHE with a scan rate of 20 mV s−1.
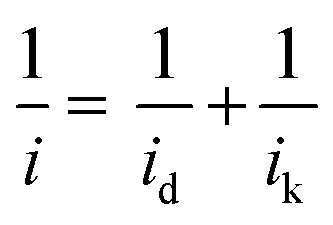 | (1) |
The Pt/C catalyst degradation studies were completed by cycling between 0.6 and 1.0 V vs. RHE with a scan rate of 50 mV s−1 in 0.1 M HClO4 electrolyte solution for 6000 cycles at room temperature, and periodically reverted to a cyclic voltammogram between 0.05 and 1.2 V vs. RHE with a scan rate of 20 mV s−1 to investigate the changes to the HUPD region. The reference electrode used for the durability measurement was Hg/HgSO4 with a gold counter electrode.
Results and discussion
Experimental setup
Images of the experimental setup is shown in Fig. 1, here a stainless-steel reactor48 with open-close valves (CV1/2) on either side is placed into a tubular furnace. One end of the reactor is filled with quartz wool, so the contents of the reactor are contained in the reactor while argon flows through it. The gas line exiting that end of the reactor is connected to one of the open/close valves and then runs into a beaker of water to monitor gas flow. The Pt precursor and support materials are then loaded into the reactor, and the other end of the reactor is connected to an argon line at 2 bar through another open/close valve. The manual control valves are open during purging (∼1 h) during the ramp to 100 °C and hold at 100 °C. Following the initial purge period, the inlet and exit valves are closed and the reactor is pressurised to 2 bar Ar.The proposed mechanism of MOCD
39 and a decomposition temperature range of 210–264 °C.52,53
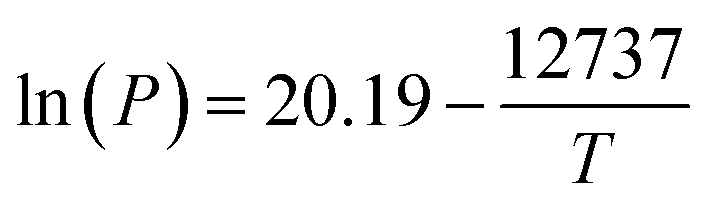 | (2) |
737) are reported for Pt(acac)2.52 Between 200–350 °C, the vapourization pressure of Pt(acac)2 is calculated to be 0.001–0.79 bar. The additional pressure due to vaporised (acac) ligands ranges from 0.37–11.1 bar for 2–60 wt% Pt catalysts, if all of the ligand vaporises and the ideal gas law is assumed. | ||
| Fig. 2 Thermogravimetric analysis (top) and differential thermal analysis (bottom) on Pt(acac)2 under air or N2. Under air: a temperature ramp of 5 °C min−1 from 20–800 °C; under N2: a ramp of 4 °C min−1 from 20–350 °C. | ||
The metal loading contribution from TGA is 44.1 wt%, measured from the remaining mass, assumed to be Pt metal. The Pt mass contribution is lower than the expected 46.3 wt% Pt in Pt(acac)2 (shown in the dashed line, Fig. 2). The discrepancy of ∼2% is attributed to water adsorption or other organic impurities in the sample. Accordingly, since the furnace was operated at 350 °C in an Ar atmosphere, the Pt/C catalyst produced is expected to be free from organic ligand contamination.
| Transition stage | Value | Determination |
|---|---|---|
| a Calculated using the Clausius–Clapeyron relation in eqn (2).b Decomposition range from TGA under N2. | ||
| Melting point | 250 °C | 53 |
| Vaporisation pressure | 0.001–0.79 bar | This worka |
| Decomposition temperature | 200–320 °C | This workb |
| Operating conditions | 350 °C, 4.6–15.3 bar | This work |
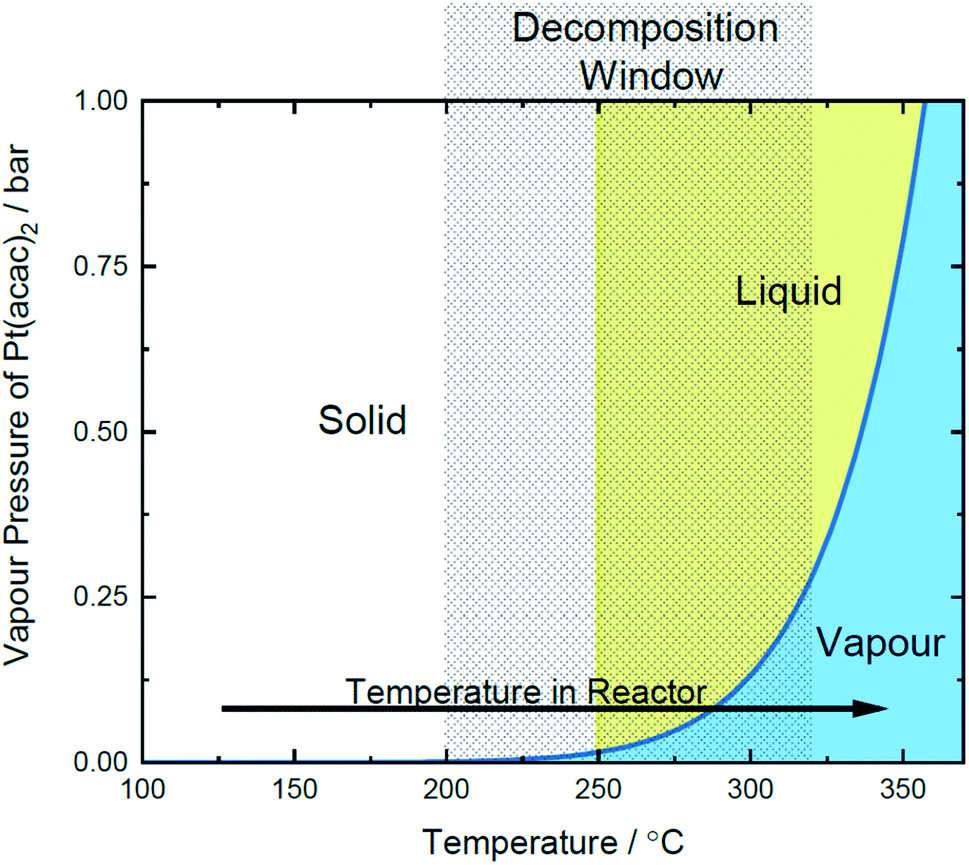 | ||
| Fig. 3 Proposed Pt(acac)2 phase diagram, plotted with the decomposition range and temperature excursion of the reactor. | ||
The proposed mechanism for the MOCD process (illustrated in Fig. 4), whereby metal deposition occurs from the solid/liquid/vapour phase to produce nanoparticles, is modified from the CVD process. Additionally, the reactor is a closed system whereby the precursors cannot be transported out of the system. Firstly, the proposed mechanism consists of (1) a physical van der Waals interaction between the precursor and the substrate, (2) excitation of the precursor by interactions with the atoms in the substrate from thermal energy until sufficient energy is obtained to begin to break chemical bonds in the precursor and (3) chemisorption of the precursor reactive intermediates occurs onto the surface of the substrate. (4) Further decomposition of the precursor reactive intermediates is proposed to occur from the solid/liquid/vapour phase. (5) Surface diffusion to growth sites and nucleation occurs, however, the system is closed and thus some of the remaining ligand fragment by-products will be transported away from the substrate but some may remain. In contrast to the MOCVD method, here it is proposed that some ligand fragments (H(acac), C2H4, CO, and other CxHyOz compounds) either cap the Pt nanoparticles to inhibit nanoparticle growth into Pt thin films or the ligand fragments adsorb onto the substrate and inhibit Pt nanoparticle growth by blocking surface diffusion.
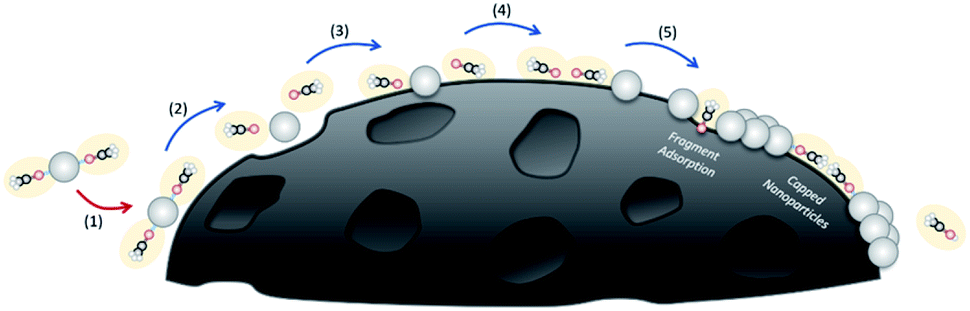 | ||
| Fig. 4 Proposed Pt(acac)2 deposition mechanism onto a support material, starting with decomposition of Pt(acac)2, adsorption of Pt, nucleation of Pt and finally, desorption of the acetyl acetone ligand. | ||
 | ||
| Fig. 5 Transmission Electron Microscope images of different Pt and Pt alloy nanoparticles deposited onto different support materials (as indicated on each image) using the MOCD deposition mechanism. | ||
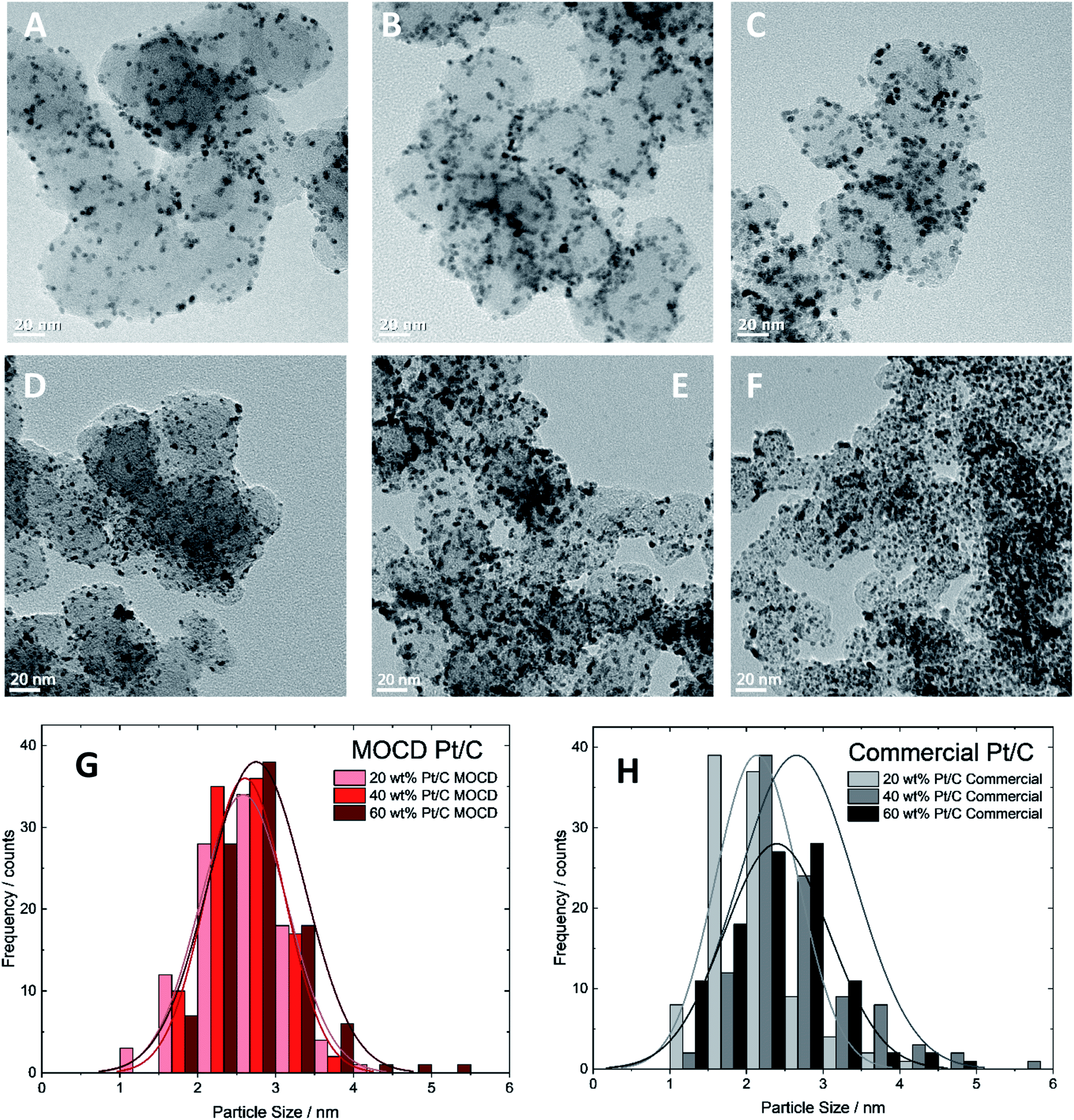 | ||
| Fig. 6 Transmission Electron Microscope images of MOCD prepared catalysts (A) 20 wt% Pt/C, (B) 40 wt% Pt/C, (C) 60 wt% Pt/C, and commercial catalysts (D) 20 wt% Pt/C, (E) 40 wt% Pt/C, (F) 60 wt% Pt/C, (G) MOCD Pt/C catalyst particle size distribution and (H) commercial Pt/C catalyst particle size distribution. | ||
| Catalyst | Metal loading/wt% | MOCD TEM particle size/nm | Literature reference Pt-M particle size/nm |
|---|---|---|---|
| a Commercial Pt/C catalysts used as a reference for MOCD Pt/C catalysts. | |||
| Pt/C | 20–60 | 2.6–2.7 | 2.1–2.6a |
| PtRu/C | 50 | 2.0 ± 0.4 | 1.7–4.3 (ref. 54 and 55) |
| PtCo/C | 40 | 1.5 ± 0.4 | 1.2–6.6 (ref. 56–58) |
| Pt/SiC | 2 | 2.2 ± 0.6 | 3.0–8.0 (ref. 16, 59–61) |
| Pt/B:SiC | 10 | 3.4 ± 0.6 | 3.0–8.0 (ref. 16, 59–61) |
| Pt/LaB6 | 8 | 5.8 ± 1.8 | — |
| Pt/TiB2 | 8 | 6.2 ± 2.0 | 3.4 (ref. 12) |
| Pt/BC | 3–13 | 2.3–2.910 | 1.6–4.7 (ref. 29 and 62) |
| Pt/B:B4C | 2 | 3.4 ± 1.0 | 1.6–4.7 (ref. 29 and 62) |
| Pt/TiN | 8 | 3.9 ± 1.2 | 3.0–6.2 (ref. 63–65) |
| Pt/Sb:SnO2 | 5–20 | 2.3–3.466 | 2.9–3.0 (ref. 67 and 68) |
| Pt/NF | 16 | 2.2 ± 0.7 | 2.0–3.0 (ref. 69 and 70) |
The Pt nanoparticle sizes reported of 2.1–2.6 nm, and particle size distributions on carbon are similar to commercial Pt/C catalysts of 2.4–2.7 nm (Fig. 6 and Table 3) and the similarities in physical properties are further discussed below under, ‘Transmission electron microscopy and X-ray diffraction’. A range of Pt loadings on Pt/C were synthesised in order to fully physically and electrochemically characterise the catalysts and compare to commercial Pt/C catalysts.
| Catalyst | TEM particle size/nm | XRD crystallite size/nm |
|---|---|---|
| MOCD 20 wt% Pt/C | 2.6 ± 0.6 | 3.67 ± 0.3 |
| MOCD 40 wt% Pt/C | 2.6 ± 0.5 | 3.60 ± 0.1 |
| MOCD 60 wt% Pt/C | 2.7 ± 0.6 | 4.08 ± 0.2 |
| Commercial 20 wt% Pt/C | 2.1 ± 0.5 | 3.37 ± 0.2 |
| Commercial 40 wt% Pt/C | 2.7 ± 0.7 | 3.49 ± 0.2 |
| Commercial 60 wt% Pt/C | 2.4 ± 0.7 | 5.83 ± 0.6 |
The PtRu/C catalyst, typically used as anode catalysts for H2 PEMFCs or Direct Methanol Fuel Cells (DMFCs) due to their CO tolerance, was synthesised using the MOCD process and produced metal nanoparticles of ∼2 nm. These PtRu particle sizes and particle size distributions are in line with previously reported values using the conventional impregnation method with NaBH4.54,55 Further electrochemical characterisations of PtRu/C catalysts using this method were previously reported by some of us48 which showed the operating temperature was able to control the degree of Pt–Ru alloying and PtRu particle size. Moreover, PtCo/C, an active alloy for the oxygen reduction reaction (the cathode reaction in H2 PEMFCs and DMFCs), was synthesised with metal particle sizes of 1.5 nm, which is in line and/or smaller than previously reported 1.2–6.6 nm PtCo/C catalysts synthesised using different methods. The different methods used to produce PtCo/C in these studies were conventional impregnation with NaBH4 (ref. 56) and isopropanol57 as reducing agents, and a reverse micelle method58 which involved reduction of metal precursors with NaBH4 in the presence of a surfactant, cosurfactant and oil phase. The PtCo/C particle size distribution using the MOCD method is small, spanning 0–3 nm, which is similar to PtCo/C particle sizes reported via the impregnation with isopropanol,57 but significantly smaller than the PtCo/C particles produced via the reverse micelle method (3–7 nm) and impregnation with NaBH4 (2–10 nm).58
The Pt nanoparticle sizes deposited on alternative support materials in this work are smaller or similar to literature values reported by other Pt deposition methods, with the exception of Pt/TiB2. For the carbide support materials, Pt nanoparticle sizes of 3–8 nm were previously reported for Pt/SiC, using the polyol16,60 and conventional impregnation methods (hypophosphite61 and lithium formate59 as reducing agents), which is larger than reported in this work of 2.2–3.4 nm. The particle size distribution using the MOCD method (Fig. S1†) is 2–5 nm, which is comparable to literature.16 Moreover, Pt/BC and Pt/B:B4C catalysts in this work obtain 2.3–3.4 nm Pt nanoparticles (particle size distributions between 1–4 nm for Pt/BC and 1–7 nm for Pt/B:B4C), of similar range to 1.6–4.7 nm reported in literature of Pt particles prepared by the polyol method29 and reduction of Pt(NH3)2(NO2)2 precursor in H2.62 The ORR activities for the Pt/BC catalysts were reported to show similar Pt particles and shapes to commercial Pt/C catalysts, but with significantly higher ORR activities, due to electronic metal–support interactions.10,11
Previous studies on boride support materials reported Pt particle sizes of 3.4 nm for Pt/TiB2,12 prepared by a colloidal method, which is smaller than the Pt particle size reported in this work of 6.2 nm, likewise, the particle size distribution reported was smaller (1–6 nm) than reported in this work of 2–11 nm. However, to our knowledge Pt/LaB6 catalysts have not yet been reported in literature, using the MOCD method, the average particle size is 5.8 nm (Table 2), with a particle size distribution of 2–11 nm (Fig. S1†). Furthermore, Pt/TiN presented in this work (average of 3.9 nm and particle size distribution of 0–8 nm) is akin to literature of 3.0–6.2 nm, prepared by the polyol63,64 and colloidal methods.65 Reported Pt sizes of 2.9–3 nm for Pt/Sb:SnO2 in literature, from particles prepared using the polyol method67 and modified impregnation with NaBH4,68 are similar to Pt nanoparticles reported using the MOCD method.66
Finally, Pt was deposited on a ceramic-carbon nanofiber support material yielding Pt particle sizes of ∼2.2 nm, which are comparable to some literature values of 2.0–3.0 nm using modified polyol methods.69,70 The particle size distribution using the MOCD method is narrower (0–4 nm) than reported in literature by the modified polyol method (0–6 nm).69
Characterisation of MOCD Pt/C catalysts
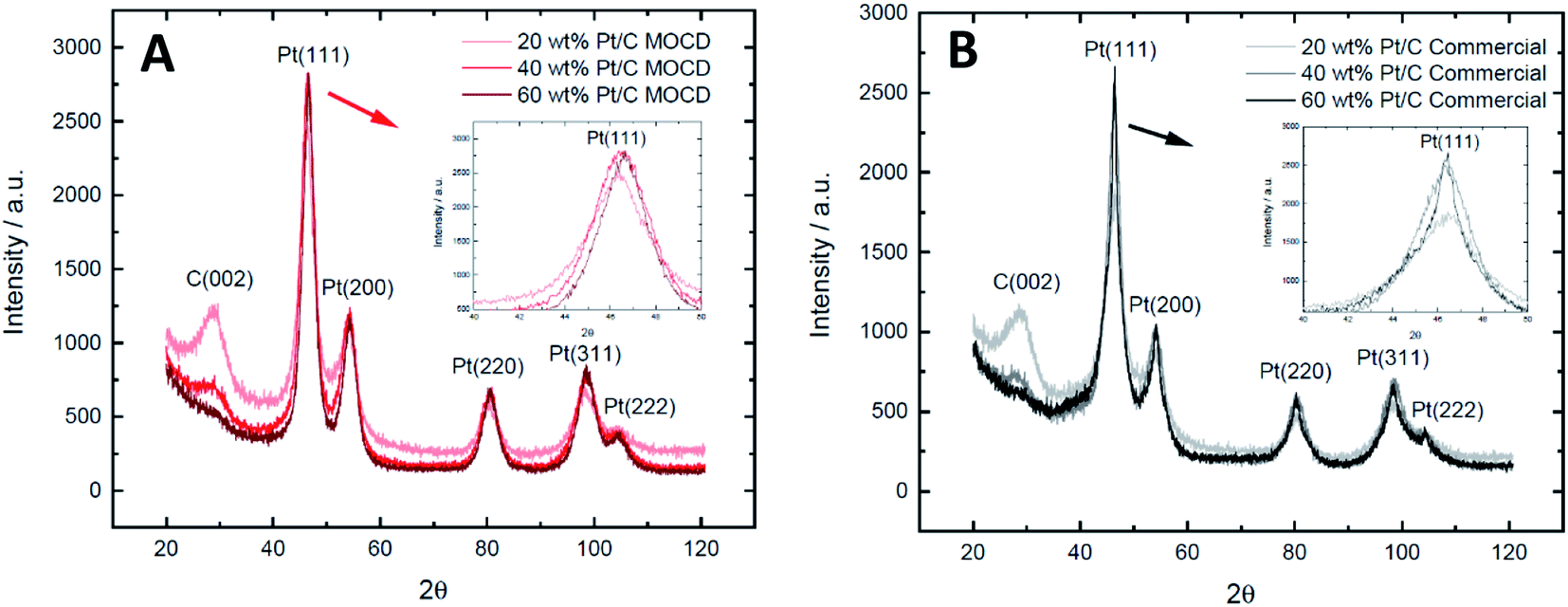 | ||
| Fig. 7 X-ray diffraction patterns of the (A) 20, 40 and 60 wt% Pt/C MOCD catalysts and (B) 20, 40 and 60 wt% Pt/C commercial catalysts, showing the carbon (002) and Pt(111), (200), (220), (311) and (222) peaks with an inset of Pt(111) for each set of catalysts. | ||
These XRD crystallite sizes may be compared to previously reported Pt/C catalysts using the MOCD method by some of the authors,72 Pt crystallite sizes of 3.4–4.6 nm for 20, 40 and 60 wt% Pt/C catalysts were reported, which are similar to the XRD crystallite sizes shown here of ∼3.6–4.1 nm. This indicates good reproducibility of the technique between batches when using the MOCD method for Pt deposition.
One issue observed with the MOCVD method is carbon contamination of the metal thin film layers by the fragments of the ligand during deposition.40 In order to confirm that carbon was not a significant contaminant in the Pt nanoparticles produced by the MOCD method, the Pt d-spacing as determined by XRD, was compared to the commercial Pt/C catalysts (Table S1†). This shows that the MOCD prepared Pt/C catalysts have d-spacing's of 1.38–1.39 Å which is the same as the commercial catalysts, thus carbon is likely not incorporated into the Pt lattice and not a contaminant in the prepared Pt nanoparticles.
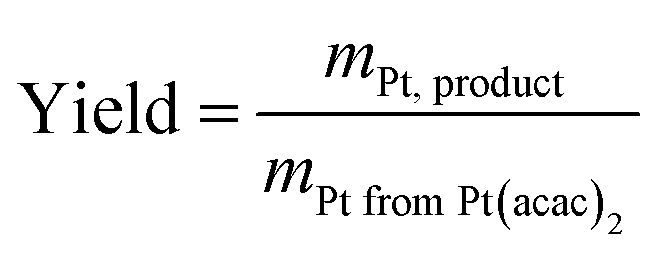 | (3) |
| Catalyst | Intended Pt loading/wt% | Measured Pt loading/wt% | Yield/% |
|---|---|---|---|
| MOCD 20 wt% Pt/C | 20 | 17.7 | 88.4 |
| MOCD 40 wt% Pt/C | 40 | 35.9 | 89.7 |
| MOCD 60 wt% Pt/C | 60 | 53.5 | 89.2 |
| Catalyst | ECSA/m2 gPt−1 | Mass activity/A gPt−1 | Specific activity/μA cmPt−1 |
|---|---|---|---|
| MOCD 20 wt% Pt/C | 75.4 ± 3.2 | 426 ± 33 | 564 ± 26 |
| MOCD 40 wt% Pt/C | 62.7 ± 0.5 | 339 ± 52 | 541 ± 78 |
| MOCD 60 wt% Pt/C | 42.6 ± 0.8 | 206 ± 19 | 485 ± 52 |
| Commercial 20 wt% Pt/C | 76.6 ± 3.6 | 271 ± 36 | 354 ± 46 |
| Commercial 40 wt% Pt/C | 52.2 ± 3.2 | 328 ± 43 | 627 ± 66 |
| Commercial 60 wt% Pt/C | 65.8 ± 3.3 | 256 ± 15 | 390 ± 29 |
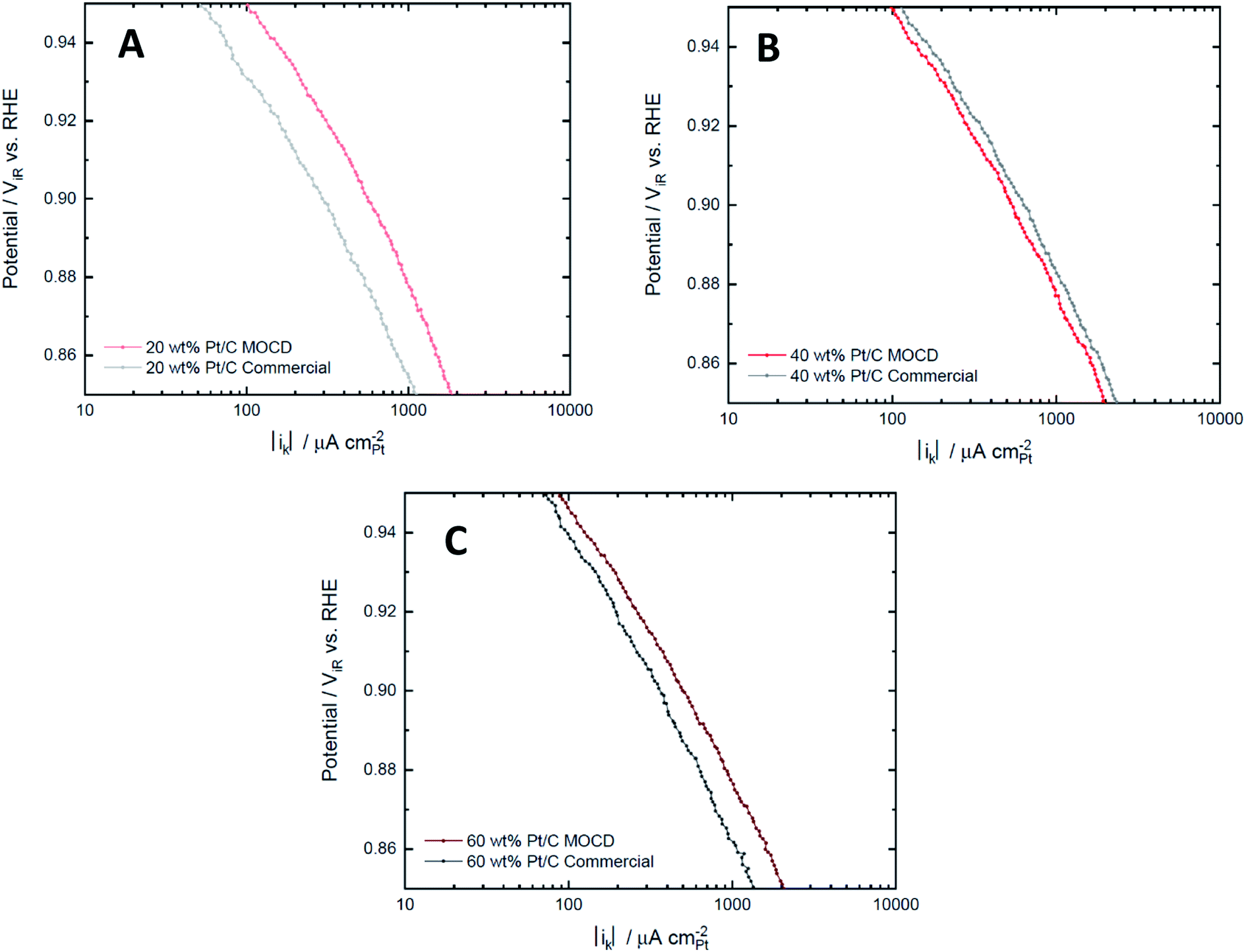 | ||
| Fig. 8 Tafel plots of ORR specific activity for (A) 20 wt%, (B) 40 wt% and (C) 60 wt% MOCD and commercial Pt/C catalysts (HiSPEC 3000 for 20 wt% Pt/C) measured in O2 saturated 0.1 M HClO4 electrolyte at room temperature with a scan rate of 20 mV s−1. | ||
Pt particle size has a significant effect on the ORR activity as larger Pt particle sizes are more active for ORR due to the shape dependence of the Pt facets and edges ratio, with larger particles having a greater proportion of surface area associated with the more ORR active facets. This leads to an increase in specific activity but lower mass activity as the Pt utilisation is decreased with increasing Pt particle size72,76 at 0.9 V. Additionally, the Pt loading effects the H2O2 desorption–readsorption reaction mechanism during the ORR,72 thus low Pt loading catalysts will produce more H2O2 (two electron process) than higher loadings. The particle size and Pt loading effects are prevalent in the ORR mass and specific activities between catalysts with the same Pt loading reported in Table 5. The 40 wt% Pt/C MOCD catalyst has a higher ECSA which indicates smaller Pt particle sizes, leading to a comparably higher mass activity and lower specific activity compared to the commercial 40 wt% Pt/C catalyst. Likewise, the ECSA of the 60 wt% Pt/C MOCD catalyst is lower than the commercial 60 wt% Pt/C catalyst which indicates larger Pt particle sizes, leading to a comparably lower mass activity and higher specific activity compared to the commercial 60 wt% Pt/C catalyst. The 20 wt% catalysts, however, deviate from this trend as the 20 wt% Pt/C MOCD prepared catalyst has higher mass and specific ORR activities compared to the 20 wt% Pt/C commercial catalyst, likely due to the low loading and small Pt particle sizes of the commercial catalyst leading to production of hydrogen peroxide, favouring the two electron oxygen reduction mechanism over the preferred four electron reduction mechanism.72 An electrochemical characterisation with a second commercial catalyst (20 wt% Pt/C from Premetek Co.) reported in Table S5,† demonstrates similar ORR activities between the 20 wt% MOCD prepared and 20 wt% Pt/C (Premetek Co.). The CO Stripping voltammograms (Fig. S4†) for the 20 wt% catalysts also show similarities in peak height and position between the 20 wt% MOCD prepared and 20 wt% Pt/C Premetek Co. catalyst, indicating similar particle sizes and Pt facets. Finally, the Tafel plots of the different Pt loadings for the MOCD prepared and commercial catalysts reported in Fig. 8 show similar ORR specific activities between the MOCD and commercial catalysts, with the exception of 20 wt% Pt/C commercial since the MOCD catalyst has a significantly higher activity.
Table 6 reports the comparison between the Pt/C MOCD prepared catalysts and commercial catalysts, showing the ORR mass activities and specific activities of the MOCD catalysts range 80–157% and 86–159% of the commercial catalysts' activity, respectively. Therefore, it can be seen that this simple one-step MOCD method can be used to produce electrochemically active Pt/C catalysts which perform similarly to or better than commercial Pt/C catalysts for oxygen reduction. Additionally, the durability after potential cycling from 0.6–1.0 V (displayed in Fig. S5†), shows similar ECSAs between the MOCD and commercial Pt/C catalysts after 6000 cycles. The potential range of 0.6–1 V isolates the Pt chemical and electrochemical stability,77 indicating further that the Pt/C MOCD and commercial catalysts are similar in chemical and electrochemical response.
| Catalyst |
|
|
|
|---|---|---|---|
| 20 wt% Pt/C | 98.4 | 157 | 159 |
| 40 wt% Pt/C | 120 | 103 | 86.3 |
| 60 wt% Pt/C | 64.7 | 80.4 | 124 |

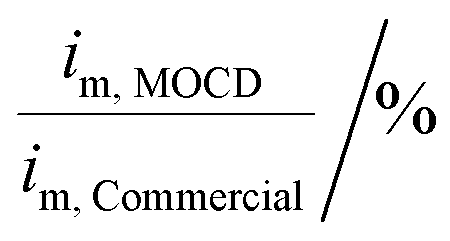
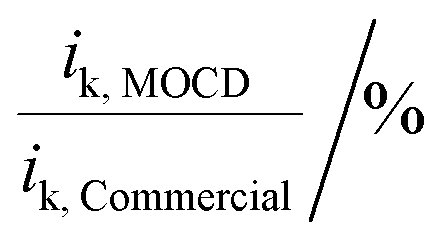
Moreover, the versatility and reproducibility of the MOCD method allows for novel Pt catalysts and electrocatalysts to be developed in a simple and reliable manner.
Conclusion
The experimental setup and method of the MOCD catalyst preparation is described, utilising a stainless steel reactor in a tubular furnace. The operating temperature of the furnace is 350 °C and argon gas is fed into the reactor with a pressure of 2 bar at room temperature, which results in an operating pressure of 4.6–15.3 bar, if the ideal gas law is assumed and the additional ligand pressure is included. This Pt deposition method is advantageous since it is an ergonomic, one-step method, compared to wet chemistry which is typically five steps. The MOCD method may also be easily scaled up to larger reactors and batch sizes. Additionally, the use of Pt(acac)2 as a precursor does not introduce chloride species into the procedure, which may act as a contaminant in the final Pt/C product.The experimental setup and deposition method is modified from the MOCVD/OMCVD method wherein which deposition occurs from the vapour phase under vacuum in order to produce Pt thin-films on a substrate. By operating under higher pressure rather than a vacuum and in a closed reactor the Pt(acac)2 precursor will decompose from a mixture of solid/liquid/vapour phases. In contrast to the MOCVD/OMCVD method, small well dispersed Pt nanoparticles with particle sizes of 1.5–6.2 nm are prepared. It is postulated that Pt(acac)2 decomposition and deposition from the liquid and vapour phases allows for the good dispersion on the support material.
A mechanism is proposed for the MOCD process, this consists of (1) a physical van der Waals interaction between the precursor and the substrate, (2) excitation of the precursor by interactions with the atoms in the substrate and thermal energy until sufficient energy is obtained to break chemical bonds in the precursor and (3) chemisorption of the precursor reactive intermediates onto the surface of the substrate. (4) Decomposition and adsorption of the precursor reactive intermediates from the solid/liquid/vapour phase. (5) Surface diffusion to growth sites and nucleation occurs. Since the system is closed, the remaining ligand fragment by-products cannot be transported away from the substrate as they would be in traditional MOCVD. Here in the MOCD method, it is proposed that ligand fragments (H(acac), C2H4, CO, and other CxHyOz compounds) either cap the Pt nanoparticles to inhibit nanoparticle growth or the ligand fragments adsorb onto the substrate and inhibit Pt nanoparticle growth by blocking surface diffusion.
The MOCD deposition method was used to deposit Pt nanoparticles onto a range of support materials, specifically carbon, silicon carbide (doped and undoped), boron carbide (doped and undoped), lanthanum hexaboride, titanium diboride, titanium nitride and a ceramic/carbon nanofiber. Furthermore, a Pt alloy catalysts were also prepared and all MOCD prepared catalysts produced well dispersed Pt/Pt alloy nanoparticle sizes (1.5–6.2 nm) similar to commercial catalysts and to other literature for Pt supported on alternative materials (carbides, borides, nitrides etc.). Thus, the flexibility of the MOCD technique is demonstrated to prepare, in a one-step method, a number of Pt and Pt alloy catalysts supported on various materials with a reliable nanoparticle size range. The reproducibility of the MOCD technique is shown in the number of different supported Pt/Pt alloy catalysts produced by the technique, which have similar Pt particle sizes, particle size distributions and particle shapes. Additionally, the crystallite sizes of Pt/C catalysts are consistent with previously reported work by some of the authors, indicating reproducibility between batches.
Three Pt/C catalysts with different Pt loadings of 20, 40 and 60 wt% were physically and electrochemically characterised and compared to commercial Pt/C catalysts with similar loadings. A high Pt yield of 90–92% of the nominal loading was found for the Pt/C MOCD synthesised catalysts, demonstrating that little precursor is lost during preparation method. While slight differences in Pt particle sizes were observed from TEM and XRD between the MOCD and commercial catalysts, both sets of Pt catalysts' average particle size ranged from 2–3 nm. The first coordination number modelled from ex situ EXAFS was predominantly influenced by the Pt particle size and resembled this in slightly larger first coordination numbers for Pt/C catalysts with larger Pt nanoparticle sizes. Additionally, a shape identifier from ex situ EXAFS, the ratio of the third to first Pt coordination numbers, demonstrated that the MOCD prepared and commercial Pt/C catalyst were similar in shape. ORR mass activities and specific activities reported are 200–430 A gPt−1 and 485–565 μA cmPt−1, respectively, for the MOCD catalysts which equates to ∼80–160% of the ORR mass and specific activities measured on the commercial catalysts. These ORR activities demonstrate that the MOCD Pt deposition technique produces well dispersed, electrochemically active Pt catalysts which are comparable to commercial standards.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Royal Society in the form of a Royal Society-Newton Advanced Fellowship (P. L.; grant no. NA140367). C. J. acknowledges the University of Cape Town for financial support through a UCT PhD Mobility Grant. D. K. and G. T. S. thank the EPSRC H2FC SUPERGEN (grant no. EP/J016454/1) for financial support. G. T. S. thanks the HySA/Catalysis Programme for a postdoctoral fellowship. D. K. acknowledges support from STFC (ST/K00171X/1 and ST/N002385/1). P. L., N. M., T. K., C. S. and S. M. T. acknowledge support from the HySA/Catalysis Programme.References
- J. Zhang, PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications, Springer London, 2008 Search PubMed.
- B. G. Pollet, S. S. Kocha and I. Staffell, Current status of automotive fuel cells for sustainable transport, Curr. Opin. Electrochem., 2019, 16, 90–95 CrossRef CAS.
- J. Durst, C. Simon, A. Siebel, P. J. Rheinländer, T. Schuler, M. Hanzlik, J. Herranz, F. Hasché and H. A. Gasteiger, Hydrogen Oxidation and Evolution Reaction (HOR/HER) on Pt Electrodes in Acid vs. Alkaline Electrolytes: Mechanism, Activity and Particle Size Effects, ECS Trans., 2014, 64, 1069–1080 CrossRef CAS.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, The stability of Pt–M (M = first row transition metal) alloy catalysts and its effect on the activity in low temperature fuel cells: a literature review and tests on a Pt–Co catalyst, J. Power Sources, 2006, 160, 957–968 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Alloys of platinum and early transition metals as oxygen reduction electrocatalysts, Nat. Chem., 2009, 1, 552–556 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs. Alkaline Electrolytes, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- K. Fishel, G. Qian, G. Eisman and B. C. Benicewicz, Electrochemical Hydrogen Pumping, in High Temperature Polymer Electrolyte Membrane Fuel Cells: Approaches, Status, and Perspectives, ed. Q. Li, D. Aili, H. A. Hjuler and J. O. Jensen, Springer International Publishing, Cham, 2016; pp. 527–540 Search PubMed.
- C. Jackson, G. T. Smith, D. W. Inwood, A. S. Leach, P. S. Whalley, M. Callisti, T. Polcar, A. E. Russell, P. Levecque and D. Kramer, Electronic metal–support interaction enhanced oxygen reduction activity and stability of boron carbide supported platinum, Nat. Commun., 2017, 8, 15802 CrossRef CAS.
- C. Jackson, G. T. Smith, M. Markiewicz, D. W. Inwood, A. S. Leach, P. S. Whalley, A. R. Kucernak, A. E. Russell, D. Kramer and P. B. J. Levecque, Support induced charge transfer effects on electrochemical characteristics of Pt nanoparticle electrocatalysts, J. Electroanal. Chem., 2018, 819, 163–170 CrossRef CAS.
- S. Yin, S. Mu, H. Lv, N. Cheng, M. Pan and Z. Fu, A highly stable catalyst for PEM fuel cell based on durable titanium diboride support and polymer stabilization, Appl. Catal., B, 2010, 93, 233–240 CrossRef CAS.
- T. Ioroi, Z. Siroma, N. Fujiwara, S. Yamazaki and K. Yasuda, Sub-stoichiometric titanium oxide-supported platinum electrocatalyst for polymer electrolyte fuel cells, Electrochem. Commun., 2005, 7, 183–188 CrossRef CAS.
- S. Huang, P. Ganesan, S. Park and B. N. Popov, Development of a Titanium Dioxide-Supported Platinum Catalyst with Ultrahigh Stability for Polymer Electrolyte Membrane Fuel Cell Applications, J. Am. Chem. Soc., 2009, 131, 13898–13899 CrossRef CAS.
- S. Huang, P. Ganesan and B. N. Popov, Electrocatalytic activity and stability of niobium-doped titanium oxide supported platinum catalyst for polymer electrolyte membrane fuel cells, Appl. Catal., B, 2010, 96, 224–231 CrossRef CAS.
- H. Lv, S. Mu, N. Cheng and M. Pan, Nano-silicon carbide supported catalysts for PEM fuel cells with high electrochemical stability and improved performance by addition of carbon, Appl. Catal., B, 2010, 100, 190–196 CrossRef CAS.
- T. Li, J. Qian, Q. Zhou, J. Lin and Y. Zheng, A pyrene-modified cobalt salophen complex immobilized on multiwalled carbon nanotubes acting as a precursor for efficient electrocatalytic water oxidation, Dalton Trans., 2017, 46, 13020–13026 RSC.
- X. Wang, Z. Ma, L. Chai, L. Xu, Z. Zhu, Y. Hu, J. Qian and S. Huang, MOF derived N-doped carbon coated CoP particle/carbon nanotube composite for efficient oxygen evolution reaction, Carbon, 2019, 141, 643–651 CrossRef CAS.
- S. Mandegarzad, J. B. Raoof, S. R. Hosseini and R. Ojani, Cu-Pt bimetallic nanoparticles supported metal organic framework-derived nanoporous carbon as a catalyst for hydrogen evolution reaction, Electrochim. Acta, 2016, 190, 729–736 CrossRef CAS.
- S. Zhang, Z. Xia, T. Ni, Z. Zhang, Y. Ma and Y. Qu, Strong electronic metal–support interaction of Pt/CeO2 enables efficient and selective hydrogenation of quinolines at room temperature, J. Catal., 2018, 359, 101–111 CrossRef CAS.
- M. Sharma, N. Jung and S. J. Yoo, Toward High-Performance Pt-Based Nanocatalysts for Oxygen Reduction Reaction through Organic–Inorganic Hybrid Concepts, Chem. Mater., 2018, 30, 2–24 CrossRef CAS.
- V. C. Anitha, R. Zazpe, M. Krbal, J. Yoo, H. Sopha, J. Prikryl, G. Cha, S. Slang, P. Schmuki and J. M. Macak, Anodic TiO2 nanotubes decorated by Pt nanoparticles using ALD: an efficient electrocatalyst for methanol oxidation, J. Catal., 2018, 365, 86–93 CrossRef CAS.
- P. S. Murphin Kumar, V. K. Ponnusamy, K. R. Deepthi, G. Kumar, A. Pugazhendhi, H. Abe, S. Thiripuranthagan, U. Pal and S. K. Krishnan, Controlled synthesis of Pt nanoparticle supported TiO2 nanorods as efficient and stable electrocatalysts for the oxygen reduction reaction, J. Mater. Chem. A, 2018, 6, 23435–23444 RSC.
- E. Antolini and E. R. Gonzalez, Ceramic materials as supports for low-temperature fuel cell catalysts, Solid State Ionics, 2009, 180, 746–763 CrossRef CAS.
- L. Xiong and A. Manthiram, Synthesis and characterization of methanol tolerant Pt/TiOx/C nanocomposites for oxygen reduction in direct methanol fuel Cells, Electrochim. Acta, 2004, 49, 4163–4170 CrossRef CAS.
- P. Kim, J. B. Joo, W. Kim, J. Kim, I. K. Song and J. Yi, NaBH4-assisted ethylene glycol reduction for preparation of carbon-supported Pt catalyst for methanol electro-oxidation, J. Power Sources, 2006, 160, 987–990 CrossRef CAS.
- S. E. Eklund and D. E. Cliffel, Synthesis and Catalytic Properties of Soluble Platinum Nanoparticles Protected by a Thiol Monolayer, Langmuir, 2004, 20, 6012–6018 CrossRef CAS PubMed.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, A review of anode catalysis in the direct methanol fuel cell, J. Power Sources, 2006, 155, 95–110 CrossRef CAS.
- H. Lv, T. Peng, P. Wu, M. Pan and S. Mu, Nano-boron carbide supported platinum catalysts with much enhanced methanol oxidation activity and CO tolerance, J. Mater. Chem., 2012, 22, 9155–9160 RSC.
- Z. Liu, L. Hong, M. P. Tham, T. H. Lim and H. Jiang, Nanostructured Pt/C and Pd/C catalysts for direct formic acid fuel cells, J. Power Sources, 2006, 161, 831–835 CrossRef CAS.
- S. Song, Y. Wang and P. K. Shen, Pulse-microwave assisted polyol synthesis of highly dispersed high loading Pt/C electrocatalyst for oxygen reduction reaction, J. Power Sources, 2007, 170, 46–49 CrossRef CAS.
- Z. Liu, X. Y. Ling, X. Su and J. Y. Lee, Carbon-Supported Pt and PtRu Nanoparticles as Catalysts for a Direct Methanol Fuel Cell, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS.
- AnonymousChapter 1 Overview of Chemical Vapour Deposition, in Chemical Vapour Deposition: Precursors, Processes and Applications, ed. A. C. Jones and M. L. Hitchman, The Royal Society of Chemistry, 2009, pp. 1–36 Search PubMed.
- J. R. V. Garcia and T. Goto, Chemical Vapor Deposition of Iridium, Platinum, Rhodium and Palladium, Mater. Trans., 2003, 44, 1717–1728 CrossRef CAS.
- I. K. Igumenov, N. V. Gelfond, P. S. Galkin, N. B. Morozova, N. E. Fedotova, G. I. Zharkova, V. I. Shipachev, E. F. Reznikova, A. D. Ryabtsev, N. P. Kotsupalo, V. I. Titarenko, Y. P. Dikov, V. V. Distler and M. I. Buleev, Corrosion testing of platinum metals CVD coated titanium anodes in seawater-simulated solutions, Desalination, 2001, 136, 273–280 CrossRef CAS.
- T. Goto, R. Vargas and T. Hirai, Preparation of iridium and platinum films by MOCVD and their properties, J. Phys. IV, 1993, 03, C3–C304 CrossRef.
- G. Malandrino, R. L. Nigro and I. L. Fragalà, MOCVD of Platinum (100) Films on Random Hastelloy C276, Chem. Vap. Deposition, 1999, 5, 59–61 CrossRef CAS.
- S. Barison, M. Fabrizio, G. Carta, G. Rossetto, P. Zanella, D. Barreca and E. Tondello, Nanocrystalline Pt thin films obtained via metal organic chemical vapor deposition on quartz and CaF2 substrates: an investigation of their chemico-physical properties, Thin Solid Films, 2002, 405, 81–86 CrossRef CAS.
- C. Thurier and P. Doppelt, Platinum OMCVD processes and precursor chemistry, Coord. Chem. Rev., 2008, 252, 155–169 CrossRef CAS.
- I. K. Igumenov, P. P. Semyannikov, S. V. Trubin, N. B. Morozova, N. V. Gelfond, A. V. Mischenko and J. A. Norman, Approach to control deposition of ultra thin films from metal organic precursors: Ru deposition, Surf. Coat. Technol., 2007, 201, 9003–9008 CrossRef CAS.
- I. K. Igumenov, A. E. Turgambaeva and P. P. Semyannikov, General aspects of surface chemistry of metal β-diketonates, J. Phys. IV, 2001, 11, Pr3-515 Search PubMed.
- A. A. Kulbakov, A. B. Kuriganova, M. Allix, A. Rakhmatullin, N. Smirnova, O. A. Maslova and I. N. Leontyev, Non-isothermal decomposition of platinum acetylacetonate as a cost-efficient and Size-Controlled Synthesis of Pt/C nanoparticles, Catal. Commun., 2018, 117, 14–18 CrossRef CAS.
- C. Jackson, SiC and B4C as Electrocatalyst Support Materials for Low Temperature Fuel Cells, University of Cape Town, 2017 Search PubMed.
- T. Khoza, Evaluation of metal nitrides and borides as alternative electrocatalyst support materials for polymer electrolyte fuel cells, University of Cape Town, 2014 Search PubMed.
- C. September, Preparation and characterisation of inorganic nanostructured support materials for polymer electrolyte fuel cells, University of Cape Town, 2015 Search PubMed.
- I. Yanase, R. Ogawara and H. Kobayashi, Synthesis of boron carbide powder from polyvinyl borate precursor, Mater. Lett., 2009, 63, 91–93 CrossRef CAS.
- Y. Sugahara, Y. Takeda, K. Kuroda and C. Kato, The preparation of boron-doped silicon carbide powder by the carbothermal reduction of oxides derived from the hydrolyzed methyltriethoxysilane, J. Non-Cryst. Solids, 1988, 100, 542–546 CrossRef CAS.
- C. Jackson, O. Conrad and P. Levecque, Systematic Study of Pt-Ru/C Catalysts Prepared by Chemical Deposition for Direct Methanol Fuel Cells, Electrocatalysis, 2017, 8, 224–234 CrossRef CAS.
- B. Ravel and M. Newville, \it ATHENA \it ARTEMIS \it HEPHAESTUS: data analysis for X-ray absorption spectroscopy using \it IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, A View from the Inside: Complexity in the Atomic Scale Ordering of Supported Metal Nanoparticles, J. Phys. Chem. B, 2001, 105, 12689–12703 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley Textbooks, 2nd edn, 2000 Search PubMed.
- N. B. Morozova, G. I. Zharkova, P. P. Semyannikov, S. V. Sysoev, I. K. Igumenov, N. E. Fedotova and N. V. Gelfond, Vapor pressure of precursors for CVD on the base of platinum group metals, J. Phys. IV, 2001, 11, Pr3–616 Search PubMed.
- S. Yoda, A. Hasegawa, H. Suda, Y. Uchimaru, K. Haraya, T. Tsuji and K. Otake, Preparation of a Platinum and Palladium/Polyimide Nanocomposite Film as a Precursor of Metal-Doped Carbon Molecular Sieve Membrane via Supercritical Impregnation, Chem. Mater., 2004, 16, 2363–2368 CrossRef CAS.
- W. Xu, T. Lu, C. Liu and W. Xing, Nanostructured PtRu/C as Anode Catalysts Prepared in a Pseudomicroemulsion with Ionic Surfactant for Direct Methanol Fuel Cell, J. Phys. Chem. B, 2005, 109, 14325–14330 CrossRef CAS PubMed.
- Q. Lu, B. Yang, L. Zhuang and J. Lu, Anodic Activation of PtRu/C Catalysts for Methanol Oxidation, J. Phys. Chem. B, 2005, 109, 1715–1722 CrossRef CAS PubMed.
- M. K. Jeon, Y. Zhang and P. J. McGinn, A comparative study of PtCo, PtCr, and PtCoCr catalysts for oxygen electro-reduction reaction, Electrochim. Acta, 2010, 55, 5318–5325 CrossRef CAS.
- S. Hsu, C. Liu, H. Chen, T. Chen, C. Lai, C. Lee, J. Lee, T. Chan, L. Tsai and K. Wang, The effect of Mn addition on the promotion of oxygen reduction reaction performance for PtCo/C catalysts, Electrochim. Acta, 2013, 105, 180–187 CrossRef CAS.
- Y. Qian, W. Wen, P. A. Adcock, Z. Jiang, N. Hakim, M. S. Saha and S. Mukerjee, PtM/C Catalyst Prepared Using Reverse Micelle Method for Oxygen Reduction Reaction in PEM Fuel Cells, J. Phys. Chem. C, 2008, 112, 1146–1157 CrossRef CAS.
- N. N. Vershinin, V. A. Bakaev, V. I. Berestenko, O. N. Efimov, E. N. Kurkin and E. N. Kabachkov, Synthesis and properties of a platinum catalyst supported on plasma chemical silicon carbide, High Energy Chem., 2017, 51, 46–50 CrossRef CAS.
- S. M. Andersen and M. J. Larsen, Performance of the electrode based on silicon carbide supported platinum catalyst for proton exchange membrane fuel cells, J. Electroanal. Chem., 2017, 791, 175–184 CrossRef CAS.
- S. Suzuki, T. Onodera, J. Kawaji, T. Mizukami, M. Morishima and K. Yamaga, Improvement in stability of carbon support for platinum catalyst by applying silicon carbide coating, J. Power Sources, 2013, 223, 79–85 CrossRef CAS.
- W. T. Grubb and D. W. McKee, Boron Carbide, a New Substrate for Fuel Cell Electrocatalysts, Nature, 1966, 210, 192–194 CrossRef CAS.
- H. Nan, D. Dang and X. L. Tian, Structural engineering of robust titanium nitride as effective platinum support for the oxygen reduction reaction, J. Mater. Chem. A, 2018, 6, 6065–6073 RSC.
- Y. Zheng, J. Zhang, H. Zhan, D. Sun, D. Dang and X. L. Tian, Porous and three dimensional titanium nitride supported platinum as an electrocatalyst for oxygen reduction reaction, Electrochem. Commun., 2018, 91, 31–35 CrossRef CAS.
- K. Kakinuma, Y. Wakasugi, M. Uchida, T. Kamino, H. Uchida, S. Deki and M. Watanabe, Preparation of titanium nitride-supported platinum catalysts with well controlled morphology and their properties relevant to polymer electrolyte fuel cells, Electrochim. Acta, 2012, 77, 279–284 CrossRef CAS.
- R. Mohamed, T. Binninger, P. J. Kooyman, A. Hoell, E. Fabbri, A. Patru, A. Heinritz, T. J. Schmidt and P. Levecque, Facile deposition of Pt nanoparticles on Sb-doped SnO2 support with outstanding active surface area for the oxygen reduction reaction, Catal. Sci. Technol., 2018, 8, 2672–2685 RSC.
- M. Dou, M. Hou, F. Wang, D. Liang, Q. Zhao, Z. Shao and B. Yi, Sb-Doped SnO2 Supported Platinum Catalyst with High Stability for Proton Exchange Membrane Fuel Cells, J. Electrochem. Soc., 2014, 161, F1231–F1236 CrossRef.
- N. R. Elezovic, V. R. Radmilovic, J. Kovac, B. M. Babic, L. Gaijic-Krstajic and N. V. Krstajic, Pt nanoparticles on tin oxide based support as a beneficial catalyst for oxygen reduction in alkaline solutions, RSC Adv., 2015, 5, 15923–15929 RSC.
- Y. Ma, S. Jiang, G. Jian, H. Tao, L. Yu, X. Wang, X. Wang, J. Zhu, Z. Hu and Y. Chen, CNx nanofibers converted from polypyrrole nanowires as platinum support for methanol oxidation, Energy Environ. Sci., 2009, 2, 224–229 RSC.
- W. Li, M. Waje, Z. Chen, P. Larsen and Y. Yan, Platinum nanopaticles supported on stacked-cup carbon nanofibers as electrocatalysts for proton exchange membrane fuel cell, Carbon, 2010, 48, 995–1003 CrossRef CAS.
- K. O'Connell and J. R. Regalbuto, High Sensitivity Silicon Slit Detectors for 1 nm Powder XRD Size Detection Limit, Catal. Lett., 2015, 145, 777–783 CrossRef.
- E. Fabbri, S. Taylor, A. Rabis, P. Levecque, O. Conrad, R. Kötz and T. J. Schmidt, The Effect of Platinum Nanoparticle Distribution on Oxygen Electroreduction Activity and Selectivity, ChemCatChem, 2014, 6, 1410–1418 CrossRef CAS.
- A. Jentys, Estimation of mean size and shape of small metal particles by EXAFS, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC.
- Y. Garsany, I. L. Singer and K. Swider-Lyons, Impact of film drying procedures on RDE characterization of Pt/VC electrocatalysts, J. Electroanal. Chem., 2011, 662, 396–406 CrossRef CAS.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, The Particle Size Effect on the Oxygen Reduction Reaction Activity of Pt Catalysts: Influence of Electrolyte and Relation to Single Crystal Models, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- K. Kinoshita, Particle Size Effects for Oxygen Reduction on Highly Dispersed Platinum in Acid Electrolytes, J. Electrochem. Soc., 1990, 137, 845–848 CrossRef CAS.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, A review of PEM fuel cell durability: Degradation mechanisms and mitigation strategies, J. Power Sources, 2008, 184, 104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03001e |
| This journal is © The Royal Society of Chemistry 2020 |