DOI:
10.1039/D0RA02994G
(Paper)
RSC Adv., 2020,
10, 16882-16885
Concise asymmetric synthesis of two natural oxacyclododecindione-type macrolactones from industrial waste†
Received
2nd April 2020
, Accepted 21st April 2020
First published on 30th April 2020
Abstract
(S)-5-Methyl-tetrahydropyran-2-one 1 is a by-product in the production of key intermediates of steroidal hormones. Asymmetric synthesis of natural oxacyclododecindione-type macrolactones 2 and 3 has been realized for the first time from (S)-1 in seven and eight steps with 37% and 27% overall yield, respectively.
Introduction
Green chemistry, which was established about two decades ago, has attracted considerable attention due to its reduction or elimination of the use and generation of hazardous substances from a chemical process.1 Most efforts have focused on waste minimization and avoiding the use of toxic or hazardous reagents and solvents, particularly in the pharmaceutical field. However, little attention has been paid on the rational reuse of industrial wastes.2 Both (R)- and (S)-5-methyl-tetrahydropyran-2-one 1 are by-products in the production of key intermediates (16-dehydropregnenolone derivatives) of steroidal hormones from steroidal sapogenins by oxidative degradation (Scheme 1).3 To produce 100 kg of 16-dehydropregnenolone derivatives may generate more than 30 kg of (R)- or (S)-1 as wastes,3b and almost all of them are destroyed by burning or other methods.4 At present, the throughput of 16-dehydropregnenolone derivatives is kiloton each year only in China. There have been only very few examples to utilize (R)- and (S)-1 as starting materials in the synthesis of bioactive compounds.5 Therefore, exploiting the synthesis application of (R)- or (S)-1 is highly welcomed.
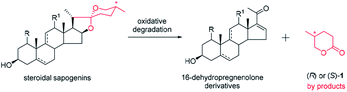 |
| | Scheme 1 Production of industrial wastes (R)- and (S)-1. | |
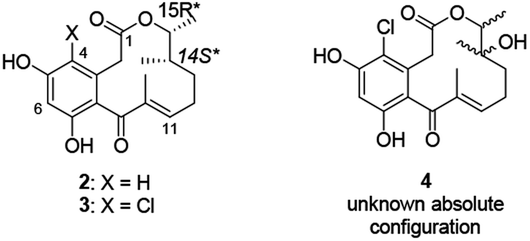
In 2008, three new 12-membered macrolactones 4-dechloro-14-deoxy-oxacyclododecindione 2, 14-deoxy-oxacylododecin-dione 3 and oxacyclododecindione 4 were isolated as fungal secondary metabolites from imperfect fungus Exserohilum rostratum by Opatz and Erkel (Fig. 1).6a They exhibit potent anti-inflammatory and anti-fibrotic activities in cell culture experiments and in an in vivo mouse model of systemic lupus erythematosus (SLE) and may serve as lead structures for therapeutics against chronic inflammatory and/or fibrotic diseases like asthma, rheumatoid arthritis, SLE or cancer.6 In 2005, Opatz attempted to finish the total synthesis of racemic 2 and 3.7 Starting from commercially available cis-2,3-epoxybutane and allyl cuprate, the total synthesis of racemic 2 (24% yield over 10 steps) and 3 (18% yield over 11 steps) was accomplished by important steps such as ring-opening reaction, witting reaction, and a late-stage intramolecular Friedel–Crafts cyclization.7a Protection and deprotection reactions are frequently used in the total synthesis of racemic 2 and 3 which made the synthesis route more complicated and led to a lower overall yield. Soon after, the absolute configuration of natural 2 and 3 was determined as (14S,15R) by X-ray crystallography.8 To date, the asymmetric synthesis of natural macrolactones 2 and 3 is still an untouched work.
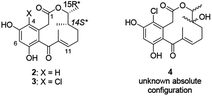 |
| | Fig. 1 Structures of the oxacyclododecindiones 2, 3 and 4. | |
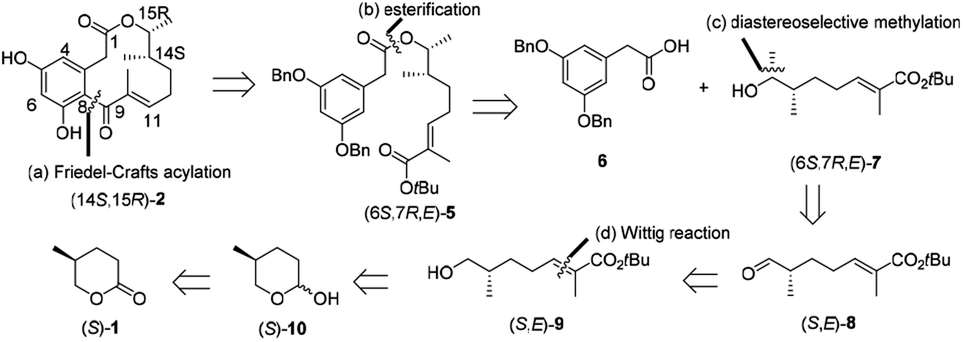
Our retrosynthetic strategy for the synthesis of (14S,15R)-2 commences with the disconnection of the C8–C9 bond, exposing tert-butyl ester (6S,7R,E)-5 (Scheme 2). We envisioned that the intramolecular Friedel–Crafts cyclization of 5 in the presence of Lewis acid could form (14S,15R)-2, meanwhile the cleavage of tert-butyl ester bond could be simultaneously achieved. Tert-butyl ester 5 could be easily generated by esterification of arylacetic acid 6 and alcohol (6S,7R,E)-7. We envisioned that the key alcohol intermediate (6S,7R,E)-7 could be obtained from aldehyde (S,E)-8 by diastereoselective methylation, while the stereogenic center at C7 could be constructed according to Kovalenko and Matiushenkov's procedure.9 (S,E)-8 could be readily prepared by a sequential reduction, Wittig reaction and oxidation from (S)-1. Based on the retrosynthetic analysis, herein, we reported the first asymmetric total synthesis of natural (14S,15R)-2 and (14S,15R)-3 starting from abundant and renewable industrial waste (S)-1.
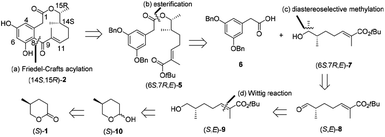 |
| | Scheme 2 Retrosynthetic analysis of (14S,15R)-2 by (a) an intramolecular Friedel–Crafts acylation, (b) an esterification, (c) a diastereoselective methylation, and (d) a Wittig reaction. | |
Results and discussion
With the synthetic blueprint in mind, our initial attention was focused on preparation of the key intermediate (6S,7R,E)-7 (Scheme 3). As depicted in Scheme 3, reduction of (S)-1 with DIBAL-H furnished the corresponding lactol (S)-10, which was subjected to a Wittig reaction with ylide 11 (ref. 10) without further purification to give (S,E)-9 in 82% yield with E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z > 95:5 selectivity. TEMPO-mediated-oxidation of (S,E)-9 employing PhI(OAc)2 as a secondary oxidant, which is compatible with the presence of olefins,11 afforded the corresponding aldehyde (S,E)-8 in 84% yield. Next, according to Kovalenko and Matiushenkov's procedure, (S,E)-8 was diastereoselectively methylated with MeTi(OiPr)3 (ref. 12) in the presence of 20 mol% of TADDOL-Ti(OiPr)2. Unfortunately, when [(S,S)-TADDOL]Ti(OiPr)2 was used as catalyst, the methylation of (S,E)-8 gave an inseparable mixture of two diastereomers (6S,7R,E)-7 and (6S,7S,E)-7 with d.r. 76:24 (the ratio of a peak of protons on C8 for (6S,7S,E)-7 at 1.17 ppm and (6S,7R,E)-7 at 1.14 ppm). To our delight, in the presence of [(R,R)-TADDOL]Ti(OiPr)2, (6S,7S,E)-7 was obtained in 94% yield with d.r. > 95:5 (the signal of (6S,7R,E)-7 was undetectable in 1H NMR). Although the configuration at C7 was opposite to that of key intermediates (6S,7R,E)-7, it could be converted by Mitsunobu reaction. Therefore, the next synthesis route was adjusted as Scheme 4.
:Z > 95:5 selectivity. TEMPO-mediated-oxidation of (S,E)-9 employing PhI(OAc)2 as a secondary oxidant, which is compatible with the presence of olefins,11 afforded the corresponding aldehyde (S,E)-8 in 84% yield. Next, according to Kovalenko and Matiushenkov's procedure, (S,E)-8 was diastereoselectively methylated with MeTi(OiPr)3 (ref. 12) in the presence of 20 mol% of TADDOL-Ti(OiPr)2. Unfortunately, when [(S,S)-TADDOL]Ti(OiPr)2 was used as catalyst, the methylation of (S,E)-8 gave an inseparable mixture of two diastereomers (6S,7R,E)-7 and (6S,7S,E)-7 with d.r. 76:24 (the ratio of a peak of protons on C8 for (6S,7S,E)-7 at 1.17 ppm and (6S,7R,E)-7 at 1.14 ppm). To our delight, in the presence of [(R,R)-TADDOL]Ti(OiPr)2, (6S,7S,E)-7 was obtained in 94% yield with d.r. > 95:5 (the signal of (6S,7R,E)-7 was undetectable in 1H NMR). Although the configuration at C7 was opposite to that of key intermediates (6S,7R,E)-7, it could be converted by Mitsunobu reaction. Therefore, the next synthesis route was adjusted as Scheme 4.
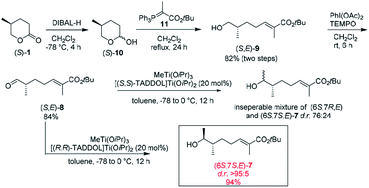 |
| | Scheme 3 Synthesis of key intermediate (6S,7S,E)-7. | |
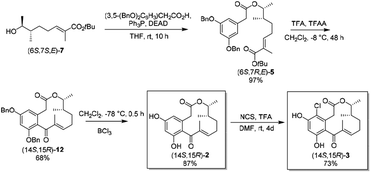 |
| | Scheme 4 Synthesis of (14S,15R)-2 and (14S,15R)-3 from (6S,7S,E)-7. | |
Reaction of (6S,7S,E)-7 with 3,5-bis(benzyloxy)phenylacetic acid under Mitsunobu reaction conditions gave ester (6S,7R,E)-5 in 97% yield. As we expected, when the diluted solution of (6S,7R,E)-5 in CH2Cl2 was treated with a mixture of trifluoroacetic anhydride and trifluoroacetic acid, the cleavage of the tert-butyl ester bond and intramolecular Friedel–Crafts acylation took place simultaneously to give the desired lactone (14S,15R)-12 in 68% yield. Finally, deprotection of the benzyl ethers with boron trichloride afforded (14S,15R)-2 in 87% yield, which was further converted to (14S,15R)-3 (in 73% yield) by chlorination with N-chlorosuccinimide. 1H and 13C NMR spectra of both synthetic (14S,15R)-2 and (14S,15R)-3 were in good agreement with the data of the natural products.8
Conclusions
In summary, concise and efficient asymmetric total synthesis of natural 4-dechloro-14-deoxy-oxacyclododecindione 2 and 14-deoxy-oxacylododecindione 3 has been accomplished for the first time. Using industrial waste (S)-5-methyl-tetrahydropyran-2-one 1 as starting material, (14S,15R)-2 and 3 were obtained in seven and eight steps with 37% and 27% overall yield, respectively. The chirality of C14 originates from the starting material, and the stereogenic center at C15 is stereospecifically constructed by highly diastereoselective methylation and Mitsunobu reaction.
Experimental section
General methods
Unless otherwise noted, all reactions sensitive to air or moisture were performed under nitrogen atmosphere. All commercially available chemicals including solvents were used without further purification. (S)-5-Methyl-tetrahydropyran-2-one (S)-1 was prepared from sarsasapogenin according to Tian's procedure.3b NMR spectra were recorded on Bruker 400 spectrometer. 1H NMR and 13C NMR spectra were measured at 400 MHz and 101 MHz. High resolution mass measurements (HRMS) were obtained on Bruker smart apex II. Optical rotations were measured on the PerkinElmer 343 polarimeter.
(S,E)-tert-Butyl 7-hydroxy-2,6-dimethylhept-2-enoate (S,E)-9. To a stirred solution of (S)-1 (3.00 g, 26.32 mmol) in dry CH2Cl2 (20 mL) was added DIBAL-H (1.0 M in hexane, 26.3 mL, 26.3 mmol) at −78 °C. After 4 h stirring at the same temperature, saturated Rochelle salt (15 mL) was added and further stirred for 6 h at room temperature. The mixture was extracted with CH2Cl2, dried over anhydrous Na2SO4, and evaporated to give the corresponding lactol (S)-10 as a yellow oil (2.51 g), which was used directly without purification. The crude product was added to the solution of tert-butyl 2-(triphenylphosphoranylidene)propanoate (12.52 g, 32.07 mol) in CH2Cl2 (20 mL). After being refluxed for 24 h, the solvent was removed in vacuo. Purification by flash chromatography on silica gel (petroleum ether:EtOAc = 5:1) yielded (S,E)-9 (4.91 g, 82%). [α]20D = −11.1 (c 0.31, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 6.65 (t, J = 1.2 Hz, 1H), 3.54–3.44 (m, 2H), 2.33–2.05 (m, 2H), 1.79 (s, 3H), 1.68–1.54 (m, 2H), 1.48 (s, 9H), 1.32–1.26 (m, 1H), 0.95 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 167.6, 140.9, 129.2, 80.0, 68.1, 35.4, 32.0, 28.1, 26.1, 16.4, 12.4. HRMS (ESI): calcd for [C13H24O3 + Na+]: 251.1618, found: 251.1620.
(6S,7S,E)-tert-Butyl-7-hydroxy-2,6-dimethyloct-2-enoate (6S,7S,E)-7. Step 1: a solution of alcohol (S,E)-9 (3.00 g, 13.16 mmol), iodobenzene diacetate (4.6 g, 14.28 mmol) and 2,2,6,6-tetramethylpiperidinooxy (205 mg, 1.31 mmol) in CH2Cl2 (26 mL, 0.5 M) was stirred at room temperature. After stirring for 3 h, the reaction mixture is washed successively with an aqueous sodium thiosulfate solution and brine. The organic solution is dried over anhydrous Na2SO4, and concentrated. Purification by flash chromatography on silica gel (petroleum ether:EtOAc = 10:1) yielded the (S,E)-8 (2.50 g, 84%) as a light red oil which was used immediately. Step 2: a solution of ((4R,5R)-2,2-dimethyl-1,3-dioxolane-4,5-diyl)bis(di-phenyl-methanol) [(R,R)-TADDOL] (488 mg, 1.05 mmol) and Ti(OiPr)4 (446 mg, 1.57 mmol) in toluene (8 mL) was heated at 110 °C for 3 h under an argon atmosphere. The solvent was removed in vacuo, after which the residue was diluted with dry toluene (28 mL) in −78 °C mixed with MeTi(OiPr)3 (1.88 g, 7.84 mmol) and stirred at the same temperature for 0.5 h. The aldehyde (S,E)-8 (1.18 g, 5.23 mmol) in toluene (3 mL) was added dropwise to the resulting stirred solution at −78 °C. The mixture was stirred for 12 h at −78 °C and then gradually warmed to 0 °C for 12 h, diluted with saturated aqueous NH4Cl (5 mL), and filtered. The filter cake was washed with CH2Cl2. The organic phase was separated and washed with saturated aqueous NaHCO3, dried over anhydrous Na2SO4. The solvent was removed in vacuo. Purification by flash chromatography on silica gel (petroleum ether:EtOAc = 10:1) yielded (6S,7S,E)-7 (1.20 g, 94%) with d.r. > 95:5. [α]20D = −18.5 (c 1.27, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 6.65 (td, J = 7.4, 1.4 Hz, 1H), 3.77–3.71 (m, 1H), 2.22–2.11 (m, 2H), 1.79 (s, 3H), 1.62–1.56 (m, 1H), 1.49 (s, 9H), 1.32–1.25 (m, 2H), 1.17 (d, J = 6.4 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 167.6, 141.0, 129.2, 80.0, 71.2, 39.4, 31.5, 28.2, 26.5, 20.3, 14.0, 12.4. HRMS (ESI): calcd for [C36H44O6 + Na+]: 265.1774, found: 265.1775.
(6S,7R,E)-tert-Butyl-7-(2-(3,5-bis(benzyloxy)phenyl)acetoxy)-2,6-dimethyloct-2-enoate (6S,7R,E)-5. To a solution of (6R,7R,E)-7 (100 mg, 0.41 mmol), 3,5-bis(benzyloxy)phenylacetic acid (216 mg, 0.62 mmol), and triphenylphosphine (163 mg, 0.62 mmol) in THF (1.2 mL) was added dropwise a THF (1 mL) solution of diisopropyl azodicarboxylate (125 mg, 0.62 mmol). After stirring for 10 h at room temperature, the solvent was removed in vacuo. Purification by column chromatography on silica gel (petroleum ether:EtOAc = 20:1) yielded (6R,7S,E)-5 (230 mg, 97%) as a colorless oil. [α]20D = −5.6 (c 0.60, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.49–7.29 (m, 10H), 6.65–6.49 (m, 4H), 5.02 (s, 4H), 4.89–4.76 (m, 1H), 3.53 (s, 2H), 2.26–1.98 (m, 2H), 1.77 (s, 3H), 1.73–1.60 (m, 1H), 1.47 (s, 9H), 1.27–1.15 (m, 2H), 1.13 (d, J = 6.4 Hz, 3H), 0.88 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 170.9, 167.5, 160.0, 140.6, 136.9, 136.4, 129.4, 128.6, 128.0, 127.5, 108.5, 100.84, 80.0, 74.5, 70.0, 42.0, 37.1, 31.3, 28.1, 26.2, 16.0, 14.6, 12.4. HRMS (ESI): calcd for [C36H44O6 + Na+]: 595.3030, found: 595.3014.
(14S,15R)-5,7-Di-O-benzyl-4-dechloro-14-deoxyoxacyclodode-cindione (14S,15R)-12. (6S,7R,E)-5 (928 mg, 1.62 mmol), trifluoroacetic acid (167 mL) and trifluoroacetic anhydride (83.5 mL) were dissolved in CH2Cl2 (2100 mL) and cooled to −8 C. The reaction mixture stood at −8 °C for 48 h. The solvent was removed in vacuo. Purification by flash chromatography on silica gel (petroleum ether:EtOAc = 15:1) yielded (14S,15R)-12 (545 mg, 68%) as a colorless oil. [α]20D = +36.6 (c 0.20, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.65–7.07 (m, 10H), 6.51 (d, J = 1.3 Hz, 3H), 5.17–4.90 (m, 4H), 4.59 (dq, J = 12.7, 6.2 Hz, 1H), 3.31 (t, J = 13.5 Hz, 2H), 2.42 (s, 1H), 2.11 (d, J = 20.3 Hz, 1H), 1.93 (s, 3H), 1.69–1.60 (m, 1H), 1.53–1.31 (m, 2H), 1.09 (d, J = 4.6 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 199.3, 170.4, 159.9, 156.6, 152.9, 136.9, 136.7, 133.3, 128.8, 128.6, 128.2, 127.8, 127.8, 127.7, 126.9, 124.3, 107.7, 100.4, 77.3, 70.4, 70.3, 53.6, 39.7, 19.2, 10.5. HRMS (ESI): calcd for [C32H34O5 + Na+]: 521.2298, found: 521.2294.
(14S,15R)-4-Dechloro-14-deoxyoxacyclododecindione (14S,15R)-2. A solution of (14S,15R)-12 (460 mg, 0.923 mmol) in CH2Cl2 (138 mL) was cooled to −78 °C. Boron trichloride (1.0 M in hexane, 9.3 mL, 9.3 mmol) was added and the mixture was stirred at that temperature. After 30 min the reaction mixture was quenched with saturated aqueous NaHCO3 (50 mL) and warmed up to room temperature. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4 and filtered. The solvent was removed in vacuo. Purification by flash chromatography on silica gel (petroleum ether:EtOAc = 2:1) yielded (14S,15R)-2 (256 mg, 87%) as a white solid. [α]20D = +41.5 (c 0.3, MeOH) lit. [α]20D = +29.1 (c 0.40, MeOH). 1H NMR (400 MHz, MeOD): δ 6.52 (s, 1H), 6.30 (s, 1H), 6.26 (s, 1H), 4.67–4.50 (m, 1H), 3.23 (t, J = 16.3 Hz, 2H), 2.48–2.42 (m, 1H), 2.25–2.21 (m, 1H), 1.88 (s, 3H), 1.70 (dd, J = 14.1, 7.8 Hz, 1H), 1.56 (dd, J = 13.7, 6.9 Hz, 1H), 1.49–1.37 (m, 1H), 1.13 (d, J = 6.1 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, MeOD): δ 203.1, 172.1, 160.2, 157.5, 154.1, 138.1, 134.8, 120.9, 109.6, 102.5, 78.2, 40.8 40.4, 34.3, 29.7, 19.5, 19.4, 10.6. HRMS (ESI): calcd for [C18H22O5 + Na+]: 341.1359, found: 341.0360.
(14S,15R)-4-Deoxyoxacyclododecindione (14S,15R)-3. Lactone (14S,15R)-2 (120 mg, 0.38 mmol) and N-chlorosuccinimide (51 mg, 0.38 mmol) were dissolved in dry DMF (15 mL) and trifluoroacetic acid (41 μL, 0.57 mmol) was added. The reaction mixture was stirred at room temperature for 4 days. The solvent was removed in vacuo. Purification by flash chromatography on silica gel (petroleum ether:EtOAc = 2:1) yielded (14R,15R)-2 (98 mg, 73%) as a white solid. (14S,15R)-3: [α]20D = +18.6 (c 0.30, MeOH) lit. [α]20D = +9.6 (c 0.34, CD3OD). 1H NMR (400 MHz, CD3CN): δ 7.44 (s, 2H), 6.65 (d, J = 7.8 Hz, 1H), 6.50 (s, 1H), 4.64–4.57 (m, 1H), 3.54 (d, J = 17.1 Hz, 1H), 3.23 (d, J = 17.2 Hz, 1H), 2.53 (dd, J = 18.1, 11.9 Hz, 1H), 2.05–1.97 (m, 1H), 1.83 (s, 3H), 1.69–1.57 (m, 1H), 1.55–1.50 (m, 1H), 1.31–1.21 (m, 1H), 1.07 (d, J = 6.2 Hz, 3H), 0.90 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CD3CN): δ 200.0, 169.6, 154.9, 154.8, 153.0, 137.8, 133.4, 123.3, 114.2, 103.9, 77.6, 40.0, 39.4, 34.8, 29.7, 19.7, 19.6, 10.8. HRMS (ESI): calcd for [C18H21ClO5 + Na+]: 375.0970, found: 375.0953.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (21272271) for financial support and Chengdu Zhenhua Bio-Tech Co., Ltd for supplying the waste liquids of producing 16-dehydropregnenolone acetate from diosgenin.
References
-
(a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed;
(b) P. J. Dunn, A. S. Wells and M. T. Williams, Green Chemistry in the Pharmaceutical Industry, Wiley-VCH, Weinheim, 2010 CrossRef;
(c) P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452 RSC;
(d) J. Clark, R. Sheldon, C. Raston, M. Poliakoffd and W. Leitner, Green Chem., 2014, 16, 18–23 RSC;
(e) D. Kralisch, D. Ott and D. Gericke, Green Chem., 2015, 17, 123–145 RSC.
-
(a) Catalytic Process Development for Renewable Materials, ed. P. Imhof and J. C. van der Wal, Wiley-VCH, Weinheim, 2013 Search PubMed;
(b) P. Gallezot, Green Chem., 2007, 9, 295–302 RSC;
(c) R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- Oxidative degraduation of diosgenin, tigogenin and rockogenin, etc. generates (R)-1, while sarsasapogenin and yamogenin give (S)-1
(a) A. Goswami, R. Kotoky, R. C. Rastogi and A. C. Ghosh, Org. Process Res. Dev., 2003, 7, 306–308 CrossRef CAS;
(b) W. Tian, X. Xu, S. Liu, J. Shen and X. Wu, US Pat. 2006/0166955, 2006.
- ChengguZhenhua Bio-Tech Co., Ltd who produce more than 400 tons of 16-dehydropregnenolone acetate each year from diosgenin by oxidative degradation, but never recover any (R)-1.
- A few examples to utilize (R)- and (S)-1 as starting materials in the synthesis of bioactive compounds have been reported.
(a) T. Suzuki, K. Usui, Y. Miyake, M. Namikoshi and M. Nakada, Org. Lett., 2004, 6, 553–556 CrossRef CAS PubMed;
(b) S. Yu, X. Pan, X. Lin and D. Ma, Angew. Chem., Int. Ed., 2005, 44, 135–138 CrossRef CAS PubMed;
(c) Z. Wang, Q. Xu, W. Tian and X. Pan, Tetrahedron Lett., 2007, 48, 7549–7551 CrossRef CAS;
(d) K. Takahashi, K. Komine, Y. Yokoi, J. Ishihara and S. Hatakeyama, J. Org. Chem., 2012, 77, 7364–7370 CrossRef CAS PubMed;
(e) S. Zhang, H. Dong, J. Gui and W. Tian, Tetrahedron Lett., 2012, 53, 1882–1884 CrossRef CAS;
(f) J. Cao and P. Perlmutter, Org. Lett., 2013, 15, 4327–4329 CrossRef CAS PubMed;
(g) J. Shen, Y. Shi and W. Tian, Chin. J. Chem., 2015, 33, 683–685 CrossRef CAS.
-
(a) G. Erkel, H. Belahmer, A. Serwe, T. Anke, H. Kunz, H. Kolshorn, J. C. Liermann and T. Opatz, J. Antibiot., 2008, 61, 285–290 CrossRef CAS PubMed;
(b) J. Richter, L. P. Sandjo, J. C. Liermann, T. Opatz and G. Erkel, Bioorg. Med. Chem., 2015, 23, 556–563 CrossRef CAS PubMed.
-
(a) J. Tauber, M. Rohr, T. Walter, G. Erkel and T. Opatz, Org. Biomol. Chem., 2015, 13, 7813–7821 RSC;
(b) J. Tauber, K. Rudolph, M. Rohr, G. Erkel and T. Opatz, Eur. J. Org. Chem., 2015, 3587–3608 CrossRef CAS.
- J. Tauber, M. Rohr, T. Walter, D. Schollmeyer, K. Rahn-Hotze, G. Erkel and T. Opatz, Org. Biomol. Chem., 2016, 14, 3695–3698 RSC.
- V. Kovalenko and E. Matiushenkov, Tetrahedron: Asymmetry, 2012, 23, 1393–1399 CrossRef CAS.
- J.-L. Giner, Tetrahedron Lett., 2002, 43, 5457–5459 CrossRef CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- A. deMeijere, H. Winsel and B. Stecker, Org. Synth., 2005, 81, 14–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02994g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Weiping Chen
* and
Weiping Chen